
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and functionalization of 3-bromo-2-(2-chlorovinyl)benzothiophenes as molecular tools†
Guangkuan Zhao,
Mouad Alami* and
Olivier Provot *
*
BioCIS, Equipe Labellisée Ligue Contre le Cancer, Univ Paris-Sud, CNRS, University Paris Saclay, 92290, Châtenay-Malabry, France. E-mail: olivier.provot@u-psud.fr; mouad.alami@u-psud.fr
First published on 27th September 2017
Abstract
An efficient bromocyclization process of ortho-substituted arylmethyl sulfide promoted by N-methyl-pyrrolidin-2-one hydrotribromide led to the synthesis of 3-bromo-2-(2-(di)chlorovinyl)benzothiophene as a polyhalogenated platform. Various arylations on the C3 atom of such di-substituted benzothiophenes and further functionalizations at the chlorine atoms of the benzothiophenes afforded efficient and rapid access to a small library of stereo-defined 2,3-disubstituted benzothiophenes.
Introduction
2,3-Disubstituted benzothiophenes have been well studied by the scientific community in past decades, mainly for their numerous biological properties.1 For example, raloxifene (Evista™), an oral selective estrogen receptor modulator, is prescribed in the prevention and treatment of osteoporosis and is also given for postmenopausal women who are at high risk for invasive breast cancers.2 In contrast to the synthesis of 2,3-diarylbenzothiophenes, which is well reported,3 the preparation of their stereo-defined 2-vinylogous analogues 1a–c is poorly documented (Scheme 1).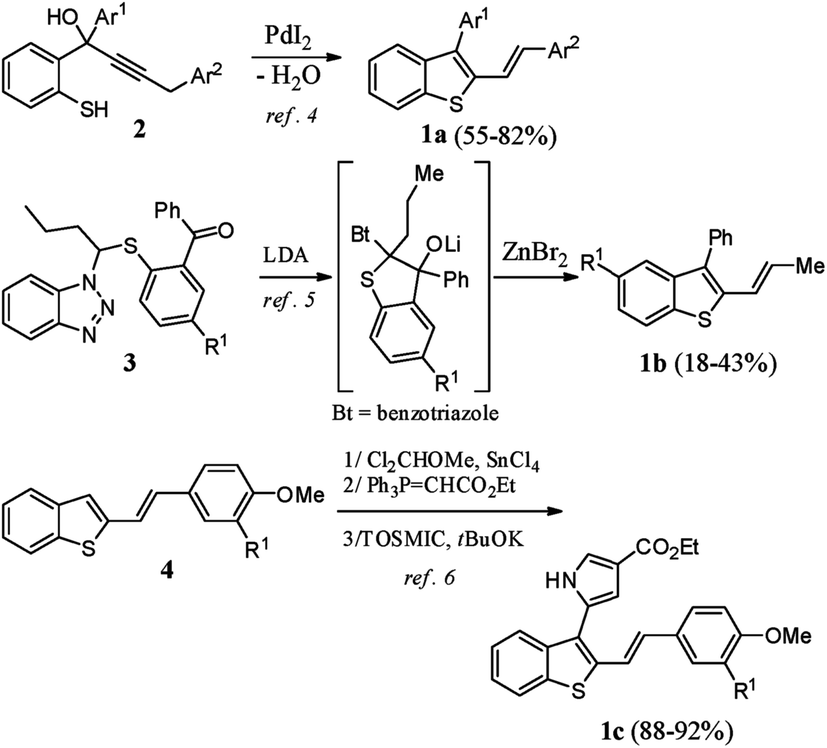 | ||
| Scheme 1 Previous syntheses of 3-aryl-2-alkenyl-benzothiophenes 1a–c. | ||
To our knowledge, these derivatives having (E)-double bonds have been prepared using three different strategies: (i) the Pd-catalyzed heterocyclodehydration of acyclic precursor 2,4 (ii) reaction of 3 with LDA followed by rearrangement in the presence of ZnBr2,5 and (iii) C3-functionalization of 2-substituted benzothiophene 4 through a three-step sequence involving a Rieche formylation, a Wittig reaction, and a pyrrole ring construction under van Leusen reaction conditions.6
In a continuation of our work dedicated to the synthesis of functionalized heterocycles,7 we described a new method to prepare a variety of stereo-defined polyhalogenated platforms 6 through the N-methyl-pyrrolidin-2-one hydrotribromide (MPHT)-promoted bromocyclization of (Z)- and (E)-chloroenynes 5 and subsequent site-selective Suzuki–Miyaura coupling reactions of 6 to prepare various 2,3-disubstituted benzothiophenes 1 (Scheme 2).
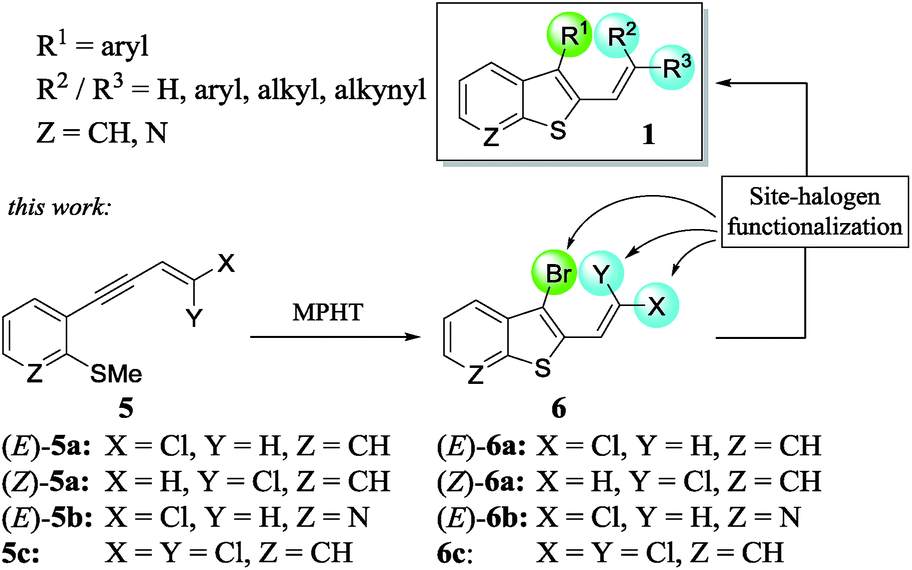 | ||
| Scheme 2 Synthesis of polyhalogenated platforms 6. | ||
We choose a bromocyclization strategy instead of Larock's iodo heteroannulation8 since the site-selective Suzuki–Miyaura coupling of a C–Br vs. C–Cl bond is more challenging from our point of view than the coupling of C–I vs. C–Cl.
Results and discussion
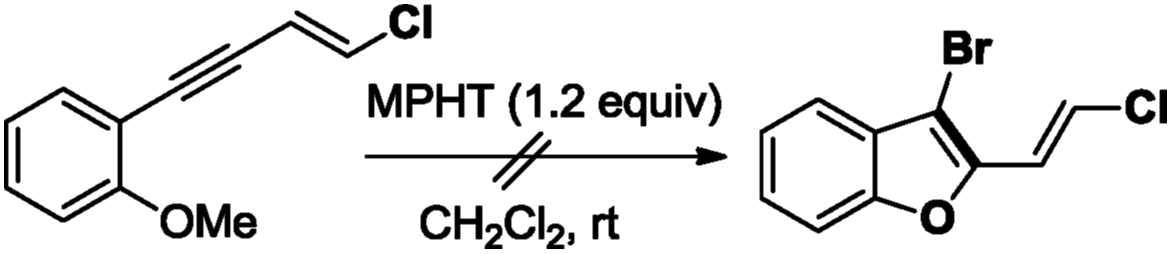
The required 1,3-chloroenynes (E)-5a with an ortho nucleophilic methyl sulfide was prepared via Pd-catalyzed Sonogashira–Linstrumelle coupling reaction.9 We were pleased to observe that in the presence of MPHT, a mild and easy-to-handle brominating agent discovered in our lab,10 (E)-5a11 underwent bromocyclization12 at rt in CH2Cl2 to provide the desired 2,3-disubstituted benzothiophene platform (E)-6a in a good (88%) yield13 (Scheme 3).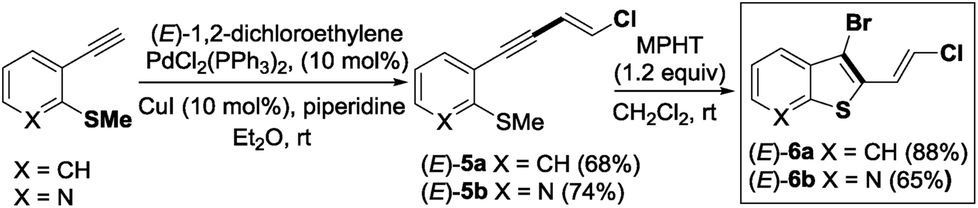 | ||
| Scheme 3 MPHT promoted the cyclization of 1,3-chloro-enyne (E)-5a,b. | ||
The scope of this bromocyclization was also demonstrated by the synthesis of (E)-6b (Scheme 3), which is suitable for the preparation of 7-aza-benzothiophene-containing scaffolds found in drug discovery.14
Next, we focus our attention on the identification of efficient experimental conditions for site-selective Suzuki–Miyaura coupling reactions between 3-bromobenzothiophene (E)-6a and arylboronic acid 7 (1.3 equiv.) as coupling partners (Table 1). Initially, to compare the reactivity of boronic acids towards a 2-vinylchlorine moity vs. a probably more reactive 3-bromine atom on a benzothiophene scaffold, we tested the conditions used for the coupling of chloroenynes15 using Pd(PPh3)4 (5 mol%) as the catalyst, K2CO3 (2 equiv.) as the base, and toluene/MeOH (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the solvent at 90 °C. However, no selectivity was observed, and the expected C-3 monoarylated benzothiophene (E)-8a was isolated in 17% yield accompanied with (E)-9 (6%) and significant amounts (39%) of the diarylated product (E)-1aa (entry 1). This result clearly highlighted that the selective introduction of an aryl substituent at the C-3 position of (E)-6a is far from trivial, although the C–Br bond is more reactive than the C–Cl bond. It should be noted that 2,3-disubstituted benzothiophene derivatives (E)-8a, (E)-9 and (E)-1aa can be easily separated by column chromatography on silica gel.
:1) as the solvent at 90 °C. However, no selectivity was observed, and the expected C-3 monoarylated benzothiophene (E)-8a was isolated in 17% yield accompanied with (E)-9 (6%) and significant amounts (39%) of the diarylated product (E)-1aa (entry 1). This result clearly highlighted that the selective introduction of an aryl substituent at the C-3 position of (E)-6a is far from trivial, although the C–Br bond is more reactive than the C–Cl bond. It should be noted that 2,3-disubstituted benzothiophene derivatives (E)-8a, (E)-9 and (E)-1aa can be easily separated by column chromatography on silica gel.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | [Pd] | x | Base | Time (h) | Solvent | Yieldb of 8a (%) | Yieldb of 9 (%) | Yieldb of 1aa (%) |
| a Conditions: (E)-6a (1 mmol), 7 (1.3 mmol), [Pd] (0.05 mmol or 0.1 mmol), base (2 equiv.) and solvent (18 mL) were heated in a sealed tube at 90 °C for time indicated in the table under argon atmosphere.b Yield of isolated product.c (E)-6a was recovered unchanged.d 45% of (E)-6a were recovered.e A complex mixture of unidentified products was obtained.f Replacing [PdCl(dbma)(IMes)] by Pd-PEPPSI™-IPr furnished 8a in a slightly lower yield of 82%. | ||||||||
| 1 | Pd(PPh3)4 | 5 | K2CO3 | 7 | Toluene/MeOH (2:1) |
17 | 6 | 39 |
| 2 | PdCl2(PPh3)2 | 5 | K2CO3 | 7 | Toluene/MeOH (2:1) |
0 | 0 | 0 |
| 3 | PdCl2(dppf) | 5 | K2CO3 | 7 | Toluene/MeOH (2:1) |
0c | 0 | 0 |
| 4 | Pd(dba)2 | 5 | K2CO3 | 7 | Toluene/MeOH (2:1) |
0c | 0 | 0 |
| 5 | [PdCl(dbma)(IMes)] | 5 | K2CO3 | 7 | Toluene/MeOH (2:1) |
38d | 0 | 3 |
| 6 | [PdCl(dbma)(IMes)] | 5 | K2CO3 | 24 | Toluene/MeOH (2:1) |
85 | 0 | 10 |
| 7 | [PdCl(dbma)(IMes)]f | 10 | K2CO3 | 24 | Toluene/MeOH (2:1) | 90 | 0 | 7 |
| 8 | [PdCl(dbma)(IMes)] | 10 | K3PO4 | 24 | Toluene/MeOH (2:1) |
85 | 0 | 8 |
| 9 | [PdCl(dbma)(IMes)] | 10 | LiOt-Bu | 24 | Toluene/MeOH (2:1) |
—e | — | — |
| 10 | [PdCl(dbma)(IMes)] | 10 | NEt3 | 24 | Toluene/MeOH (2:1) |
0c | 0 | 0 |
| 11 | [PdCl(dbma)(IMes)] | 10 | KOAc | 24 | Toluene/MeOH (2:1) |
0c | 0 | 0 |
| 12 | [PdCl(dbma)(IMes)] | 10 | K2CO3 | 24 | DMF | 0c | 0 | 0 |
| 13 | [PdCl(dbma)(IMes)] | 10 | K2CO3 | 24 | THF | 0c | 0 | 0 |
| 14 | [PdCl(dbma)(IMes)] | 10 | K2CO3 | 24 | MeOH | 72 | 0 | 17 |
| 15 | [PdCl(dbma)(IMes)] | 10 | K2CO3 | 24 | Toluene | 21 | 0 | 4 |
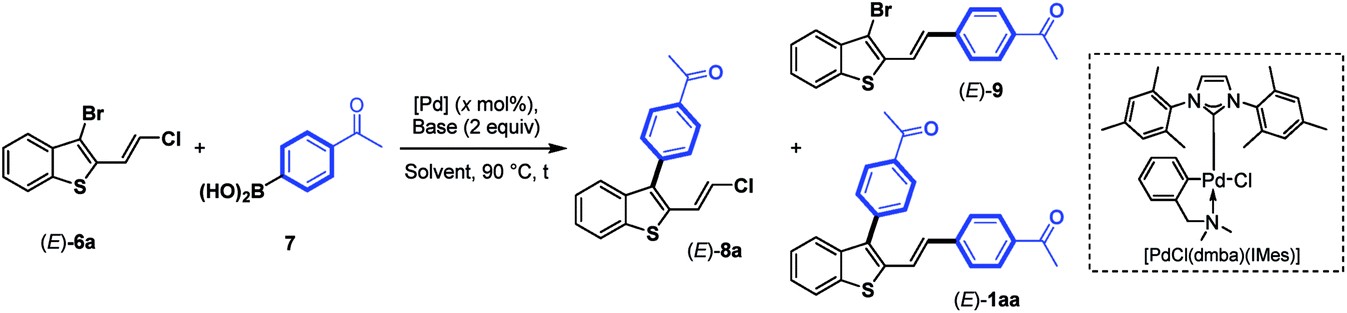
Next, we continued our study by exploring the influence of the palladium- and ligand-controlled site-selective Suzuki–Miyaura cross couplings. No reaction occurred by replacing Pd(PPh3)4 with other palladium sources such as PdCl2(PPh3)2, PdCl2(dppf) and Pd(dba)2 (entries 2–4), even when increasing the amount of the catalyst from 5 to 20 mol%. By using the N-heterocyclic carbene palladacycle precatalyst [PdCl(dmba)(IMes)] (Pd–NHC) developed in 2008 by Ying16 for Heck- and Suzuki-coupling reactions, we were pleased to observe the formation of the desired C-3 monoarylated benzothiophene (E)-8a in a moderate but promising yield of 38% after 7 h of reaction (entry 5). In this case, (E)-9 was not detected, and a trace amount (3%) of the diarylated product (E)-1aa was isolated along with significant amounts (45%) of unreacted (E)-6a. Increasing the reaction time from 7 to 24 h (entry 6) improved the yield of (E)-8a from 38 to 85%, but also increased the quantity of the diarylated product (E)-1aa from 3% to 10%. Finally, using 10 mol% of this Pd–NHC precatalyst with 2 equiv. of K2CO3 in a hot mixture of toluene/MeOH (2:1) for 24 h led to the selective C-3 monoarylation of (E)-6a, thus providing (E)-8a in 90% yield together with 7% of (E)-1aa (entry 7). Due to the σ-donation and steric bulk around the metal, this Pd complex with a carbene ligand instead of phosphine ligands facilitates the oxidative addition and the reductive elimination in the palladacycle. Thereby, the selectivity between a C–Br vs. a C–Cl bond was increased in the presence of a boronic acid. This result was confirmed by replacing [PdCl(dmba)(IMes)] by PEPPSI™-IPr precatalyst, and the reaction furnished mainly the mono-coupling product (E)-8a (82% yield). The effect of the base was next investigated, and K3PO4 gave a similar result to K2CO3 (entry 8). All other bases were unsuccessful in achieving efficient coupling reaction, leading to a complex mixture of by-products when using LiOt-Bu (entry 9) or to unchanged starting material (E)-6a in the presence of NEt3 or KOAc (entries 10 and 11). The effect of the solvent was studied, but no improvement was noted when toluene/MeOH was replaced by DMF, THF, MeOH or toluene (entries 12–15). A mixture of toluene associated with MeOH was found to be the best solvent combination, likely for solubility reasons. The conditions used in entry 7 ([PdCl(dmba)(IMes)] (10 mol%), K2CO3 (2 equiv.), and arylboronic acid (1.3 equiv.) in toluene/MeOH (2:1) in a sealed tube at 90 °C for 24 h) were then used for other coupling reactions using a variety of boronic acids to demonstrate the versatility and the chemoselectivity of the present protocol (Table 2). As expected, using the experimental conditions depicted in entry 7 of Table 1, a variety of arylboronic acids17 bearing electron-donating and electron-withdrawing groups were introduced at the C-3 position of (E)-6a,b, leading to (E)-8a–g in good to excellent yields (59–90%).
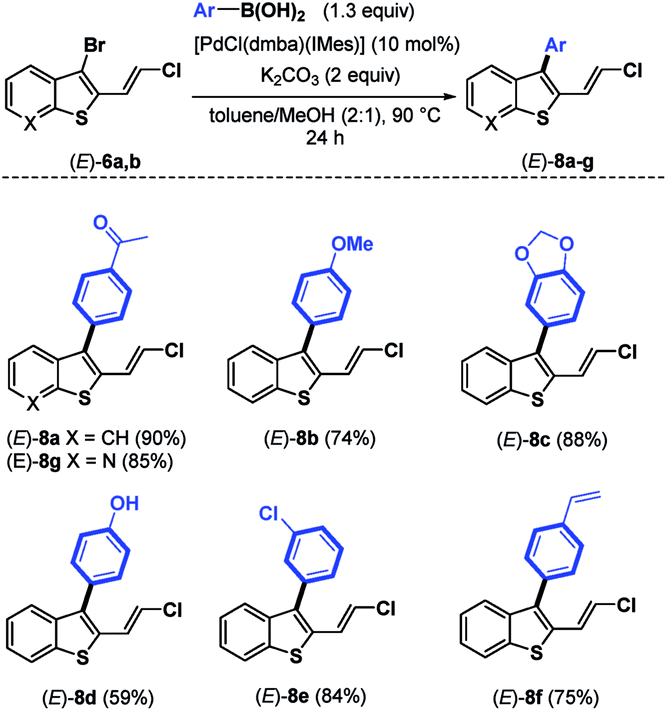 |
As the next logical extension, Suzuki–Miyaura coupling reactions at the remaining C–Cl bond of benzothiophene compounds (E)-8a–d were attempted under the previous conditions reported for the couplings of arylboronic acids with chloroenynes.15 In the presence of Pd(PPh3)4 (5 mol%), K2CO3 (2 equiv.), and arylboronic acid (1.2 equiv.) in a hot mixture of toluene/MeOH, we were pleased to observe the successful replacement of the chlorine atom by various aromatic and heteroaromatic rings (Table 3). The reactions proceeded in good yields (75–92%) with electron-poor and electron-rich arylboronic acids used as coupling partners for (E)-3-aryl-2-(2-chlorovinyl)benzothiophenes (E)-8a–d. It should be noted that using Pd(PPh3)4 (10 mol%), K2CO3 (2 equiv.) and 4-acetylboronic acid in a slight excess (2.2 equiv.) furnished (E)-1aa (see Table 1), in which the boronic acid replaced both the bromine and chlorine atoms of (E)-6a (83%).
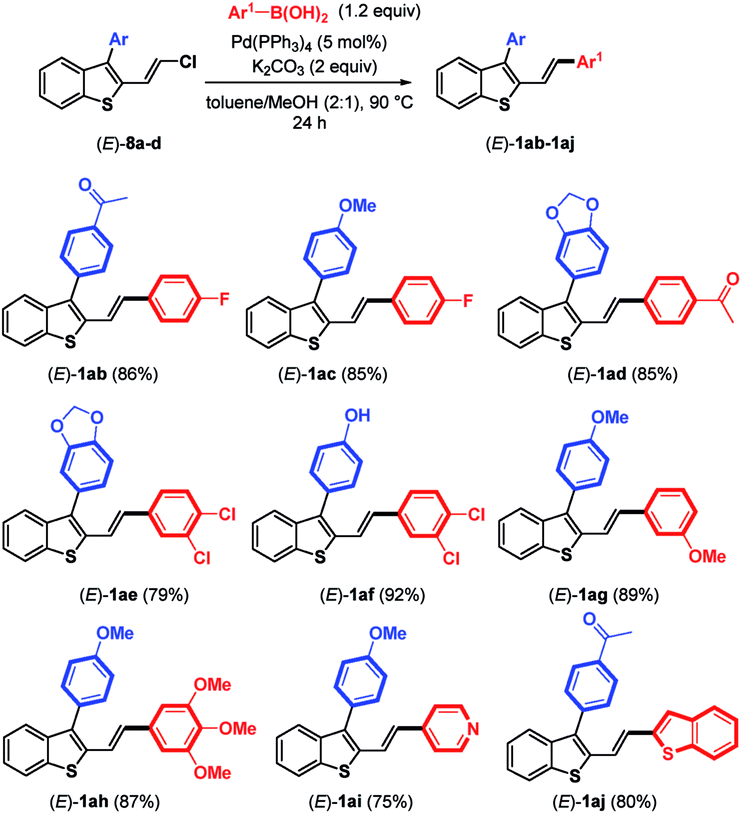 |
Because the reaction conditions for the two-step Suzuki–Miyaura couplings are similar (K2CO3, toluene/MeOH, 90 °C), we investigated whether the two-step coupling reactions could be carried out in a one-pot fashion directly from (E)-6a, avoiding the isolation of the monocoupling products (E)-8. Reactions were conducted using K2CO3 (2 equiv.) in toluene/MeOH (2:1) as the solvent at 90 °C. In the first step, (4-acetylphenyl)boronic acid (1.3 equiv.) reacted with (E)-6a. When consumption of the substrate was complete according to TLC, Pd(PPh3)4 (5 mol%) and 4-fluoroboronic acid were added to the reaction mixture. Accordingly, we were pleased to isolate (E)-1ab containing two different aryl groups in a good overall yield of 68% (Scheme 4). One can also note that the one-pot synthesis of (E)-1ak (65%) was successfully achieved from (E)-6a by inverting the addition order of boronic acids (first 4-fluoroboronic acid followed by 4-acetylboronic acid).
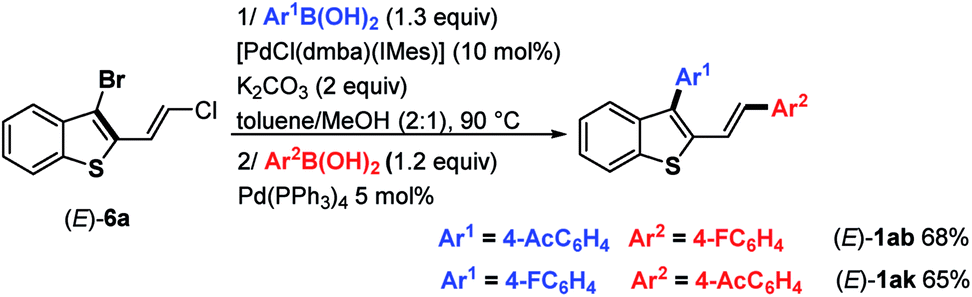 | ||
| Scheme 4 One-pot synthesis of (E)-1ab and (E)-1ak from (E)-6a. | ||
However, it should be noted that attempts to achieve one-pot coupling in the presence of only one catalyst [PdCl(dmba)(IMes)] (10–20 mol%) furnished (E)-1ab in low yield (28%) along with (E)-8a (42%) after 48 h of reaction, clearly indicating that the Pd–NHC precatalyst was not efficient to introduce an aryl substituent on the C–Cl bond of (E)-8a.
2,3-Disubstituted benzothiophenes (E)-1ak were also accessed by inversing the MPHT-bromo-cyclization process followed by the arylation of the resulting 3-bromoposition of the thiophene backbone (Scheme 5).
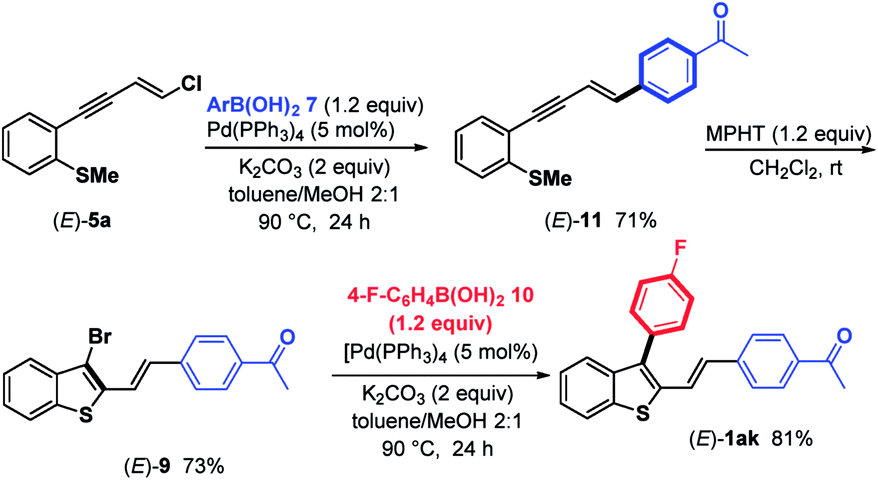 | ||
| Scheme 5 Synthesis of (E)-1ak. | ||
In detail, chloroenyne (E)-5a was initially coupled with (4-acetylphenyl)boronic acid 7 in the presence of Pd(PPh3)4 (5 mol%), K2CO3 (2 equiv.) in toluene/MeOH (2:1) at 90 °C to give the expected 1,4-diarylenyne (E)-11 in 71% yield.15 (E)-11 then undergoes MPHT-promoted bromo-cyclization to furnish the 3-bromobenzothiophene (E)-9 (73%). Further Suzuki–Miyaura coupling between (E)-9 and (4-fluoro-phenyl)boronic acid 10 in the presence of Pd(PPh3)4 (5 mol%) led to (E)-1ak in a good (81%) yield. The comparison of the two strategies, bromocyclization/Suzuki/Suzuki to prepare (E)-1ab–aj and Suzuki/bromocyclization/Suzuki to obtain (E)-1ak, shows that they are equivalent in terms of overall yield and ease of implementation.
Next, the reactivity of 3-aryl-2-(2-chlorovinyl)benzothiophene (E)-8b in Pd-catalyzed couplings was examined to demonstrate the usefulness of such molecular tools. Gratifyingly, the use of PdCl2(PhCN)2 (5 mol%) and CuI (10 mol%) as the catalysts in piperidine at 60 °C (ref. 18) allowed the coupling to proceed efficiently between (E)-8b and ortho-substituted arylalkynes to provide (E)-enynes 12 and 13 in good yields (Scheme 6).
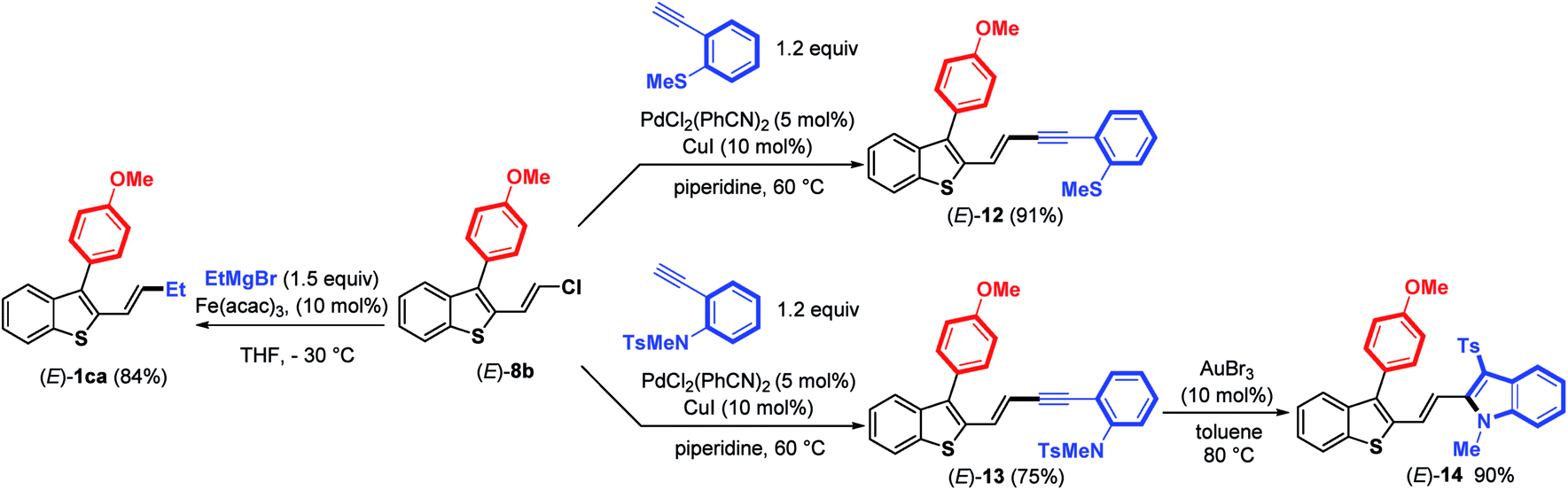 | ||
| Scheme 6 Coupling reactions of vinylchloride (E)-8b with terminal ortho-substituted arylalkynes and EtMgBr. | ||
Taking advantage of the structure of enyne (E)-13 having an ortho-N-tosyl-N-methylaniline function, we achieved a gold-catalyzed cyclization19 to give, after tosyl migration, 2-alkenyl-3-sulfonylindole (E)-14 in 90% yield. We further demonstrated that the C–Cl bond in (E)-8b can react with a Grignard reagent (EtMgBr) in the presence of a catalytic amount of Fe(acac)3 (ref. 20) to introduce an alkyl substituent, thus providing 2-benzothiophene (E)-1ca in a good (84%) yield with no trace of (E)-double bond isomerization. Altogether, these results clearly highlight that it is possible to create various C–C bonds through the coupling of alkyl Grignard reagents (Csp3), arylboronic acids (Csp2) or terminal alkynes (Csp) with the C–Cl bond of benzothiophenes (E)-8, demonstrating the synthetic potential of these molecular tools.
Due to the biological interest in benzothiophenes containing a (Z)-double bond21 at the C-2 position as tubulin polymerization inhibitors, we investigated the preparation of (Z)-6a and explored its coupling to provide stereoselectively (Z)-2,3-disubstituted benzothiophene derivatives (Scheme 7). Thus, by replacing piperidine with nBuNH2 as the base,9 we were able to synthesize the expected chloroenyne (Z)-5a (56%) through the Sonogashira–Linstrumelle coupling of (2-ethynylphenyl)(methyl)-sulfane with (Z)-1,2-dichloroethylene in the presence of catalytic amounts of PdCl2(PPh3)2 and CuI.
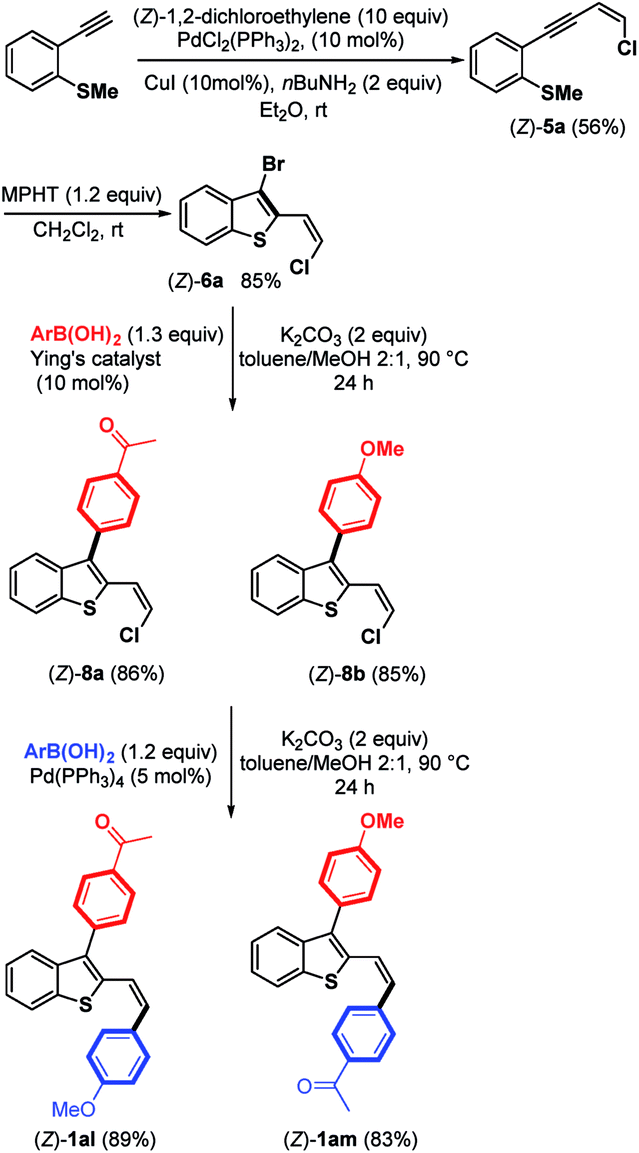 | ||
| Scheme 7 Bromocyclization of (Z)-5a into (Z)-6a and subsequent Suzuki–Miyaura coupling reactions. | ||
Interestingly, the reaction conditions used for the synthesis of (E)-6a (MPHT, CH2Cl2) also allowed us to selectively prepare (Z)-6a (85%) through the bromo-cyclization of (Z)-5a (Scheme 7).
Further Suzuki–Miyaura monoarylation reactions of (Z)-6a with electron-poor phenylboronic acid 7 or the electron-rich (4-methoxyphenyl)boronic acid in the presence of Pd–NHC16 led to the expected coupling products (Z)-8a and (Z)-8b in good yields of 86% and 85%, respectively. Further arylation of the (Z)-Csp2–Cl bond can be accomplished by arylboronic acids in the presence of Pd(PPh3)4 as a palladium source. Push-pull benzothiophene derivatives (Z)-1al and (Z)-1am were synthesized in excellent yields from (Z)-8a and (Z)-8b, respectively, with no trace of (Z)-double bond isomerization (Scheme 7).
The scope of this strategy involving bromocyclization followed by Pd-catalyzed coupling reactions was also extended to the synthesis of polyhalogenated platform 6c, suitable for the elaboration of benzothiophenes containing a trisubstituted double bond at the C-2 position (Scheme 8).
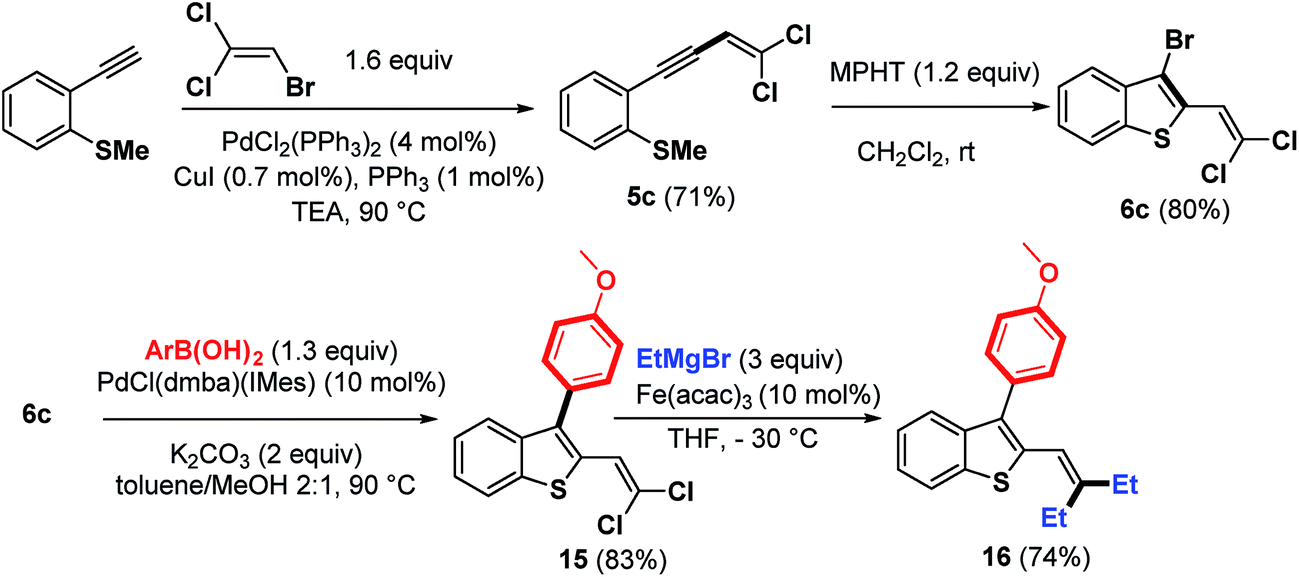 | ||
| Scheme 8 Synthesis of substituted-3-bromobenzothiophenes 6c and its functionalization. | ||
As expected, the bromocyclization process of 5c promoted by MPHT furnished 6c in 80% yield. An illustration of its synthetic potential is shown in Scheme 8. Trihalogenated benzothiophene 6c was first monoarylated at the C-3 position using Pd–NHC precatalyst as a palladium source to give 15 (83%). This product was then coupled with EtMgBr (3 equiv.) in the presence of a catalytic amount of Fe(acac)3 (ref. 20) at −30 °C to give benzothiophene 16 in 74% yield.
Finally, under the mild conditions (MPHT 1.2 equiv., CH2Cl2, rt) described above for the bromocyclization of chloroenynes 5 having an ortho-methyl sulfide group on the aromatic moity, we successfully transformed the ortho-SeMe-aryl chloro enyne (E)-18 into a 2,3-disubstituted benzoselenophene 19 (93%), which is possibly useful for further chemical transformations (Scheme 9).
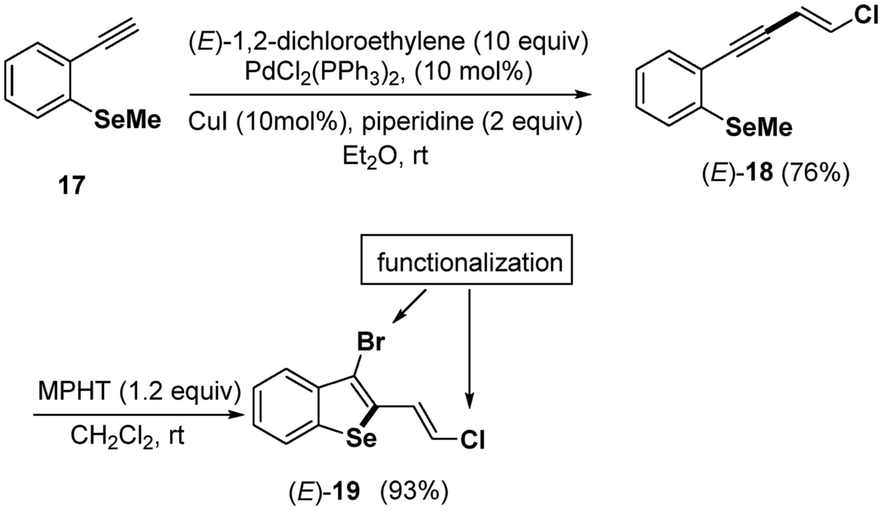 | ||
| Scheme 9 Synthesis of (E)-19. | ||
Efforts are in progress in our lab to develop satisfactory coupling conditions at both the bromine and chlorine atoms of (E)-19 and (Z)-19 benzoselenophenes.
Conclusion
In summary, we have demonstrated that MPHT is a mild and efficient brominating agent useful for the room-temperature bromocyclization of ortho-alkynylaryl methyl sulfides 5. The resulting 3-bromo-2-(2-(di)chlorovinyl)benzothiophenes 6 may serve as di- or tri-halogenated benzothiophene platforms useful for chemoselective and successive coupling reactions (Suzuki, Sonogashira, etc.) leading to rapid and convergent access to a series of 2,3-disubstituted benzothiophenes. We have also demonstrated that it is possible to access to these benzothiophene targets using a complementary strategy involving the arylation of stereo-defined chloroenynes 5a followed by bromocyclization and a second C-3 functionalization on the resulting 3-bromo-benzothiophene (arylation, alkynylation, or alkylation). Finally, we have shown that this bromocyclization process is also efficient for arylselenomethyl ether 18, which was transformed into the novel and potentially functionalizable 2-chlorovinyl-3-bromobenzo selenophene platform 19. We believe that these novel methodologies will find broad applications in synthetic organic chemistry and in pharmaceutical sciences.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support of this project by CNRS, University Paris Sud, and by La Ligue Nationale Contre le Cancer through an Equipe Labellisée 2014 grant. Guangkuan Zhao thanks the CSC (Chinese Scholarship Council) for its Ph.D. funding. Our laboratory BioCIS-UMR 8076 is a member of the Laboratory of Excellence LERMIT supported by a grant from the Agence Nationale de la Recherche (ANR-10-LABX-33).Notes and references
- See for examples: (a) L. H. Li, M. C. Mathieu, D. Denis, A. G. Therien and Z. Y. Wang, Bioorg. Med. Chem. Lett., 2011, 21, 734–737 CrossRef CAS PubMed; (b) L. H. C. Berthelette, A. Chateauneuf, M. Ouellet, C. F. Sturino and Z. Y. Wang, Bioorg. Med. Chem. Lett., 2010, 20, 7440–7443 CrossRef PubMed; (c) R. Romagnoli, P. G. Baraldi, C. L. Cara, E. Hamel, G. Basso, R. Bortolozzi and G. Viola, Eur. J. Med. Chem., 2010, 45, 5781–5791 CrossRef CAS PubMed; (d) L. N. Li, L. Chang, S. Pellet-Rostaing, F. Liger, M. Lemaire, R. Buchet and Y. Q. Wu, Bioorg. Med. Chem., 2009, 17, 7290–7300 CrossRef CAS PubMed; (e) M. Aleksić, B. Bertoša, R. Nhili, L. Uzelac, I. Jarak, S. Depauw, M. H. David-Cordonnier, M. Kralj, S. Tomić and G. Karminski-Zamola, J. Med. Chem., 2012, 55, 5044–5060 CrossRef PubMed; (f) C. Yang, K. Cross, G. J. Myatt, P. E. Blower and J. F. Rathman, J. Med. Chem., 2004, 47, 5984–5994 CrossRef CAS PubMed.
- F. X. Li, J. L. Dou, L. J. Wei, S. X. Li and J. T. Liu, Cancer Chemother. Pharmacol., 2016, 77, 895–903 CrossRef CAS PubMed.
- For recent papers concerning the synthesis of 2,3-diarylbenzothiophenes, see for examples: (a) D. Y. Wan, Y. D. Yang, X. Y. Liu, M. L. Li, S. L. Zha and J. S. You, Eur. J. Org. Chem., 2016, 55–59 CrossRef CAS; (b) T. Yamauchi, F. Shibahara and T. Murai, Tetrahedron Lett., 2016, 57, 2945–2948 CrossRef CAS; (c) Y. Masuya, M. Tobisu and N. Chatani, Org. Lett., 2016, 18, 4312–4315 CrossRef CAS PubMed; (d) I. Smari, H. Ben Ammar, B. Ben Hassine, J. F. Soulé and H. Doucet, Synthesis, 2015, 47, 3354–3362 CrossRef CAS; (e) K. Nobushige, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1188–1191 CrossRef CAS PubMed; (f) L. Q. Zhao, C. Bruneau and H. Doucet, Tetrahedron, 2013, 69, 7082–7089 CrossRef CAS; (g) J. Yin, Y. Zhou, T. Lei and J. Pei, Angew. Chem., Int. Ed., 2011, 50, 6320–6323 CrossRef CAS PubMed; (h) C. C. McAtee, P. S. Riehl and C. S. Schindler, J. Am. Chem. Soc., 2017, 139, 2960–2963 CrossRef CAS PubMed; (i) P. R. Likhar, S. M. Salian, S. Roy, M. L. Kantam, B. Sridhar, K. V. Mohan and B. Jagadeesh, Organometallics, 2009, 28, 3966–3969 CrossRef CAS; (j) M. S. Subhas, S. S. Racharlawar, B. Sridhar, P. K. Kennady, P. R. Likhar, M. L. Kantam and S. K. Bhargava, Org. Biomol. Chem., 2010, 8, 3001–3010 RSC.
- B. Gabriele, R. Mancuso, E. Lupinacci, L. Veltri, G. Salerno and C. Carfagna, J. Org. Chem., 2011, 76, 8277–8286 CrossRef CAS PubMed.
- A. R. Katritzky, K. Kirichenko, D. Hür, X. M. Zhao, Y. Ji and P. J. Steel, ARKIVOC, 2004, 6, 27–44 Search PubMed.
- P. Raju and A. K. Mohanakrishnan, Eur. J. Org. Chem., 2016, 4361–4371 CrossRef CAS.
- (a) M. Jacubert, O. Provot, J. F. Peyrat, A. Hamze, J. D. Brion and M. Alami, Tetrahedron, 2010, 66, 3775–3787 CrossRef CAS; (b) B. Tréguier, E. Rasolofonjatovo, A. Hamze, O. Provot, J. Wdzieczac-Bakala, J. Dubois and M. Alami, Eur. J. Org. Chem., 2011, 4868–4876 Search PubMed; (c) G. Le Bras, A. Hamze, S. Messaoudi, O. Provot, P. B. Le Calvez, J. D. Brion and M. Alami, Synthesis, 2008, 1607–1611 CAS; (d) G. K. Zhao, L. Z. Yuan, M. Roudier, J. F. Peyrat, A. Hamze, J. D. Brion, O. Provot and M. Alami, Synthesis, 2016, 48, 3382–3392 CrossRef CAS.
- For a recent review on the synthesis of iodoheterocycles by iodocyclization, see: (a) B. Gabriele, R. Mancuso and R. C. Larock, Curr. Org. Chem., 2014, 18, 341–358 CrossRef CAS; (b) B. Godoi, R. F. Schumacher and G. Zeni, Chem. Rev., 2011, 111, 2937–2980 CrossRef CAS PubMed and references herein; (c) T. Kesharwani, S. A. Worlikar and R. C. Larock, J. Org. Chem., 2006, 71, 2307–2312 CrossRef CAS PubMed; (d) D. W. Yue and R. C. Larock, J. Org. Chem., 2002, 67, 1905–1909 CrossRef CAS PubMed; (e) C. H. Cho, D. I. I. Jung, B. Neuenwander and R. C. Larock, ACS Comb. Sci., 2011, 13, 501–510 CrossRef CAS PubMed; (f) S. Mehta, J. P. Waldo and R. C. Larock, J. Org. Chem., 2009, 74, 1141–1147 CrossRef CAS PubMed; (g) N. A. Danalkina, A. E. Kulyashova, A. F. Khlebnikov, S. Brase and I. A. Balova, J. Org. Chem., 2014, 79, 9018–9045 CrossRef PubMed; (h) S. Mehta and R. C. Larock, J. Org. Chem., 2010, 75, 1652–1658 CrossRef CAS PubMed; (i) C. H. Cho, B. Neuenswander and R. C. Larock, J. Comb. Chem., 2010, 12, 278–285 CrossRef CAS PubMed; (j) C. H. Cho, D. I. I. Jung and R. C. Larock, Tetrahedron Lett., 2010, 51, 6485–6488 CrossRef CAS PubMed; (k) G. Ferrara, T. Jin, M. Akhtaruzzaman, A. Islam, L. Y. Han and H. Yamamoto, Tetrahedron Lett., 2012, 53, 1946–1950 CrossRef CAS; (l) N. A. Danilkina, S. Bräse and I. A. Balova, Synlett, 2011, 4, 517–520 Search PubMed.
- For the Pd-coupling of (Z)-1,2-dichloroethylene with terminal alkynes, we have demonstrated that nBuNH2 is superior to piperidine as base, see: (a) M. Alami, S. Gueugnot, E. Domingues and G. Linstrumelle, Tetrahedron, 1995, 51, 1209–1220 CrossRef CAS; (b) M. Alami, J. F. Peyrat and J. D. Brion, Synthesis, 2000, 1499–1518 CrossRef CAS.
- (a) J. F. Berrien, O. Provot, D. Joseph and A. Bekaert, J. Chem. Educ., 2004, 81, 1348–1349 CrossRef CAS; (b) A. Bekaert, O. Provot, O. Rasolojahona, M. Alami and J. D. Brion, Tetrahedron Lett., 2005, 46, 4187–4191 CrossRef CAS.
- Switching from (E)-5a to substrate (E)-1-(4-chlorobut-3-en-1-yn-1-yl)-2-methoxybenzene having a less nucleophile ortho OMe substituent, no bromocyclization occurred in the presence of MPHT to give the expected 3-bromo-2-chlorovinyl-benzofuran derivative
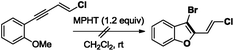 .
. - It is also possible to prepare a 3-iodo-2-(chlorovinyl)benzothiophene using I2 and chloroenyne (E)-5a (82%).
- When NBS (1.2 equiv.) was used in place of MPHT, (E)-6a was obtained in a poor yield of 20%. Using Br2, the reaction furnished (E)-6a in 80%. Solid MPHT is more convenient to handle than Br2 particularly when small quantities were used (1.2 equiv., 61 μL).
- B. E. Sleebs, A. Levit, I. P. Street, H. Falk, T. Hammonds, A. C. Wong, M. D. Charles, M. F. Olson and J. B. Baell, Med. Chem. Commun., 2011, 2, 977–981 RSC.
- A. Tikad, A. Hamze, O. Provot, J. D. Brion and M. Alami, Eur. J. Org. Chem., 2010, 725–731 CrossRef CAS.
- (a) E. A. B. Kantchev, G. R. Peh, C. Zhang and J. Y. Ying, Org. Lett., 2008, 10, 3949–3952 CrossRef CAS PubMed; (b) G. R. Peh, E. A. B. Kantchev, J. C. Er and J. Y. Ying, Chem.–Eur. J., 2010, 14, 4010–4017 CrossRef PubMed.
- Experimental conditions found to prepare 3-arylated benzothiophenes (E)-8a–g (see Table 1, entry 7) failed with heterocyclic boronic acids such as 3-pyridineboronic acid, 4-pyridine boronic acid and benzo[b]thiophen-2-ylboronic acid and starting material (E)-6a was recovered unchanged.
- (a) M. Alami and G. Linstrumelle, Tetrahedron Lett., 1991, 32, 6109–6112 CrossRef CAS; (b) M. Alami, B. Crousse and F. Ferri, J. Organomet. Chem., 2001, 624, 114–123 CrossRef CAS.
- I. Nakamura, U. Yamagishi, D. Song, S. Konta and Y. Yamamoto, Chem.–Asian J., 2008, 3, 285–295 CrossRef CAS PubMed.
- (a) G. Cahiez and H. Avedissian, Synthesis, 1998, 1199–1205 CrossRef CAS; (b) M. Seck, X. Frank, R. Hocquemiller, B. Figadère, J. F. Peyrat, O. Provot, J. D. Brion and M. Alami, Tetrahedron Lett., 2004, 45, 1881–1884 CrossRef CAS; (c) M. Dos Santos, X. Frank, R. Hocquemiller, B. Figadère, J. F. Peyrat, O. Provot, J. D. Brion and M. Alami, Synlett, 2004, 15, 2697–2700 Search PubMed.
- T. T. B. Nguyen, T. Lomberget, N. C. Tran, E. Colomb, L. Nachtergaele, S. Thoret, J. Dubois, J. Guillaume, R. Abdayem, M. Haftek and R. Barret, ACS Med. Chem. Lett., 2012, 22, 7227–7231 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07340b |
| This journal is © The Royal Society of Chemistry 2017 |