
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Grafting polycarbonate onto graphene nanosheets: synthesis and characterization of high performance polycarbonate–graphene nanocomposites for ESD/EMI applications†
Sang-Hwa Yoon ab and
Hee-Tae Jung*a
ab and
Hee-Tae Jung*a
aDepartment of Chemical and Biomolecular Engineering (BK-21 Plus), KAIST Institute for Nanocentury, Korea Advanced Institute of Science and Technology, 291 Daehak-ro, Yuseong-gu, Daejeon 305-701, Republic of Korea. E-mail: heetae@kaist.ac.kr
bLotte Chemical Research Institute, 115 Gajeongbuk-ro, Yuseong-gu, Daejeon 305-726, Korea
First published on 26th September 2017
Abstract
We have developed high performance polycarbonate–graphene nanocomposites with outstanding mechanical and electrical properties by grafting polycarbonate (PC) onto graphene nanosheets (PC-g-MGNS). The PC-g-MGNS nanocomposites were synthesized by grafting hydroxyl-terminated PC onto methylene diphenyl diisocyanate-functionalized graphene nanosheets (MGNS); the MGNS were synthesized via the alcoholysis reaction between 4,4′-methylene diphenyl diisocyanate (MDI) and chemically reduced graphene nanosheets (GNS). The resulting PC-g-MGNS nanocomposites exhibit dispersions of graphene in the PC matrix that are much better than those of PC–graphene nanocomposites prepared via simple mixing in a solvent (s-PC/GNS) and provide highly enhanced mechanical strength and electrical conductivity. For instance, the PC-g-MGNS nanocomposite containing 3.0 wt% graphene exhibits an ∼20% increase in tensile strength, an ∼23% increase in Young's modulus, and three orders of magnitude greater electrical conductivity when compared with the equivalent s-PC/GNS nanocomposite.
Introduction
The excellent opto-electronic,1 mechanical,2 and thermal properties3 of graphene mean that it has various applications in electrodes,4,5 batteries,6,7 supercapacitors,8 sensors,9 membranes,10 and polymer nanocomposites.11 Graphene-reinforced polymer nanocomposites have attracted considerable attention because of their recent commercial applications in electrostatic discharge (ESD),12 electromagnetic interference (EMI) shielding,13,14 thermal interface materials (TIMs),15 and high strength equipment.16 The addition of graphene considerably improves the mechanical,17 thermal,18 and electrical properties19 of the matrix polymer. In particular, the fabrication of polycarbonate/graphene nanocomposites is desirable because of the transparency, high mechanical strength, and good thermal and dimensional stability of the matrix polymer. The high tensile modulus (E ∼ 2.4 GPa), high glass transition temperature (Tg ∼ 147 °C), and high heat deflection temperature (HDT ∼ 140 °C at 0.45 MPa) of polycarbonate (PC) are very useful in high temperature applications such as lenses for automotive headlamps,20 heat sinks for light emitting diodes (LEDs),21 medical devices,22 and ESD23 and EMI shielding24 applications. The presence of a uniform dispersion of graphene nanosheets (GNS) in a PC matrix can enhance its mechanical and electrical properties at low filler loadings above those of matrixes containing other conventional additives such as carbon black and carbon fibers, primarily due to the high surface area of graphenes.25As in other polymer composites, however, the poor dispersion of graphenes in PC has critically restricted its applications as a conductive filler. Pristine graphene with a high specific surface area irreversibly aggregates or restacks in PC matrixes because the van der Waals forces and π–π stacking interactions between graphene layers are much stronger than the interfacial interaction between graphene and PC.11,26,27 Conventional melt28,29 and solvent30,31 mixing methods have been used to disperse graphenes in PC matrixes. For example, thermally reduced graphite oxides and PC melts were mixed in a twin-screw extruder at 280 °C, and found to exhibit an 80% improvement in the Young's modulus and an electrical resistivity of 3.2 × 103 Ω cm at a loading of 10 wt%.28 Further, liquid-phase-exfoliated single- and few-layer graphenes and PC were mixed in 1,3-dioxolane.31 The Young's modulus of the nanocomposite obtained via solvent mixing was found to be enhanced up to 26% at a loading of 1.0 wt% with respect to that of pristine PC, and its electrical conductivity was found to be 10−3 S m−1 at a loading of 10 wt%.
In this study, we report a new approach to the highly efficient dispersion of graphene nanosheets (GNS) into PC matrixes in which polycarbonate is grafted onto the graphene nanosheets (PC-g-MGNS). Methylene diphenyl diisocyanate-functionalized graphene nanosheets (MGNS) were prepared via the alcoholysis reaction between the hydroxyl groups of graphene and the isocyanate groups of 4,4′-methylene diphenyl diisocyanate (MDI). Then, the free isocyanate groups of MGNS were reacted with the hydroxyl end groups of polycarbonate with various MGNS loadings to fabricate PC-g-MGNS nanocomposites. The tensile strength and Young's modulus of the PC-g-MGNS nanocomposites containing 3 wt% graphene were found to be improved by 20.5% and 22.7% respectively, and the electrical conductivity was improved by three orders of magnitude over those of PC–graphene nanocomposites with the same graphene loading prepared via simple solvent mixing (s-PC/GNS). Thus, this approach provides an effective route for fabricating high performance PC–graphene nanocomposites with potential ESD and EMI applications.
Experimental
Materials
Natural graphite (FP-99.95% pure) was purchased from Graphit Kropfmühl. Potassium permanganate (KMnO4, 99.3%) was purchased from Junsei Chemical. Sulfuric acid (H2SO4, 98%) was purchased from Daejung Chemicals. Hydrogen peroxide solution (H2O2, 30–35%) and hydrochloric acid (HCl, 35–37%) were purchased from Samchun Chemical. Hydrazine monohydrate (N2H4, 64–65%, 98%), 4,4′-methylene diphenyl diisocyanate (MDI, 98%), anhydrous N,N-dimethylformamide (DMF, 99.8%), anhydrous chloroform (99%), and triethylamine (TEA, 99.5%) were purchased from Sigma-Aldrich. Polycarbonate (PC-1220S) was provided by Lotte Chemical. All chemicals were used as received.Preparation of graphene oxide (GO) and graphene nanosheets (GNS)
Graphene oxide (GO) was prepared from natural graphite by using a modified Hummers method.32,33 Typically, 2.0 g of natural graphite and 50 mL of sulfuric acid were initially stirred uniformly in a 500 mL round bottom flask with an ice bath. Then, 7.0 g of potassium permanganate was added very slowly to the mixture with continuous stirring. The solution was heated to 35 °C in a water bath and continuously stirred for 2 h. The flask was then placed in the ice bath and deionized water (400 mL) was added gradually. After stirring for 15 min, the reaction was terminated by the addition of 30% H2O2 (300 mL). The product was filtered and washed with 10% hydrochloric acid and deionized water sequentially. GO was obtained after drying in a vacuum oven. To prepare graphene nanosheets (GNS) by performing the chemical reduction of GO, 100 mg of GO was dispersed in 100 mL of water with sonication until the solution became clear yellow-brown. Hydrazine hydrate (1 μL per 3 mg of GO) was then added with stirring and the solution was heated in an oil bath at 100 °C under a water-cooled condenser. After reduction for 24 h, a precipitated black solid was isolated by filtration and washed with water. Then, the filtered graphene nanosheets were dried in a vacuum oven for 24 h.Synthesis of MDI-functionalized graphene nanosheets (MGNS)
The synthesis of the MDI-functionalized graphene nanosheets (MGNS) was as follows. 20.0 mg of chemically reduced GNS was dispersed in 30.0 mL DMF with the assistance of ultrasonication for 45 min. Then, 0.2 g of MDI was added into the above GNS solution and stirred for 4 h at 70 °C in a three-necked flask protected by N2. The product was filtered and washed with additional DMF to remove the unreacted MDI, and then dried in a vacuum oven.Grafting of polycarbonate onto MGNS (PC-g-MGNS)
A desired amount of MGNS was dispersed in 200 mL of chloroform with ultrasonication for 60 min. Then, 20.0 g of PC was dissolved in the MGNS solution with vigorous stirring. Afterwards, 1.38 mL of TEA was introduced into the PC/GNS solution at 40 °C in a three-necked flask protected by N2. The mixture was constantly stirred for an additional 4 h and precipitated in excess acetone. The precipitate was isolated with filtration and washed several times with water to remove TEA and acetone. The resulting powder sample was placed in a vacuum oven at 120 °C for 12 h. 1H-NMR (CDCl3, 500 MHz): δ 7.42 (t, 2H, j = 7.8 Hz, f), 7.26 (d, 4H, j = 7.3 Hz, d), 7.18 (d, 4H, j = 8.3 Hz, c), 6.71 (d, 2H, j = 5.9 Hz, g), 6.51 (s, 1H, a), 3.91 (s, 2H, b), 1.69 (s, 6H, e).As a control, solvent-mixed PC–graphene nanocomposites (s-PC/GNS) were prepared following a process similar to that used for the PC-g-MGNS nanocomposites. A desired amount of GNS was dispersed in 200 mL of chloroform by performing ultrasonication for 60 min. Then, 20.0 g of PC was dissolved in the GNS solution under vigorous stirring. The mixture was precipitated in excess acetone. The precipitate was isolated by filtration and washed several times with water to remove acetone. The resulting powder sample was placed in a vacuum oven at 120 °C.
Characterization
The introduction of chemical bonds and functional groups into the graphene nanosheets was confirmed by performing Fourier transform infrared spectroscopy (FT-IR) and X-ray photoelectron spectroscopy (XPS). The absorption bands between 650 and 4000 cm−1 were examined with a Nicolet iN10 MX FT-IR spectrometer (Thermo Scientific). XPS was carried out on an Axis Nova (Kratos Analytical) by using a monochromatic (Al-Kα) X-ray source. Structural changes in graphene were identified by using Raman spectroscopy with a 514 nm laser source (Aramis, Horiba Jobin Yvon). The size and thickness of the GNS and MGNS were measured by using atomic force microscopy (AFM: XE-100, Park Systems). The AFM samples were prepared by depositing GNS and MGNS in NMP solution onto oxidized silicon substrates. Thermogravimetric analysis (TGA) was carried out on a Q500 (TA Instruments). The samples were scanned from 20 to 800 °C at a heating rate of 20 °C min−1 under a nitrogen atmosphere. The 1H-NMR spectra were recorded on a DD2 500 MHz NMR spectrometer (Agilent). Chloroform and tetramethylsilane (TMS) were used as the solvent and the chemical shift reference respectively. To measure the mechanical properties of the nanocomposites, s-PC/GNS and PC-g-MGNS were molded at 300 °C into bar specimens by using a mini-laboratory injection molder (IM 12, Xplore Instruments). The tensile strength, elongation at break, and Young's modulus of each nanocomposite were determined from 5 samples with a 5566 Universal Testing Machine (Instron). Tensile tests were carried out in accordance with ASTM D638 and 10 mm min−1 of crosshead movement. The glass transition temperatures (Tg) of pristine PC and PC–graphene nanocomposite samples were measured using differential scanning calorimetry (DSC: Pyris Diamond DSC, PerkinElmer). The samples were heated to 300 °C, which was maintained for 2 min, then cooled to 50 °C, and heated again to 300 °C. The heating and cooling rates were 10 °C min−1 in all cases. The electrical conductivities were determined in terms of the electrical resistivities of compression-molded films by using a four-point probe meter (Loresta-EP MCP-T360, direct current source, Mitsubishi Chemical Analytech). In order to obtain scanning electron microscopy (SEM) images of graphene and polycarbonate, a scanning electron microscope was used (SM 701, TOPCON). SEM images were obtained of a fracture surface of each sample in order to investigate the variation in the interaction between graphene and polycarbonate with the graphene content.Results and discussion
Synthesis and properties of MGNS
The chemical structures and synthetic routes for the PC-g-MGNS are presented in Schemes 1 and 2. To graft PC onto graphene nanosheets, we first prepared the MDI-functionalized graphene nanosheets (MGNS), as shown in Scheme 1. The synthesis of the MGNS was carried out via the alcoholysis reaction of the isocyanate groups of MDI with the hydroxyl groups of graphene, which generates carbamate ester linkages.34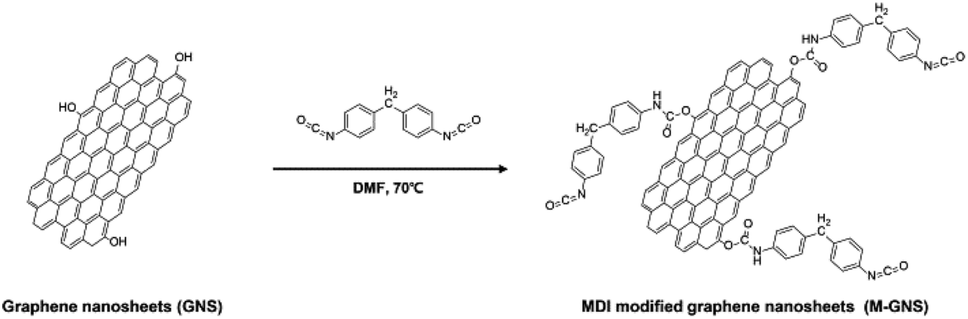 | ||
| Scheme 1 The synthesis of MDI-functionalized graphene nanosheets (MGNS). | ||
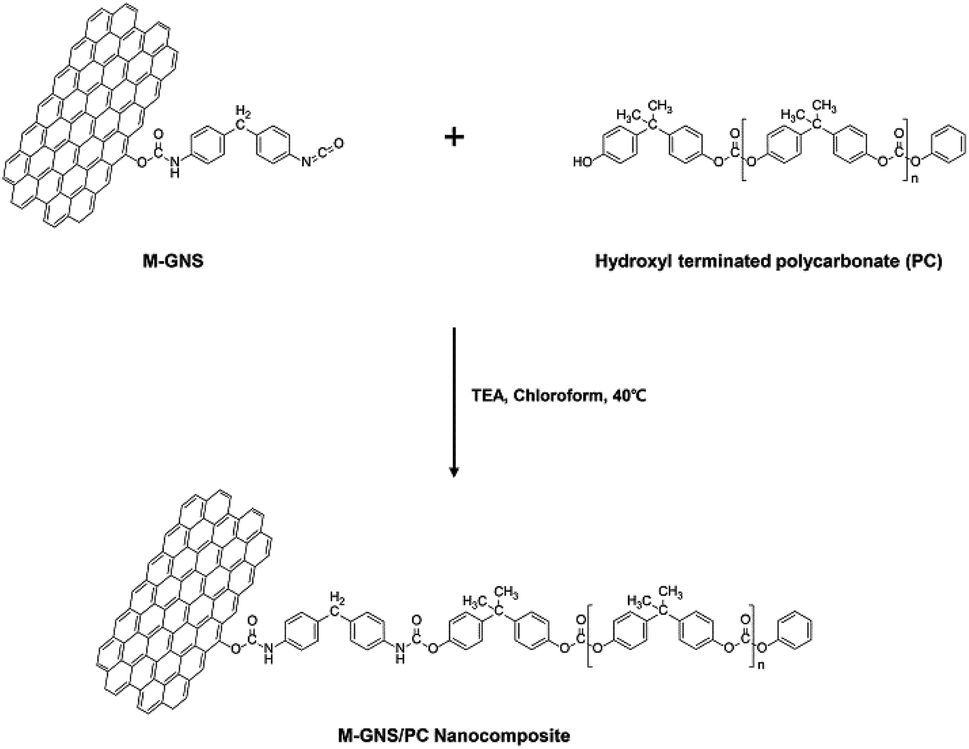 | ||
| Scheme 2 Fabrication process for the PC-g-MGNS nanocomposites. | ||
The successful synthesis of the MGNS was confirmed with FT-IR and XPS. The absorption peaks in the FT-IR spectrum of GO at 3430 cm−1, 1732 cm−1, 1614 cm−1, 1411 cm−1, 1220 cm−1 and 1042 cm−1 in Fig. 1a are assigned to the OH (hydroxyl), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (carboxyl), CC (aromatic), C–OH (carboxyl), C–O (epoxy) and C–O (alkoxy) stretching vibrations, respectively.35 After the chemical reduction of GO to graphene, the FT-IR spectrum contains no characteristic absorption peaks due to the overlapping of the strong absorption bands of the graphene nanosheets.36 After the MDI functionalization of graphene, the CO stretching vibration at 1646 cm−1 appears, which is assigned to the carbonyl stretching vibration of the carbamate ester.37 The new bands at 3331 cm−1 and 1507 cm−1 also originate from the carbamate ester and correspond to the N–H stretching vibration and the N–H out-of-plane bending vibration with CNH deformation respectively.37,38 These results confirm the formation of carbamate ester linkages between GNS and MDI as a result of the alcoholysis reaction. Further, the characteristic absorption peaks of the –NCO group and p-phenylene at 2167 cm−1 and 815 cm−1, respectively, are clearly evident in the spectrum, providing additional evidence that the functionalization of the graphene with MDI was successful.
O (carboxyl), CC (aromatic), C–OH (carboxyl), C–O (epoxy) and C–O (alkoxy) stretching vibrations, respectively.35 After the chemical reduction of GO to graphene, the FT-IR spectrum contains no characteristic absorption peaks due to the overlapping of the strong absorption bands of the graphene nanosheets.36 After the MDI functionalization of graphene, the CO stretching vibration at 1646 cm−1 appears, which is assigned to the carbonyl stretching vibration of the carbamate ester.37 The new bands at 3331 cm−1 and 1507 cm−1 also originate from the carbamate ester and correspond to the N–H stretching vibration and the N–H out-of-plane bending vibration with CNH deformation respectively.37,38 These results confirm the formation of carbamate ester linkages between GNS and MDI as a result of the alcoholysis reaction. Further, the characteristic absorption peaks of the –NCO group and p-phenylene at 2167 cm−1 and 815 cm−1, respectively, are clearly evident in the spectrum, providing additional evidence that the functionalization of the graphene with MDI was successful.
 | ||
| Fig. 1 (a) FT-IR spectra of GO, GNS, and MGNS. High resolution C1s XPS spectra of (b) GNS and (c) MGNS. (d) Raman spectra of GO, GNS, and MGNS. | ||
The XPS spectra verify the preparations of GO (Fig. S1†), GNS (Fig. 1b) and MGNS (Fig. 1c). After the chemical reduction of GO, the oxygen containing functionalities of GNS become weak compared to those of GO (Fig. S1† and 1b). The C1s peak of GNS can be fitted to five component peaks, located at 284.4 eV (C–C/C–H), 285.3 eV (C–N), 286.2 eV (C–O/C–OH), 288.1 eV (CO), and 290.1 eV (OC–OH).26 The characteristic peak at 285.3 eV arises from the C–N bond in the pyrazole group, which arises during the chemical reduction process.39,40 After the GNS are functionalized with MDI molecules, there are obvious changes in binding energy and intensity in the C1s spectra of the MGNS (Fig. 1c). The intensity of the C–N peak at 285.3 eV is significantly increased, which can be attributed to the formation of carbamate ester linkages between GNS and MDI. Moreover, the intensity of the C–O/C–OH (hydroxyl/epoxy, 286.1 eV) peak is considerably lower, demonstrating that the hydroxyl group of GNS is converted to a carbamate ester linkage between GNS and MDI. Therefore, the FT-IR and XPS results confirm that MGNS are successfully synthesized by the alcoholysis reaction of isocyanate groups of MDI with hydroxyl group of GNS.
Raman spectroscopy was carried out to verify the structural changes from GO to GNS and then to MGNS. In Fig. 1d, the G-band of GO is broadened and up-shifted to 1602 cm−1 when compared to that of pure graphite (1580 cm−1, Fig. S2†);41 this band corresponds to the first-order scattering of the E2g phonon of sp2 C atoms.42 In addition, a D-band at 1350 cm−1 is observed in the spectrum of GO that was not observed in the spectrum of pure graphite, consistent with a decrease in the size of the in-plane sp2 domains.26 After chemical reduction with hydrazine, the G-band of GNS is down-shifted to 1589 cm−1, which arises due to the recovery of the hexagonal network of carbon atoms.43 Further, for the GNS the ratio of the intensities of the D-band and G-band (ID/IG) increases to ∼1.19, which indicates that the chemical reduction process results in graphitic domains that are smaller than those of GO.26 The location of the G-band (1587 cm−1) and the ID/IG (1.21) for the MGNS are very similar to those of the GNS. These results indicate that the graphitic structure is maintained after functionalization, as expected because functionalization occurs at hydroxyl groups situated at the edges of the graphene nanosheets.44
Fig. 2a shows digital photographs of the GNS and MGNS dispersions in various organic solvents after ultrasonication and standing for 24 h. Indeed, the MGNS must be soluble in good solvents for PC in order to fabricate homogeneous PC-g-MGNS nanocomposites. Pristine graphene commonly aggregates and precipitates in good solvents for PC, including dimethylformamide (DMF), methylene chloride (MC), and chloroform. In contrast, the MGNS are dispersed very well in DMF and chloroform and do not precipitate within 24 h, because the presence of carbamate ester and isocyanate groups change the surface properties of graphene, increasing the solubility.
 | ||
| Fig. 2 (a) Digital photographs of dispersions of GNS and MGNS in DMF, MC, and chloroform after standing for 24 h. AFM images and thickness profiles of (b) GNS and (c) MGNS. | ||
To assess the exfoliation of the graphenes, AFM images and height profiles of the GNS and MGNS were obtained (Fig. 2b and c). The lateral dimension and thickness of the chemically exfoliated GNS were measured to be 3.8 μm and 0.64 nm respectively, consistent with previous reports.45–47 After MDI functionalization, there is no obvious change in lateral dimension of the graphene (3.5 μm). However, the thickness of the MGNS was much higher than that of GNS (∼2.5 nm) due to the MDI molecules that are chemically attached to graphene. A similar phenomenon was observed previously.48
Synthesis and characterization of PC-g-MGNS
We used hydroxyl-functionalized PC, which was prepared using bisphenol A (BPA) and diphenyl carbonate (DPC),49 to enable covalent bonding with the MGNS. As shown in Scheme 2, the hydroxyl end groups of PC react with the free isocyanate groups of the MGNS in the presence of triethylamine (TEA) in chloroform at 40 °C. Tertiary amines are the most widely used catalysts for the preparation of carbamate ester linkages between isocyanates and alcohols.50 Two mechanisms have been proposed for the tertiary amine catalyzed reactions of isocyanates with alcohols: (a) formation of an amine–isocyanate complex, followed by attack by the alcohol,51 (b) formation of an amine–alcohol complex, followed by reaction with the isocyanate.52Fig. 3a shows FT-IR spectra of pristine PC and the PC-g-MGNS nanocomposites. The PC spectrum contains four main peaks: –CH (aliphatic) stretching at 2923 cm−1, CO (ketone) stretching at 1767 cm−1, CC (aromatic) stretching at 1501 cm−1, and C–O–C (ether) stretching at 1211 and 1004 cm−1. After the chemical grafting of PC onto MGNS, the characteristic peaks of PC are still dominant in the FT-IR spectra of all nanocomposites we prepared in this study. However, there is a new peak due to the N–H stretching vibration at 3369 cm−1, which increases gradually in intensity with increases in the MGNS content. This increase is attributed to the original carbamate ester linkages in the MGNS and the new carbamate ester linkages formed by reaction between free isocyanate groups on the MGNS and the hydroxyl end groups of PC, as shown in Scheme 2.
 | ||
| Fig. 3 (a) FT-IR spectra and (b) 1H NMR spectra of pristine PC and PC-g-MGNS nanocomposites with various MGNS contents: 0.1 wt%, 0.5 wt%, 1.0 wt%, and 3.0 wt%. (c) Hydroxyl end group contents of pristine PC, s-PC/GNS, and PC-g-MGNS for various GNS and MGNS contents: 0.1 wt%, 0.5 wt%, 1.0 wt%, and 3.0 wt%. | ||
The NMR spectra provide further evidence for covalent bonding between the MGNS and PC. The NMR peaks at 7.18–7.29 ppm and 1.71 ppm are attributed to the protons of aromatic and methyl groups respectively in the PC main chains (Fig. S3a†). The protons in the phenoxy and phenolic end groups in pristine PC give rise to peaks at 7.43 ppm and 6.70 ppm respectively. In the case of the PC-g-MGNS nanocomposites (Fig. S3b†), new peaks at 6.52 ppm and 3.92 ppm are present that are assigned to carbamate ester linkages (Ar–OCONH–Ar) and methylene (Ar–CH2–Ar) of reacted MDI respectively. The intensities of these peaks increase with increases in the MGNS content from 0.1 wt% to 3.0 wt% (Fig. 3b). Conversely, the relative intensity of the peak due to terminal aromatic protons at 6.71 ppm decreases sharply with increases in the MGNS content, which indicates that the hydroxyl end groups in PC have successfully reacted with free isocyanate groups in the MGNS.
In order to quantify the number of PC chains grafted onto the graphene nanosheets, we calculated the number of unreacted hydroxyl end groups of PC by analyzing the NMR spectrum.53 The number of hydroxyl end groups in a nanocomposite can be quantitatively determined from the intensity in the NMR spectrum of the peak due to aromatic protons located in hydroxyl groups relative to that of aromatic protons located in phenoxy groups.53 In Fig. 3c, the hydroxyl contents of the s-PC/GNS nanocomposites decreases gradually with increases in the GNS content. When 3.0 wt% GNS is incorporated into PC, the hydroxyl end group content of the s-PC/GNS nanocomposites is lowered to 27.6%. This result suggests that the hydroxyl end groups in PC have reacted with other functional groups such as carboxyl groups in the GNS. As for the PC-g-MGNS nanocomposites, the decrease in the hydroxyl content is much sharper due to the excellent reactivity between isocyanate and hydroxyl groups in the presence of a catalyst.50 The hydroxyl end group content of the PC-g-MGNS nanocomposites decreases to 1.7% with a MGNS loading of 3.0 wt%, which indicates that most hydroxyl end groups in PC have successfully reacted with MGNS.
Mechanical and electrical properties of the PC-g-MGNS nanocomposites
The variations in the tensile properties of the s-PC/GNS and PC-g-MGNS nanocomposites with GNS and MGNS loading are shown in Fig. 4 and Table 1. The concentrations of GNS and MGNS in the nanocomposites are in the range 0 to 3.0 wt%. In Fig. 4a, the tensile strength of the s-PC/GNS nanocomposites (−), which were prepared via the conventional solvent mixing of neat PC and GNS in chloroform, increases for GNS loadings up to ∼1.0 wt%, then decreases upon a further graphene loading increase. In detail, the tensile strength of the s-PC/GNS nanocomposites increases gradually to 70.1 MPa for GNS loadings up to 1.0 wt%, then decreases to 67.2 MPa at a GNS loading of 3.0 wt%. In the case of the PC-g-MGNS nanocomposites (−), however, the tensile modulus dramatically increases for MGNS loadings up to 1.0 wt%, and then increases more gradually with a further increase in the MGNS content (see also Fig. S4†). Very interestingly, the tensile strength of the PC-g-MGNS nanocomposite containing 3.0 wt% MGNS is 81.0 MPa, which is 20.5% higher than that of s-PC/GNS with an equivalent GNS loading. This result suggests that graphene severely aggregates at high concentrations in the s-PC/GNS nanocomposites and as a result has little effect on the tensile strength of the composites at higher loadings. In contrast, grafting PC chains onto graphene significantly influences the tensile strength because the graphene is well-dispersed in the matrix polymer. Note also that the elongation at break of the s-PC/GNS nanocomposites (−−) decreases with increases in the GNS content. Further, the elongation at break of the PC-g-MGNS nanocomposite (−−) with a low MGNS content (∼0.5 wt%) is slightly lower than that of the s-PC/GNS nanocomposite with the equivalent GNS loading. However, for high MGNS contents (1.0 wt% and above), the elongation at break for the PC-g-MGNS nanocomposites decreases more significantly than for the s-PC/GNS nanocomposites. For instance, the value of the elongation at break decreases to 2% for the composite with 3.0 wt% MGNS from 102% for pristine PC. The strong interfacial interaction between PC and graphene in the nanocomposites results in a compact structure and ultimately restricts the orientations of the polymer chains when the composite is stretched.17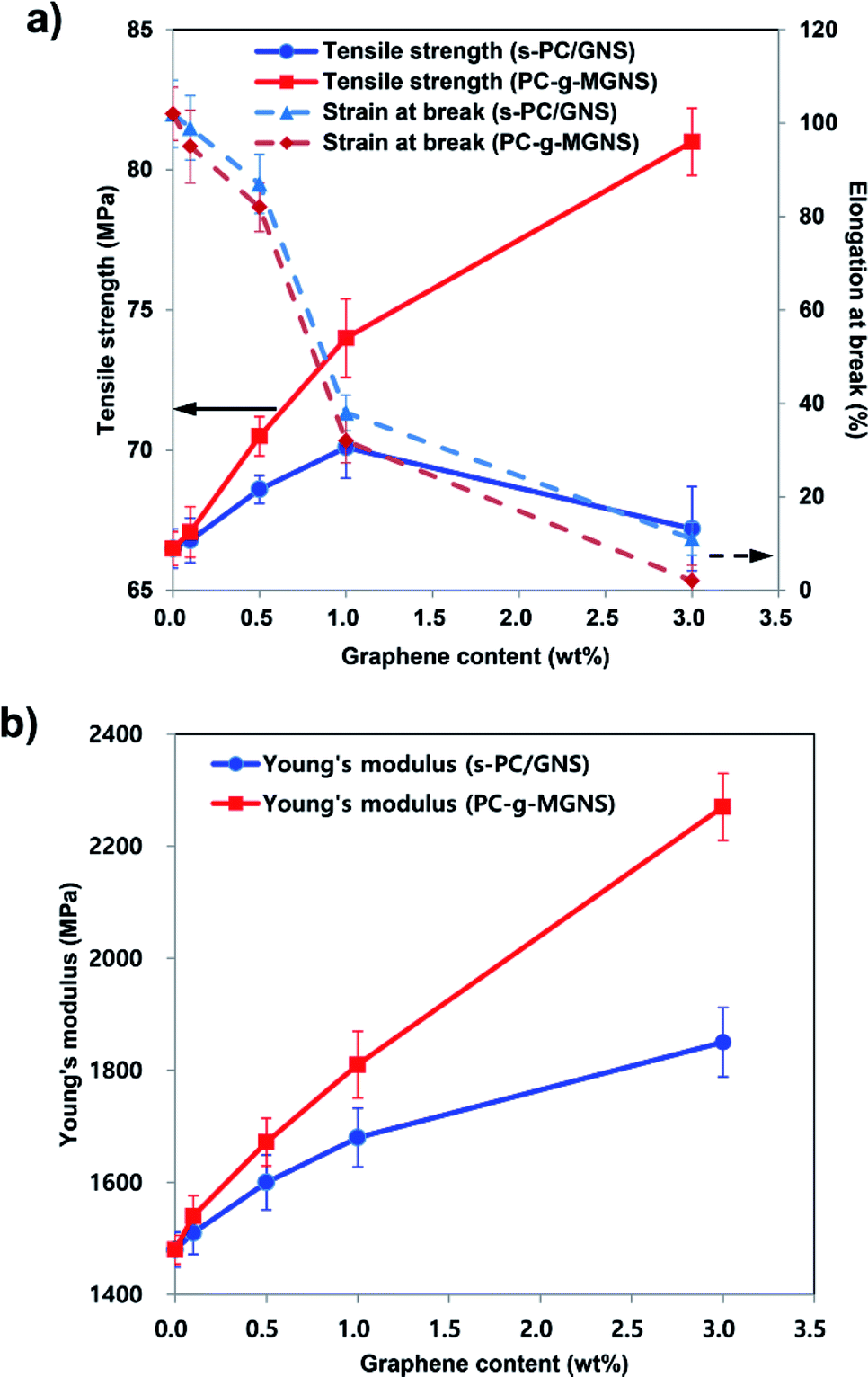 | ||
| Fig. 4 (a) The variations in the tensile strength, elongation at break, and (b) Young's modulus of pristine PC, s-PC/GNS, and PC-g-MGNS with increases in the GNS and MGNS contents: 0.1 wt%, 0.5 wt%, 1.0 wt%, and 3.0 wt%. | ||
| Filler content | Tensile strength, yield (MPa) | Young's modulus (MPa) | Strain at break (%) | Electrical resistivity (Ω cm) | ||||
|---|---|---|---|---|---|---|---|---|
| s-PC/GNS | PC-g-MGNS | s-PC/GNS | PC-g-MGNS | s-PC/GNS | PC-g-MGNS | s-PC/GNS | PC-g-MGNS | |
| 0 wt% | 66.5 | 66.5 | 1480 | 1480 | 102 | 102 | 5.8 × 1017 | 5.8 × 1017 |
| 0.1 wt% | 66.8 | 67.1 | 1510 | 1540 | 99 | 95 | 4.5 × 1017 | 3.1 × 1017 |
| 0.5 wt% | 68.6 | 70.5 | 1600 | 1670 | 87 | 82 | 9.2 × 1016 | 4.2 × 1011 |
| 1.0 wt% | 70.1 | 74.0 | 1680 | 1810 | 38 | 32 | 1.5 × 108 | 2.9 × 103 |
| 3.0 wt% | 67.2 | 81.0 | 1850 | 2270 | 11 | 2 | 2.2 × 106 | 8.8 |
The Young's modulus of the s-PC/GNS nanocomposites (−) significantly increases up to a GNS loading of ∼1.0 wt%, followed by a slight increase at a higher GNS loading (Fig. 4b). The Young's modulus of the PC-g-MGNS nanocomposites (−) increases almost linearly as the MGNS content increases, and is significantly higher than that of the s-PC/GNS nanocomposites for equivalent GNS contents. For example, the Young's modulus of the PC-g-MGNS nanocomposite containing 3.0 wt% MGNS is 2270 MPa, which is 22.7% higher than that of the s-PC/GNS nanocomposite with the equivalent GNS loading. This significant enhancement of the mechanical properties of the PC-g-MGNS nanocomposites is mainly due to the improvements in the interfacial interaction and the compatibility between graphene and the PC matrix that result from grafting the polymer chains onto graphene nanosheets. For a similar reason, the glass transition temperature (Tg) of the PC-g-MGNS nanocomposites gradually increases as the MGNS loading increases from 0 to 3.0 wt%, whereas that of the s-PC/GNS nanocomposites does not increase for GNS loadings above 1.0 wt% (Fig. S5 and Table S1†).
Fig. 5 and Table 1 present the variations in the electrical resistivities of s-PC/GNS (−) and PC-g-MGNS (−) nanocomposites with GNS and MGNS loadings. The electrical resistivity of the s-PC/GNS nanocomposites gradually decreases for GNS loadings in the range 0 to 0.5 wt%, followed by a dramatic reduction at higher GNS loadings. In the case of the PC-g-MGNS nanocomposites, at 0.1 wt% MGNS the electrical resistivity is slightly lower than that of pristine PC. The electrical resistivity then decreases dramatically over a very narrow range of MGNS concentrations up to 1.0 wt% and further decreases to 8.8 Ω cm at a loading of 3.0 wt%. This stepwise decrease in the electrical resistivity is known as an electrical percolation threshold.54 At a certain graphene loading, the graphene network forms a conductive path that leads to a sudden increase in the electrical conductivity. The electrical percolation threshold of PC-g-MGNS is low compared to that of s-PC/GNS, because the PC chains grafted onto the MGNS enhance the dispersion of graphene in the polycarbonate matrix, which results in the formation of an effective conductive pathway at a lower graphene loading. In general, for ESD and EMI applications the electrical resistivities must be in the range 104 to 1011 Ω cm and below 104 Ω cm respectively.55,56 Thus, the electrical resistivity of PC-g-MGNS fully satisfies the ESD/EMI ranges for MGNS loadings below 1.0 wt%.
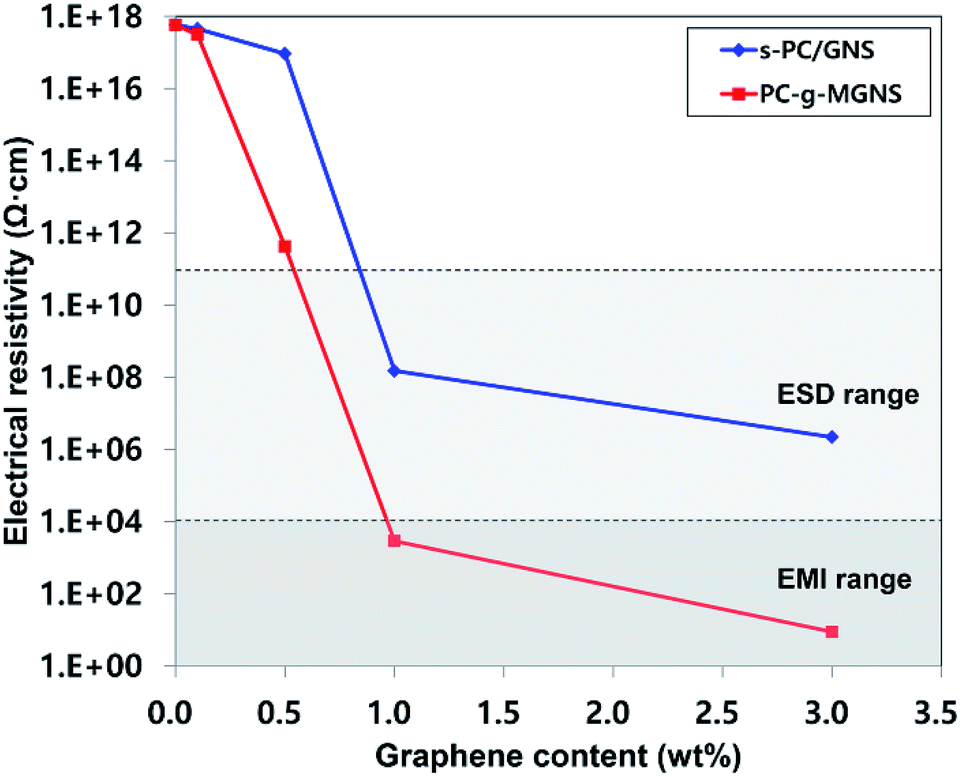 | ||
| Fig. 5 The variations in the electrical resistivities of pristine PC, s-PC/GNS, and PC-g-MGNS with their GNS and MGNS contents: 0.1 wt%, 0.5 wt%, 1.0 wt%, and 3.0 wt%. | ||
The excellent mechanical and electrical properties of the PC-g-MGNS nanocomposites are attributed to the high level of dispersion of graphene nanosheets in the PC matrix. In order to verify the dispersion of nanosheets in the PC-g-MGNS nanocomposites, FE-SEM images were obtained. Fig. 6 shows cross-sectional FE-SEM micrographs of the s-PC/GNS and PC-g-MGNS nanocomposites. As the content of GNS is increased from 0.1 to 3.0 wt%, many graphene aggregates (marked by blue arrows) are evident in the s-PC/GNS nanocomposites even at low GNS contents because the interaction between graphene and the polymer is weaker than the van der Waals forces and π–π stacking interactions between the graphene layers.8,25 When the loading is high, consecutively connected graphenes can be seen and the graphene nanosheets are connected to form a conductive pathway. In contrast, individual PC-grafted graphene nanosheets (marked by red arrows) are evident in the PC-g-MGNS nanocomposite, and there are no graphene aggregates. Moreover, all the PC-g-MGNS nanocomposites have dense and smooth fracture surfaces even at high MGNS loadings, whereas the s-PC/GNS nanocomposites have irregular and rough fracture surfaces. The homogeneous dispersion of graphene in the PC-g-MGNS nanocomposites is ascribed to the chemically grafting of the PC chains onto the graphene nanosheets, which results in enhanced mechanical and electrical properties.
 | ||
| Fig. 6 SEM images of fracture surfaces of s-PC/GNS nanocomposites with various GNS contents: (a) 0.1 wt%, (b) 0.5 wt%, (c) 1.0 wt%, and (d) 3.0 wt%, and of PC-g-MGNS nanocomposites with various MGNS contents: (e) 0.1 wt%, (f) 0.5 wt%, (g) 1.0 wt%, and (h) 3.0 wt%. Blue and red arrows indicate aggregated and individual graphene nanosheets, respectively. | ||
Conclusions
In summary, we have prepared PC-g-MGNS nanocomposites via the functionalization of graphene with MDI and subsequent grafting with PC. The PC-g-MGNS nanocomposites exhibit a 20.5% increase in tensile strength and a 22.7% increase in Young's modulus over those of conventional solvent-mixed PC–graphene nanocomposites because of the enhanced interfacial interaction between PC and graphene. Moreover, the PC-g-MGNS nanocomposites exhibit significant improvements in their electrical properties because effective conductive paths form in the PC-g-MGNS even at very low graphene loadings (∼1.0 wt%). Microscopy images clearly show that these properties are due to the superior dispersion of the graphene nanosheets in the PC matrix over that in the s-PC/GNS nanocomposites. In contrast to previous methods, this approach provides an effective means of fabricating graphene–polymer nanocomposites with enhanced mechanical and electrical properties for ESD/EMI applications.Conflicts of interest
There are no conflicts to declare.References
- Y. Zhang, Y.-W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201–204 CrossRef CAS PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- B. H. Min, D. W. Kim, K. H. Kim, H. O. Choi, S. W. Jang and H.-T. Jung, Carbon, 2014, 80, 446–452 CrossRef CAS.
- M. Wu, J. Y. Jo, S. J. Kim, Y. Kang, H.-T. Jung and H.-K. Jung, RSC Adv., 2016, 6, 23467–23470 RSC.
- K. H. Kim, Y.-S. Jun, J. A. Gerbec, K. A. See, G. D. Stucky and H.-T. Jung, Carbon, 2014, 69, 543–551 CrossRef CAS.
- Y. Wang, Z. Shi, Y. Huang, Y. Ma, C. Wang, M. Chen and Y. Chen, J. Phys. Chem. C, 2009, 113, 13103–13107 CAS.
- J. D. Fowler, M. J. Allen, V. C. Tung, Y. Yang, R. B. Kaner and B. H. Weiller, ACS Nano, 2009, 3, 301–306 CrossRef CAS PubMed.
- D. W. Kim, J. Choi, D. Kim and H.-T. Jung, J. Mater. Chem. A, 2016, 4, 17773–17781 CAS.
- S. Stankovich, D. A. Dikin, G. H. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- R. Verdejo, M. M. Bernal, L. J. Romasanta and M. A. Lopez-Manchado, J. Mater. Chem., 2011, 21, 3301–3310 RSC.
- J. Liang, Y. Wang, Y. Huang, Y. Ma, Z. Liu, J. Cai, C. Zhang, H. Gao and Y. Chen, Carbon, 2009, 47, 922–925 CrossRef CAS.
- H.-B. Zhang, Q. Yan, W.-G. Zheng, Z. He and Z.-Z. Yu, ACS Appl. Mater. Interfaces, 2011, 3, 918–924 CAS.
- K. M. Shahil and A. A. Balandin, Solid State Commun., 2012, 152, 1331–1340 CrossRef CAS.
- R. J. Young and M. Liu, J. Mater. Sci., 2016, 51, 3861–3867 CrossRef CAS.
- X. Zhao, Q. Zhang, D. Chen and P. Lu, Macromolecules, 2010, 43, 2357–2363 CrossRef CAS.
- Z. Tang, H. Kang, Z. Shen, B. Guo, L. Zhang and D. Jia, Macromolecules, 2012, 45, 3444–3451 CrossRef CAS.
- N. Liu, F. Luo, H. Wu, Y. Liu, C. Zhang and J. Chen, Adv. Funct. Mater., 2008, 18, 1518–1525 CrossRef CAS.
- M. Nichols and C. Peters, Polym. Degrad. Stab., 2002, 75, 439–446 CrossRef CAS.
- A. R. Betz and D. Attinger, Int. J. Heat Mass Transfer, 2010, 53, 3683–3691 CrossRef CAS.
- Q. Wang and T. J. Webster, J. Biomed. Mater. Res., Part A, 2012, 100, 3205–3210 CrossRef PubMed.
- S. Kumar, B. Lively, L. Sun, B. Li and W. Zhong, Carbon, 2010, 48, 3846–3857 CrossRef CAS.
- L. Monnereau, L. Urbanczyk, J.-M. Thomassin, T. Pardoen, C. Bailly, I. Huynen, C. Jérôme and C. Detrembleur, Polymer, 2015, 59, 117–123 CrossRef CAS.
- L. Wang, J. Hong and G. Chen, Polym. Eng. Sci., 2010, 50, 2176–2181 CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- J. Yan, T. Wei, B. Shao, F. Ma, Z. Fan, M. Zhang, C. Zheng, Y. Shang, W. Qian and F. Wei, Carbon, 2010, 48, 1731–1737 CrossRef CAS.
- P. Steurer, R. Wissert, R. Thomann and R. Mülhaupt, Macromol. Rapid Commun., 2009, 30, 316–327 CrossRef CAS PubMed.
- B. Shen, W. Zhai, M. Tao, D. Lu and W. Zheng, Compos. Sci. Technol., 2013, 86, 109–116 CrossRef CAS.
- M. Yoonessi and J. R. Gaier, ACS Nano, 2010, 4, 7211–7220 CrossRef CAS PubMed.
- E. Lago, P. S. Toth, G. Pugliese, V. Pellegrini and F. Bonaccorso, RSC Adv., 2016, 6, 97931–97940 RSC.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- K. H. Kim, M. Yang, K. M. Cho, Y.-S. Jun, S. B. Lee and H.-T. Jung, Sci. Rep., 2013, 3, 3251 CrossRef PubMed.
- G. Raspoet, M. T. Nguyen, M. McGarraghy and A. F. Hegarty, J. Org. Chem., 1998, 63, 6878–6885 CrossRef CAS PubMed.
- T. Yang, L.-h. Liu, J.-w. Liu, M.-L. Chen and J.-H. Wang, J. Mater. Chem., 2012, 22, 21909–21916 RSC.
- D. Li, M. B. Mueller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS.
- M. Kumar, J. S. Chung, B.-S. Kong, E. J. Kim and S. H. Hur, Mater. Lett., 2013, 106, 319–321 CrossRef CAS.
- R. Waltman, J. Pacansky and C. Bates Jr, Chem. Mater., 1993, 5, 1799–1804 CrossRef CAS.
- S. Park, Y. Hu, J. O. Hwang, E.-S. Lee, L. B. Casabianca, W. Cai, J. R. Potts, H.-W. Ha, S. Chen and J. Oh, Nat. Commun., 2012, 3, 638 CrossRef PubMed.
- R. Voggu, B. Das, C. S. Rout and C. Rao, J. Phys.: Condens. Matter, 2008, 20, 472204 CrossRef.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 73 Search PubMed.
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2009, 114, 832–842 Search PubMed.
- A. Gupta, G. Chen, P. Joshi, S. Tadigadapa and P. Eklund, Nano Lett., 2006, 6, 2667–2673 CrossRef CAS PubMed.
- Z. Chen, Y.-M. Lin, M. J. Rooks and P. Avouris, Phys. E, 2007, 40, 228–232 CrossRef CAS.
- C. Casiraghi, A. Hartschuh, E. Lidorikis, H. Qian, H. Harutyunyan, T. Gokus, K. Novoselov and A. Ferrari, Nano Lett., 2007, 7, 2711–2717 CrossRef CAS PubMed.
- L. C. Silva, G. G. Silva, P. M. Ajayan and B. G. Soares, J. Mater. Sci., 2015, 50, 6407–6419 CrossRef CAS.
- B. G. Woo, K. Y. Choi, K. H. Song and S. H. Lee, J. Appl. Polym. Sci., 2001, 80, 1253–1266 CrossRef CAS.
- J. Burkus, J. Org. Chem., 1961, 26, 779–782 CrossRef CAS.
- J. W. Baker and J. Holdsworth, J. Chem. Soc., 1947, 713–726 RSC.
- A. Farkas and P. F. Strohm, Ind. Eng. Chem. Fundam., 1965, 4, 32–38 CAS.
- M. Mennicken, R. Nagelsdiek, H. Keul and H. Höcker, Macromol. Chem. Phys., 2004, 205, 143–153 CrossRef CAS.
- D. Stauffer and A. Aharony, Introduction to percolation theory, CRC press, 1994 Search PubMed.
- S. Dhawan, N. Singh and S. Venkatachalam, Synth. Met., 2002, 129, 261–267 CrossRef CAS.
- C. Klason, D. H. MCQueen and J. Kubát, Macromolecular Symposia, Wiley Online Library, 1996, pp. 247–260 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07537e |
| This journal is © The Royal Society of Chemistry 2017 |