
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcise synthesis of the pyruvic acid acetal containing pentasaccharide repeating unit of the cell wall O-antigen of Escherichia coli O156†
Anshupriya Si and
Anup Kumar Misra *
*
Bose Institute, Division of Molecular Medicine, P-1/12, C.I.T. Scheme VII M, Kolkata 700054, India. E-mail: akmisra69@gmail.com; Fax: +91-33-2355-3886
First published on 26th October 2017
Abstract
An elegant convergent synthetic strategy has been developed for the preparation of the 4,6-O-(R)-pyruvate acetal containing pentasaccharide repeating unit of the cell wall O-antigen of Escherichia coli O156 using stereoselective [2 + 3] block glycosylation. Stereoselective 1,2-cis glycosylation of the judiciously functionalized monosaccharide intermediates led to the formation of the desired pentasaccharide in satisfactory yield.
Introduction
Diarrhoeal epidemic due to bacterial infections is a serious health concern worldwide.1 A significant number of people are suffering from gastrointestinal infections in developing countries where adequate sanitation is lacking.2 Diarrhoea is one of the leading causes of death in children, elderly people and patients with insufficient immunity.3,4 Enteric infections and colitis are also commonly found in developed countries due to the consumption of uncooked or semi cooked foods.5 Enteropathogenic Escherichia coli (E. coli) strains are the predominant bacteria causing gastrointestinal disorder among the various microorganisms responsible for the diarrhoea.6 E. coli is a commensal organism present in the gastrointestinal microflora in human.7 In an immunocompromised state of the host they become virulent and cause a number of infections such as diarrhoea,8 urinary tract infection,9 hemorrhagic colitis (HC),10 hemolytic uremic syndrome (HUS),11 septicaemia12 etc. E. coli strains causing enteric infections are classified into several subclasses based on their mode of infections,13 which include (a) enteropathogenic E. coli (EPEC); (b) enterohemorrhagic E. coli (EHEC); (c) enteroinvasive E. coli (EIEC); (d) enterotoxigenic E. coli (ETEC); (e) enteroaggregative E. coli (EAEC); (f) diffusely adherent E. coli (DAEC). EHEC strains are also termed as Shiga-toxin producing E. coli (STEC) because of their capability to secrete Shiga-like toxin during the early stage of infections.14 E. coli strains belong to the subclass EHEC or STEC have been associated with a number of deadly gastrointestinal outbreaks in the developed countries.15 One of the frequently found EHEC strain is E. coli O157, which was the causative agent for several diarrhoeal outbreaks in Europe and America.16 Besides, E. coli O157, several E. coli strains associated with diarrheal diseases have been identified and characterized which belong to the EHEC category such as E. coli O4, O26, O55, O103, O111, O145, O150, O156 etc.17 The cell walls of the pathogenic bacteria are highly associated with their virulent properties. The O-polysaccharides or O-antigens, a component of bacterial cell wall endotoxins play vital role at the initial stage of bacterial infections to the host.18 Therefore, the O-antigen of the bacterial cell wall is useful tool for the development of therapeutics for the eradication of the bacterial infections. Conventionally, several polysaccharide vaccines have been developed in the past using cell wall polysaccharides of pathogenic bacterial strains.19 Later the polysaccharide vaccines have been replaced by more efficacious glycoconjugate vaccines based on the cell wall O-antigens.20 Duan et al.21 reported the structure of the pyruvic acid acetal containing pentasaccharide repeating unit of the cell wall O-antigen of E. coli O156, which belongs to STEC class and responsible for a number of EHEC associated diseases in humans. Therefore, it would be pertinent to develop vaccine candidates against E. coli O156 using the O-antigen. However, isolation of polysaccharides from the natural bacterial sources is quite troublesome and suffers from a number of shortcomings such as, handling of live bacterial strain, cannot produce substantial quantities of polysaccharides at a time, lack of adequate purity and free from biological impurities, lack of homogeneity in the isolated polysaccharide fragments etc. Therefore, it is preferable to develop concise synthetic strategies for the chemical synthesis of the oligosaccharide repeating units with precise structures devoid of above mentioned shortcomings.22 Recently, glycoconjugate vaccines developed using synthetic oligosaccharides have been proved to be equally or better immunogenic than the conventional polysaccharide conjugate vaccines.23 The synthetic oligosaccharides can be conveniently functionalized according to the requirement of the conjugation with appropriate proteins to furnish glycoconjugates with higher better homogeneity. In this context, it was decided to develop a convergent synthetic strategy for the synthesis of the pyruvic acid acetal containing pentasaccharide repeating unit of the O-antigen of E. coli O156 as its 2-aminoethyl glycoside. The 2-aminoethyl group at the reducing end of the pentasaccharide could be useful in conjugating the glycan moiety with an appropriate protein or aglycon to prepare glycoconjugate derivatives. A concise chemical synthesis of the pentasaccharide repeating unit of the O-antigen of E. coli O156 is reported herein.Results and discussion
The target pentasaccharide containing a pyruvic acid acetal has been synthesized as its 2-aminoethyl glycoside using a stereoselective convergent [2 + 3] block glycosylation strategy. Presence of a number of 1,2-cis glycosidic linkages present pentasaccharide poses extra challenges for its synthesis. The key features of the synthetic strategy are (a) stereoselective 1,2-cis glycosylation using either thioglycoside or glycosyl trichloroacetimidate derivatives as glycosyl donors; (b) use of perchloric acid supported over silica gel (HClO4–SiO2)24 as a solid acid catalyst; (c) activation of thioglycosides using a combination of N-iodosuccinimide (NIS) and HClO4–SiO2;25 (d) activation of glycosyl trichloroacetimidate using HClO4–SiO2;26 (e) preparation of pyruvate acetal using pyruvate dithioacetal;27 (f) achievements of 1,2-cis glycosylated products in high yield. A set of suitably functionalized monosaccharide intermediates 2,28 3,29 4,30 5 (ref. 31) and 6 (ref. 32) were prepared from the commercially available reducing sugars using literature reported reaction methodologies (Fig. 1).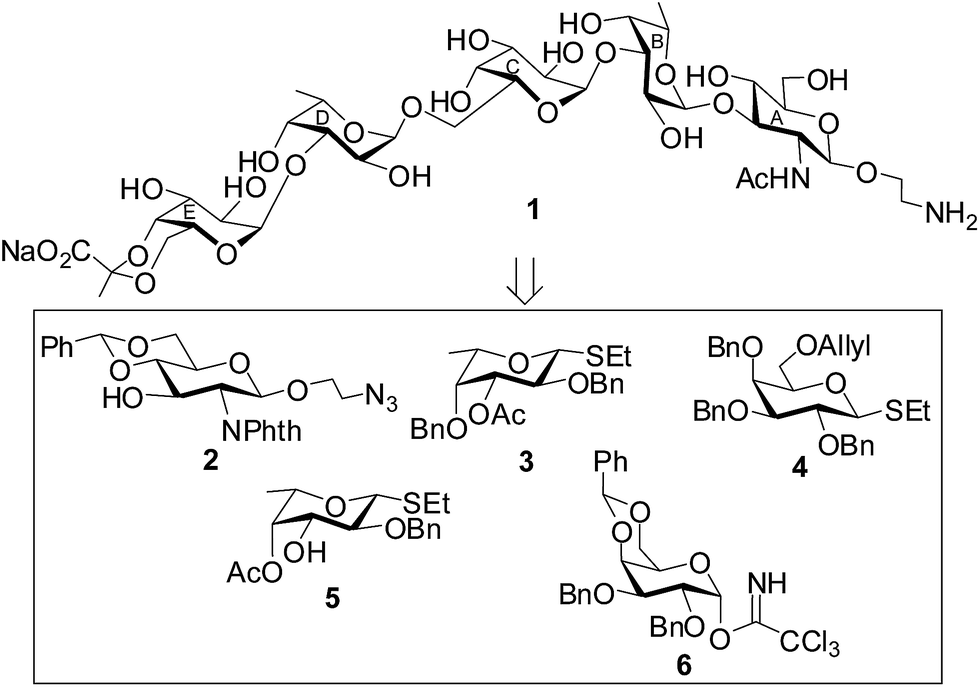 | ||
| Fig. 1 Structure of the synthesized pentasaccharide containing pyruvic acid acetal and its synthetic intermediates. | ||
Stereoselective 1,2-cis-glycosylation of compound 2 and L-fucosyl thioglycoside 3 in the presence of a combination25 of NIS and HClO4–SiO2 in a mixed solvent of CH2Cl2–Et2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) furnished disaccharide derivative 7 in 70% yield, which was characterized by its NMR spectral analysis [signals at δ 5.34 (d, J = 8.5 Hz, H-1A), 4.84 (d, J = 3.5 Hz, H-1B) in 1H NMR and δ 99.1 (C-1A), 98.7 (C-1B) in 13C NMR spectra]. Treatment of compound 7 with sodium methoxide33 resulted in the formation of the disaccharide acceptor 8 after de-O-acetylation in 93% yield. Stereoselective 1,2-cis glycosylation of compound 8 with D-galactosyl thioglycoside 4 using NIS and HClO4–SiO2 combination25 in a mixed solvent of CH2Cl2–Et2O (1:3) furnished trisaccharide derivative 9 in 76% yield together with the minor quantity (∼5%) of its 1,2-trans glycoside, which was separated by column chromatography. The newly formed glycosyl linkage in compound 9 was confirmed by its NMR spectral analysis [signals at δ 5.33 (d, J = 8.5 Hz, H-1A), 4.85 (d, J = 3.5 Hz, H-1B), 4.65 (d, J = 3.0 Hz, H-1C) in 1H NMR and δ 99.5 (C-1C), 99.0 (C-1A), 98.4 (C-1B) in 13C NMR spectra]. Treatment of compound 9 with acetic anhydride in the presence of HClO4–SiO2 resulted in the formation of compound 10 in 88% yield by direct conversion of the benzylidene acetal into di-O-acetylated derivative.34 The allyl ether of compound 10 was removed by the treatment with palladium chloride35 to furnish trisaccharide acceptor 11 in 72% yield. NMR spectral analysis of compound 11 supported its formation [signals at δ 5.44 (d, J = 8.0 Hz, H-1A), 4.50 (d, J = 3.0 Hz, H-1B), 4.30 (d, J = 3.0 Hz, H-1C) in 1H NMR and δ 101.3 (C-1C), 98.6 (C-1A), 95.9 (C-1B) in 13C NMR spectra] (Scheme 1).
:5) furnished disaccharide derivative 7 in 70% yield, which was characterized by its NMR spectral analysis [signals at δ 5.34 (d, J = 8.5 Hz, H-1A), 4.84 (d, J = 3.5 Hz, H-1B) in 1H NMR and δ 99.1 (C-1A), 98.7 (C-1B) in 13C NMR spectra]. Treatment of compound 7 with sodium methoxide33 resulted in the formation of the disaccharide acceptor 8 after de-O-acetylation in 93% yield. Stereoselective 1,2-cis glycosylation of compound 8 with D-galactosyl thioglycoside 4 using NIS and HClO4–SiO2 combination25 in a mixed solvent of CH2Cl2–Et2O (1:3) furnished trisaccharide derivative 9 in 76% yield together with the minor quantity (∼5%) of its 1,2-trans glycoside, which was separated by column chromatography. The newly formed glycosyl linkage in compound 9 was confirmed by its NMR spectral analysis [signals at δ 5.33 (d, J = 8.5 Hz, H-1A), 4.85 (d, J = 3.5 Hz, H-1B), 4.65 (d, J = 3.0 Hz, H-1C) in 1H NMR and δ 99.5 (C-1C), 99.0 (C-1A), 98.4 (C-1B) in 13C NMR spectra]. Treatment of compound 9 with acetic anhydride in the presence of HClO4–SiO2 resulted in the formation of compound 10 in 88% yield by direct conversion of the benzylidene acetal into di-O-acetylated derivative.34 The allyl ether of compound 10 was removed by the treatment with palladium chloride35 to furnish trisaccharide acceptor 11 in 72% yield. NMR spectral analysis of compound 11 supported its formation [signals at δ 5.44 (d, J = 8.0 Hz, H-1A), 4.50 (d, J = 3.0 Hz, H-1B), 4.30 (d, J = 3.0 Hz, H-1C) in 1H NMR and δ 101.3 (C-1C), 98.6 (C-1A), 95.9 (C-1B) in 13C NMR spectra] (Scheme 1).
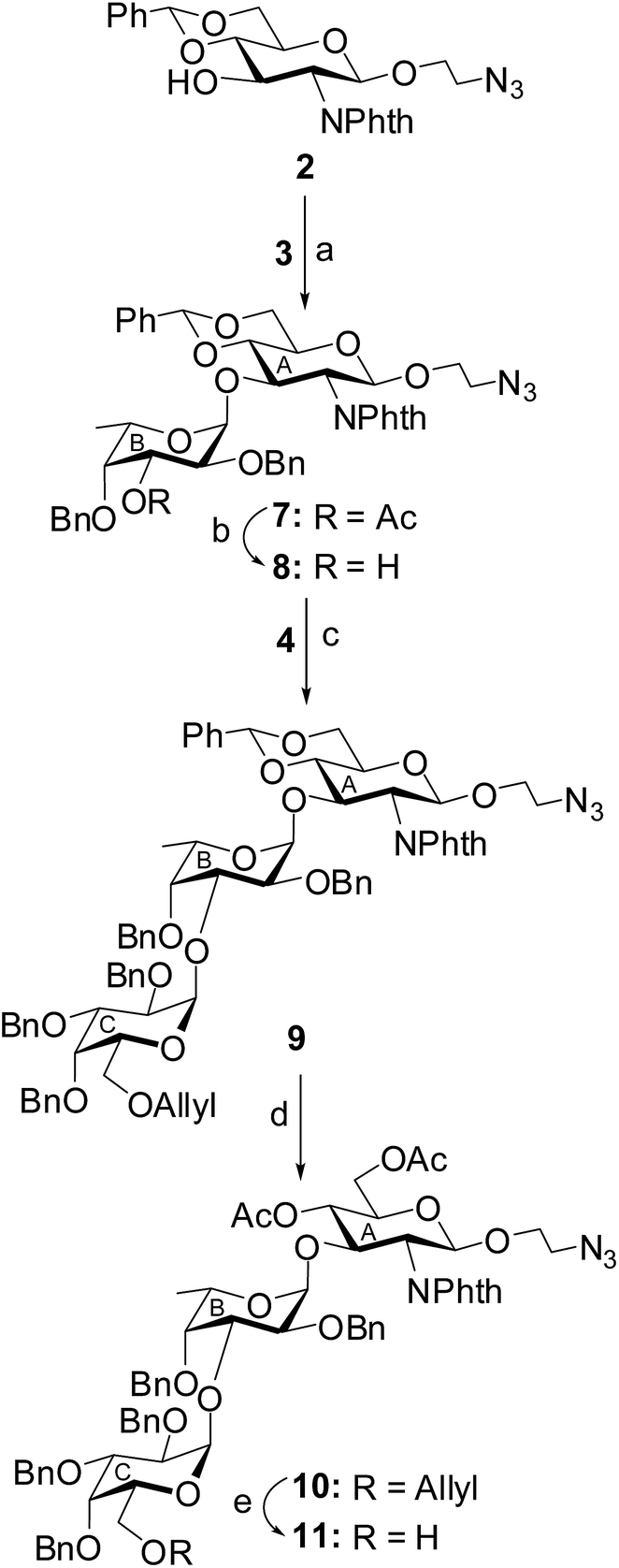 | ||
| Scheme 1 Reagents: (a) NIS, HClO4–SiO2, MS 4 Å, CH2Cl2–Et2O (1:5, v/v), 0 °C, 15 min, 70%; (b) CH3ONa, CH3OH, room temperature, 3 h, 93%; (c) NIS, HClO4–SiO2, MS 4 Å, CH2Cl2–Et2O (1:3, v/v), −10 °C, 25 min, 76%; (d) acetic anhydride, HClO4–SiO2, room temperature, 20 min, 88%; (e) PdCl2, CH3OH, 0 °C, 1 h, 76%. | ||
In another experiment, stereoselective orthogonal 1,2-cis-glycosylation of L-fucosyl thioglycoside acceptor 5 with D-galactosyl trichloroacetimidate donor 6 in the presence of HClO4–SiO2 (ref. 26) in CH2Cl2–Et2O (1:3) resulted in the formation of compound 12 in 74% yield. The anomeric thioether present in the acceptor was unaffected under the reaction condition maintaining the orthogonality36 of the reaction. The formation the 1,2-cis-glycosylated disaccharide thioglycoside derivative 12 was confirmed from its NMR spectral analysis [signals at δ 5.44 (d, J = 3.5 Hz, H-1E), 4.44 (d, J = 9.5 Hz, H-1D) in 1H NMR and δ 100.0 (C-1E), 85.2 (C-1D) in 13C NMR spectra] (Scheme 2).
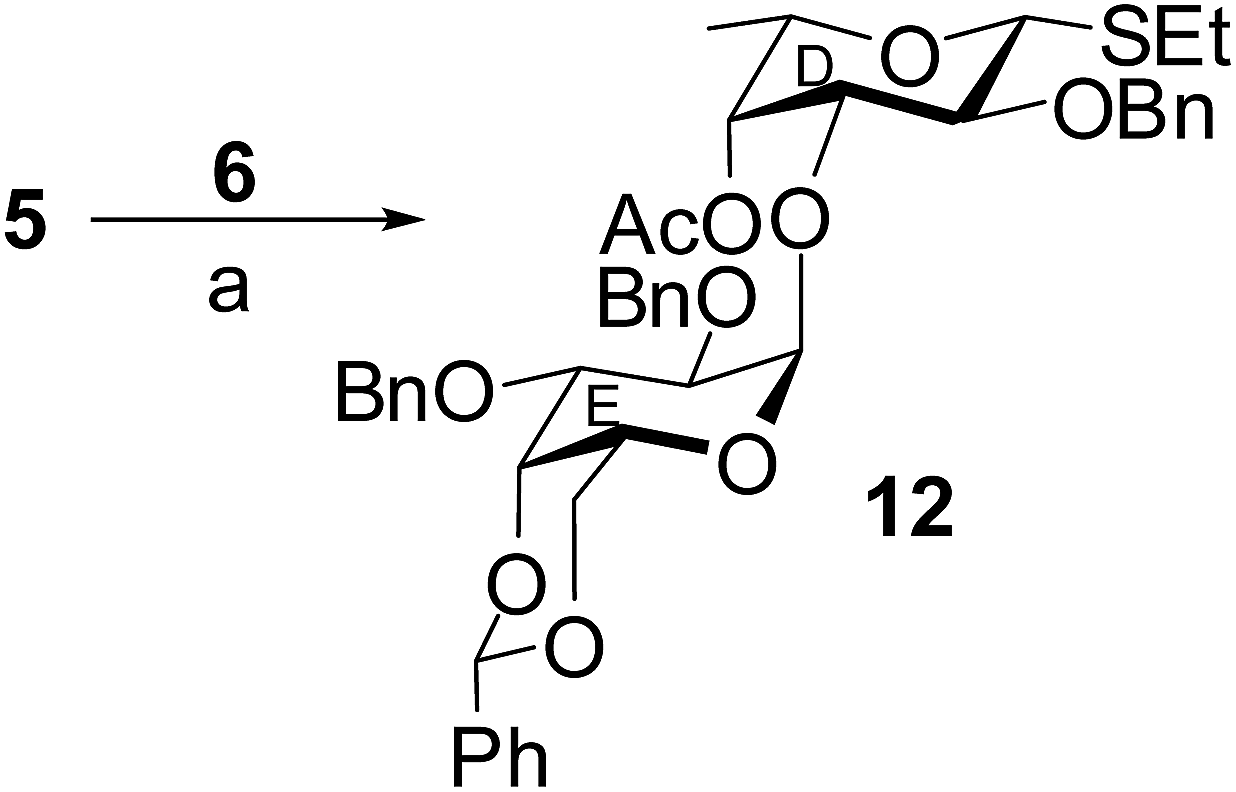 | ||
| Scheme 2 Reagents: (a) HClO4–SiO2, MS 4 Å, CH2Cl2–Et2O (1:3, v/v), −10 °C, 2 h, 74%. | ||
The disaccharide thioglycoside donor 12 and trisaccharide acceptor 11 was allowed to couple in a 1,2-cis stereoselective manner in the presence of a combination25 of NIS and HClO4–SiO2 in CH2Cl2–Et2O (1:4) to furnish the pentasaccharide derivative 13 in 72% yield. The formation of compound 13 was confirmed from its NMR spectral analysis [signals at δ 5.57 (d, J = 3.5 Hz, H-1D), 5.42 (d, J = 7.5 Hz, H-1A), 4.86 (d, J = 3.0 Hz, H-1E), 4.65 (d, J = 3.0 Hz, H-1B), 4.23 (d, J = 3.0 Hz, H-1C) in 1H NMR and δ 102.4 (JC-1/H-1 = 168 Hz, C-1C), 99.03 (JC-1/H-1 = 170 Hz, C-1D), 98.3 (JC-1/H-1 = 168 Hz, C-1E), 98.2 (JC-1/H-1 = 170 Hz, C-1B), 97.5 (JC-1/H-1 = 158 Hz, C-1A) 13C NMR spectra]. The presence of four α-glycosyl linkages and one β-glycosyl linkage in the molecule was also unambiguously confirmed from the C-1/H-1 coupling constants (JC-1/H-1) of the monosaccharide moieties in 1H coupled 13C NMR spectrum.37,38 The benzylidene acetal in the terminal D-galactosyl moiety in compound 13 was smoothly removed by the treatment34 with HClO4–SiO2 at room temperature to give the diol derivative 14 in 82% yield. Compound 14 was allowed to react with methyl 2,2-di(ethylthio)propionate39 in the presence of a combination of NIS and triflic acid (TfOH)27 to furnish compound 15 in 68% yield containing the desired 4,6-(R)-pyruvate acetal in the D-galactosyl moiety, which was confirmed from its NMR spectral analysis [signals at δ 5.55 (d, J = 3.5 Hz, H-1D), 5.42 (d, J = 8.0 Hz, H-1A), 4.89 (d, J = 3.0 Hz, H-1E), 4.67 (d, J = 3.5 Hz, H-1B), 4.28 (d, J = 3.5 Hz, H-1C) in 1H NMR and δ 102.4 (C-1C), 99.08 (C-1D), 98.8 (CCH3), 98.3 (C-1E), 98.1 (C-1B), 97.5 (C-1A), 26.0 (CCH3) in 13C NMR spectra]. Appearance of the methyl carbon of the pyruvate acetal at δ 26 ppm in 13C NMR spectrum confirmed the formation of the 4,6-(R)-pyruvate acetal.40 Finally, compound 15 was subjected to a sequence of reactions involving (a) treatment with ethylenediamine to remove the N-phthaloyl group;41 (b) acetylation of the newly generated amine; (c) removal of the benzyl ethers using hydrogenolysis over Pd(OH)2–C;42 (d) removal of the O-acetyl group followed by hydrolysis of the methyl ester in the pyruvate moiety using sodium methoxide to furnish the desired pentasaccharide 1 in 54% over all yield. The NMR spectral analysis of compound 1 supported the formation of the desired compound [signals at δ 5.08 (d, J = 3.5 Hz, H-1E), 5.05 (d, J = 4.0 Hz, H-1C), 4.89 (d, J = 3.5 Hz, H-1B), 4.78 (d, J = 4.0 Hz, H-1D), 4.41 (d, J = 8.5 Hz, H-1A) in 1H NMR and δ 101.5 (C-1A), 100.7 (2C, C-1C, C-1E), 100.6 (CCOOH), 99.8 (C-1D), 98.4 (C-1B) in 13C NMR spectra] (Scheme 3).
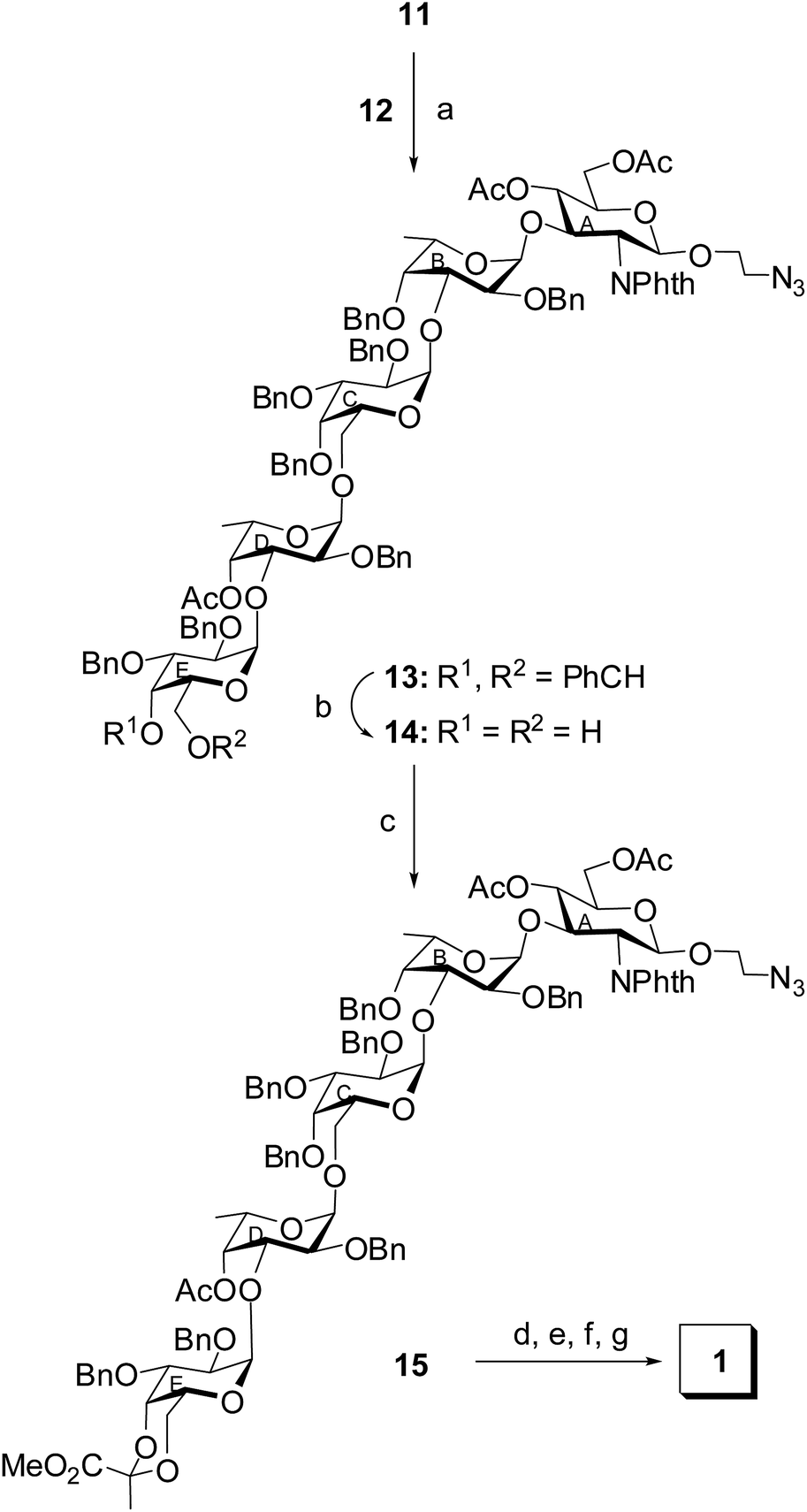 | ||
| Scheme 3 Reagents: (a) NIS, HClO4–SiO2, MS 4 Å, CH2Cl2–Et2O (1:4, v/v), −10 °C, 30 min, 72%; (b) HClO4–SiO2, CH3CN, room temperature, 20 min, 82%; (c) methyl 2,2-di(ethylthio)propionate, NIS, TfOH, 0 °C, 30 min, 68%; (d) (CH2NH2)2, nBuOH, 90 °C, 10 h; (e) acetic anhydride, pyridine, room temperature, 1 h; (f) H2, 20% Pd(OH)2–C, CH3OH, room temperature, 15 h; (g) CH3ONa, CH3OH, room temperature, 3 h, then H2O, 8 h, 54% in four steps. | ||
Experimental
General methods
All reactions were monitored by thin layer chromatography over silica gel coated TLC plates. The spots on TLC were visualized by warming ceric sulphate (2% Ce(SO4)2 in 2 N H2SO4) sprayed plates in hot plate. Silica gel 230–400 mesh was used for column chromatography. NMR spectra were recorded on Bruker Avance 500 MHz using CDCl3 as solvent and TMS as internal reference unless stated otherwise. Chemical shift value is expressed in δ ppm. The complete assignment of proton and carbon spectra was carried out by using a standard set of NMR experiments, e.g. 1H NMR, 13C NMR, 13C DEPT 135, 2D COSY and 2D HSQC etc. MALDI-MS were recorded on a Bruker Daltonics mass spectrometer. Optical rotations were recorded in a Jasco P-2000 spectrometer. Commercially available grades of organic solvents of adequate purity are used in all reactions. HClO4–SiO2 was prepared following the procedure reported by Chakraborti et al.242-Azidoethyl O-(3-O-acetyl-2,4-di-O-benzyl-α-L-fucopyranosyl)-(1 → 3)-2-N-phthalimido-4,6-O-benzylidene-2-deoxy-β-D-glucopyranoside (7)
To a solution of compound 2 (1.5 g, 3.22 mmol) and compound 3 (1.5 g, 3.48 mmol) in anhydrous CH2Cl2–Et2O (18 mL, 1:5 v/v) was added MS 4 Å (2 g) and it was cooled to 0 °C under argon. To the cold reaction mixture were added NIS (945 mg, 4.19 mmol) and HClO4–SiO2 (40 mg) and it was allowed to stir at same temperature for 15 min. The reaction mixture was filtered and washed with CH2Cl2 (100 mL). The combined filtrate was successively washed with 5% Na2S2O3 (50 mL), satd aq. NaHCO3 (50 mL) and water (50 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified over SiO2 using hexane–EtOAc (2:1) as eluant to furnish pure compound 7 (1.9 g, 70%). Colorless oil; [α]25D −50 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.68–6.94 (m, 15H, Ar-H), 5.56 (s, 1H, PhCH), 5.34 (d, J = 8.5 Hz, 1H, H-1A), 4.97 (dd, J = 10.5, 3.0 Hz, 1H, H-3B), 4.84 (d, J = 3.5 Hz, 1H, H-1B), 4.72 (t, J = 9.0 Hz, 1H, H-3A), 4.46–4.40 (m, 4H, 2PhCH, H-2A, H-6aA), 4.14–4.11 (m, 2H, PhCH, H-5B), 4.01 (m, 1H, OCH), 3.97 (d, J = 11.5 Hz, 1H, PhCH), 3.87 (t, J = 10.0 Hz, 1H, H-6bA), 3.76 (t, J = 9.0 Hz, 1H, H-4A), 3.74–3.70 (m, 2H, H-2C, H-5A), 3.68–3.63 (m, 1H, OCH), 3.61 (d, J = 2.0 Hz, 1H, H-4B), 3.41–3.35 (m, 1H, NCH), 3.23–3.19 (m, 1H, NCH), 1.64 (s, 3H, COCH3), 0.74–0.73 (m, 3H, CCH3); 13C NMR (125 MHz, CDCl3): δ 170.1 (COCH3), 168.2, 167.4 (PhthCO), 138.7–123.5 (Ar-C), 101.8 (PhCH), 99.1 (C-1A), 98.7 (C-1B), 81.6 (C-4A), 78.6 (C-4B), 77.3 (PhCH), 75.6 (C-2B), 75.5 (C-3A), 74.7 (C-3B), 73.5 (PhCH), 68.7 (OCH), 68.5 (C-6A), 66.6 (C-5A), 66.5 (C-5B), 55.6 (C-2A), 50.5 (NCH), 21.1 (COCH3), 16.3 (CCH3); MALDI-MS: 857.3 [M + Na]+; anal. calcd for C45H46N4O12 (834.86): C, 64.74; H, 5.55%; found: C, 64.60; H, 5.70%.
2-Azidoethyl O-(2,4-di-O-benzyl-α-L-fucopyranosyl)-(1 → 3)-2-N-phthalimido-4,6-O-benzylidene-2-deoxy-β-D-glucopyranoside (8)
A solution of compound 7 (1.7 g, 2.04 mmol) in 0.1 M CH3ONa in CH3OH (20 mL) was allowed to stir at room temperature for 3 h. The reaction mixture was neutralized with Dowex 50W X8 (H+) resin, filtered and concentrated under reduced pressure. The crude mass was passed through a short pad of SiO2 using hexane–EtOAc (1:1) as eluant to give pure compound 8 (1.5 g, 93%). White solid; mp 174–175 °C [EtOH]; [α]25D −97 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.75–6.97 (m, 15H, Ar-H), 5.55 (s, 1H, PhCH), 5.36 (d, J = 3.0 Hz, H-1B), 4.70 (t, J = 9.0 Hz, 1H, H-3A), 4.57 (d, J = 11.5 Hz, 1H, PhCH), 4.49 (d, J = 11.5 Hz, 1H, PhCH), 4.40 (m, 1H, H-6aA), 4.39 (t, J = 9.0 Hz, 1H, H-2A), 4.30 (d, J = 12.5 Hz, 1H, PhCH), 4.12–4.09 (m, 1H, H-5B), 4.01–3.95 (m, 1H, H-6bA), 3.87 (d, J = 10.0 Hz, 1H, PhCH), 3.85–3.79 (m, 2H, H-3B, OCH), 3.75–3.60 (m, 3H, H-4A, H-5A, OCH), 3.45 (d, J = 3.0 Hz, 1H, H-4B), 3.43 (s, 1H, H-2B), 3.73–3.33 (m, 1H, NCH), 3.21–3.17 (m, 1H, NCH), 0.87–0.85 (m, 3H, CCH3); 13C NMR (125 MHz, CDCl3): δ 168.2, 167.4 (PhthCO), 134.0–125.3 (Ar-C), 101.6 (PhCH), 99.0 (C-1A), 97.8 (C-1B), 81.6 (C-4A), 79.9 (C-4B), 78.6 (C-2B), 77.3 (PhCH), 76.2 (C-3A), 75.5 (PhCH), 70.1 (C-3B), 68.7 (OCH2), 68.6 (C-6A), 67.0 (C-5A), 66.4 (C-5B), 55.7 (C-2A), 50.5 (NCH2), 15.8 (CCH3); MALDI-MS: 815.3 [M + Na]+; anal. calcd for C43H44N4O11 (792.83): C, 65.14; H, 5.59%; found: C, 64.95; H, 5.75%.
2-Azidoethyl O-(6-O-allyl-2,3,4-tri-O-benzyl-α-D-galactopyranosyl)-(1 → 3)-(2,4-di-O-benzyl-α-L-fucopyranosyl)-(1 → 3)-2-N-phthalimido-4,6-O-benzylidene-2-deoxy-β-D-glucopyranoside (9)
A solution of compound 8 (1.4 g, 1.77 mmol), compound 4 (1.1 g, 2.06 mmol) and MS 4 Å (2 g) in anhydrous CH2Cl2–Et2O (15 mL, 1:3 v/v) was cooled to −10 °C under argon. NIS (560 mg, 2.5 mmol) and HClO4–SiO2 (20 mg) were added to the cold reaction mixture and it was allowed to stir at same temperature for 25 min. The reaction mixture was filtered and washed with CH2Cl2 (100 mL). The combined filtrate was successively washed with 5% Na2S2O3 (50 mL), satd aq. NaHCO3 (50 mL) and water (50 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified over SiO2 using hexane–EtOAc (3:1) as eluant to furnish pure compound 9 (1.7 g, 76%). Colorless oil; [α]25D +30 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.70–6.84 (m, 30H, Ar-H), 5.90–5.80 (m, 1H, OCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.53 (s, 1H, PhCH), 5.33 (d, J = 8.5 Hz, 1H, H-1A), 5.28–5.15 (m, 2H, CHCH2), 4.92 (d, J = 11.5 Hz, 1H, PhCH), 4.85 (d, J = 3.5 Hz, 1H, H-1B), 4.72 (d, J = 11.5 Hz, 1H, PhCH), 4.65 (d, J = 3.0 Hz, 1H, H-1C), 4.63–4.59 (m, 2H, H-3A, OCH2–CH), 4.56–4.51 (m, 5H, 5PhCH), 4.41–4.37 (m, 2H, H-6aA, OCH2–CH), 4.35 (t, J = 10.5 Hz, 1H, H-2A), 4.27 (d, J = 12.5 Hz, 1H, PhCH), 4.08–4.03 (m, 1H, H-5B), 4.02–3.92 (m, 3H, PhCH, H-3B, H-6bA), 3.91–3.80 (m, 2H, H-2B, OCH), 3.78–3.60 (m, 7H, H-2C, H-3C, H-4A, H-4C, H-5A, H-5C, OCH), 3.59–3.51 (m, 1H, H-6aC), 3.50 (s, 1H, H-4B), 3.49–3.41 (m, 1H, H-6bC), 3.38–3.31 (m, 1H, NCH), 0.85–0.84 (m, 3H, CCH3); 13C NMR (125 MHz, CDCl3): δ 167.8, 168.4 (PhthCO), 138.9–125.5 (Ar-C, CH2CH), 117.0 (CH2CH), 101.4 (PhCH), 99.5 (C-1C), 99.0 (C-1A), 98.4 (C-1B), 81.9 (C-4A), 81.6 (C-4B), 78.6 (C-4C), 75.8 (C-2B), 75.5 (PhCH), 74.9 (3C, C-2C, C-3C, C-5C), 73.1 (C-3A), 72.9 (PhCH), 72.7 (PhCH), 72.4 (PhCH), 72.3 (OCH2–CHCH2), 70.4 (C-3B), 69.4 (C-6C), 68.7 (OCH2), 68.5 (C-6A), 68.1 (C-5A), 67.5 (C-5B), 55.6 (C-2A), 50.5 (NCH2), 16.3 (CCH3); MALDI-MS: 1287.5 [M + Na]+; anal. calcd for C73H76N4O16 (1265.40): C, 69.29; H, 6.05%; found: C, 69.10; H, 6.20%.
CH2), 5.53 (s, 1H, PhCH), 5.33 (d, J = 8.5 Hz, 1H, H-1A), 5.28–5.15 (m, 2H, CHCH2), 4.92 (d, J = 11.5 Hz, 1H, PhCH), 4.85 (d, J = 3.5 Hz, 1H, H-1B), 4.72 (d, J = 11.5 Hz, 1H, PhCH), 4.65 (d, J = 3.0 Hz, 1H, H-1C), 4.63–4.59 (m, 2H, H-3A, OCH2–CH), 4.56–4.51 (m, 5H, 5PhCH), 4.41–4.37 (m, 2H, H-6aA, OCH2–CH), 4.35 (t, J = 10.5 Hz, 1H, H-2A), 4.27 (d, J = 12.5 Hz, 1H, PhCH), 4.08–4.03 (m, 1H, H-5B), 4.02–3.92 (m, 3H, PhCH, H-3B, H-6bA), 3.91–3.80 (m, 2H, H-2B, OCH), 3.78–3.60 (m, 7H, H-2C, H-3C, H-4A, H-4C, H-5A, H-5C, OCH), 3.59–3.51 (m, 1H, H-6aC), 3.50 (s, 1H, H-4B), 3.49–3.41 (m, 1H, H-6bC), 3.38–3.31 (m, 1H, NCH), 0.85–0.84 (m, 3H, CCH3); 13C NMR (125 MHz, CDCl3): δ 167.8, 168.4 (PhthCO), 138.9–125.5 (Ar-C, CH2CH), 117.0 (CH2CH), 101.4 (PhCH), 99.5 (C-1C), 99.0 (C-1A), 98.4 (C-1B), 81.9 (C-4A), 81.6 (C-4B), 78.6 (C-4C), 75.8 (C-2B), 75.5 (PhCH), 74.9 (3C, C-2C, C-3C, C-5C), 73.1 (C-3A), 72.9 (PhCH), 72.7 (PhCH), 72.4 (PhCH), 72.3 (OCH2–CHCH2), 70.4 (C-3B), 69.4 (C-6C), 68.7 (OCH2), 68.5 (C-6A), 68.1 (C-5A), 67.5 (C-5B), 55.6 (C-2A), 50.5 (NCH2), 16.3 (CCH3); MALDI-MS: 1287.5 [M + Na]+; anal. calcd for C73H76N4O16 (1265.40): C, 69.29; H, 6.05%; found: C, 69.10; H, 6.20%.
2-Azidoethyl O-(2,3,4-tri-O-benzyl-α-D-galactopyranosyl)-(1 → 3)-(2,4-di-O-benzyl-α-L-fucopyranosyl)-(1 → 3)-4,6-di-O-acetyl-2-N-phthalimido-2-deoxy-β-D-glucopyranoside (11)
To a solution of compound 9 (1.7 g, 1.34 mmol) in acetic anhydride (5 mL) was added HClO4–SiO2 (200 mg) and the reaction mixture was stirred at room temperature for 20 min. The reaction mixture was filtered and washed with EtOAc (50 mL). The combined filtrate was concentrated and co-evaporated with toluene (3 × 20 mL) under reduced pressure. The crude product was passed through a short pad of SiO2 using hexane–EtOAc (2:1) as eluant to give pure compound 10 (1.49 g, 88%). To a solution of compound 10 (1.4 g, 1.11 mmol) in dry CH3OH (25 mL) was added PdCl2 (100 mg, 0.56 mmol) and the reaction mixture was stirred at 0 °C for 1 h. Then the reaction mixture was filtered through a Celite bed and washed with CH3OH (50 mL). The combined filtrate was concentrated under reduced pressure and the crude product was purified over SiO2 using hexane–EtOAc (1:1) as eluant to give pure compound 11 (980 mg, 72%). Yellowish solid; mp 154–155 °C [EtOH]; [α]25D +100 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.71–6.95 (m, 25H, Ar-H), 5.44 (d, J = 8.0 Hz, 1H, H-1A), 5.03 (d, J = 10.0 Hz, 1H, H-4A), 4.99 (d, J = 11.5 Hz, 1H, PhCH), 4.75 (d, J = 11.5 Hz, 1H, PhCH), 4.73 (d, J = 11.5 Hz, 1H, PhCH), 4.63 (d, J = 11.5 Hz, 1H, PhCH), 4.60 (d, J = 11.5 Hz, 1H, PhCH), 4.53 (d, J = 12.0 Hz, 1H, PhCH), 4.50 (d, J = 3.0 Hz, 1H, H-1B), 4.42 (d, J = 12.0 Hz, 1H, PhCH), 4.40 (d, J = 11.5 Hz, 1H, PhCH), 4.30 (d, J = 3.0 Hz, 1H, H-1C), 4.28–4.25 (m, 2H, H-2A, H-4C), 4.20–4.01 (m, 6H, 2PhCH, H-3A, H-6abA, OCH), 3.98–3.90 (m, 1H, H-5B), 3.85–3.78 (m, 2H, H-2B, H-3C), 3.71–3.69 (m, 3H, H-5A, H-5C, OCH), 3.68–3.58 (m, 2H, H-2C, H-3B), 3.55–3.39 (m, 4H, H-4B, H-6abC, NCH), 3.20–3.15 (m, 1H, NCH), 2.1, 1.9 (s, 6H, 2COCH3), 1.01–1.00 (m, 3H, CCH3); 13C NMR (125 MHz, CDCl3): δ 170.8, 169.7 (COCH3), 168.2, 167.4 (PhthCO), 138.6–122.3 (Ar-C), 101.3 (C-1C), 98.6 (C-1A), 95.9 (C-1B), 80.9 (C-4B), 78.7 (C-4C), 78.4 (C-2B), 76.4 (C-2C), 76.3 (C-3C), 75.8 (PhCH), 75.5 (C-5C), 74.3 (PhCH), 73.6 (PhCH), 72.9 (PhCH), 72.7 (PhCH), 72.5 (C-3A), 72.1 (C-3B), 71.2 (C-5A), 71.1 (C-4A), 68.6 (OCH2), 68.4 (C-5B), 54.8 (C-2A), 50.4 (NCH2), 21.2, 20.9 (2COCH3), 15.7 (CCH3); MALDI-MS: 1243.4 [M + Na]+; anal. calcd for C67H72N4O18 (1221.30): C, 65.89; H, 5.94%; found: C, 65.74; H, 6.06%.
Ethyl (2,3-di-O-benzyl-4,6-O-benzylidene-α-D-galactopyranosyl)-(1 → 3)-4-O-acetyl-2-O-benzyl-1-thio-β-L-fucopyranoside (12)
To a solution of compound 5 (500 mg, 1.47 mmol) and compound 6 (1.1 g, 1.86 mmol) in anhydrous CH2Cl2–Et2O (10 mL, 1:3 v/v) was added MS 4 Å (4 g) and the reaction mixture was allowed to stir at room temperature for 10 min under argon then cooled to −10 °C. To the cooled reaction mixture was added HClO4–SiO2 (50 mg) and it was allowed to stir at the same temperature for 2 h. The reaction was quenched with Et3N (0.1 mL), filtered and washed with CH2Cl2 (100 mL). The organic layer was washed with satd aq. NaHCO3 (50 mL) and water (50 mL), dried (Na2SO4) and concentrated. The crude product was purified over SiO2 using hexane–EtOAc (2:1) as eluant to give pure compound 12 (840 mg, 74%). White solid; 109–110 °C [EtOH]; [α]25D +44.3 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.55–7.12 (m, 20H, Ar-H), 5.52 (s, 1H, PhCH), 5.44 (d, J = 3.5 Hz, 1H, H-1E), 5.12 (d, J = 3.5 Hz, 1H, H-4D), 4.90 (d, J = 12.0 Hz, 1H, PhCH), 4.83–4.71 (4d, J = 12 Hz, 4H, 4PhCH), 4.57 (d, 1H, J = 12.5 Hz, PhCH), 4.44 (d, J = 9.5 Hz, 1H, H-1D), 4.27 (d, J = 2.5 Hz, 1H, H-4E), 4.25 (d, J = 12.5 Hz, 1H, H-6aE), 4.12 (dd, J = 9.0, 3.0 Hz, 1H, H-2E), 4.09 (d, J = 11.5 Hz, H-6bE), 3.98–3.91 (m, 2H, H-3D, H-3E), 3.86 (s, 1H, H-5E), 3.78–3.73 (m, 1H, H-5D), 3.71 (t, J = 8.5 Hz, 1H each, H-2D), 2.85–2.70 (m, 2H, SCH2CH3), 2.06 (s, 3H, COCH3), 1.35 (t, J = 7.5 Hz, 3H, SCH2CH3), 1.19–1.74 (m, 3H, CCH3); 13C NMR (125 MHz, CDCl3): δ 171.3 (COCH3), 138.6–126.4 (Ar-C), 100.9 (PhCH), 100.0 (C-1E), 85.2 (C-1D), 78.3 (C-2D), 77.6 (C-3D), 75.6 (C-3E), 75.1 (PhCH), 74.8 (C-2E), 74.5 (C-4E), 73.8 (PhCH), 73.0 (C-5D), 72.9 (C-4D), 71.4 (PhCH), 69.6 (PhCH), 68.9 (C-6E), 63.4 (C-5E), 25.6 (SCH2CH3), 22.7 (COCH3), 16.6 (CCH3), 16.1 (SCH2CH3); MALDI-MS: 793.3 [M + Na]+; anal. calcd for C44H50O10S (770.92): C, 68.55; H, 6.54%; found: C, 68.40; H, 6.70%.
2-Azidoethyl O-(2,3-di-O-benzyl-4,6-O-benzylidene-α-D-galactopyranosyl)-(1 → 3)-(4-O-acetyl-2-O-benzyl-α-L-fucopyranosyl)-(1 → 6)-(2,3,4-tri-O-benzyl-α-D-galactopyranosyl)-(1 → 3)-(2,4-di-O-benzyl-α-L-fucopyranosyl)-(1 → 3)-4,6-di-O-acetyl-2-N-phthalimido-2-deoxy-β-D-glucopyranoside (13)
A solution of compound 11 (900 mg, 0.72 mmol), compound 12 (660 mg, 0.86 mmol) and MS 4 Å (1 g) in anhydrous CH2Cl2–Et2O (15 mL, 1:4 v/v) was cooled to −10 °C under argon. NIS (230 mg, 1.03 mmol) and HClO4–SiO2 (10 mg) were added to the cold reaction mixture and it was allowed to stir at same temperature for 30 min. The reaction mixture was filtered and washed with CH2Cl2 (50 mL). The combined filtrate was successively washed with 5% Na2S2O3 (25 mL), satd aq. NaHCO3 (25 mL) and water (25 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified over SiO2 using hexane–EtOAc (3:2) as eluant to give pure compound 13 (1.0 g, 72%). White solid; mp 167–168 °C [EtOH]; [α]25D +46 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.92–6.87 (m, 45H, Ar-H), 5.57 (d, J = 3.5 Hz, 1H, H-1D), 5.50 (s, 1H, PhCH), 5.42 (d, J = 7.5 Hz, 1H, H-1A), 5.12 (d, J = 12.0 Hz, 1H, PhCH), 5.02 (d, J = 3.0 Hz, 1H, H-4D), 4.95 (t, J = 9.0 Hz each, 1H, H-4A), 4.86 (d, J = 3.0 Hz, 1H, H-1E), 4.65 (d, J = 3.0 Hz, 1H, H-1B), 4.74–4.42 (m, 11H, 9PhCH, H-6abA), 4.38–4.03 (m, 13H, H-2A, H-2D, H-3A, H-3D, H-4E, H-6aE, H-6bE, 6PhCH), 4.23 (d, J = 3.0 Hz, 1H, H-1C), 4.09–3.98 (m, 1H, OCH), 3.95–3.78 (m, 10H, H-2B, H-2E, H-3B, H-3C, H-3E, H-4C, H-5B, H-5C, H-5D, H-6aC), 3.79–3.63 (m, 2H, H-5A, OCH), 3.58–3.50 (m, 2H, H-2C, H-6bC), 3.41–3.33 (m, 2H, H-4B, NCH), 3.25–3.15 (m, 2H, H-5E, NCH), 2.08, 2.02, 1.90 (3s, 9H, 3COCH3), 0.92–0.98 (m, 6H, 2CCH3); 13C NMR (125 MHz, CDCl3): δ 171.3, 170.8, 169.8 (3COCH3), 168.2, 167.4 (PhthCO), 139.2–122.7 (Ar-C), 102.4 (C-1C), 101.1 (PhCH), 99.03 (C-1D), 98.3 (C-1E), 98.2 (C-1B), 97.5 (C-1A), 81.9 (C-4B), 79.4 (C-4C), 79.0 (C-2B), 76.3 (2C, C-2C, C-2D), 75.9 (PhCH), 75.3 (C-3C), 75.1 (C-5C), 74.9 (C-4E), 74.7 (C-3D), 74.5 (2C, C-2E, C-3E), 73.3 (C-3B), 72.8 (PhCH), 72.7 (PhCH), 72.5 (PhCH), 72.1 (C-3A), 71.9 (PhCH), 71.8 (C-4D), 71.4 (PhCH), 71.1 (C-5D), 69.6 (C-6E), 69.3 (C-4A), 68.6 (OCH2), 68.3 (C-5A), 66.2 (C-6C), 64.9 (C-5E), 63.3 (C-5B), 62.3 (C-6A), 54.8 (C-2A), 50.4 (NCH2), 21.3, 20.9 (2C) (3COCH3), 15.9, 15.8 (2CCH3); MALDI-MS: 1951.8 [M + Na]+; anal. calcd for C109H116N4O28 (1930.09): C, 67.83; H, 6.06%; found: C, 67.70; H, 6.20%.
2-Azidoethyl O-(2,3-di-O-benzyl-4,6-O-[(R)-(1-carboxymethyl)ethylidene]-α-D-galactopyranosyl)-(1 → 3)-(4-O-acetyl-2-O-benzyl-α-L-fucopyranosyl)-(1 → 6)-(2,3,4-tri-O-benzyl-α-D-galactopyranosyl)-(1 → 3)-(2,4-di-O-benzyl-α-L-fucopyranosyl)-(1 → 3)-4,6-di-O-acetyl-2-N-phthalimido-2-deoxy-β-D-glucopyranoside (15)
To a solution of compound 13 (900 mg, 0.47 mmol) in CH3CN (15 mL) was added HClO4–SiO2 (100 mg) and it was stirred at room temperature for 20 min. The reaction mixture was filtered and washed with EtOAc (50 mL). The solvents were removed under reduced pressure and the crude product was purified over SiO2 using hexane–EtOAc (1:1) as eluant to give pure compound 14 (705 mg, 82%). To a solution of compound 14 (500 mg, 0.27 mmol) in dry CH3CN (15 mL) was added methyl 2,2-di(ethylthio)propionate (170 mg, 0.81 mmol) and NIS (460 mg, 2.03 mmol) and the mixture was stirred at 0 °C under argon for 10 min. To the cold reaction mixture was added TfOH (15 μL) and it was stirred at 0 °C for a further 20 min. After completion of the reaction (TLC), 10% aq. Na2S2O3 (50 mL) was added to the reaction mixture and the solvents were removed and the crude mass was diluted with CH2Cl2 (50 mL). The organic solution was washed with satd aq. NaHCO3 (50 mL) and water (50 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified over SiO2 using hexane–EtOAc (3:1) to give the pure compound 15 (360 mg, 68%). White solid; mp 144–145 °C [EtOH]; [α]25D +22.8 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.92–6.93 (m, 40H, Ar-H), 5.55 (d, J = 3.5 Hz, 1H, H-1D), 5.42 (d, J = 8.0 Hz, 1H, H-1A), 5.15 (d, J = 12.0 Hz, 1H, PhCH), 5.13–4.95 (m, 2H, H-4A, H-4D), 4.89 (d, J = 3.0 Hz, 1H, H-1E), 4.87 (d, J = 12.0 Hz, 1H, PhCH), 4.79–4.45 (m, 9H, 9PhCH), 4.67 (d, J = 3.5 Hz, 1H, H-1B), 4.41–4.30 (m, 7H, H-2A, H-3A, H-4E, H-6aE, 3PhCH), 4.28 (d, J = 3.5 Hz, 1H, H-1C), 4.25–4.13 (m, 5H, H-2D, H-3D, H-6bE, 2PhCH), 4.08–3.92 (m, 8H, H-2B, H-2E, H-3B, H-4C, H-6abA, H-6aC, OCH), 3.90 (s, 3H, CO2CH3), 3.88–3.78 (m, 5H, H-3C, H-3E, H-5B, H-5C, H-5D), 3.76–3.68 (m, 2H, H-5A, OCH), 3.62–3.55 (m, 2H, H-2C, H-6bC), 3.47–3.38 (m, 2H, NCH, H-4B), 3.28–3.19 (m, 2H, NCH, H-5E), 2.08, 2.00, 1.95 (3s, 9H, 3COCH3), 1.61 (s, 3H, CCH3), 0.97–0.89 (m, 6H, 2CCH3); 13C NMR (125 MHz, CDCl3): δ 171.3, 170.8, 170.7 (3COCH3), 169.8 (CO2CH3), 168.2, 167.4 (PhthCO), 139.2–122 (Ar-C), 102.4 (C-1C), 99.08 (C-1D), 98.8 (CCH3), 98.3 (C-1E), 98.1 (C-1B), 97.5 (C-1A), 81.9 (C-4B), 79.4 (C-4C), 78.9 (C-2B), 76.3 (2C, C-2C, C-2D), 75.9 (PhCH), 75.3 (2C, C-3C, C-5C), 75.0 (C-4E), 74.9 (PhCH), 74.7 (C-3D), 74.5 (2C, C-2E, C-3E), 74.0 (PhCH), 73.3 (C-3B), 72.8 (2C, 2PhCH), 72.7 (PhCH), 72.1 (C-3A), 71.9 (PhCH), 71.1 (C-4D), 70.5 (PhCH), 69.4 (C-5D), 68.6 (C-6E), 68.3 (C-4A), 66.1 (OCH2), 65.8 (C-6C), 64.6 (C-5A), 64.9 (C-6A), 63.3 (C-5E), 62.3 (C-5B), 54.8 (C-2A), 50.4 (NCH2), 26.0 (CCH3), 21.2, 20.9, 20.8 (3COCH3), 15.9, 15.8 (2CCH3); MALDI-MS: 1947.7 [M + Na]+; anal. calcd for C106H116N4O30 (1926.06): C, 66.10; H, 6.07%; found: C, 66.00; H, 6.20%.
2-Aminoethyl O-(4,6-O-[(R)-(1-sodium carboxylate)ethylidene]-α-D-galactopyranosyl)-(1 → 3)-(α-L-fucopyranosyl)-(1 → 6)-(α-D-galactopyranosyl)-(1 → 3)-(α-L-fucopyranosyl)-(1 → 3)-2-acetamido-2-deoxy-β-D-glucopyranoside (1)
To a solution of compound 15 (300 mg, 0.23 mmol) in nbutanol (15 mL) was added ethylene diamine (0.3 mL) and the mixture was stirred at 90 °C for 10 h. The solvents were removed under reduced pressure, the crude product was dissolved in acetic anhydride (2 mL) and pyridine (2 mL) and the solution was kept at room temperature for 1 h. The solvents were removed under reduced pressure to give the acetylated product that was passed through a short pad of silica gel with EtOAc (50 mL) as eluent. To a solution of the acetylated product in CH3OH (10 mL) was added 20% Pd(OH)2–C (100 mg) and the reaction mixture was stirred under a positive pressure of hydrogen at room temperature for 15 h. The reaction mixture was filtered through a Celite bed, washed with CH3OH (30 mL) and concentrated. A solution of the crude product in 0.1 M CH3ONa (10 mL) was stirred at room temperature for 3 h, then few drops of water was added and further stirred for another 8 h. The reaction mixture was neutralized with Dowex 50W X8 (H+), filtered, and concentrated to give the crude product that was purified by gel chromatography using Sephadex LH-20 using CH3OH–H2O (3:1) as eluant to give 1 (120 mg, 54%). White powder; [α]25D +10.2 (c 1.0, H2O); 1H NMR (500 MHz, D2O): δ 5.08 (d, J = 3.5 Hz, 1H, H-1E), 5.05 (d, J = 4.0 Hz, 1H, H-1C), 4.89 (d, J = 3.5 Hz, 1H, H-1B), 4.78 (d, J = 4.0 Hz, 1H, H-1D), 4.41 (d, J = 8.5 Hz, 1H, H-1A), 4.18 (d, J = 7.0 Hz, 1H, H-5B), 4.12 (t, J = 6.0 Hz each, 1H, H-5C), 4.09 (d, J = 3.5 Hz, 1H, H-4E), 4.00–3.85 (m, 4H, H-3E, H-4C, H-4B, H-5D), 3.84–3.78 (m, 5H, H-2D, H-2E, H-3D, H-5E, OCH), 3.77–3.70 (m, 10H, H-2A, H-2B, H-2C, H-3B, H-3C, H-4D, H-6abE, H-6aA, H-6aC), 3.69–3.58 (m, 2H, H-4A, OCH), 3.57–3.50 (m, 1H, H-3A), 3.48–3.32 (m, 3H, H-5A, H-6bA, H-6bC), 3.12–3.0 (m, 2H, NCH2), 1.88 (s, 3H, NHCOCH3), 1.43 (s, 3H, CCH3), 1.03–1.01 (m, 6H, 2CCH3); 13C NMR (125 MHz, D2O): δ 176.09 (COOH), 174.0 (NHCOCH3), 101.5 (C-1A), 100.7 (2C, C-1C, C-1E), 100.6 (CCOOH), 99.8 (C-1D), 98.4 (C-1B), 80.0 (C-3A), 78.9 (C-3B), 78.2 (C-4D), 75.8 (C-5A), 71.7 (2C, C-3D, C-4E), 71.4 (2C, C-4B, C-4C), 69.7 (C-5C), 69.4 (C-4A), 69.2 (C-3C), 68.6 (C-2C), 68.5 (C-2E), 68.4 (C-3E), 67.9 (C-2D), 66.8 (2C, C-5B, C-5D), 66.6 (C-2B), 66.5 (C-6C), 65.9 (C-6E), 64.9 (C-6A), 62.9 (C-5E), 60.6 (OCH2), 55.0 (C-2A), 39.4 (NCH2), 25.1 (CCH3), 22.2 (COCH3), 15.5, 15.2 (CCH3); MALDI-MS: 972.3 [M]+; anal. calcd for C37H61N2NaO26 (972.86): C, 45.68; H, 6.32%; found: C, 45.50; H, 6.45%.
Conclusions
In conclusion, a concise convergent strategy has been developed for the synthesis of the pyruvate acetal containing pentasaccharide repeating unit of the cell wall O-antigen of E. coli O156 in excellent yield. Application of stereoselective [2 + 3] block glycosylation, achievements of several α-glycosidic linkages, use of HClO4–SiO2, a cheap and stable solid acid in the glycosylation, preparation of pyruvic acid acetal using pyruvate dithioacetal under glycosylation condition, attachment of 2-aminoethyl linker at the reducing end for easy connectivity with an appropriate protein or aglycone are important features of the synthetic scheme. All glycosylation steps are high yielding with excellent stereoselectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. S. thanks CSIR, New Delhi for providing Senior Research Fellowship. The work is supported by SERB, New Delhi (Project No. EMR/2015/000282 dated 17.09.2015) (AKM) and Bose Institute.Notes and references
- J. T. Watson, M. Gayer and M. A. Connolly, Emerging Infect. Dis., 2007, 13, 1–5 CrossRef PubMed.
- V. Curtis, S. Cairncross and R. Yonli, Trop. Med. Int. Health, 2000, 5, 22–32 CrossRef CAS PubMed.
- G. Gavazzi, F. Herrmann and K.-H. Krause, Clin. Infect. Dis., 2004, 39, 83–91 CrossRef PubMed.
- O. Müller and M. Krawinkel, Can. Med. Assoc. J., 2005, 173, 279–286 CrossRef PubMed.
- B. M. Lund and S. J. O'Brien, Foodborne Pathog. Dis., 2011, 8, 961–973 CrossRef PubMed.
- F. Jafari, L. Shokrzadeh, M. Hamidian, S. Salmanzadeh-Ahrabi and M. R. Ali, Jpn. J. Infect. Dis., 2008, 61, 269–273 Search PubMed.
- G. O. Canny and B. A. McCormick, Infect. Immun., 2008, 76, 3360–3373 CrossRef CAS PubMed.
- J. P. Nataro and J. B. Kaper, Clin. Microbiol. Rev., 1998, 11, 142–201 CAS.
- B. Köves and B. Wullt, Eur. Urol., Suppl., 2016, 15, 88–94 CrossRef.
- J. M. Rhodes, Gut, 2007, 56, 610–612 CrossRef CAS PubMed.
- R. E. Besser, S. M. Lett, J. T. Weber, M. P. Doyle, T. J. Barrett, J. G. Wells and P. M. Griffin, J. Am. Med. Assoc., 1993, 269, 2217–2220 CrossRef CAS PubMed.
- F. E. Preston, R. G. Malia, M. J. Sworn and E. K. Blackburn, J. Clin. Pathol., 1973, 26, 120–125 CrossRef CAS PubMed.
- M. A. Croxen, R. J. Law, R. Scholz, K. M. Keeney, M. Wlodarska and B. B. Finlay, Clin. Microbiol. Rev., 2013, 26, 822–880 CrossRef CAS PubMed.
- J. C. Paton and A. W. Paton, Clin. Microbiol. Rev., 1998, 11, 450–479 CAS.
- J. Y. Lim, J. W. Yoon and C. J. Hovde, J. Microbiol. Biotechnol., 2010, 20, 5–14 CAS.
- N. Allocati, M. Masulli, M. F. Alexeyev and C. D. Ilio, Int. J. Environ. Res. Public Health, 2013, 10, 6235–6254 CrossRef PubMed.
- J. M. Bosilevac and M. Koohmaraie, Appl. Environ. Microbiol., 2011, 77, 2103–2112 CrossRef CAS PubMed.
- I. Lerouge and J. Vanderleyden, FEMS Microbiol. Rev., 2002, 26, 17–47 CrossRef CAS PubMed.
- (a) Carbohydrate based vaccines, ed. R. Roy, Oxford University Press, Oxford, 2008 Search PubMed; (b) J. J. Mond, A. Lees and C. M. Snapper, Annu. Rev. Immunol., 1995, 13, 655–692 CrossRef CAS PubMed.
- (a) R. D. Astronomo and D. R. Burton, Nat. Rev. Drug Discovery, 2010, 9, 308–324 CrossRef CAS PubMed; (b) M. Cavallari and G. De Libero, Vaccines, 2017, 5, 4, DOI:10.3390/vaccines5010004.
- Z. Duan, S. N. Senchenkova, X. Guo, A. V. Perepelov, A. S. Shashkov, B. Liu and Y. A. Knirel, Carbohydr. Res., 2016, 430, 24–28 CrossRef CAS PubMed.
- A. Chakkumkal, S. Benjamin, L. P. Claney and P. H. Seeberger, Chem. Biol., 2014, 21, 38–50 CrossRef PubMed.
- (a) V. Pozsgay, Curr. Top. Med. Chem., 2008, 8, 126–140 CrossRef CAS PubMed; (b) A. Hölemann and P. H. Seeberger, Curr. Opin. Biotechnol., 2004, 15, 522–555 CrossRef PubMed.
- A. K. Chakraborti and R. Gulhane, Chem. Commun., 2003, 1896–1897 RSC.
- C. Mukherjee and A. K. Misra, Synthesis, 2007, 683–692 CAS.
- B. Mukhopadhyay, S. V. Maurer, N. Rudolph, R. M. van Well, D. A. Russell and R. A. Field, J. Org. Chem., 2005, 70, 9059–9062 CrossRef CAS PubMed.
- G. Agnihotri and A. K. Misra, Tetrahedron Lett., 2006, 47, 8493–8497 CrossRef CAS.
- P. K. Mandal, Synthesis, 2015, 47, 836–844 CrossRef CAS.
- T. Ziegler, Carbohydr. Res., 1994, 262, 195–212 CrossRef CAS PubMed.
- J. Lindberg, P. Stralfors and P. Konradsson, Tetrahedron, 2002, 58, 4245–4254 CrossRef CAS.
- Y. Hua, Y. Du, G. Yu and S. Chu, Carbohydr. Res., 2004, 339, 2083–2090 CrossRef CAS PubMed.
- S. Figueroa-Perez and R. R. Schmidt, Carbohydr. Res., 2000, 328, 95–102 CrossRef CAS PubMed.
- G. Zemplen, Ber. Dtsch. Chem. Ges., 1926, 59, 1254–1266 CrossRef.
- G. Agnihotri and A. K. Misra, Tetrahedron Lett., 2006, 47, 3653–3658 CrossRef CAS.
- T. Ogawa and H. Yamamoto, Agric. Biol. Chem., 1985, 49, 475–482 CAS.
- O. Kanie, Y. Ito and T. Ogawa, J. Am. Chem. Soc., 1994, 116, 12073–12074 CrossRef CAS.
- K. Bock and C. Pederson, J. Chem. Soc., Perkin Trans. 2, 1974, 293–297 RSC.
- D. Crich and H. Li, J. Org. Chem., 2002, 67, 4640–4646 CrossRef CAS PubMed.
- A. Liptak, I. Bajza, J. Kerekgyarto, J. Hajko and L. Szilagyi, Carbohydr. Res., 1994, 253, 111–120 CrossRef.
- P. A. J. Gorin, M. Mazurek, H. S. Duarte, M. Iacomini and J. H. Duarte, Carbohydr. Res., 1982, 100, l–15 CrossRef.
- J. S. Debenham, R. Madsen, C. Roberts and B. Fraser-Reid, J. Am. Chem. Soc., 1995, 117, 3302–3303 CrossRef CAS.
- W. M. Pearlman, Tetrahedron Lett., 1967, 8, 1663–1664 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of the NMR spectra of compounds 1, 7, 8, 9, 11, 12, 13, 15. See DOI: 10.1039/c7ra07567g |
| This journal is © The Royal Society of Chemistry 2017 |