
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and structural analysis of aryloxo-modified trinuclear half-titanocenes, and their use as catalyst precursors for ethylene polymerisation†
Qing Yan,
Ken Tsutsumi and
Kotohiro Nomura *
*
Department of Chemistry, Tokyo Metropolitan University, 1-1 Minami Osawa, Hachioji, Tokyo 192-0397, Japan. E-mail: ktnomura@tmu.ac.jp
First published on 24th August 2017
Abstract
A series of trinuclear half-titanocenes, [Cp′TiX2{(O-2,4-R2C6H2)-6-CH2}]3N [X = Cl, R = Me, Cp′ = Cp (1); X = Cl, R = tBu, Cp′ = Cp (4), Cp* (5), tBuC5H4 (6), 1,2,4-Me3C5H2 (7); X = Me, Cp′ = Cp*, R = Me (8), tBu (9)] and the related bimetallic complexes, [Cp′TiCl2{(O-2,4-Me2C6H2)-6-CH2}][Cp′TiCl{(O-2,4-Me2C6H2)-6-CH2}2]N [Cp′ = Cp* (2), 1,2,4-Me3C5H2 (3)], have been prepared and identified. Structures of 1–5, 7 and 9 were determined by X-ray crystallography, and all complexes fold with distorted tetrahedral geometries around titanium. These complexes (2–9) are stable in solution except the Cp analogue (1), which presents as a mixture of the trinuclear analogue (1) and the (proposed) binuclear analogue, CpTiCl3, and CpTi[{(O-2,4-Me2C6H2)-6-CH2}]3N in solution; there is an equilibrium between 1 and the binuclear analogue (and CpTiCl3) depending on the temperature, solvent and concentration. The Cp* analogues (2, 8, 9), exhibited high catalytic activities for ethylene polymerisation in the presence of MAO cocatalyst, affording ultrahigh molecular weight polymers with uniform molecular weight distributions in most cases. [Cp*TiMe2{(O-2,4-Me2C6H2)-6-CH2}]3N (8) showed the higher catalytic activities than the related mononuclear analogue, Cp*TiCl2(O-2-R-4,6-Me3C6H2) (R = Me, tBu); the activity by 8 in the presence of AliBu3–[Ph3C][B(C6F5)4] cocatalysts was higher than that in the presence of MAO.
Introduction
Transition metal catalysed olefin polymerisation is an important key reaction in the production of polyolefins [such as HDPE (high density polyethylene), LLDPE (linear low density polyethylene), isotactic polypropylene and elastomers etc.] in the chemical industry, and the market capacity still increases every year. Since the discovery of Ziegler-Natta catalysts, the design of efficient molecular catalysts has been considered as a key subject in terms of the synthesis of new polymers as well as of better control of molecular weights, compositions in the copolymerisations, and the development of more efficient processes.1–5 Nowadays, many examples such as metallocenes,1a–d,g linked half-metallocenes (constrained geometry type),1e modified half-titanocenes,4,5 and the other transition metal complex catalysts, the so called non-metallocene type,1f,3b,c,i–k have been reported as so called ‘single-site catalysts’ which facilitate the coordination and insertion polymerisation with uniform catalytically active species.1–7 These metal complexes are generally activated by different types of cocatalysts [such as Al alkyls, methylaluminoxane (MAO), borates etc.]6,7 in the polymerisation to afford cationic alkyl species, which are the proposed catalytically active species in this catalysis.Certain bimetallic olefin polymerisation catalysts (exemplified in Chart 1) have been known to exhibit unique characteristics in catalytic activity, polymer microstructure (molecular weight, chain branching, monomer sequences, regioregularity) compared to the monomeric analogues in olefin polymerisation8,9 due to so called metal–metal cooperative effects.3d,h,7a,8,9 This ‘bimetallic effect’ has been considered as due to their improved activation-transport (trapping/activation for subsequent comonomer incorporation on another metal center),3d,h,8 intermolecular chain transfer and/or catalyst–cocatalyst nuclearity effect on the multi-nuclear reaction sphere,3d,h,7a,8,9 and should be considered to provide another possibility of the new catalyst design.
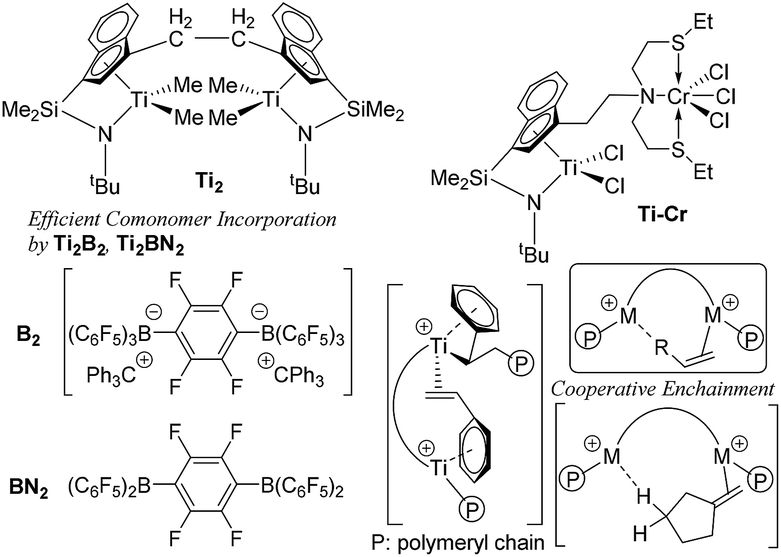 | ||
| Chart 1 Typical bimetallic catalysts for efficient metal–metal cooperative effect.3d,h,8,9 | ||
Modified (nonbridged) half-titanocenes containing anionic ancillary donor ligand (Y) of type, Cp′TiX2(Y) [Cp′ = cyclopentadienyl; X = halogen, alkyl; Y = aryloxo (Chart 2), ketimide, imidazolin-2-iminato etc.],4,5 have been known to display promising characteristics especially in ethylene copolymerisation with sterically encumbered olefins, styrene, cyclic olefins.4 It has been demonstrated that both the activity and the commoner incorporation (reactivity) can be tuned by the ligand modification.4,10 It has thus been simply assumed that design of multi-metallic half-titanocenes would facilitate activation-transport, intermolecular chain transfer on the multi-nuclear reaction sphere, which would lead to the better catalytic activity and/or better comonomer incorporation. As titanium complexes containing amine triphenolate ligands (titanatranes), TiX[(O-2,4-R2C6H2)-6-CH2]3N (Chart 2 left), displays unique catalysts for ethylene polymerisation,11 we thus explored synthesis and structural analysis of a series of trinuclear half-titanocenes of the type, [Cp′TiX2{(O-2,4-R2C6H2)-6-CH2}]3N [Chart 2 right, Cp′ = Cp, tBuC5H4, 1,2,4-Me3C5H2, C5Me5 (Cp*); X = Cl, Me; R = Me, tBu], and their use as the catalyst precursors for ethylene polymerisation.12 Through this study, we wish to demonstrate some unique characteristics as the olefin polymerisation catalysts which are different from the monomeric analogues.
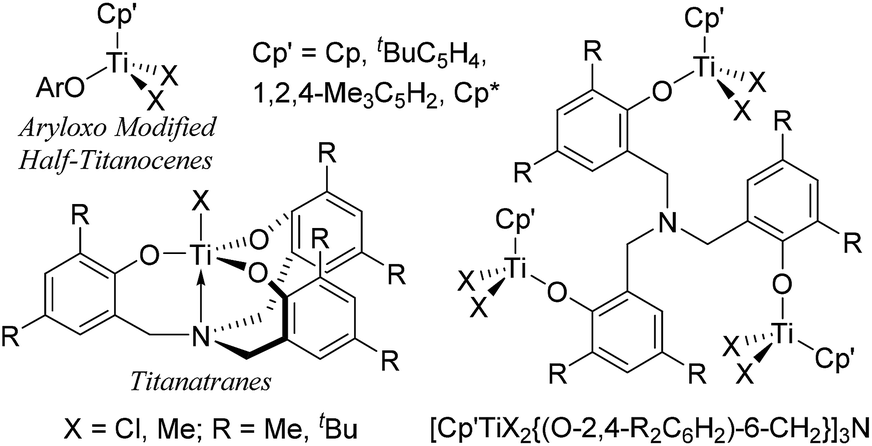 | ||
| Chart 2 Reported aryloxo-modified half-titanocenes, titanatranes, and the trinuclear half-titanocenes prepared in this study. | ||
Results and discussion
1. Synthesis, structural analysis of trinuclear/dinuclear aryloxo-modified half-titanocenes
Trinuclear [CpTiCl2{(O-2,4-Me2C6H2)-6-CH2}]3N (1) could be prepared by reaction of 3 equiv. of CpTiCl3 with [(LiO-2,4-Me2C6H2)-6-CH2]3N in Et2O in high yield (80%, Scheme 1). This is an analogous procedure for synthesis of aryloxo-modified half-titanocenes, Cp′TiCl2(OAr),10i,13,14 and the complex could be identified by 1H NMR spectrum, elemental analysis, and the structure was determined by X-ray crystallography (shown below, Fig. 2). In contrast, it turned out that the similar reactions with (1,2,4-Me3C5H2)TiCl3 and Cp*TiCl3, in place of CpTiCl3, afforded a mixture of several complexes, and the isolated complexes were bimetallic [Cp′TiCl2{(O-2,4-Me2C6H2)-6-CH2}][Cp′TiCl{(O-2,4-Me2C6H2)-6-CH2}2]N [Cp′ = Cp* (2), 1,2,4-Me3C5H2 (3)] determined by X-ray crystallographic analyses (shown below, Fig. 3). Complexes 2 and 3 were also identified by NMR spectra and elemental analysis. The major products observed in 1H NMR spectra were 2 or 3 (and the trichloride), even though the reaction was conducted with 5 equiv. of Cp*TiCl3 or (1,2,4-Me3C5H2)TiCl3 in Et2O.15 Complex 2 was also the major product observed in the 1H NMR spectrum, when the reaction was conducted in toluene (shown in the ESI†).15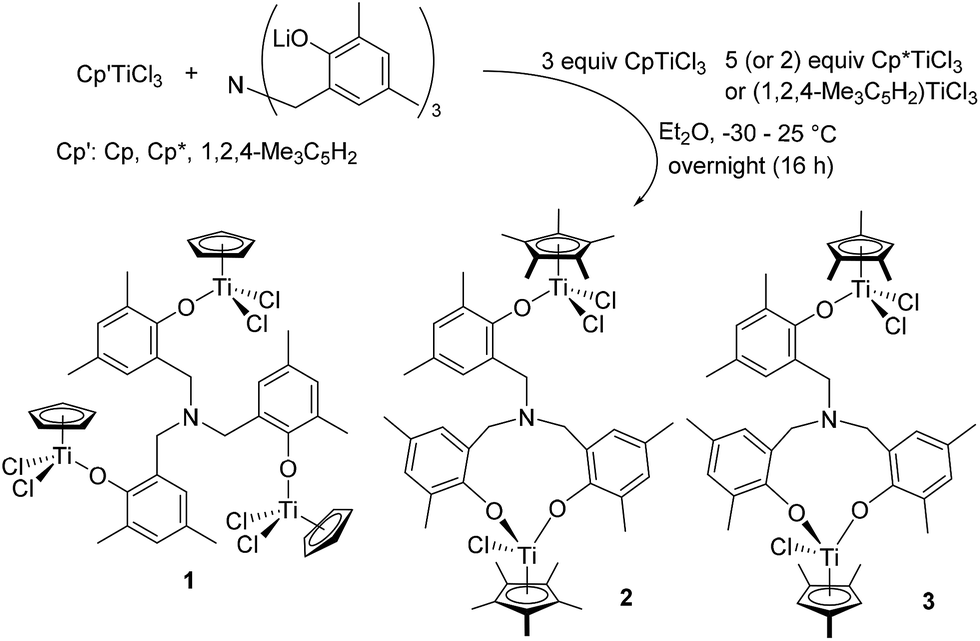 | ||
| Scheme 1 Reaction of Cp′TiCl3 (Cp′ = Cp, Cp*, and 1,2,4-Me3C5H2) with [(LiO-2,4-Me2C6H2)-6-CH2]3N in Et2O. | ||
It turned out that the Cp analogue (1) was unstable in solution at room temperature monitored by 1H NMR spectra (Fig. 1).15 The toluene-d8 solution containing 1 at −60 °C showed a simple NMR spectrum (marked with +) assigned as 1 (Fig. 1i, 1 1.08 × 10−3 M), when the NMR sample (in toluene-d8) was prepared at −30 °C (in the drybox) and kept cool until the measurement. However, the spectrum at −30 °C (Fig. 1a and j) showed resonances ascribed to 1 in addition to the others (including CpTiCl3), as mentioned below. Upon increasing at −40 °C and 0 °C (Fig. 1c and d), resonance ascribed to CpTiCl3 (marked with ▼) was appeared in addition to resonances ascribed to (probably) bimetallic species (marked with ○ estimated by the resonances by 2 and 3, two doublets at 3.7–4.0 ppm and two singlets at 5.9–6.1 ppm). The intensities ascribed to CpTiCl3 (and assumed bimetallic species) increased at 0 °C. The other resonances ascribed to titanatrane, CpTi[{(O-2,4-Me2C6H2)-6-CH2}3N]16 (marked with #) were also appeared upon increasing at 25 °C with increasing the intensities ascribed to both CpTiCl3 and the assumed bimetallic species (Fig. 1e). Upon cooling, the intensity of resonances ascribed to CpTiCl3 etc. decreased and the solution at −60 °C showed the original spectrum assigned to 1 in addition of CpTiCl3 and CpTi[{(O-2,4-Me2C6H2)-6-CH2}3N] with ratio of ca. 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45:20 (Fig. 1h), which would be well suggest that the titanatrane was formed by dissociation of CpTiCl3 from 1. The similar spectra were observed when the sample of 1 was prepared with rather low concentration (Fig. 1 right), although the spectrum measured at −60 °C showed simple resonances ascribed to 1 (Fig. 1i).
:45:20 (Fig. 1h), which would be well suggest that the titanatrane was formed by dissociation of CpTiCl3 from 1. The similar spectra were observed when the sample of 1 was prepared with rather low concentration (Fig. 1 right), although the spectrum measured at −60 °C showed simple resonances ascribed to 1 (Fig. 1i).
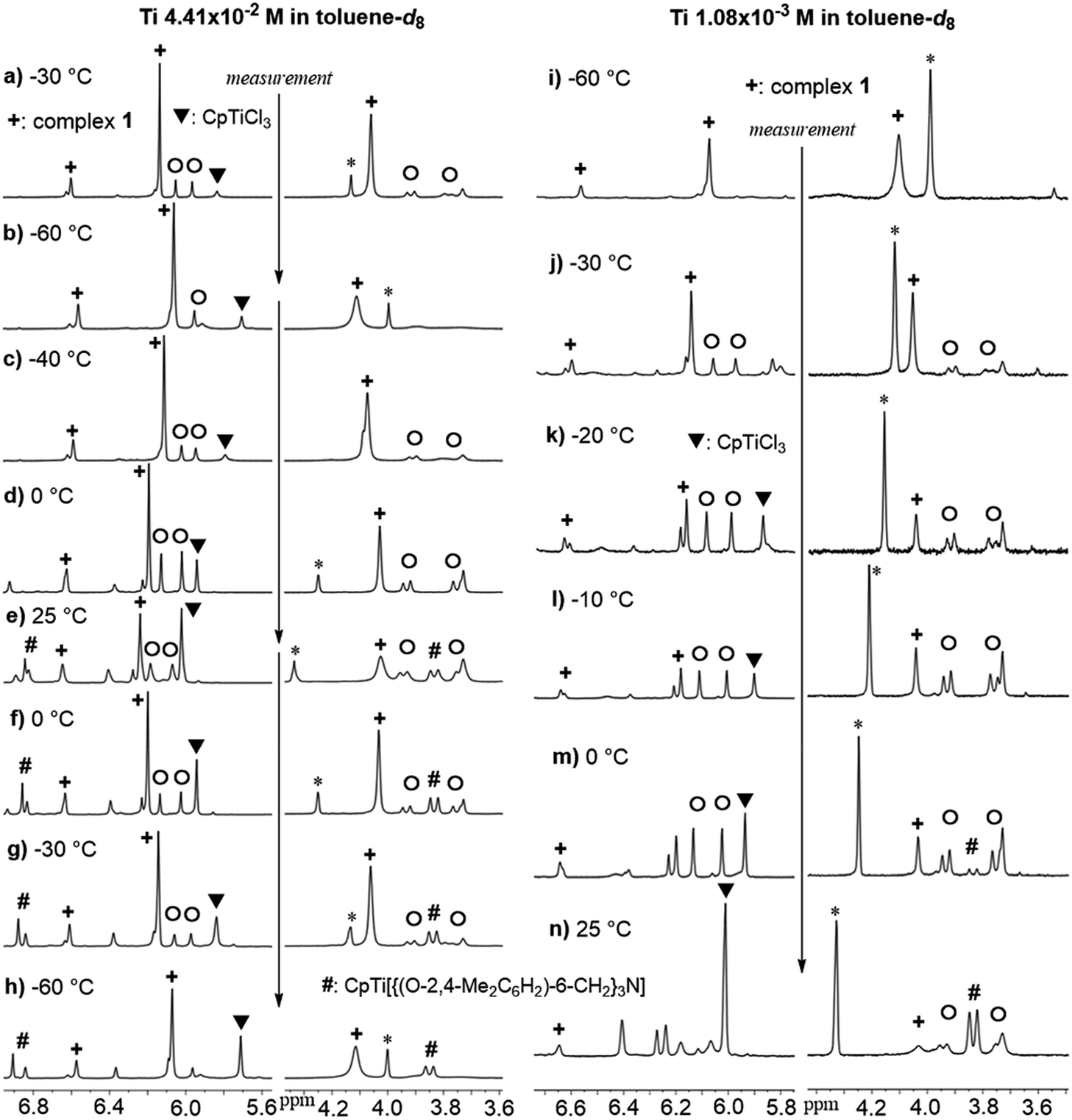 | ||
| Fig. 1 Variable Temperature (VT) 1H NMR spectra of toluene-d8 solution containing [CpTiCl2{(O-2,4-Me2C6H2)-6-CH2}]3N (1). Left: VT NMR spectra with rather high Ti concentration (Ti 4.41 × 10−2 M). Right: The VT NMR spectra with rather low Ti concentration (Ti 1.08 × 10−3 M). The more data are shown in the ESI.†15 Complex 1 (+), CpTiCl3 (▼), CpTi[{(O-2,4-Me2C6H2)-6-CH2}3N] (#), assumed bimetallic species (○). *Impurity. | ||
Moreover, the intensity of resonances ascribed to CpTiCl3 (and those in assumed bimetallic species) increased upon increasing the temperature (from −30 °C through −10 °C, Fig. 1j–l), along with decreasing the intensities of resonances ascribed to 1.15 Resonances ascribed to titanatrane were observed at 0 °C and the intensities increased at 25 °C. It was thus revealed from these spectra that the ratios (1, CpTiCl3, titanatrane) were also affected by the Ti concentrations. It was also revealed that the ratio was also affected by the solvent (shown in the ESI†).15 The integration ratios of CpTiCl3 and the assumed bimetallic species were relatively close to 1:1, and the ratios were affected by the temperature measured and were reversible, except once formed titanatrane, CpTi[{(O-2,4-Me2C6H2)-6-CH2}3N].15,16
These results suggest a presence of an equilibrium between 1 and the assumed bimetallic species, probably [CpTiCl2{(O-2,4-Me2C6H2)-6-CH2}][CpTiCl{(O-2,4-Me2C6H2)-6-CH2}2]N, with dissociation of CpTiCl3, as depicted in Scheme 2. The results also suggest that the further reaction could afford the titanatrane, CpTi[{(O-2,4-Me2C6H2)-6-CH2}3N], with further dissociation of CpTiCl3 that is very stable in solution (Scheme 2). This assumption would be able to explain why the bimetallic 2 and 3 were isolated in the reactions of Cp′TiCl3 (Cp′ = Cp*, 1,2,4-Me3C5H2, Scheme 1). It also turned out that once formed 2 and 3 are, in contrast, relatively stable even in toluene-d8 upon heating at 80 °C for 30 min (shown in the ESI†).15
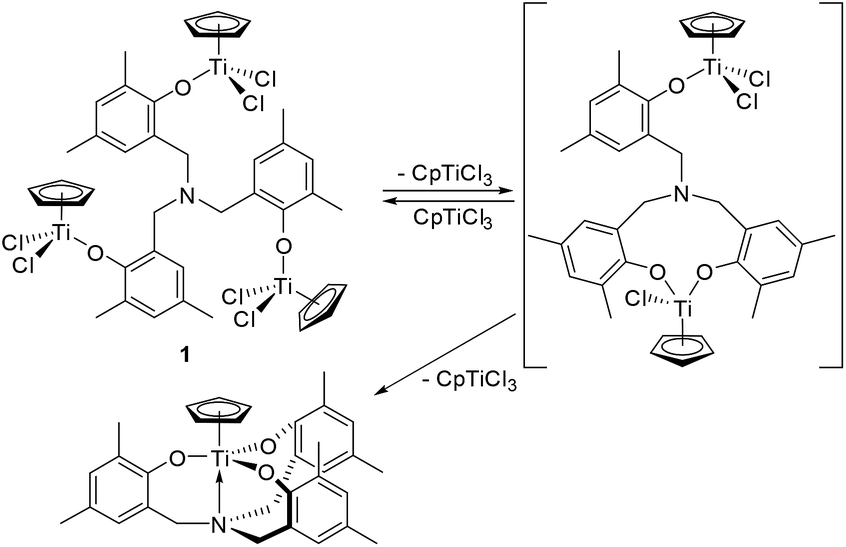 | ||
| Scheme 2 Proposed equilibrium in solution containing 1 (observed in NMR spectra, Fig. 1). | ||
It turned out that reaction of Cp′TiCl3 (Cp′ = Cp, Cp*, tBuC5H4, 1,2,4-Me3C5H2) with [(LiO-2,4-tBu2C6H2)-6-CH2]3N in Et2O afforded the corresponding trinuclear complexes, [Cp′TiCl2{(O-2,4-tBu2C6H2)-6-CH2}]3N [Cp′ = Cp (4), Cp* (5), tBuC5H4 (6), 1,2,4-Me3C5H2 (7), Scheme 3], and the resultant complexes could be identified by NMR spectra and elemental analysis; their structures (4, 5, 7) were determined by X-ray crystallography (shown below, Fig. 2). In contrast to the methyl analogue (1), the tert-Bu complex (4) is relatively stable and no significant differences in the 1H NMR spectra (in toluene-d8) were observed upon heating at 80 °C for 30 min (shown in the ESI, Fig. S2–S6†).15 No additional resonances were observed when the toluene-d8 solution containing the Cp* analogue (5) was heated at 80 °C for 30 min (Fig. S2–S7, in the ESI†),15 whereas certain additional resonances (but very small amount) were observed when the toluene-d8 solutions containing the tBuC5H4 analogue (6) and the 1,2,4-Me3C5H2 analogue (7) were heated under the same conditions (Fig. S2–S9†).15 The observed difference in the stability may be somewhat related to the stability and uniformity of the catalytically active species as described below.
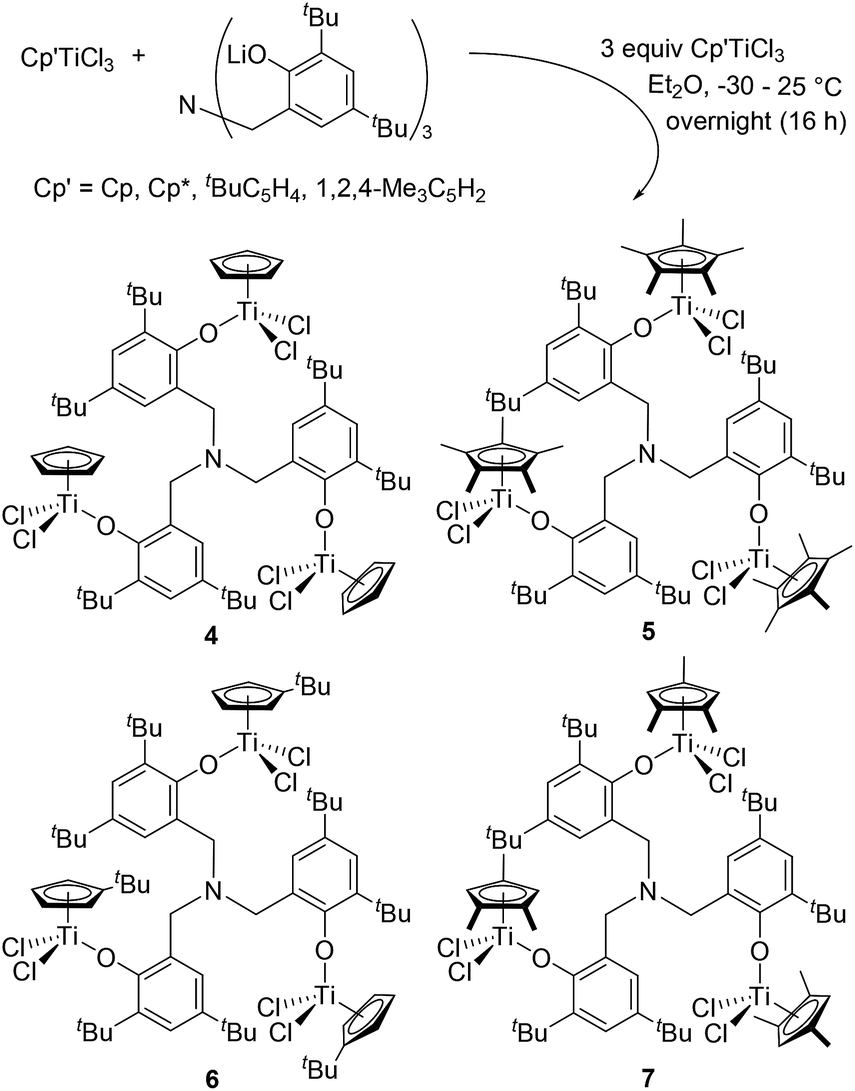 | ||
| Scheme 3 Synthesis of [Cp′TiCl2{(O-2,4-tBu2C6H2)-6-CH2}]3N. | ||
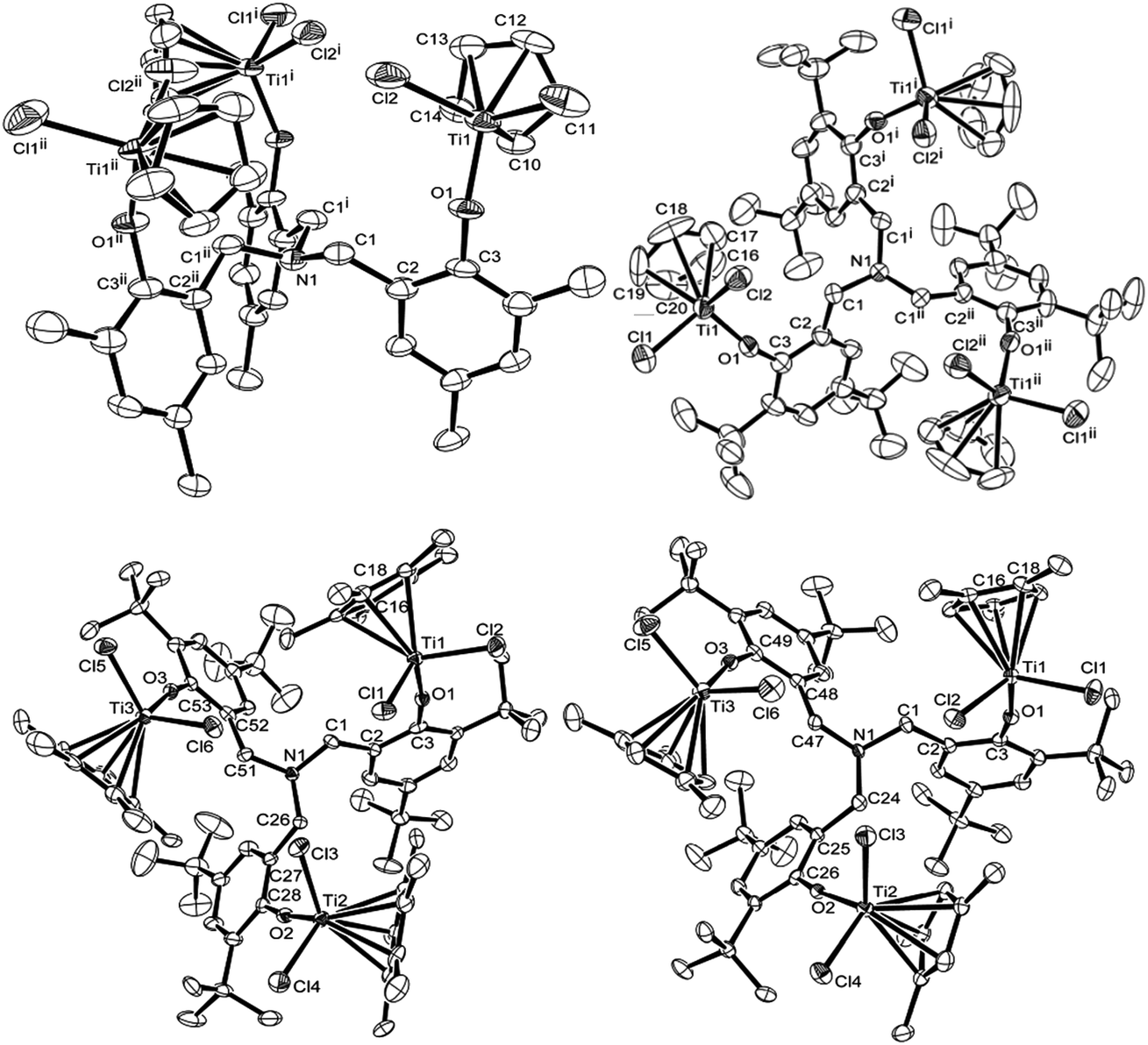 | ||
| Fig. 2 ORTEP drawings for [CpTiCl2{(O-2,4-Me2C6H2)-6-CH2}]3N (1, top left), [Cp′TiCl2{(O-2,4-tBu2C6H2)-6-CH2}]3N [Cp′ = Cp (4, top right), Cp* (5, bottom left), 1,2,4-Me3C5H2 (7, bottom right)]. Thermal ellipsoids are drawn at the 50% probability level, and H atoms are omitted for clarity.17 | ||
Fig. 2 shows ORTEP drawings for the trinuclear half-titanocenes, [CpTiCl2{(O-2,4-Me2C6H2)-6-CH2}]3N (1) and [Cp′TiCl2{(O-2,4-tBu2C6H2)-6-CH2}]3N [Cp′ = Cp (4), Cp* (5), 1,2,4-Me3C5H2 (7)] determined by X-ray crystallography,17 and the selected bond distances and the angles are summarised in Table 1. The complex 1 folds distorted tetrahedral geometry around titanium, as observed in the mononuclear half-titanocenes.10i,14 No significant differences in the bond distances and angles were observed in each titanium complex. The Ti–Cl bond distances in 1 [2.252(3), 2.249(2) Å] are slightly shorter than those in the mononuclear analogues, CpTiCl2(O-2,6-Et2C6H3)14d and CpTiCl2(O-2,6-iPr2C6H3)14c [2.262(1)–2.2710(10) Å], but within the range of those in the Cp analogues containing chlorinated phenoxides like CpTiCl2(OC6Cl5), [2.2492(17), 2.261(3) Å].10i The Ti–O bond distance [1.788(4) Å] is slightly longer than those in CpTiCl2(O-2,6-Et2C6H3)14d and CpTiCl2(O-2,6-iPr2C6H3)14c [1.7781(18), 1.760(4) Å, respectively] but shorter than those in CpTiCl2(OC6Cl5) [1.814(4) Å] and CpTiCl2(O-2,6-Cl2C6H3) [1.8091(12) Å];10i the bond distance is, however, apparently shorter than that (1.978 Å) for Ti–O (in CF3SO3) bond distance in Cp*TiMe(CF3SO3)(O-2,6-iPr2C6H3)14e due to the π-donation from oxygen to titanium. The Ti–O–C(3) (in phenyl) bond angle is 149.9(6)°, which is smaller than those in CpTiCl2(O-2,6-Et2C6H3)14d and CpTiCl2(O-2,6-iPr2C6H3)14c [161.37(17), 163.0(4)°, respectively] and those in the Cp analogues containing chlorinated phenoxides [151.5(3)–152.96(10)°].10i The Cl(1)–Ti–Cl(2) bond angle [103.41(11)°] is within the range of the reported Cp analogues [101.75(2)–104.23(7)°].10i,14c,d
| Complex Cp′:R | 1 Cp:Me | 4 Cp:tBu | 5 Cp*:tBu | 7 1,2,4-Me3C5H2:tBu | CpTiCl2(O-2,6-Et2C6H3)a | CpTiCl2(O-2,6-iPr2C6H3)b | Cp*TiCl2(O-2,6-tBu2C6H3)c |
|---|---|---|---|---|---|---|---|
| a Cited from ref. 14d.b Cited from ref. 14c.c Cited from ref. 14f. | |||||||
| Bond distances (Å) | |||||||
| Ti(1)–Cl(1) | 2.252(3) | 2.2649(19) | 2.2616(14) | 2.2615(12) | 2.2680(10) | 2.262(1) | 2.2674(10) |
| Ti(1)–Cl(2) | 2.249(2) | 2.2361(16) | 2.2702(14) | 2.2475(8) | 2.2710(10) | 2.262(1) | 2.2674(10) |
| Ti(1)–O(1) | 1.788(4) | 1.774(3) | 1.790(3) | 1.779(2) | 1.7781(18) | 1.760(4) | 1.804(2) |
| Ti–C in Cp′ | 2.330(9); 2.356(9), Ti–C(10); –C(12) | 2.339(8); 2.336(5), Ti–C(16); –C(18) | 2.348(4); 2.415(5), Ti–C(16); –C(18) | 2.343(3); 2.393(3), Ti–C(16); –C(18) | 2.351(3); 2.325(3), Ti–C(1); –C(3) | 2.282(8); 2.325(5), Ti–C(1); –C(3) | 2.359(4); 2.370(3), Ti–C(1); –C(3) |
|
|||||||
| Bond angles (°) | |||||||
| Cl(1)–Ti(1)–Cl(2) | 103.41(11) | 101.52(6) | 101.11(5) | 101.14(4) | 102.24(3) | 104.23(7) | 98.10(4) |
| Cl(1)–Ti–O(1) | 104.50(19) | 104.37(10) | 103.02(10) | 103.89(8) | 103.35(6) | 102.53(9) | 103.22(6) |
| Cl(2)–Ti–O(1) | 102.58(15) | 103.17(12) | 103.27(10) | 104.09(8) | 101.97(7) | 102.53(9) | 103.22(6) |
| Ti(1)–O–C(3) | 149.9(6) | 157.6(2) | 169.3(3) | 165.4(2) | 161.37(17), Ti–O–C(11) | 163.0(4), Ti–O–C(6) | 155.5(2), Ti–O–C(7) |
| C(1)–N(1)–C(1)i | 110.6(5) | 112.5(2) | 110.5(3), C(1)–N(1)–C(26) | 110.1(2), C(1)–N(1)–C(24) | |||
| N(1)–C(1)–C(2) | 112.8(6) | 112.3(4) | 111.9(3) | 112.5(3) | |||
The complexes 4, 5 and 7 also fold a distorted tetrahedral geometry around titanium. No significant differences in the both bond distances and angles in each titanium complex were observed in the Cp analogue (4), whereas slight differences in both the distances and the angles were observed in the Cp* analogue (5) and the 1,2,4-Me3C5H2 analogue (7), probably due to a steric bulk.17 It seems likely that the Ti–Cl bond distances in the Cp* analogue [5, 2.2546(15)–2.2702(14) Å] is slightly longer than those in the Cp [4, 2.2361(16), 2.2649(19) Å] and the 1,2,4-Me3C5H2 analogues [7, 2.2376(11)–2.2679(10) Å];17 the Ti–O bond distances in 5 [1.789(3)–1.792(3) Å] are slightly longer than those in 4 [1.774(3) Å] but within the range of those in 7 [1.779(2)–1.797(2) Å]. The Ti–O–C(3) in phenyl angles increased in the order: 157.6(2)° (4, Cp′ = Cp) < 163.0(2)–166.7(2) (7, 1,2,4-Me3C5H2) < 167.9(3)–169.8(3)° (5, Cp*). Moreover, the Ti–O–C bond angle in 5 is larger than that in Cp*TiCl2(O-2,6-tBu2C6H3) [155.5(2)°],14c probably due to less steric bulk. Since the observed difference in the Ti–O–C bond angle reflects the degree of O–Ti π-donation, this may explain the observed difference in the Cp* analogue (5) especially compared to the Cp analogue (4). It also seems likely that both the C(1)–N–C(1) [or C(1)–N–C(26), C(1)–N–C(24)] and N–C(1)–C(2) angles are slightly influenced by the cyclopentadienyl fragment (Table 1). As observed in the mononuclear half-titanocenes,10i,14 it was thus revealed that these bond distances and angles are influenced by the ligand substituent.
Fig. 3 shows ORTEP drawings for the binuclear complexes, [Cp′TiCl2{(O-2,4-Me2C6H2)-6-CH2}][Cp′TiCl{(O-2,4-Me2C6H2)-6-CH2}2]N [Cp′ = Cp* (2), 1,2,4-Me3C5H2 (3)], determined by X-ray crystallography. These complexes fold a distorted tetrahedral geometry around each titanium metal center. In the Cp* analogue (2), the Ti(1)–Cl(1), Ti(1)–Cl(2), Ti(1)–O(1) bond distances [monophenoxide, 2.2830(16), 2.2787(16), 1.776(3) Å, respectively] are shorter than those in the bis(phenoxide) [Ti(2)–Cl(3), Ti(2)–O(2), Ti(2)–O(3): 2.3140(15), 1.804(3), 1.806(3) Å, respectively], whereas there were no significant differences in the Ti–O–C (in phenyl) angles were observed [Ti(1)–O(1)–C(3), Ti(2)–O(2)–C(22), Ti(2)–O(3)–C(31): 172.4(3), 173.9(3), 174.0(3)°, respectively]. Therefore, the observed difference in the bond distances would be due to degree of π-donation from oxygen to titanium. The C(1)–N–C(20) bond angle [112.2(3)°] was rather larger than those in the others [C(1)–N–C(29), C(20)–N–C(29): 110.3(3), 110.0(3)°], whereas no significant differences were observed in N–C(1)–C(2), N–C(20)–C(21), and N–C(29)–C(30) bond angles [113.5(5), 113.8(3), 112.9(3)°, respectively].
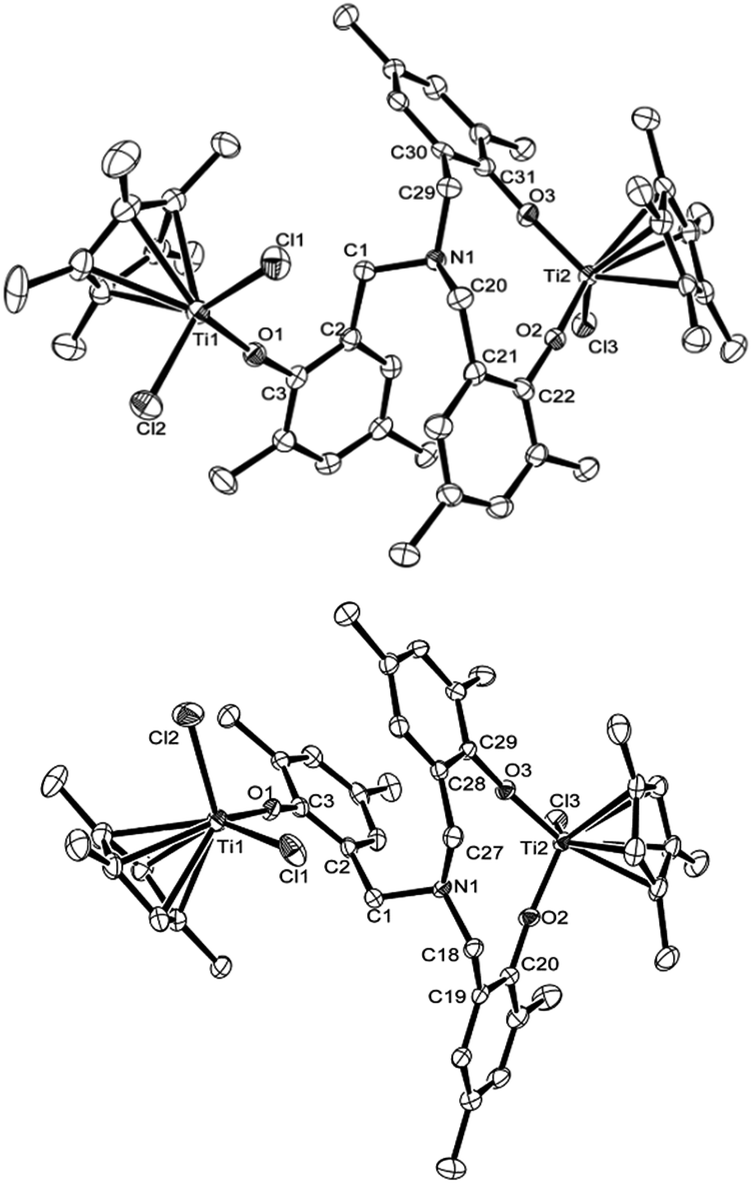 | ||
| Fig. 3 ORTEP drawings for [Cp′TiCl2{(O-2,4-Me2C6H2)-6-CH2}][Cp′TiCl{(O-2,4-Me2C6H2)-6-CH2}2]N [Cp′ = Cp* (2, left), 1,2,4-Me3C5H2 (3, right)]. Thermal ellipsoids are drawn at the 50% probability level, and H atoms are omitted for clarity.17 Selected bond distances (Å) and angles (°) in 2: Ti(1)–Cl(1) 2.2830(16), Ti(1)–Cl(2) 2.2787(16), Ti(1)–O(1) 1.776(3), Ti(2)–Cl(3) 2.3140(15), Ti(2)–O(2) 1.804(3), Ti(2)–O(3) 1.806(3), N(1)–C(1) 1.461(5), N(1)–C(20) 1.471(5), N(1)–C(29) 1.474(5) Å; Cl(1)–Ti(1)–Cl(2) 103.17(5), Cl(1)–Ti(1)–O(1) 102.65(10), Cl(2)–Ti(1)–O(1) 102.46(10), Cl(3)–Ti(2)–O(2) 100.78(10), Cl(3)–Ti(2)–O(3) 100.50(11), O(2)–Ti(2)–O(3) 108.14(12), Ti(1)–O(1)–C(3) 172.4(3), Ti(2)–O(2)–C(22) 173.9(3), Ti(2)–O(3)–C(31) 174.0(3), C(1)–N(1)–C(20) 112.2(3), C(1)–N(1)–C(29) 110.3(3), C(20)–N(1)–C(29) 110.0(3), N(1)–C(1)–C(2) 113.5(3), N(1)–C(20)–C(21) 113.8(3), N(1)–C(29)–C(30) 112.9(3)°. Selected bond distances (Å) and angles (°) in 3: Ti(1)–Cl(1) 2.2703(11), Ti(1)–Cl(2) 2.2655(11), Ti(1)–O(1) 1.771(2), Ti(2)–Cl(3) 2.3046(13), Ti(2)–O(2) 1.802(2), Ti(2)–O(3) 1.793(2), N(1)–C(1) 1.472(4), N(1)–C(18) 1.460(4), N(1)–C(27) 1.463(5) Å; Cl(1)–Ti(1)–Cl(2) 101.44(5), Cl(1)–Ti(1)–O(1) 101.03(8), Cl(2)–Ti(1)–O(1) 104.74(8), Cl(3)–Ti(2)–O(2) 100.36(10), Cl(3)–Ti(2)–O(3) 102.06(9), O(2)–Ti(2)–O(3) 106.52(10), Ti(1)–O(1)–C(3) 167.6(2), Ti(2)–O(2)–C(20) 170.0(3), Ti(2)–O(3)–C(29) 168.4(3), C(1)–N(1)–C(18) 111.8(2), C(1)–N(1)–C(27) 112.8(2), C(18)–N(1)–C(27) 110.6(3), N(1)–C(1)–C(2) 112.3(2), N(1)–C(18)–C(19) 114.1(3), N(1)–C(27)–C(28) 114.5(3)°. | ||
Structure in the 1,2,4-Me3C5H2 analogue (3) is similar to that in the Cp* analogue (2); the Ti(1)–Cl(1), Ti(1)–Cl(2), Ti(1)–O(1) bond distances [monophenoxide, 2.2703(11), 2.2655(11), 1.771(2) Å, respectively] are shorter than those in the bis(phenoxide) [Ti(2)–Cl(3), Ti(2)–O(2), Ti(2)–O(3): 2.3046(13), 1.802(2), 1.793(2) Å, respectively], whereas no significant differences in the Ti–O–C (in phenyl) bond angles were observed [Ti(1)–O(1)–C(3), Ti(2)–O(2)–C(20), Ti(2)–O(3)–C(29): 167.6(2), 170.3(3), 168.4(3)°, respectively]. The Ti–O–C bond angles in the Cp* analogue [2: 172.4(3)–174.0(3)°] were larger than those in the 1,2,4-Me3C5H2 analogue [3: 167.6(2)–170.0(3)°].
As shown in Scheme 4, [Cp*TiMe2{(O-2,4-Me2C6H2)-6-CH2}]3N (8) could be prepared from Cp*TiMe3, prepared in situ by treating Cp*TiCl3 with MeLi in toluene, by reacting with [(HO-2,4-Me2C6H2)-6-CH2]3N, although, as described above, attempted isolation of the dichloride analogue afforded the bimetallic complex, [Cp*TiCl2{(O-2,4-Me2C6H2)-6-CH2}]–[Cp*TiCl{(O-2,4-Me2C6H2)-6-CH2}2]N (2). It was also revealed that the reaction of [Cp*TiCl2{(O-2,4-tBu2C6H2)-6-CH2}]3N (5) with MeMgBr afforded the corresponding dimethyl complex (9), although the similar attempt for obtainment of 9 through one-pot synthesis from Cp*TiCl3 (at 25 °C overnight) failed and recovered both Cp*TiMe3 and [(HO-2,4-tBu2C6H2)-6-CH2]3N (shown in the ESI†).15 These complexes (8 and 9) were identified by NMR spectra and elemental analysis, and the structure of 9 could be determined by X-ray crystallographic analysis (shown in Fig. 4).
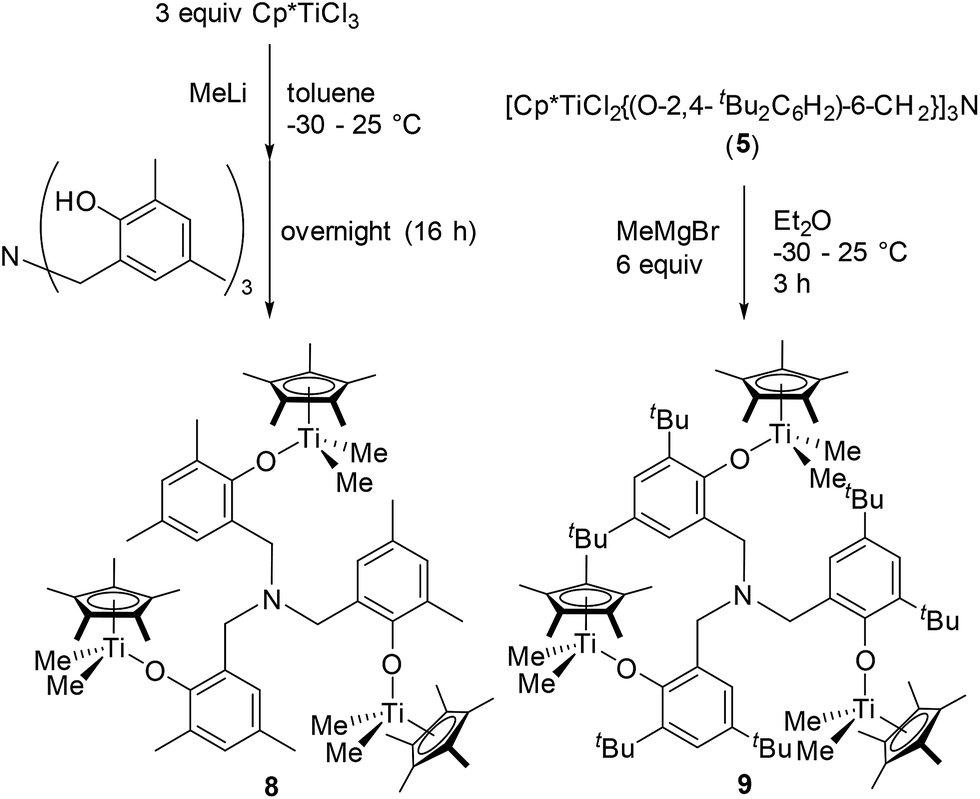 | ||
| Scheme 4 Synthesis of [Cp*TiMe2{(O-2,4-R2C6H2)-6-CH2}]3N [R = Me (8), tBu (9)]. | ||
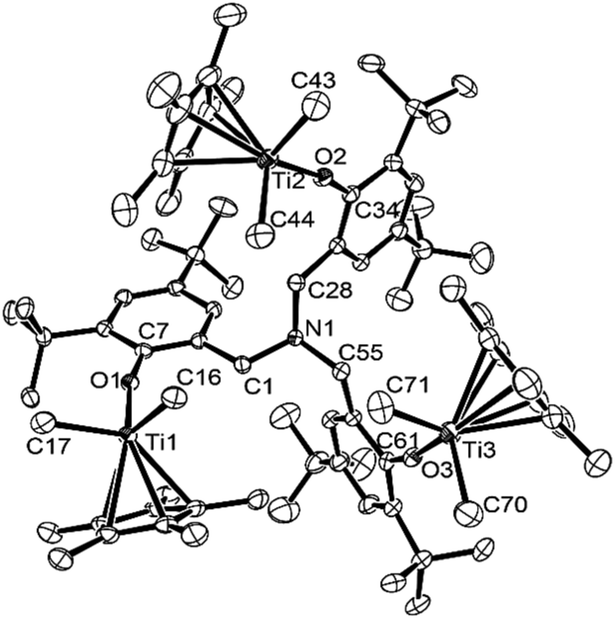 | ||
| Fig. 4 ORTEP drawing for [Cp*TiMe2{(O-2,4-tBu2C6H2)-6-CH2}]3N (9). Thermal ellipsoids are drawn at the 50% probability level, and H atoms are omitted for clarity.17 Selected bond distances (Å): Ti(1)–O(1) 1.8221(16), Ti(2)–O(2) 1.8230(17), Ti(3)–O(3) 1.8149(17), Ti(1)–C(16) 2.122(2), Ti(1)–C(17) 2.133(3), Ti(2)–C(43) 2.122(3), Ti(2)–C(44) 2.136(3), Ti(3)–C(70) 2.129(4), Ti(3)–C(71) 2.128(3), N(1)–C(1) 1.462(3), N(1)–C(28) 1.467(3), N(1)–C(55) 1.456(3). Selected bond angle (°): C(16)–Ti(1)–C(17) 95.63(10), C(43)–Ti(2)–C(44) 96.79(12), C(70)–Ti(3)–C(71) 95.20(13), Ti(1)–O(1)–C(7) 165.63(15), Ti(2)–O(2)–C(34) 165.28(15), Ti(3)–O(3)–C(61) 166.02(16). | ||
Complex 9 folds a distorted tetrahedral geometry around each titanium, and no significant differences in Ti–O, Ti–C (in methyl) bond distances, C(methyl)–Ti–C(methyl), Ti–O–C (in phenyl) angles etc. were observed in the three titanium complexes in 9. The Ti–O bond distances in 9 [1.8149(17)–1.8230(17) Å] are longer than those in the dichloride analogue [5: 1.789(3)–1.792(3) Å], and Ti–O–C (in phenyl) bond angles in 9 [165.28(15)–166.02(16)°] are smaller than those in 5 [167.9(3)–169.8(3)°]. Moreover, the bond angles formed by two methyl group and titanium in 9 [95.20(13)–96.79(12)] are smaller than those formed by two chloride and the titanium in 5 [101.11(5)–101.85(6)].17 These are observed trend between the dichloride and the dimethyl complex, exemplified by Cp*TiX2(O-iPr2C6H3) [Ti–O, Ti–O–C (phenyl), X(1)–Ti–X(2): 1.772(3) Å, 173.0(3)°, 103.4(5)°, respectively (X = Cl); 1.790(2) Å, 168.7°, 99.8(1)° (X = Me)],14e and rather small bond angles in 9, described above, compared to those in 5 would be explained due to a steric bulk (rather large methyl group compared to Cl).
2. Ethylene polymerisation by [Cp′TiCl2{(O-2,4-R2C6H2)-6-CH2}]3N [R = Me, Cp′ = Cp (1); R = tBu, Cp′ = Cp (4), Cp* (5), tBuC5H4 (6), 1,2,4-Me3C5H2 (7)], [Cp′TiCl2{(O-2,4-Me2C6H2)-6-CH2}][Cp′TiCl{(O-2,4-Me2C6H2)-6-CH2}2]N [Cp′ = Cp* (2), 1,2,4-Me3C5H2 (3)], and [Cp*TiMe2{(O-2,4-R2C6H2)-6-CH2}]3N [R = Me (8), tBu (9)]
Table 2 summarises results in ethylene polymerisation at 25 °C in toluene using [Cp′TiCl2{(O-2,4-R2C6H2)-6-CH2}]3N [R = Me, Cp′ = Cp (1); R = tBu, Cp′ = Cp (4), Cp* (5), tBuC5H4 (6), 1,2,4-Me3C5H2 (7)], [Cp′TiCl2{(O-2,4-Me2C6H2)-6-CH2}][Cp′TiCl{(O-2,4-Me2C6H2)-6-CH2}2]N [Cp′ = Cp* (2), 1,2,4-Me3C5H2 (3)], and [Cp*TiMe2{(O-2,4-R2C6H2)-6-CH2}]3N [R = Me (8), tBu (9)] in the presence of methyl-aluminoxane (MAO) cocatalyst. MAO white solid, prepared by removing toluene and AlMe3 from the commercially available sample [TMAO, 9.5 wt% (Al) toluene solution, Tosoh Finechem Co.], was employed because use of this MAO was effective in the ethylene (co)polymerisation for obtainment of (co)polymers with uniform molecular weight distributions.10,14b,c,f| Run | Catalyst (μmol) | MAO/mmol | Time/min | Polymer/mg | Activityb | Mnc × 10−4 | Mw/Mnc |
|---|---|---|---|---|---|---|---|
| a Conditions: toluene total 30 mL, d-MAO (prepared by removing toluene and AlMe3 from the ordinary MAO).b Activity in kg PE per mol Ti per h.c GPC data in o-dichlorobenzene vs. polystyrene standards. | |||||||
| 1 | 1 (0.50) | 3.0 | 10 | 80 | 320 | 341 | 2.13 |
| 2 | 2 (0.03) | 3.0 | 10 | 107 | 10700 |
309, 3.91 | 2.11, 1.95 |
| 3 | 2 (0.03) | 3.0 | 5 | 78 | 15600 |
368, 3.09 | 2.67, 2.19 |
| 4 | 2 (0.03) | 3.0 | 5 | 75 | 15000 |
300, 1.93 | 2.29, 2.58 |
| 5 | 3 (0.30) | 3.0 | 10 | 116 | 1160 | 368, 0.96 | 1.72, 1.82 |
| 6 | 4 (0.50) | 2.0 | 10 | 120 | 480 | 220 | 2.13 |
| 7 | 4 (0.50) | 2.0 | 10 | 124 | 496 | 239 | 2.21 |
| 8 | 4 (0.50) | 3.0 | 10 | 186 | 744 | 204 | 2.44 |
| 9 | 4 (0.50) | 3.0 | 10 | 180 | 720 | 202 | 2.48 |
| 10 | 4 (0.50) | 4.0 | 10 | 140 | 560 | 238 | 2.64 |
| 11 | 4 (0.50) | 4.0 | 10 | 136 | 544 | 232 | 2.66 |
| 12 | 5 (0.50) | 3.0 | 10 | 107 | 428 | 172 | 2.90 |
| 13 | 6 (0.50) | 3.0 | 10 | 44 | 176 | 277 | 2.26 |
| 14 | 7 (0.25) | 3.0 | 10 | 75 | 600 | 93.1, 1.12 | 3.95, 2.11 |
| 15 | 7 (0.25) | 3.0 | 5 | 26 | 416 | 238, 0.97 | 2.75, 2.05 |
| 16 | 8 (0.01) | 3.0 | 10 | 84 | 16800 |
214 | 2.20 |
| 17 | 9 (0.10) | 3.0 | 10 | 453 | 9060 | 354 | 2.24 |
| 18 | 9 (0.01) | 3.0 | 10 | 58 | 11600 |
128 | 3.04 |
It turned out that the Cp analogues (1, 4) exhibited low catalytic activities affording ultrahigh molecular weight polymers with uniform molecular weight distributions (Mn = 2.02–3.58 × 106, Mw/Mn = 2.06–2.66, runs 1, 6–11 and runs S1 in Table S1 in the ESI†).18 It also turned out that the activities at 25 °C by the 1,2,4-Me3C5H2 analogues (3 and 7) and the tBuC5H4 analogue (6) were low and the resultant polymers prepared by 3 and 7 possessed bimodal molecular weight distributions (runs 5, 14, 15, runs S3, S6 and S7†), although slight improvement in the activity was observed by the bimetallic complex (3) compared to 1. In contrast, it was revealed that the bimetallic Cp* analogue (2) and the Cp*-dimethyl analogues (8, 9) exhibited higher catalytic activities (9060–16800 kg PE per mol Ti per h, runs 2–4, 16–18) than the others, whereas the activity by the related Cp*-dichloride analogue (5) was low (384–428 kg PE per mol Ti per h, runs 12, S4†). The resultant polymers prepared by 8 and 9 possessed ultrahigh molecular weights with unimodal molecular weight distributions (Mn = 1.21–3.68 × 106, Mw/Mn = 2.16–3.16, runs 16–18, runs S9–S11 in the ESI†),18 whereas the resultant polymers prepared by 2 possessed a mixture of ultrahigh and low molecular weights (runs 2–4, S2†) which might suggest a presence of two catalytically active species in situ. The observed polymerisation results are reproducible as shown in runs 6–11 as well as shown in the ESI.†18
It was revealed that the activities (on the basis of polymer yields) by [Cp′TiCl2{(O-2,4-tBu2C6H2)-6-CH2}]3N [Cp′ = Cp (4), Cp* (5), tBuC5H4 (6), 1,2,4-Me3C5H2 (7)] increased at 50 °C, and the resultant polymers by both the Cp analogue (4) and tBuC5H4 analogue (6) possessed ultrahigh molecular weights with unimodal molecular weight distributions (Mn = 2.08–2.28 × 106, Mw/Mn = 2.04–2.48, runs 19, 20, S12, S13, Table 3 and ESI†).18 Although notable improvement in the activity was observed when the polymerisation by 5 was conducted at 50 °C (run 24), the catalyst was short lived at 80 °C (runs 25, 26) and the activity decreased at 100 °C (run 27).18 Moreover, the activity by 2 decreased upon increasing the polymerisation temperature (runs 22 and 23).18 The dimethyl analogues (8, 9) also showed the similar trends, and the activities decreased at 50 °C probably due to partial decomposition of the catalytically active species (runs 16, 17, 28–31, S21–S24†).18
| Run | Catalyst (μmol) | Temp./°C | Time/min | Polymer/mg | Activityb | Mnc × 10−4 | Mw/Mnc |
|---|---|---|---|---|---|---|---|
| a Conditions: toluene total 30 mL, d-MAO 3.0 mmol, ethylene 4 atm.b Activity in kg PE per mol Ti per h.c GPC data in o-dichlorobenzene vs. polystyrene standards. | |||||||
| 8 | 4 (0.50) | 25 | 10 | 186 | 744 | 204 | 2.44 |
| 19 | 4 (0.10) | 50 | 10 | 74 | 1480 | 215 | 2.04 |
| 13 | 6 (0.50) | 25 | 10 | 44 | 176 | 277 | 2.26 |
| 20 | 6 (0.30) | 50 | 10 | 58 | 387 | 226 | 2.48 |
| 14 | 7 (0.25) | 25 | 10 | 75 | 600 | 93.1, 1.12 | 3.95, 2.11 |
| 21 | 7 (0.10) | 50 | 10 | 43 | 860 | 220, 0.67 | 2.15, 1.62 |
| 2 | 2 (0.03) | 25 | 10 | 107 | 10700 |
309, 3.91 | 2.11, 1.95 |
| 22 | 2 (0.05) | 50 | 10 | 140 | 8400 | 220, 1.44 | 1.67, 2.13 |
| 23 | 2 (0.05) | 80 | 10 | 65 | 3900 | 173, 0.95 | 1.47, 1.81 |
| 12 | 5 (0.50) | 25 | 10 | 107 | 428 | 172 | 2.90 |
| 24 | 5 (0.10) | 50 | 10 | 158 | 3160 | 113 | 2.85 |
| 25 | 5 (0.10) | 80 | 10 | 150 | 3000 | 26.4, 0.83 | 3.08, 1.63 |
| 26 | 5 (0.10) | 80 | 5 | 121 | 4840 | 37.7, 0.88 | 4.85, 1.64 |
| 27 | 5 (0.10) | 100 | 10 | 68 | 1360 | 112, 0.61 | 1.92, 1.68 |
| 16 | 8 (0.01) | 25 | 10 | 84 | 16800 |
214 | 2.20 |
| 28 | 8 (0.02) | 50 | 10 | 128 | 12800 |
215, 1.44 | 1.49, 2.06 |
| 29 | 8 (0.02) | 50 | 5 | 110 | 22000 |
277, 2.09 | 2.33, 1.79 |
| 30 | 8 (0.04) | 80 | 10 | 84 | 4200 | 180, 0.96 | 1.53, 1.69 |
| 17 | 9 (0.10) | 25 | 10 | 453 | 9060 | 354 | 2.24 |
| 18 | 9 (0.01) | 25 | 10 | 58 | 11600 |
128 | 3.04 |
| 31 | 9 (0.05) | 50 | 10 | 107 | 4280 | 126, 2.44 | 3.94, 1.53 |
Ethylene polymerisations using the mononuclear analogues, CpTiCl2(O-2-R-4,6-Me2C6H2) [R = Me (10), tBu (11)], and Cp*TiCl2(O-2-R-4,6-Me2C6H2) [R = Me (12), tBu (13)],14b,c were also conducted under the same conditions for comparison. Complexes 10 and 11 were prepared according the analogous procedure for synthesis of the Cp-aryloxo analogues (shown in the Experimental section).10i,14b,c The results are summarised in Table 4. The selected results by the trinuclear analogues, [Cp′TiCl2{(O-2,4-R2C6H2)-6-CH2}]3N [R = Me, Cp′ = Cp (1); R = tBu, Cp′ = Cp (4), Cp* (5)] and [Cp*TiMe2{(O-2,4-R2C6H2)-6-CH2}]3N [R = Me (8), tBu (9)], and bimetallic [Cp*TiCl2{(O-2,4-Me2C6H2)-6-CH2}][Cp*TiCl{(O-2,4-Me2C6H2)-6-CH2}2]N (2) are also placed for comparison.
| Run | Catalyst (μmol) | Cp′ | R | Temp./°C | Polymer/mg | Activityb | Mnc × 10−4 | Mw/Mnc |
|---|---|---|---|---|---|---|---|---|
| a Conditions: toluene total 30 mL, d-MAO 3.0 mmol, ethylene 4 atm, 10 min.b Activity in kg PE per mol Ti per h.c GPC data in o-dichlorobenzene vs. polystyrene standards. | ||||||||
| 1 | 1 (0.50) | Cp | Me | 25 | 80 | 320 | 341 | 2.13 |
| 32 | 10 (0.50) | Cp | Me | 25 | 110 | 1320 | 305 | 2.15 |
| 8 | 4 (0.50) | Cp | tBu | 25 | 186 | 744 | 204 | 2.44 |
| 19 | 4 (0.10) | Cp | tBu | 50 | 74 | 1480 | 215 | 2.04 |
| 33 | 11 (0.50) | Cp | tBu | 25 | 128 | 1540 | 30.3 | 7.69 |
| 34 | 11 (0.50) | Cp | tBu | 50 | 144 | 1730 | 78.4 | 4.35 |
| 2 | 2 (0.03) | Cp* | Me | 25 | 107 | 10700 |
309, 3.91 | 2.11, 1.95 |
| 16 | 8 (0.01) | Cp* | Me | 25 | 84 | 16800 |
214 | 2.20 |
| 28 | 8 (0.02) | Cp* | Me | 50 | 128 | 12800 |
215, 1.44 | 1.49, 2.06 |
| 35 | 12 (0.50) | Cp* | Me | 25 | 430 | 5160 | 208 | 2.09 |
| 36 | 12 (0.10) | Cp* | Me | 25 | 173 | 10400 |
139 | 2.71 |
| 12 | 5 (0.50) | Cp* | tBu | 25 | 107 | 428 | 172 | 2.90 |
| 24 | 5 (0.10) | Cp* | tBu | 50 | 158 | 3160 | 113 | 2.85 |
| 17 | 9 (0.10) | Cp* | tBu | 25 | 453 | 9060 | 354 | 2.24 |
| 18 | 9 (0.01) | Cp* | tBu | 25 | 58 | 11600 |
128 | 3.04 |
| 31 | 9 (0.05) | Cp* | tBu | 50 | 107 | 4280 | 126, 2.44 | 3.94, 1.53 |
| 37 | 13 (0.50) | Cp* | tBu | 25 | 189 | 2270 | 26.0 | 9.33 |
| 38 | 13 (0.50) | Cp* | tBu | 50 | 199 | 2390 | 121 | 2.92 |
It turned out that the activity by the Cp analogue, [CpTiCl2{(O-2,4-Me2C6H2)-6-CH2}]3N (1), was lower than that by CpTiCl2(O-2,4,6-Me3C6H2) (10) (runs 1, 32),18 probably due to disproportionation of 1 (generating CpTiCl3 exhibiting negligible activity)14c in the toluene solution as shown in Fig. 1. It also turned out that the activities by 4 were lower than those by the related mononuclear analogue (11) [runs 8, 19 (by 4) vs. runs 33, 34 (by 11)]. However, the resultant polymers prepared by 4 possessed unimodal molecular weight distributions suggesting a possibility that the polymerisation proceeded with uniform catalytically active species, whereas the resultant polymers prepared by 11 possessed the broad distributions (strongly suggesting a possibility that several catalytically active species were present in situ).
Note that the activities by the Cp* trinuclear analogue, [Cp*TiMe2{(O-2,4-Me2C6H2)-6-CH2}]3N (8) showed higher catalytic activities than the related mononuclear analogue, Cp*TiCl2(O-2,4,6-Me3C6H2) (12) [runs 16, 28 (by 8) vs. runs 35, 36 (by 12)].18 The activities by 2 and 8 were higher than those by Cp*TiCl2(O-2-tBu-4,6-Me2C6H2) (13) conducted under the same conditions (runs 2, 16, 28 vs. runs 37, 38). Although the activity by [Cp*TiCl2{(O-2,4-tBu2C6H2)-6-CH2}]3N (5) at 25 °C was low, the activity increased at 50 °C (runs 12, 24), whereas no significant increase in the activity by 13 were observed at 50 °C (runs 37, 38).18 Moreover, the activities by the dimethyl analogue, [Cp*TiMe2{(O-2,4-tBu2C6H2)-6-CH2}]3N (9) were higher than those by 13 (runs 17, 31 vs. runs 37, 38) conducted under the same conditions. Therefore, we believe that these would be unique characteristics observed by placement of three titanium complexes in tris(phenoxy)amine ligand.
It was revealed that the activities in the ethylene polymerisation by [Cp*TiX2{(O-2,4-R2C6H2)-6-CH2}]3N [R = tBu, X = Cl (5); R = Me, X = Me (8), Table 5] in the presence of AliBu3–[Ph3C][B(C6F5)4] cocatalysts were higher than those in the presence of MAO under the optimized conditions (run 39 vs. run 12; run 43 vs. run 16).18 Similar trends were observed in the mononuclear analogues (12 and 13), whereas the activity by 9 in the presence of AliBu3–[Ph3C][B(C6F5)4] cocatalysts was lower than that in the presence of MAO (run 47 vs. run 17). The trinuclear complex (8) exhibited the highest catalytic activities affording ultrahigh molecular weight polymer with uniform molecular weight distribution (27600 kg PE per mol Ti per h, run 43, Mn = 1.15 × 106, Mw/Mn = 3.15). The Mn values in addition to the activity by 8 was affected by the Al/Ti molar ratios (runs 40–44), and the polymerisations at 0 °C also afforded polymers with uniform molecular weight distributions without significant changes in the Mn values (runs 45, 46). These might suggest that the major chain transfer would not be the chain transfer to Al in this catalysis.
| Run | Catalyst (μmol) | Al/Tib | Temp./°C | Polymer/mg | Activityc | Mnd × 10−4 | Mw/Mnd |
|---|---|---|---|---|---|---|---|
| a Conditions: toluene total 30 mL, AliBu3, [Ph3C][B(C6F5)4] B/Ti = 1.5 (molar ratio), 4 atm, 10 min.b Molar ratio.c Activity in kg PE per mol Ti per h.d GPC data in o-dichlorobenzene vs. polystyrene standards. | |||||||
| 39 | 5 (0.02) | 700 | 25 | 71 | 7100 | 71.6 | 3.37 |
| 40 | 8 (0.02) | 100 | 25 | Trace | |||
| 41 | 8 (0.01) | 300 | 25 | 60 | 12000 |
71.6 | 3.78 |
| 42 | 8 (0.01) | 500 | 25 | 84 | 16800 |
88.1 | 3.79 |
| 43 | 8 (0.01) | 700 | 25 | 138 | 27600 |
115 | 3.15 |
| 44 | 8 (0.01) | 900 | 25 | 36 | 7200 | 70.8 | 3.33 |
| 45 | 8 (0.01) | 700 | 0 | 61 | 12200 |
77.7 | 2.70 |
| 46 | 8 (0.01) | 700 | 0 | 70 | 14000 |
78.4 | 2.70 |
| 47 | 9 (0.02) | 700 | 25 | 20 | 2000 | 78.5 | 4.21 |
| 48 | 12 (0.03) | 700 | 25 | 109 | 21800 |
63.7 | 4.17 |
| 49 | 13 (0.06) | 700 | 25 | 85 | 8500 | 54.9 | 2.82 |
Concluding remarks
A series of trinuclear half-titanocenes, [Cp′TiCl2{(O-2,4-R2C6H2)-6-CH2}]3N [R = Me, Cp′ = Cp (1); R = tBu, Cp′ = Cp (4), Cp* (5), tBuC5H4 (6), 1,2,4-Me3C5H2 (7)] and [Cp*TiMe2{(O-2,4-R2C6H2)-6-CH2}]3N [R = Me (8), tBu (9)], and the bimetallic complexes, [Cp′TiCl2{(O-2,4-Me2C6H2)-6-CH2}][Cp′TiCl{(O-2,4-Me2C6H2)-6-CH2}2]N [Cp′ = Cp* (2), 1,2,4-Me3C5H2 (3)], have been prepared and identified. Structures of 1–5, 7 and 9 were determined by X-ray crystallography, and all complexes fold distorted tetrahedral geometries around titanium and Ti–O–C (phenyl) bond angles were affected by ligand substituents (on cyclopentadienyl, phenyl and halogen or methyl). It was revealed that these complexes (2–9) are stable in solution (25–80 °C) except the Cp analogue, [CpTiCl2{(O-2,4-Me2C6H2)-6-CH2}]3N (1), that was unstable in solution and present as a mixture of the trinuclear complex (1), (proposed) binuclear analogue and CpTiCl3 and titanatrane, CpTiCl[(O-2,4-Me2C6H2)-6-CH2]3N; an equilibrium between 1 and the binuclear species (and CpTiCl3) was observed depending upon temperature and concentration.These complexes (2–9) exhibited from moderate to high catalytic activities for ethylene polymerisation in the presence of MAO cocatalyst, affording ultrahigh molecular weight polymers with uniform molecular weight distributions in most cases, especially in both the Cp and the Cp* analogues; different polymerisation behaviours compared to the related mononuclear analogues observed. In particular, the Cp* trinuclear analogue, [Cp*TiMe2{(O-2,4-Me2C6H2)-6-CH2}]3N (8) showed higher catalytic activities than the related mononuclear analogue, Cp*TiCl2(O-2-R-4,6-Me2C6H2) [R = Me (12), tBu (13)] conducted under the same conditions. The activities in the presence of AliBu3–[Ph3C][B(C6F5)4] cocatalysts were higher than those in the presence of MAO in most cases, although the Mn values in the resultant polymers decreased compared to those in the presence of MAO cocatalyst with rather broad molecular weight distributions. We thus believe that information observed here should be promising as an effect of integration of the catalytically active species, and would introduce a new possibility for design of efficient catalysts for olefin polymerisation.
Experimental section
1. General procedures
All experiments were carried out under a nitrogen atmosphere in a Vacuum Atmospheres drybox unless otherwise specified. All chemicals used were of reagent grade and were purified by the standard purification procedures. Anhydrous grades of toluene, n-hexane, and dichloromethane (Kanto Kagaku Co. Ltd) was transferred into a bottle containing molecular sieves (mixture of 3A and 4A 1/16, and 13X) in the drybox, and was used without further purification. Ethylene for polymerisation was of polymerisation grade (purity > 99.9%; Sumitomo Seika Co., Ltd.) and was used as received. Cp*TiCl2(O-2-R-4,6-Me2C6H2) [R = Me (12), tBu (13)] were prepared according to the reported procedure.14b,c Toluene and AlMe3 in the commercially available methylaluminoxane [TMAO, 9.5 wt% (Al) toluene solution, Tosoh Finechem Co.] were removed under reduced pressure (at ca. 50 °C for removing toluene, AlMe3, and then heated at >100 °C for 1 h for completion) in the drybox to give white solids.14cElemental analyses were performed by using EAI CE-440 CHN/O/S Elemental Analyzer (Exeter Analytical, Inc.). All 1H and 13C NMR spectra were recorded on a Bruker AV500 spectrometer (500.13 MHz for 1H, 125.77 MHz for 13C). All spectra were obtained in the solvent indicated at 25 °C unless otherwise noted. Chemical shifts are given in ppm and are referenced to SiMe4 (δ 0.00 ppm, 1H, 13C). Coupling constants are given in Hz. Molecular weights and molecular weight distributions for the resultant polymers were measured by gel permeation chromatography (Tosoh HLC-8121GPC/HT) using a RI-8022 detector (for high temperature; Tosoh Co.) with a polystyrene gel column (TSK gel GMHHR-H HT × 2, 30 cm × 7.8 mm i.d.), ranging from <102 to <2.8 × 108 MW at 140 °C using o-dichlorobenzene containing 0.05 w/v% 2,6-di-tert-butyl-p-cresol as the solvent. The molecular weight was calculated by a standard procedure based on the calibration with standard polystyrene samples.
2. Polymerisation of ethylene
Reactions with ethylene were conducted in a 100 mL scale stainless steel autoclave, and the typical reaction procedure is as follows. Toluene (29.0 mL) and prescribed amount of MAO were added into the autoclave in the drybox. The reaction apparatus was then filled with ethylene (1 atm), and the prescribed amount of complex in toluene (1.0 mL) was added into the autoclave. The reaction apparatus was then immediately pressurized to 3 atm (total 4 atm), and the mixture was magnetically stirred for 5 or 10 min (ethylene pressure was kept constant during the reaction). After the above procedure, the mixture in the autoclave was poured into MeOH containing HCl, and the resultant polymer (white precipitate) was collected on a filter paper by filtration and was adequately washed with MeOH. The resultant polymer was then dried in vacuo at 60 °C for 2 h.3. Crystallographic analysis
All measurements were made on a Rigaku XtaLAB P200 diffractometer using multi-layer mirror monochromated Mo-Kα radiation. The crystal collection parameters are listed in Tables S5 and S6 (ESI†). The data were collected and processed using CrystalClear (Rigaku)19 or CrysAlisPro (Rigaku Oxford Diffraction),20 and the structure was solved by direct methods21 and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined using the riding model. All calculations were performed using the Crystal Structure22 crystallographic software package except for refinement, which was performed using SHELXL Version 2014/7.23 Details are shown in the ESI.†17 CCDC numbers for complexes 1–5, 7 and 9 are CCDC 1549581–1549587, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was partly supported by Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (JSPS, No. 15H03812). The authors express their thanks to Profs. S. Komiya, A. Inagaki, and Dr S. Sueki (Tokyo Metropolitan Univ.) for discussions, and to Tosoh Finechem Co. for donating MAO.Notes and references
- Selected reviews, accounts in 1990's, see: (a) R. F. Jordan, Adv. Organomet. Chem., 1991, 32, 325 CrossRef CAS; (b) H. H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143 CrossRef CAS; (c) W. Kaminsky and M. Arndt, Adv. Polym. Sci., 1997, 127, 144 CrossRef; (d) J. Suhm, J. Heinemann, C. Wörner, P. Müller, F. Stricker, J. Kressler, J. Okuda and R. Mülhaupt, Macromol. Symp., 1998, 129, 1 CrossRef CAS; (e) A. L. McKnight and R. M. Waymouth, Chem. Rev., 1998, 98, 2587 CrossRef CAS PubMed; (f) G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428 CrossRef CAS; (g) W. Kaminsky, J. Chem. Soc., Dalton Trans., 1998, 1413 RSC.
- Selected special issues: (a) Frontiers in Metal-Catalyzed Polymerization (special issue), J. A. Gladysz, Chem. Rev., 2000, 100, 1167 Search PubMed; (b) Metallocene complexes as catalysts for olefin polymerization, H. G. Alt, Coord. Chem. Rev., 2006, 250, 1 Search PubMed; (c) Metal-catalysed Polymerisation, B. Milani and C. Claver, Dalton Trans., 2009, 8769 Search PubMed; (d) Advances in Metal-Catalysed Polymerisation and Related Transformations, P. Mountford, Dalton Trans., 2013, 42, 8977 Search PubMed.
- Selected recent reviews/accounts, see: (a) The Lecture Notes in Chemistry 85, in Organometallic Reactions and Polymerization, ed. K. Osakada, Springer-Verlag, Berlin, 2014 Search PubMed; (b) K. Nomura and S. Zhang, Chem. Rev., 2011, 111, 2342 CrossRef CAS PubMed; (c) H. Makio, H. Terao, A. Iwashita and T. Fujita, Chem. Rev., 2011, 111, 2363 CrossRef CAS PubMed; (d) M. Delferro and T. J. Marks, Chem. Rev., 2011, 111, 2450 CrossRef CAS PubMed; (e) G. M. Miyake and E.-Y. X. Chen, Polym. Chem., 2011, 2, 2462 RSC; (f) C. Redshaw and Y. Tang, Chem. Soc. Rev., 2012, 41, 4484 RSC; (g) A. Valente, A. Mortreux, M. Visseaux and P. Zinck, Chem. Rev., 2013, 113, 3836 CrossRef CAS PubMed; (h) J. P. McInnis, M. Delferro and T. J. Marks, Acc. Chem. Res., 2014, 47, 2545 CrossRef CAS PubMed; (i) J. Klosin, P. P. Fontaine and R. Figueroa, Acc. Chem. Res., 2015, 48, 2004 CrossRef CAS PubMed; (j) S. A. Ryken and L. L. Schafer, Acc. Chem. Res., 2015, 48, 2576 CrossRef CAS PubMed; (k) B. L. Small, Acc. Chem. Res., 2015, 48, 2599 CrossRef CAS PubMed.
- (a) K. Nomura, J. Liu, S. Padmanabhan and B. Kitiyanan, J. Mol. Catal. A: Chem., 2007, 267, 1 CrossRef CAS; (b) K. Nomura, Dalton Trans., 2009, 8811 RSC; (c) K. Nomura and J. Liu, Dalton Trans., 2011, 40, 7666 RSC.
- (a) D. W. Stephan, Organometallics, 2005, 24, 2548 CrossRef CAS; (b) M. J. Ferreira and A. M. Martins, Coord. Chem. Rev., 2006, 250, 118 CrossRef CAS.
- (a) Coordination Polymerization, ed. J. C. W. Chien, Academic Press, New York, 1975 Search PubMed; (b) A. Andresen, H. G. Cordes, H. Herwig, W. Kaminsky, A. Merk, R. Mottweiler, J. Pein, H. Sinn and H. J. Vollmer, Angew. Chem., Int. Ed. Engl., 1976, 15, 630 CrossRef; (c) H. Sinn, W. Kaminsky, H.-J. Vollmer and R. Woldt, Angew. Chem., Int. Ed. Engl., 1980, 19, 390 CrossRef; (d) H. Sinn and W. Kaminsky, Adv. Organomet. Chem., 1980, 18, 99 CrossRef CAS.
- Selected review article, see: (a) A. Macchioni, Chem. Rev., 2005, 105, 2039 CrossRef CAS PubMed; (b) M. Bochmann, Organometallics, 2010, 29, 4711 CrossRef CAS; (c) W. Kaminsky, Macromolecules, 2012, 4, 3289 CrossRef; (d) S. Dagorne and C. Fliedel, in Modern Organoaluminium Reagents, Topics in Organometallic Chemistry, ed. S. Woodward and S. Dagorne, Springer-Verlag, Berlin-Heidelberg, 2013, vol. 41, p. 125 Search PubMed.
- (a) L. Li, M. V. Metz, H. Li, M.-C. Chen, T. J. Marks, L. Liable-Sands and A. L. Rheingold, J. Am. Chem. Soc., 2002, 124, 12725 CrossRef CAS PubMed; (b) H. Li, L. Li, T. J. Marks, L. Liable-Sands and A. L. Rheingold, J. Am. Chem. Soc., 2003, 125, 10788 CrossRef CAS PubMed; (c) N. Guo, L. Li and T. J. Marks, J. Am. Chem. Soc., 2004, 126, 6542 CrossRef CAS PubMed; (d) H. Li, L. Li and T. J. Marks, Angew. Chem., Int. Ed., 2004, 43, 4937 CrossRef CAS PubMed; (e) H. Li, L. Li, D. J. Schwartz, M. V. Metz, T. J. Marks, L. Liable-Sands and A. L. Rheingold, J. Am. Chem. Soc., 2005, 127, 14756 CrossRef CAS PubMed; (f) H. Li and T. J. Marks, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15295 CrossRef CAS PubMed; (g) N. Guo, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 2246 CrossRef CAS PubMed; (h) A. Motta, I. L. Fragala and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 3974 CrossRef CAS PubMed.
- (a) S. Liu, A. Motta, M. Delferro and T. J. Marks, J. Am. Chem. Soc., 2013, 135, 8830 CrossRef CAS PubMed; (b) S. Liu, A. Motta, A. R. Mouat, M. Delferro and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 10460 CrossRef CAS PubMed.
- For examples (by aryloxo-modified half-titanocene catalysts), see: (a) K. Nomura, H. Okumura, T. Komatsu and N. Naga, Macromolecules, 2002, 35, 5388 CrossRef CAS; (b) K. Nomura, M. Tsubota and M. Fujiki, Macromolecules, 2003, 36, 3797 CrossRef CAS; (c) W. Wang, M. Fujiki and K. Nomura, J. Am. Chem. Soc., 2005, 127, 4582 CrossRef CAS PubMed; (d) K. Nomura, K. Itagaki and M. Fujiki, Macromolecules, 2005, 38, 2053 CrossRef CAS; (e) K. Nomura, K. Itagaki and M. Fujiki, Macromolecules, 2005, 38, 8121 CrossRef CAS; (f) F. Z. Khan, K. Kakinuki and K. Nomura, Macromolecules, 2009, 42, 3767 CrossRef CAS; (g) K. Kakinuki, M. Fujiki and K. Nomura, Macromolecules, 2009, 42, 4585 CrossRef CAS; (h) W. Apisuk and K. Nomura, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1902 CrossRef CAS; (i) W. Zhao, Q. Yan, K. Tsutsumi and K. Nomura, Organometallics, 2016, 35, 1895 CrossRef CAS.
- (a) W. Wang, M. Fujiki and K. Nomura, Macromol. Rapid Commun., 2004, 25, 504 CrossRef CAS; (b) P. M. Gurubasavaraj and K. Nomura, Organometallics, 2010, 29, 3500 CrossRef CAS; (c) Y. Takii, P. M. Gurubasavaraj, S. Katao and K. Nomura, Organometallics, 2012, 31, 8237 CrossRef CAS; (d) K. Nomura, U. Tewasekson and Y. Takii, Organometallics, 2015, 34, 3272 CrossRef CAS.
- Synthesis of aryloxo-modified half-titanocenes supported on carbosilane dendrimers, see: (a) S. Arévalo, J. M. Benito, E. de Jesús, F. J. de la Mata, J. C. Flores and R. Gómez, J. Organomet. Chem., 2000, 602, 208 CrossRef; (b) S. Arévalo, E. de Jesús, F. J. de la Mata, J. C. Flores and R. Gómez, Organometallics, 2001, 20, 2583 CrossRef; (c) S. Arévalo, E. de Jesús, F. J. de la Mata, J. C. Flores, R. Gómez, M. P. Gómez-Sal, P. Ortega and S. Vigo, Organometallics, 2003, 22, 5109 CrossRef; (d) S. Arévalo, E. de Jesús, F. J. de la Mata, J. C. Flores, R. Gómez, M.-M. Rodrigo and S. Vigo, J. Organomet. Chem., 2005, 690, 4620 CrossRef . Ethylene polymerisation by half-titanocene supported on carbosilane dendrimer, [[CpTiCl2{O-2,6-(MeO)2C6H2-4-(CH2)3SiMe2(CH2)3}]2SiMe(CH2)3]4Si and [CpTiCl2{O-2-MeC6H3-6-(CH2)2SiMe2(CH2)3}]4Si in the presence of MAO cocatalyst. Latter complex showed the negligible activity.
- Synthesis of the other trinuclear half-titanocenes, tris-1,3,5-[Cp*Ti(X2)O]3C6H3 (X = Cl, Me). S. Arévalo, M. R. Bonillo, E. de Jesús, F. J. de la Mata, J. C. Flores, R. Gómez, P. Gómez-Sal and P. Ortega, J. Organomet. Chem., 2003, 681, 228 CrossRef.
- Examples for synthesis, structural analysis of aryloxo-modified half-titanocenes (related to this study),10i see: (a) P. Gómez-Sal, A. Martín, M. Mena, P. Royo and R. Serrano, J. Organomet. Chem., 1991, 419, 77 CrossRef; (b) K. Nomura, N. Naga, M. Miki, K. Yanagi and A. Imai, Organometallics, 1998, 17, 2152 CrossRef CAS; (c) K. Nomura, N. Naga, M. Miki and K. Yanagi, Macromolecules, 1998, 31, 7588 CrossRef CAS; (d) S. J. Sturla and S. L. Buchwald, Organometallics, 2002, 21, 739 CrossRef CAS; (e) K. Nomura and A. Fudo, Inorg. Chim. Acta, 2003, 345, 37 CrossRef CAS; (f) K. Nomura, A. Tanaka and S. Katao, J. Mol. Catal. A: Chem., 2006, 254, 197 CrossRef CAS.
- Additional experiments for reactions of Cp*TiCl3 or (1,2,4-Me3C5H2)TiCl3 with [(LiO-2,4-Me2C6H2)-6-CH2]3N, reaction of Cp*TiMe3 (generated in situ) with [(HO-2,4-tBu2C6H2)-6-CH2]3N, and additional VT 1H NMR spectra for [CpTiCl2{(O-2,4-Me2C6H2)-6-CH2}]3N (1) in toluene-d8, and 1H NMR spectra for [Cp′TiCl2{(O-2,4-Me2C6H2)-6-CH2}][Cp′TiCl{(O-2,4-Me2C6H2)-6-CH2}2]N [Cp′ = Cp* (2), 1,2,4-Me3C5H2 (3)], and [Cp′TiCl2{(O-2,4-R2C6H2)-6-CH2}]3N [R = Me, Cp′ = Cp (1); R = tBu, Cp′ = Cp (4), Cp* (5), tBuC5H4 (6), 1,2,4-Me3C5H2 (7)] upon heating in toluene-d8 at 80 °C (for 30 min) are shown in the ESI.†.
- L. Michalczyk, S. de Gala and J. W. Bruno, Organometallics, 2001, 20, 5547 CrossRef CAS.
- Structural reports for [CpTiCl2{(O-2,4-Me2C6H2)-6-CH2}]3N (1), [Cp′TiCl2{(O-2,4-tBu2C6H2)-6-CH2}]3N [Cp′ = Cp (4), Cp* (5), 1,2,4-Me3C5H2 (7)], [Cp′TiCl2{(O-2,4-Me2C6H2)-6-CH2}][Cp′TiCl{(O-2,4-Me2C6H2)-6-CH2}2]N [Cp′ = Cp* (2), 1,2,4-Me3C5H2 (3)], and [Cp*TiMe2{(O-2,4-tBu2C6H2)-6-CH2}]3N (9) are shown in the ESI.† CCDC numbers for complexes 1–5, 7 and 9 are CCDC 1549581–1549587, respectively.†.
- Additional ethylene polymerisation results including confirmation of reproducibility are shown in the ESI.†.
- CrystalClear: Data Collection and Processing Software, Rigaku Corporation, Tokyo 196-8666, Japan, 1998–2015 Search PubMed.
- CrysAlisPro: Data Collection and Processing Software, Rigaku Corporation, Tokyo 196-8666, Japan, 1998–2015 Search PubMed.
- (a) G. M. Sheldrick, SHELXT, Acta Crystallogr., Sect. A: Found. Adv., 2014, 70, C1437 Search PubMed; (b) A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, SIR92, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- Crystal Structure 4.2: Crystal Structure Analysis Package, Rigaku Corporation, Tokyo 196-8666, Japan, 2000–2015 Search PubMed.
- G. M. Sheldrick, SHELXL Version 2014/7, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: (i) Additional experiments and VT 1H NMR spectra of toluene-d8 solution of [Cp′TiCl2{(O-2,4-R2C6H2)-6-CH2}]3N [R = Me, Cp′ = Cp (1); R = tBu, Cp′ = Cp (4), Cp* (5), tBuC5H4 (6), 1,2,4-Me3C5H2 (7)], [Cp′TiCl2{(O-2,4-Me2C6H2)-6-CH2}][Cp′TiCl{(O-2,4-Me2C6H2)-6-CH2}2]N [Cp′ = Cp* (2), 1,2,4-Me3C5H2 (3)], additional ethylene polymerization results, and tables summarized in crystal collection parameters for 1–5 and 7–9, and their structures reports. CCDC 1549581–1549587. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra07581b |
| This journal is © The Royal Society of Chemistry 2017 |