
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dissipative particle dynamics and molecular dynamics simulations on mesoscale structure and proton conduction in a SPEEK/PVDF-g-PSSA membrane
Yue Ma†
,
Yuxiang Wang†,
Xuejian Deng,
Guanggang Zhou,
Sha Khalid ,
Xiaoliang Sun,
Wei Sun,
Qiong Zhou and
Guiwu Lu*
,
Xiaoliang Sun,
Wei Sun,
Qiong Zhou and
Guiwu Lu*
College of Science, China University of Petroleum, Beijing 102249, China. E-mail: lugw@cup.edu.cn; sunw@cup.edu.cn; zhouqiong_cn@163.com; Fax: +86 10 6974 4849
First published on 14th August 2017
Abstract
The proton exchange membrane fuel cell (PEMFC) is an innovative technology for the realization of next-generation power sources, in which a polymer or blend membrane is used to separate the fuel from the oxidant and to transport protons. Although a variety of membranes have been synthesized and characterized by both experimental and molecular simulation methods in the past few decades, the underlying microscopic molecular mechanism is still unclear. In this article, a blend membrane composed of SPEEK and PVDF is investigated by dissipative particle dynamics (DPD) and molecular dynamics (MD) simulation methods. The results show that the blend of SPEEK and PVDF has preferable compatibility as the sulfonation degree is about 50%, and the compatibility is improved further by adding 10% PSSA grafting additive. For both 90/10 and 80/20 blending proportions, SPEEK and PVDF-g-PSSA mixed fairly well, and a proton exchange channel network is observed. What's more, as the PVDF-g-PSSA content increases from 5% to 20%, the hydrophilic clusters, which consist of sulfonate groups, water molecules and hydronium ions, are formed and aggregate inside the SPEEK membrane. In addition, the transport properties and proton conductivities of the blend membrane are also significantly improved by the increase of the content of PVDF-g-PSSA.
1. Introduction
Proton exchange membrane fuel cell (PEMFC) technology is a promising research area, which directly translates the chemical energy existing in the fuel and oxidant into electrical energy.1,2 One of the most important parts of the PEMFC is the proton exchange membrane (PEM), which separates fuel from oxidant.3,4 A fluorinated PEM like Nafion®, possesses superior proton conductivity but also high fuel permeability and high costs. These drawbacks have limited the applications of Nafion® in PEMFCs.5,6 In recent years, various alternative PEMs such as sulfonated polyimides, sulfonated polysulfones and sulfonated poly(ether ether ketone) (SPEEK)7–11 have been extensively studied.SPEEK has been proved to be the most effective alternative to Nafion® because of its low methanol permeability, low commercial cost and excellent thermodynamic stability. However, the performance of SPEEK highly depends on the degree of sulfonation. To improve the proton conductivity, it is important to increase the sulfonation degree of SPEEK. While high sulfonation degree will also lead to brittleness, obvious reduction of methanol resistance (the proton exchange membrane needs to prevent the anode from contacting with methanol fuel) and dimensional stability.12 In addition, the low hydrolytic stability caused by high sulfonation degree also limits the application of SPEEK in PEMFCs at higher temperature.13,14
The blend membrane of SPEEK and PVDF showed higher stability and improved methanol permeability, which benefits the application of PEMFCs.15,16 However, attenuated proton conductivity always appears due to the blend of SPEEK and PVDF. And the mixture of SPEEK with PVDF may also lead to a decrease in the stability of the membrane when PVDF content is increased.17–19 The reason why these results happen is that the hydrophilic SPEEK is incompatible with the hydrophobic PVDF. Herein, it is a good idea to construct SPEEK/PVDF blend membranes to improve their miscibility and homogeneous separation. To improve the compatibility between SPEEK and PVDF, Liu20 et al. designed a new type of SPEEK/PVDF-g-PSSA blend membrane in previous experimental works.
Recently, many computational simulations have been performed to study the structure and dynamic properties of the proton exchange membranes. In water and methanol solvated fluorinated membranes such as Nafion®, the relationship between microstructure and properties was efficiently illustrated. The simulation methods, including dissipative particle dynamics (DPD) and molecular dynamics (MD), are mainly used to study the mesoscale structure and dynamical properties. Yamamoto et al. conducted the first DPD simulation of Nafion® in acid form,21 in which the conservative repulsion parameters were calculated with the mixing energy generated by fully atom molecular dynamic simulation. Jorn and Voth22 used the smoothed particle hydrodynamics method to calculate proton conductivity in Nafion®. The calculation results are in good agreement with the experimental conductivity data. On the other hand, the dynamic and static characteristics of SPEEK membranes were simulated by using MD under different hydration degree23–25 by Brunello et al. and Mahajan et al. Komarov and others also studied the conformation of SPEEK membrane by using dynamic density functional theory (DDFT) and MD methods at different degrees of sulfonation and hydration levels.26,27
The DPD and MD studies mentioned above made an important step toward modeling the segregation morphology and transport properties of SPEEK membrane. However, there has been very few simulations reported SPEEK and PVDF blend membranes. The mechanism for proton transport channel formation and mesoscopic morphology evolution in blending membrane is still unclear. It is needed to be carried out with further theoretical insights in details to illustrate how the sulfonation degree, grafting and blending ratio affect the structure and performance of SPEEK/PVDF blend membrane. Therefore, in this paper we investigated the effect of PVDF-g-PSSA on the mesoscopic morphology, diffusion dynamics in the blend membrane and transport dynamics-structure relationship at different temperatures using DPD and MD methods. The simulation results were compared with the previous experimental observations. Importantly, we also provided the theoretical insights for the structure–property relationship of the blend membranes in PEMFC.
2. Methodology
2.1 Coarse-grained model
In this paper, the DPD simulations for the SPEEK/PVDF-g-PSSA blends (Fig. 1) were performed using the Materials Studio software. Before the simulation, we used one bead to represent several groups of atoms (see Fig. 1) and, each monomer of the SPEEK are divided into bead A (sulfonated parts) and bead B (unsulfonated part), which is called coarse-grained approach. The PVDF-g-PSSA is considered to be bead C and D. Therefore, the numbers of DPD beads of SPEEK and PVDF-g-PSSA could be obtained by their respective degrees of polymerization. Table 1 show the parameters used in the simulations (all at 298 K). The adopted dissection of the SPEEK and PVDF-g-PSSA into coarse-grained beads is performed to minimize the difference in volumes between the fragments represented by different bead types. The effective volume of the beads is calculated with Gaussian 09 software. For each blend, the total number of beads was 1.92 × 105 in a simulation box 40 × 40 × 40 rc3, and the real size of the simulation box is about 36 × 36 × 36 nm3. The total simulation time is 541 ns with 4 × 105 DPD steps and the time step is 0.05. After the initial 1 × 105 DPD steps in calculation, temperature and pressure of system remained stable, so that the time of 4 × 105 DPD steps is sufficient to allow for the polymers to find their equilibrium configuration. The thermal energy kBT = 1 was used to maintain the default values of dissipation parameter 4.5 and spring constant 4.0.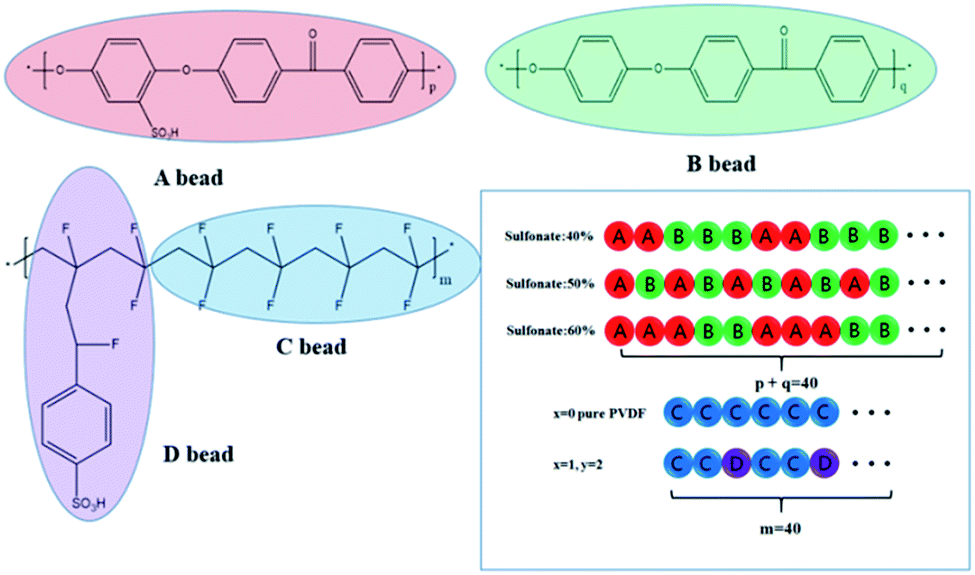 | ||
| Fig. 1 The chemical formula and coarse-grained model of SPEEK (A bead), PEEK (B bead), PVDF (C bead) and g-PSSA (D bead). The bead chains of different sulfonate degree and graft are shown in square frame. | ||
| Bead type | Fragment | Effective volume (Å3) | Solubility parameter ((J cm−3)0.5) | Conservative parameter (aij) | |||
|---|---|---|---|---|---|---|---|
| A | B | C | D | ||||
| A | C19H12O6S | 282.3 | 21.58 | 25.00 | 28.72 | 55.13 | 38.68 |
| B | C19H12O3 | 256.5 | 23.61 | 28.72 | 25.00 | 61.46 | 45.71 |
| C | [C4H6F4]2 | 261.6 | 14.15 | 55.13 | 61.46 | 25.00 | 30.37 |
| D | C13H16F4O3S | 238.3 | 17.83 | 38.68 | 45.71 | 30.37 | 25.00 |
2.2 DPD methods
Compared to conventional molecular dynamics simulation methods, DPD is a coarse-grained simulation method aiming for calculations in larger space and longer time scales.28–30 In DPD, several atoms or repeating units are bundled together to form a single group (bead), which move in phase space following Newton's motion equations. The mass values of each bead is normalized to 1 (reduced unit), and the force imposed on bead equals to the summation of three pair interactive force: conservative (FC), dissipative (FD), and random (FR) forces. The three forces are given by:
 | (1) |
In principle, the number density ρ chosen in DPD simulation is a free value, but the system with a larger ρ requires a longer computational time. Groot and Warren29 suggested that ρ = 3 and a = 25kBT are reasonable parameters for liquids. The interaction parameters of different beads aij and the Flory–Huggins interaction parameter χ are followed aij = aii + 3.50χ. There are many calculation methods can be used to calculate Flory–Huggins parameters, including the solubility parameter method, in which Flory–Huggins parameter χ could be estimated from
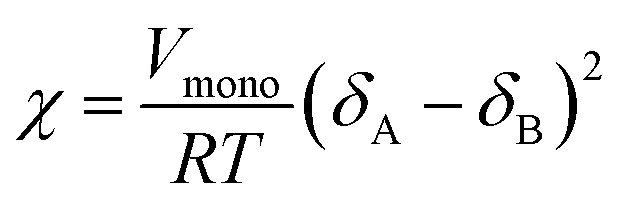 | (2) |
2.3 Molecular dynamics
For proton conduction performance of the composite membrane, we re-select the parameters and model on the atomic scale to calculate the RDFs and proton conductivity. The atomistic structure models of SPEEK/PVDF-g-PSSA blends were investigated by molecular dynamics (MD) simulations. Polymer chains were first constructed by repeating units of SPEEK and PVDF-g-PSSA, and then cubic simulation boxes with periodic boundary condition were built by using of the Amorphous Cell module. The number of repeat unites, atoms, setting initial density, and box side length are listed in Table 2. The water content was initially set as λ = 10 (λ is the ratio of the number of water molecules to sulfonate groups, n(H2O)/n(SO3−)).| SPEEK/PVDF-g-PSSA compositions | Number of SPEEK/PVDF-g-PSSA units | Number of H3O+ | Number of H2O | Initial density (g cm−3) | Box length (Å) |
|---|---|---|---|---|---|
| 100/0 | 40/0 | 40 | 400 | 1.213 | 36.04 |
| 90/10 | 36/4 | 40 | 400 | 1.232 | 35.92 |
| 80/20 | 32/8 | 40 | 400 | 1.248 | 35.81 |
MD simulations were performed according to the following steps: the system was first minimized with steepest descents and conjugate gradients minimization algorithms to relax overlapping atoms. After initial minimization, the dynamics simulation procedure was performed as follows: (1) in NVT ensemble, simulation was run at 800 K within 60 ps; (2) after then, simulation was conducted in NPT ensemble within 50 ps from 800 K to 298 K with intervals of 75 K; (3) simulation was carried on in NVT ensemble at 298 K within 60 ps; (4) the equilibrium system state was achieved at 298 K after 100 ps in NPT ensemble; (5) MD simulation under NVT ensemble at 298 K was performed in 5 ns, of which the last 1 ns trajectories were used for analysis of structural and dynamic properties of the system. The Berendsen thermostat and barostat methods were applied for all MD simulations to control the temperature and pressure at desired temperature and ambient pressure during dynamics runs. All of the above steps were performed using the COMPASS force field and the Materials Studio Program package. This force field is the first ab initio force field that its parameterization and validation have been done using condensed-phase properties and various ab initio and empirical data of various molecules. In addition to bonded and non-bonded interactions that were considered by other force fields, COMPASS also takes into account the cross coupling interactions, thus enabling accurate prediction of various properties for a wide range of materials especially for polymer molecules. Partial atomic charges of the all molecules were assigned by means of the charge equilibration method. The structure of H3O+ ( ) is from our earlier article.31
) is from our earlier article.31
3. Results and discussion
3.1 Morphology
From DPD simulations, the effect of sulfonated degree on the blends morphology is investigated, and the results are shown in Fig. 2. In general, the evolution of system morphology is qualitatively similar to the classical scenario of the percolation system formation. At low sulfonate levels (Fig. 2(a)), SPEEK and PVDF formed small clusters and the density of SPEEK (bead A) is about 1.1. The average densities of PVDF and unsulfonated part of SPEEK are 0.2 and 1.6, respectively. When the sulfonation degree increased to more than 50% (Fig. 2(b) and (c)), the isolated PVDF clusters merged into larger aggregates and mixed with SPEEK clusters which formed a three-dimensional vermicular informal network structure that leads to gradually weakened phase separation. The reasons for these phenomena are that the sulfonic acid group of SPEEK and PSSA gathered and formed three-dimensional hydrophilic wormlike structure. These results indicate that the compatibility of SPEEK and PVDF is improved, and as a result, a proton conduction channel is formed at higher sulfonated degree. When the sulfonated degree further increased to 60%, the size of PVDF clusters started to decrease, and the volume density distribution presents a multimodal fluctuation. This is due to a high degree of sulfonation affects the dimensional stability of SPEEK. From the results, we found that the blend of SPEEK and PVDF-g-PSSA has preferable compatibility as the sulfonation rate is about 50%.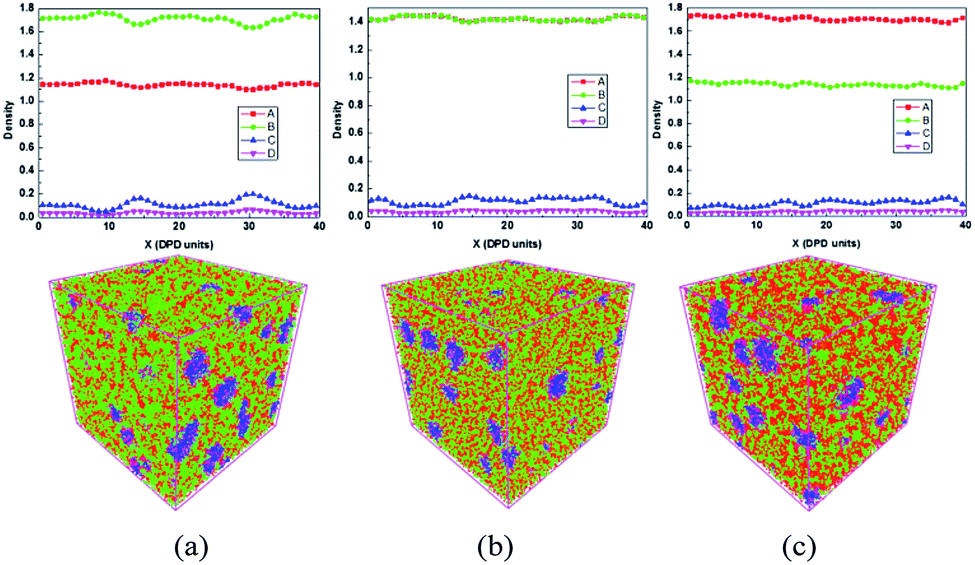 | ||
| Fig. 2 The density distribution (upper section) and the mesoscopic simulation morphology (bottom section) of 40% (a), 50% (b) and 60% (c) degree of sulfonation in blends at 300 K. The bead A, B, C and D, is shown in red, green, blue and purple, respectively (the real side length of all the cube boxes is 36 nm). | ||
Fixing SPEEK 50% sulfonated degree, we studied the graft's effect on the blends morphology, and the result is shown in Fig. 3. For non-grafting, the snapshot (Fig. 3(c)) and isopycnic surface (Fig. 3(e)) indicated that PVDF formed many dispersed spherical particles. The corresponding density distribution of the particles (Fig. 3(a)) presents a multimodal fluctuation, which suggests that SPEEK has poor compatibility with pure PVDF, as shown in the obvious phase separation. In another case, i.e. 10% grafting rate, grafted part (PSSA) has connected SPEEK and PVDF, and the presented multimodal fluctuation amplitude (Fig. 3(b)) is less than that of non-grafting (Fig. 3(a)). The density fluctuation amplitude of PVDF is 0.12 before grafting and decreased after grafting, which is about 0.07. This indicates that the compatibility of SPEEK and PVDF has improved in the case of 10% grafting, which contributes to the formation of the channel network for proton exchange.
 | ||
| Fig. 3 The density distribution (a, b), the mesoscopic simulation morphology (c, d) and isopycnic surface (e, f) of PVDF in non-grafting and grafted (grafting ratio is 10%) blends at 300 K (the real side length of all the cube boxes is 36 nm). | ||
Fixing the sulfonation degree and grafting ratio at 50% and 10%, we studied the effect of blending ratio on the mesoscopic blend membrane structure by adjusting the blending proportion of SPEEK and PVDF, and the morphologies of the blends after 4 × 105 time steps are shown in Fig. 4. By visualization, SPEEK and PVDF-g-PSSA mix fairly well in 90/10 and 80/20 blends, whereas isolated SPEEK and PVDF domains are still observed (Fig. 3(c)). As shown in Fig. 4, as the content of PVDF increased, the SPEEK clusters gradually deformed, and PVDF-g-PSSA formed irregular mesh structure. This is because the increase of PVDF content promotes the hydrophilic channel formations. The compatibility of PVDF and SPEEK is improved, which provides a proton conduction channel. In the case of 80/20 blending ratio, channel sinuosity appeared, which will affect the proton conduction efficiency, and therefore it can be concluded that the proton conductivity will decrease at a ratio of more than 20% blending proportion. We have defined the “channel sinuosity” as the ratio of “the size of a cluster composed of SPEEK beads” to “bead numbers in a cluster”. So the channel sinuosity was calculated and the results were shown in Table 3, which better exhibited the correlation between the values of channel sinuosity and the conductivity values.
 | ||
| Fig. 4 The isopycnic surface (upper section) and mesoscopic simulation morphology (bottom section) of 5, 10 and 20 wt% PVDF-g-PSSA in blends at 300 K (the real side length of all the cube boxes is 36 nm). | ||
| PVDF-g-PSSA (wt%) | 0 | 5 | 10 | 20 |
| Channel sinuosity (rc3) | — | 252 | 165 | 95 |
| Proton conductivity (mS cm−1) | 66 | — | 43 | 28 |
3.2 Compatibilizing progress
Fixing the sulfonation degree and grafting ratio, the system morphology evolution process is studied with 95/5 and 90/10 SPEEK/PVDF-g-PSSA blend ratio, respectively, and the results are shown in Fig. 5. From 100 to 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps, the morphologies of both the 95/5 and 90/10 blends evolve from a randomly disordered small sphere particle to a regular network of spherical PVDF clusters up to 6 and 9 nm in diameter respectively. The formed spherical PVDF clusters were connected by SPEEK cylindrical channels of approximately 3 nm in diameter. The polymers evolve from a homogeneous state to a heterogeneous one with ordered mesoscopic domains. Those results are in agreement with the experimental results of Liu et al.20
000 steps, the morphologies of both the 95/5 and 90/10 blends evolve from a randomly disordered small sphere particle to a regular network of spherical PVDF clusters up to 6 and 9 nm in diameter respectively. The formed spherical PVDF clusters were connected by SPEEK cylindrical channels of approximately 3 nm in diameter. The polymers evolve from a homogeneous state to a heterogeneous one with ordered mesoscopic domains. Those results are in agreement with the experimental results of Liu et al.20
 | ||
| Fig. 5 The morphological evolution process of 95/5 (upper) and 90/10 (bottom) blends at 300 K (the real side length of all the cube boxes is 36 nm). | ||
There are three stages in the phase separation process. Firstly, the phase separates rapidly from 0 to 1 × 104 steps on the nanoscopic level. Secondly, a droplet structure forms by polymer organization from 1 × 104 to 1 × 105 steps. Finally, the droplets collide and coalesce, causing the superstructure to have a special symmetry similar to that of a sphere.
We compared the morphological evolution process between the 95/5 and 90/10 blends in Fig. 5. We found that in the case of the 95/5 blend, it takes 1 × 103 steps for the binary blend to reach the final symmetry ball of PVDF, while ten times of the number of steps, 1 × 104 steps were spent for the 90/10 blend to reach that ball. However, the ultimate size of the disperse phase domains for the 90/10 blend is larger than the 95/5 blend.
3.3 Radial distribution functions (RDF)
Radial distribution function g(r) is calculated by full-atoms MD simulation methods, from which the microscopic molecular structure was characterized. This function represents the probability of finding a pair of atoms at a distance r with respect to the bulk phase in a completely random distribution.The RDFs of sulfur–sulfur pairs are shown in Fig. 6 at various blends ratio. As can be seen from Fig. 6, all the sulfonated parts of different blends ratio present similar shape and position of the first peak is at about 4.855 Å. This distance is directly related to the interactions between sulfonate group and water molecules, and the sulfonate group distance is constant in the case of same water content. It is also found that the first peak elevated with an increase of PVDF-g-PSSA content, and the addition of sulfonate groups cause that all sulfonate groups in the system formed compact and regular channel for proton transport. Moreover, the distance between first and second peak at 90/10 blends is larger than other two blends, which indicate that sulfonate groups are almost surrounded by water molecules in 90/10 blends.
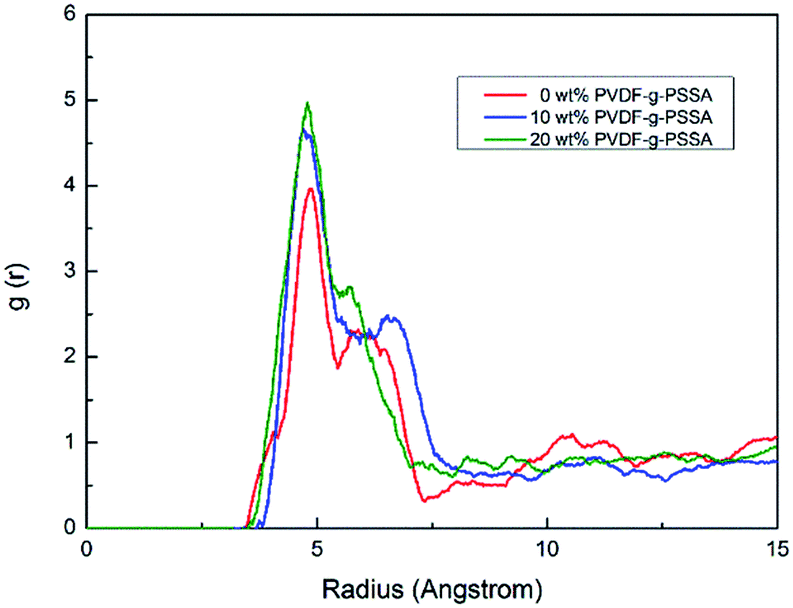 | ||
| Fig. 6 Pair correlation functions of sulfur–sulfur pairs for 0, 10, 20 wt% PVDF-g-PSSA at 298 K. | ||
Fig. 7(a) shows the RDFs between oxygen atoms of water molecules and sulfur atoms of sulfonate cluster. For all different blends ratio, there is an obvious first peak observed at a distance about 3.785 Å, which is directly related to the interactions between oxygen atoms of sulfonate group and water molecules.
 | ||
| Fig. 7 Pair correlation functions of sulfur–water (oxygen atom of water) (a) and sulfur–hydronium ion (oxygen atom of hydronium ion) (b) pairs for 0, 10 and 20% PVDF-g-PSSA at 298 K. | ||
RDFs of oxygen atoms of hydronium ions and sulfur atoms are presented in Fig. 7(b). For all different blends ratio, a wide peak is observed in the range of 3.455–3.935 Å, which is higher than that of water oxygen atoms and sulfur atoms. The difference in peak is induced by strong electrostatic non-bonded interactions between sulfonate groups and hydronium ions. It is also observed that peak height remains unchanged with increasing PVDF-g-PSSA content, due to the fact that the number of sulfonate groups and hydronium ions stay the same.
The RDFs of the oxygen atoms in a pairs of water molecules and in a water-hydronium pairs are shown in Fig. 8(a) and (b), respectively. The first peaks are observed at distances of about 2.9 and 2.75 Å, which reveals that water molecules have a tendency to aggregate with each other and with hydronium ions, resulting in the formation of hydrophilic clusters through which hydronium ions can transfer. It also shows that water molecules formed larger clusters at 90/10 blends than other two blends, from this we determined that the 90/10 blends content is more appropriate for SPEEK/PVDF-g-PSSA blends. The presence of the two peaks appearing at 5 and 7 Å is caused by Coulomb long-range interactions between water molecules and hydronium ions. Pure water molecules are electrically neutral, so it is short-range order, long-range disorder, thus only one peak displayed in Fig. 8(a). Due to the existence of positive charge in H3O+, the interaction between H2O and H3O+ is long-range Coulomb interaction, which explains the presence of the two peaks appearing at 5 and 7 Å in Fig. 8(b).
 | ||
| Fig. 8 Pair correlation functions of water–water (oxygen atom of water) (a) and water–hydronium ion (oxygen atom of water and hydronium ion) (b) pairs for 0, 10 and 20% PVDF-g-PSSA at 298 K. | ||
3.4 Dynamic property analysis
According to the MD simulation results, transport properties of hydronium ions were calculated. Diffusion coefficient D of hydronium ions was evaluated using the slope of MSD curves. The conductivity of hydronium ions is evaluated by the obtained diffusion coefficients according to the eqn (3):
 | (3) |
Fig. 9(a) shows the mean squared displacement (MSD) of hydronium ions, which reveals its diffusivity increase with increasing PVDF-g-PSSA content at room temperature. This is due to large water cluster formation at 10 wt% PVDF-g-PSSA content, through which hydronium can diffuse following the vehicular and hopping mechanisms. It must be noted that MSD statistical calculation used the displacement data after t time.
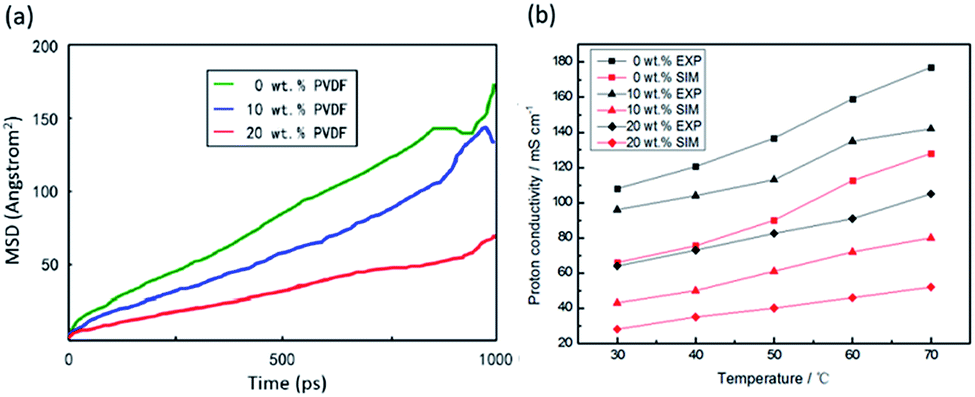 | ||
| Fig. 9 MSD of hydronium ions (a) at 0, 10, 20 wt% PVDF-g-PSSA at 313 K, and photon conductivities (b) for SPEEK/PVDF-g-PSSA blend in membranes at 303–343 K. | ||
When the t = 1000 ps, due to no available displacement data as t > 1000 ps, the beating appears in the MSD curve in Fig. 9(a). In order to increase the calculation accuracy, the slope of the linear part (t = 0–800 ps) of the MSD curve is used to calculate the diffusion coefficient.
The obtained proton conductivities using eqn (3) and corresponding experimental data from the previous work are collected in Table 4. For better comparison, the calculated and experimental ion conductivities are also plotted in Fig. 9(b). As shown in Fig. 9(b). The result of our simulation is in qualitative agreement with the experimental data. With the increase in temperature and PVDF-g-PSSA content, the conductivity is also significantly improved. However, there is a large deviation in the proton conductivity of SPEEK membranes reported in the literature with our simulation results.20 The simulated conductivities are about half of the experimental values, which is attributed to the simulation of proton diffusion inside the membrane without considering Grotthuss mechanism.32,33 Another explanation of the discrepancy between the calculated and the experimental values for the proton conductivity is the Dislocalization Body (DB) effects on the migration of the charged species inside the membrane. DB refers to a volume of bulk membrane containing both the hydrophobic and hydrophilic domains where the migrating H+ can be considered to be delocalized.34–36 The charge migrates along the interface within the hydrophobic–hydrophilic domains in the membrane. In this case, the exchange of protons between different DBs is likely the rate-limiting step of the overall conduction process. However, in our MD simulation process we didn't involve delocalization bodies, thus the discrepancy was observed between the calculated and the experimental values for the proton conductivity.
| Temperature/°C | 30 | 40 | 50 | 60 | 70 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Wt% of PVDF-g-PSSA | Proton conductivity/mS cm−1 | |||||||||
| EXP20 | SIM | EXP | SIM | EXP | SIM | EXP | SIM | EXP | SIM | |
| 0 wt% | 108 | 66 | 121 | 76 | 137 | 90 | 159 | 111 | 177 | 128 |
| 10 wt% | 96 | 43 | 104 | 50 | 113 | 61 | 135 | 72 | 142 | 80 |
| 20 wt% | 64 | 28 | 73 | 35 | 83 | 40 | 91 | 46 | 105 | 52 |
From the simulation results shown in Fig. 9 and Table 4, the proton conductivities of hydronium ions is obtained at different PVDF-g-PSSA content and different temperature, and the water content is fixed to be λ = 10. In the reported experimental data,20 however, the sample is fully moisture, and the water content is estimated to be less than λ = 20. So the effect of water content on proton conductivities is also studied in present work. Fig. 10(a) shows the model of system with λ = 10, 15 and 20 water content at room temperature, in which gray represents carbon, red represents oxygen, white represents hydrogen, yellow represents sulfur and light blue represents fluorine. As shown in the figure, with the increased of water content, water molecules in the system gradually increased. These results are in agreement with the previous study.37 Fig. 10(b) shows proton conductivity of fully wetting experimental sample and λ = 10, 15, 20 for simulations systems with 90/10 blends at room temperature. The results exhibit that the proton conductivity of λ = 15 water content is slightly lower than the experimental sample, while higher than the experiment result when λ = 20. Therefore, we estimate that the SPEEK/PVDF-g-PSSA membrane requires about λ = 15–20 water content when fully moisture.
 | ||
| Fig. 10 Model of system with λ = 10, 15, 20 water content at room temperature (a), proton conductivity of experiment and simulations system for 10 wt% PVDF-g-PSSA content (b) at 30 °C. | ||
4. Conclusions
Mesoscale DPD method has been used to investigate the compatibility of the SPEEK/PVDF-g-PSSA blends. The mesoscales morphologies demonstrate that SPEEK and PVDF-g-PSSA mix fairly well in 90/10 and 80/20 blends, whereas SPEEK and PVDF (without grafting) domains are observed in SPEEK/PVDF blend. It is revealed that the compatibility of SPEEK and PVDF is improved due to the grafting with PSSA.The fully atomistic MD simulations have been used to investigate structural and dynamical properties of the SPEEK/PVDF-g-PSSA membrane at various blends ratio and at different temperatures between 303 and 343 K. Using pair correlation functions of different atomic (sulfur, oxygen atoms of water and hydronium ions) pairs, we found that with increasing PVDF-g-PSSA content of SPEEK membrane, water clusters in the membrane are formed because of the higher water absorption of the membrane.
The mean square displacements and diffusion coefficients of the hydronium ions were calculated, from which the proton conductivity was obtained. The simulated conductivities for various blends ratio, water contents and temperatures were compared with our previous experimental results, which indicated the qualitative agreement. These results show that the transport properties of SPEEK/PVDF-g-PSSA membrane are strongly dependent on its blending proportion, microstructure and moisture condition. Thus, an increasing PVDF-g-PSSA content may affect the size and shape of the hydrophilic clusters which is consisted of sulfonate groups, water molecules and hydronium ions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (Grant no. 51303211), and outstanding young teachers' research projects of China University of Petroleum-Beijing (Grant no. 2462015YQ0603).Notes and references
- C. Song, Fuel processing for low-temperature and high-temperature fuel cells: challenges, and opportunities for sustainable development in the 21st century, Catal. Today, 2002, 77(1–2), 17–49 CrossRef CAS.
- J. W. Patrick and J. W. Patrick, Handbook of fuel cells. Fundamentals technology and applications, Focus Catalyst., 2009, 2003(4), 7 Search PubMed.
- K. D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Transport in proton conductors for fuel-cell applications: simulations, elementary reactions, and phenomenology, Chem. Rev., 2004, 104(10), 4637–4678 CrossRef CAS PubMed.
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. Mcgrath, Alternative polymer systems for proton exchange membranes (PEMs), ChemInform, 2004, 35(51), 4587–4611 CrossRef.
- M. Rikukawa and K. Sanui, Proton-conducting polymer electrolyte membranes based on hydrocarbon polymers, Prog. Polym. Sci., 2000, 25(10), 1463–1502 CrossRef CAS.
- K. D. Kreuer, On the development of proton conducting polymer membranes for hydrogen and methanol fuel cells, J. Membr. Sci., 2001, 185(1), 29–39 CrossRef CAS.
- M. Gil, X. Ji, X. Li, H. Na, J. Eric Hampsey and Y. Lu, Direct synthesis of sulfonated aromatic poly(ether ether ketone) proton exchange membranes for fuel cell applications, J. Membr. Sci., 2004, 234(1–2), 75–81 CrossRef CAS.
- S. M. J. Zaidi, S. D. Mikhailenko, G. P. Robertson, M. D. Guiver and S. Kaliaguine, Proton conducting composite membranes from polyether ether ketone and heteropolyacids for fuel cell applications, J. Membr. Sci., 2000, 173(1), 17–34 CrossRef CAS.
- A. H. Haghighi, M. M. Hasani-Sadrabadi, E. Dashtimoghadam, G. Bahlakeh, S. E. Shakeri, F. S. Majedi, S. H. Emami and H. Moaddel, Direct methanol fuel cell performance of sulfonated poly(2,6-dimethyl-1,4-phenylene oxide)–polybenzimidazole blend proton exchange membranes, Int. J. Hydrogen Energy, 2011, 36(5), 3688–3696 CrossRef CAS.
- M. M. Hasani-Sadrabadi, N. Mokarram, M. Azami, E. Dashtimoghadam, F. S. Majedi and K. I. Jacob, Preparation and characterization of nanocomposite polyelectrolyte membranes based on Nafion®; ionomer and nanocrystalline hydroxyapatite, Polymer, 2011, 52(5), 1286–1296 CrossRef CAS.
- M. M. Hasani-Sadrabadi, E. Dashtimoghadam, F. S. Majedi, K. Kabiri, N. Mokarram, M. Solati-Hashjin and H. Moaddel, Novel high-performance nanohybrid polyelectrolyte membranes based on bio-functionalized montmorillonite for fuel cell applications, Chem. Commun., 2010, 46(35), 6500–6502 RSC.
- K. D. Kreuer, On the development of proton conducting materials for technological applications, Solid State Ionics, 1997, 97(1–4), 1–15 CrossRef CAS.
- L. Li, J. Zhang and Y. Wang, Sulfonated poly(ether ether ketone) membranes for direct methanol fuel cell, J. Membr. Sci., 2003, 226(1–2), 159–167 CrossRef CAS.
- S. Xue and G. Yin, Methanol permeability in sulfonated poly(ether ether ketone) membranes: A comparison with Nafion membranes, Eur. Polym. J., 2006, 42(4), 776–785 CrossRef CAS.
- J. Wootthikanokkhan and N. Seeponkai, Methanol permeability and properties of DMFC membranes based on sulfonated PEEK/PVDF blends, J. Appl. Polym. Sci., 2006, 102(6), 5941–5947 CrossRef CAS.
- G. Sun, C. Li, Z. Wu, W. Jin, W. Chen, Q. Xin and X. Yang, Sulfonated poly(ether ether ketone)/polyvinylidene fluoride polymer blends for direct methanol fuel cells, Mater. Lett., 2006, 60(1), 44–47 CrossRef.
- X. Song and G. Yin, Proton exchange membranes based on poly(vinylidene fluoride) and sulfonated poly(ether ether ketone), Polymer, 2006, 47(14), 5044–5049 CrossRef.
- T. Y. Inan, H. Doğan, E. E. Unveren and E. Eker, Sulfonated PEEK and fluorinated polymer based blends for fuel cell applications: Investigation of the effect of type and molecular weight of the fluorinated polymers on the membrane's properties, Int. J. Hydrogen Energy, 2010, 35(21), 12038–12053 CrossRef CAS.
- H. Y. Jung and J. K. Park, Long-term performance of DMFC based on the blend membrane of sulfonated poly(ether ether ketone) and poly(vinylidene fluoride), Int. J. Hydrogen Energy, 2009, 34(9), 3915–3921 CrossRef CAS.
- X. Liu, X. Meng, J. Wu, J. Huo, L. Cui and Q. Zhou, Microstructure and properties of novel SPEEK/PVDF-g-PSSA blends for proton exchange membrane with improved compatibility, RSC Adv., 2015, 5(85), 69621–69628 RSC.
- S. Yamamoto and S. Hyodo, A Computer Simulation Study of the Mesoscopic Structure of the Polyelectrolyte Membrane Nafion, Polym. J., 2003, 35(6), 519–527 CrossRef CAS.
- R. Jorn and G. A. Voth, Mesoscale Simulation of Proton Transport in Proton Exchange Membranes, J. Phys. Chem. C, 2012, 116(19), 10476–10489 CAS.
- G. Brunello, S. G. Lee, S. S. Jang and Y. Qi, A molecular dynamics simulation study of hydrated sulfonated poly(ether ether ketone) for application to polymer electrolyte membrane fuel cells: Effect of water content, J. Renewable Sustainable Energy, 2009, 1(3), 189–200 Search PubMed.
- C. V. Mahajan and V. Ganesan, Atomistic Simulations of Structure of Solvated Sulfonated Poly(ether ether ketone) Membranes and Their Comparisons to Nafion: I. Nanophase Segregation and Hydrophilic Domains, J. Phys. Chem. B, 2010, 114(25), 8357–8366 CrossRef CAS PubMed.
- C. V. Mahajan and V. Ganesan, Atomistic Simulations of Structure of Solvated Sulfonated Poly(ether ether ketone) Membranes and Their Comparisons to Nafion: II. Structure and Transport Properties of Water, Hydronium Ions, and Methanol, J. Phys. Chem. B, 2010, 114(25), 8367–8373 CrossRef CAS PubMed.
- P. V. Komarov, I. N. Veselov, P. P. Chu, P. G. Khalatur and A. R. Khokhlov, Atomistic and mesoscale simulation of polymer electrolyte membranes based on sulfonated poly(ether ether ketone), Chem. Phys. Lett., 2010, 487(4–6), 291–296 CrossRef CAS.
- P. V. Komarov, I. N. Veselov and A. S. Pavlov, The mesoscopic modeling of the structure of a hydrated ionomeric membrane based on a sulfonated aromatic poly(ether-ether-ketone), Russ. J. Phys. Chem. A, 2009, 83(6), 984–989 CrossRef CAS.
- P. J. Hoogerbrugge, Simulating Microscopic Hydrodynamic Phenomena with Dissipative Particle Dynamics, EPL, 2007, 19(3), 155 CrossRef.
- R. D. Groot and P. B. Warren, Dissipative particle dynamics: bridging the gap between atomistic and mesoscopic simulation, J. Chem. Phys., 1997, 107(11), 4423–4435 CrossRef CAS.
- P. Español and P. Warren, Statistical Mechanics of Dissipative Particle Dynamics, Europhys. Lett., 1995, 30(4), 191 CrossRef.
- S. Meng, J. Zhang, Y. Ma, X. Li, R. Zhang, B. Liu, F. Zhang, T. Hou and G. Lu, Multi-scale simulation studies on interaction between anionic surfactants and cations, AIP Adv., 2014, 4(12), 127110 CrossRef.
- P. Y. Chen, C. P. Chiu and C. W. Hong, Molecular structure and transport dynamics in Nafion and sulfonated poly(ether ether ketone ketone) membranes, J. Power Sources, 2009, 194(2), 746–752 CrossRef CAS.
- J. Pozuelo, E. Riande, E. Saiz and V. Compañ, Molecular Dynamics Simulations of Proton Conduction in Sulfonated Poly(phenyl sulfone)s, Macromolecules, 2006, 39(25), 8862–8866 CrossRef CAS.
- G. A. Giffin, G. M. Haugen, S. J. Hamrock and N. V. Di, Interplay between structure and relaxations in perfluorosulfonic acid proton conducting membranes, J. Am. Chem. Soc., 2013, 135(2), 822–834 CrossRef CAS PubMed.
- G. Nawn, K. Vezzù, F. Bertasi, G. Pagot, G. Pace, F. Conti, E. Negro and V. Di Noto, Electric Response and Conductivity Mechanism in H3PO4-Doped Polybenzimidazole-4N-HfO2 Nanocomposite Membranes for High Temperature Fuel Cells, Electrochim. Acta, 2017, 228, 562–574 CrossRef CAS.
- T. P. Pandey, A. M. Maes, H. N. Sarode, B. D. Peters, S. Lavina, K. Vezzù, Y. Yang, S. D. Poynton, J. R. Varcoe and S. Seifert, Interplay between water uptake, ion interactions, and conductivity in an e-beam grafted poly(ethylene-co-tetrafluoroethylene) anion exchange membrane, Phys. Chem. Chem. Phys., 2014, 17(6), 4367–4378 RSC.
- N. Zhang, Z. Liu, X. Ruan, X. Yan, Y. Song, Z. Shen, X. Wu and G. He, Molecular dynamics study of confined structure and diffusion of hydrated proton in Hyfion® perfluorosulfonic acid membranes, Chem. Eng. Sci., 2017, 158, 234–244 CrossRef CAS.
Footnote |
| † These author have equal contribution. |
| This journal is © The Royal Society of Chemistry 2017 |