
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermodynamics, functional and structural characterization of inosine–uridine nucleoside hydrolase from Leishmania braziliensis†
Pedro Ferrari Dalbertoa,
Leonardo Kras Borges Martinellia,
Jose Fernando Ruggiero Bachegab,
Luis Fernando Saraiva Macedo Timmersb,
Antonio Frederico Michel Pintoa,
Adilio da Silva Daddaa,
Guilherme Oliveira Petersena,
Fernanda Teixeira Subtila,
Luiza Galinaa,
Anne Drumond Villelaa,
Kenia Pissinatea,
Pablo Machadoa,
Cristiano Valim Bizarroa,
Osmar Norberto de Souzab,
Edgar Marcelino de Carvalho Filhoc,
Luiz Augusto Basso *a and
Diogenes Santiago Santosa
*a and
Diogenes Santiago Santosa
aCentro de Pesquisas em Biologia Molecular e Funcional (CPBMF), Pontifícia Universidade Católica do Rio Grande do Sul (PUCRS), 92A building at TECNOPUC, 4592 Bento Gonçalves avenue, Porto Alegre 90650-001, RS, Brazil. E-mail: luiz.basso@pucrs.br; Tel: +55 51 3320 3629
bLaboratório de Bioinformática, Modelagem e Simulação de Biossistemas (LABIO), Pontifícia Universidade Católica do Rio Grande do Sul (PUCRS), Porto Alegre 90650-001, RS, Brazil
cHospital Universitário Professor Edgard Santos, Universidade Federal da Bahia, Salvador 40110160, BA, Brazil
First published on 17th October 2017
Abstract
Leishmaniasis is considered one of the main endemic diseases in the world, and Brazil is among the countries with the highest incidence of cutaneous and mucocutaneous forms of leishmaniasis caused mainly by Leishmania braziliensis. The first-line drugs used in the treatment of leishmaniasis have several limitations: parenteral administration, long duration of treatment, and serious toxicity. One key metabolic characteristic of these parasites is the lack of a de novo purine biosynthesis pathway, making them auxotrophic to purines. Accordingly, they rely solely on the purine salvage pathway for nucleotide synthesis. A better understanding of the purine salvage pathway can reveal details of the biology of L. braziliensis that could, in turn, be used to develop new strategies to combat this parasite. The inosine–uridine nucleoside hydrolase from L. braziliensis (LbIU-NH) plays an important role in the salvage process and is an attractive drug target as there is no similar catalytic activity in mammals. Here is described cloning, heterologous protein expression, and a three-step purification protocol that yielded homogenous recombinant protein. The determination of LbIU-NH steady-state kinetic constants for inosine, adenosine, cytidine, uridine and p-nitrophenyl β-D-ribofuranoside is also reported. These data suggest that LbIU-NH displays characteristics of a nonspecific hydrolase. The thermodynamic profile suggests that D-ribose can bind to free enzyme with favorable enthalpic (ΔH) and entropic (ΔS) contributions. Thermodynamic activation parameters (Ea, ΔG#, ΔS#, ΔH#) for the LbIU-NH-catalyzed chemical reaction, pre-steady-state kinetics, solvent kinetic isotope effects, and pH-rate profiles are also presented. In addition, the crystal structure of LbIU-NH in complex with β-D-ribose and Ca2+ at 1.5 Å resolution is described.
Introduction
The Trypanosomatidae family consists of protozoan parasites that can cause various diseases. Amongst its members, Leishmania is one of great importance, especially in Brazil, where the species L. (V.) braziliensis, L. (L.) amazonensis and L. (L.) infantum chagasi have been identified.1,2 These protozoans are flagellated parasites that can cause several dermatological and visceral manifestations in mammals, known as leishmaniasis. This disease is an antropozoonose with great veterinary and medical significance.3,4 Its transmission is mediated by a female sand fly of the Phlebotominae subfamily of the genus Lutzomyia. The mosquito is responsible for transferring the amastigote form from one mammal to another. This protozoan goes through drastic physiological changes in its host, going from the extracellular promastigote form in the mosquito's intestine to the intracellular amastigote form in macrophage's phagolysosomes of mammals.4,5 Brazil is among the countries with the highest incidence of the cutaneous and mucocutaneous forms of leishmaniasis, which are present in almost every Brazilian state. Additionally, there are several reported cases of fatal visceral leishmaniasis.6The first-line drugs employed in the treatment of leishmaniasis have several limitations including parenteral administration and the duration of treatment, which lasts for at least three weeks.7 Antimonial treatments require cautious medical attention because of deleterious side effects.8 Pentamidine and amphotericin B are used to treat resistant strains; however, these compounds are equally limited due to the need for parenteral administration and severe side effects.9 Although liposomal formulation of amphotericin B and miltefosine have been approved by the FDA, these drugs are costly, difficult to administer, and poorly stable at high temperatures, typical of endemic regions.10,11 Efforts to discover new anti-leishmanial agents are thus worth pursuing.
Leishmania rely solely on the purine salvage pathway for nucleotide synthesis.5,12–14 Nucleoside hydrolases are members of this pathway that irreversibly hydrolyze the N-glycosidic bond of ribonucleosides, forming α-D-ribose and the corresponding base.15 IU-nucleoside hydrolase is a nonspecific Ca2+-dependent enzyme that catalyzes the hydrolysis of inosine and uridine to, respectively, hypoxanthine and uracil.14,16–18 As no similar catalytic activity is present in mammals, protozoan nucleoside hydrolases are targets for the development of anti-leishmanial drugs.19,20
Several studies have reported the kinetic properties and structure of this enzyme in other Trypanosomatidae organisms such as Trypanosoma cruzi,21 Crithidia fasciculata,20,22 Leishmania major17 and Leishmania donovani.5 Even though these enzymes share a high degree of sequence similarity, they show different kinetic and structural properties. In this work, we address the open questions regarding these characteristics for IU-NH from L. braziliensis, which is the main causative agent of leishmaniasis in Brazil.
Here, it is described cloning, heterologous protein expression, and purification of tetrameric recombinant L. braziliensis IU-NH protein (LbIU-NH). Steady-state kinetics measurements showed that LbIU-NH catalyzes the hydrolysis of inosine, adenosine, cytidine, uridine and p-nitrophenyl β-D-ribofuranoside, suggesting that it is a nonspecific hydrolase enzyme. Interestingly, LbIU-NH displayed positive homotropic cooperativity for uridine. The thermodynamic parameters (ΔH, ΔS, and ΔG) for D-ribose binding to LbIU-NH were determined by isothermal titration calorimetry (ITC). The ITC data indicated an ordered mechanism, in which the free base release is followed by D-ribose dissociation to yield free enzyme. The thermodynamic activation parameters (Ea, ΔG#, ΔS#, ΔH#) were assessed. The negative value for the entropy of activation suggested loss of degrees of freedom on going from the ground state to activated state (ΔS#), whereas the positive value for the enthalpy of activation (ΔH#) indicated that more interatomic interactions are formed in the transition-state complex as compared to reactants in the ground state. Solvent kinetic deuterium isotope effect (SKIE) on V suggested participation of a solvent proton transfer that partially limits hydrolysis of inosine in a step that occurs after substrate binding, whereas SKIE on V/K pointed to a modest, if any, contribution to proton transfer step(s) between the free enzyme and substrate up to the first irreversible step. Proton inventory data demonstrated a single proton transfer during the solvent isotope-sensitive step. The pH-rate profile for kcat yielded values of 5.2 and 7.9 for, respectively, pKa and pKb, whereas a pKa value of 5.7 was found for kcat/Kinosine. The imidazole side chain of His240 was proposed as the group with pKb value of 7.9 involved in protonation of hypoxanthine leaving group. The unprotonated carboxyl group of Asp241 may be involved in either stabilization of the developing charge on the ribosyl oxocarbenium ion or proton abstraction of an enzyme-bound water molecule for nucleophilic attack on C1′ of nucleosides. The side chains of Asp10, Asp15, or Asp241 are suggested as the likely candidates with a pKa value of 5.7 that play a role in substrate binding. Pre-steady-state kinetics measurements showed no burst in product formation suggesting that release of product does not contribute to a rate-limiting step. The crystal structure at 1.53 Å resolution of LbIU-NH in complex with β-D-ribose and Ca2+ (PDB ID code 5TSQ) showed an intermolecular disulfide bond between the interface of subunits A–D (and B and C), that has not been reported for other known structures of IU-NHs. Interestingly, this disulfide bond is not required to maintain LbIU-NH quaternary structure or enzyme activity.
Experimental
Cloning and protein expression
The LbIU-NH coding gene LbrM.18.1610 was synthetized by Biomatik® and was cloned into the pET23a(+) expression vector using the NdeI and HindIII restriction enzymes. To confirm the product's identity and integrity as well as to ensure that no mutations were introduced in the cloned fragment, automatic DNA sequencing of LbrM.18.1610 gene was carried out.The recombinant plasmid pET23a(+):LbrM.18.1610 was transformed into E. coli Rosetta (DE3) electrocompetent host cells and grown on Luria-Bertani (LB) agar plates containing 50 μg mL−1 ampicillin and 34 μg mL−1 chloramphenicol. A single colony was cultivated overnight in 50 mL of LB at 180 rpm at 37 °C. Nine milliliters of the culture were inoculated into 500 mL of LB medium with the same concentrations of antibiotics, and grown at 37 °C and 180 rpm until an OD600 of 0.4, and grown further for 12 h at 30 °C with no IPTG induction. Cells were harvested by centrifugation at 8000 × g for 30 min at 4 °C and stored at −20 °C.
Protein purification
Approximately 3 g of frozen cells were resuspended in 30 mL of 50 mM Tris HCl pH 7.5 (buffer A) containing 0.2 mg mL−1 of lysozyme (Sigma-Aldrich) and gently stirred for 30 min. Cells were disrupted by sonication (10 pulses of 10 s each at 60% amplitude) and centrifuged at 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 30 min. The supernatant was incubated with 1% (w/v) of streptomycin sulfate and gently stirred for 30 min. The solution was centrifuged at 48000 × g for 30 min. The supernatant was dialyzed two times against 2 L of buffer A using a dialysis tubing with a molecular weight exclusion limit of 12000–14000 Da. This sample was centrifuged at 48000 × g for 30 min and the debris-free supernatant was loaded on a Hiprep Q-Sepharose Fast Flow anion exchange column (GE Healthcare) pre-equilibrated with buffer A. The column was washed with 3 column volumes (CV) of the same buffer, and adsorbed proteins were eluted with a linear gradient (0–100%) of 20 CV of 50 mM Tris HCl pH 7.5 containing 200 mM NaCl (buffer B) at 1 mL min−1 flow rate. Fractions containing the target protein were pooled and ammonium sulfate was added to a final concentration of 1 M, clarified by centrifugation at 48000 × g for 30 min, and the resulting supernatant was loaded on a Hiprep Butyl Sepharose High Performance aliphatic hydrophobic column (GE Healthcare) pre-equilibrated with 50 mM Tris HCl pH 7.5 containing 1 M (NH4)2SO4 (buffer C). This hydrophobic column was washed with 10 CVs of buffer C and the adsorbed material eluted with 20 CVs of a linear gradient (0–100%) of buffer A at 1 mL min−1 flow rate. The fractions containing the LbIU-NH were pooled, concentrated down to 8 mL using an Amicon ultrafiltration cell (molecular weight cutoff of 10000 Da), and loaded on a HiLoad Superdex 200 25/60 size exclusion column (GE Healthcare), which was previously equilibrated with buffer A. Proteins were isocratically eluted with 1 CV of buffer A at a flow rate of 0.3 mL min−1. FPLC was performed using the AKTA system (GE Healthcare) and all purification steps were carried out at 4 °C and sample elution monitored by UV detection at 215, 254 and 280 nm simultaneously. Protein concentration was determined by the method of BCA using bovine serum albumin as standard (Thermo Scientific Pierce™ BCA protein Assay Kit). LbIU-NH recombinant protein fractions were analyzed by SDS-PAGE.
000 × g for 30 min. The supernatant was incubated with 1% (w/v) of streptomycin sulfate and gently stirred for 30 min. The solution was centrifuged at 48000 × g for 30 min. The supernatant was dialyzed two times against 2 L of buffer A using a dialysis tubing with a molecular weight exclusion limit of 12000–14000 Da. This sample was centrifuged at 48000 × g for 30 min and the debris-free supernatant was loaded on a Hiprep Q-Sepharose Fast Flow anion exchange column (GE Healthcare) pre-equilibrated with buffer A. The column was washed with 3 column volumes (CV) of the same buffer, and adsorbed proteins were eluted with a linear gradient (0–100%) of 20 CV of 50 mM Tris HCl pH 7.5 containing 200 mM NaCl (buffer B) at 1 mL min−1 flow rate. Fractions containing the target protein were pooled and ammonium sulfate was added to a final concentration of 1 M, clarified by centrifugation at 48000 × g for 30 min, and the resulting supernatant was loaded on a Hiprep Butyl Sepharose High Performance aliphatic hydrophobic column (GE Healthcare) pre-equilibrated with 50 mM Tris HCl pH 7.5 containing 1 M (NH4)2SO4 (buffer C). This hydrophobic column was washed with 10 CVs of buffer C and the adsorbed material eluted with 20 CVs of a linear gradient (0–100%) of buffer A at 1 mL min−1 flow rate. The fractions containing the LbIU-NH were pooled, concentrated down to 8 mL using an Amicon ultrafiltration cell (molecular weight cutoff of 10000 Da), and loaded on a HiLoad Superdex 200 25/60 size exclusion column (GE Healthcare), which was previously equilibrated with buffer A. Proteins were isocratically eluted with 1 CV of buffer A at a flow rate of 0.3 mL min−1. FPLC was performed using the AKTA system (GE Healthcare) and all purification steps were carried out at 4 °C and sample elution monitored by UV detection at 215, 254 and 280 nm simultaneously. Protein concentration was determined by the method of BCA using bovine serum albumin as standard (Thermo Scientific Pierce™ BCA protein Assay Kit). LbIU-NH recombinant protein fractions were analyzed by SDS-PAGE.
LbIU-NH identification by mass spectrometry
In-gel digestion was performed according to Shevchenko.23 Tryptic digest of LbIU-NH was separated on an in-house made 20 cm reverse-phase column (5 μm ODSAQ C18, Yamamura Chemical Lab, Japan) using a nanoUPLC (nanoLC Ultra 1D plus, Eksigent, USA) and eluted directly to a nanospray ion source connected to a hybrid mass spectrometer (LTQ-XL and LTQ Orbitrap Discovery, Thermo, USA). The flow rate was set to 300 nL min−1 in a 120 minute reverse-phase gradient. The mass spectrometer was operated in a data-dependent mode, with full MS1 scan collected in the Orbitrap, with m/z range of 400–1600 at 30000 resolution. The eight most abundant ions per scan were selected to CID MS2 in the ion trap. Mass spectra were analyzed using PatternLab platform.24 MS2 spectra were searched with COMET25 using a non-redundant database containing forward and reverse E. coli DH10B reference proteome and the sequence of LbIU-NH (A4H9Q9). The validity of the peptide-spectra matches (PSMs) generated by COMET was assessed using Patternlab's module SEPro24 with a false discovery rate of 1% based on the number of decoys.
Oligomeric state determination
Determination of LbIU-NH molecular mass in solution was determined by size exclusion liquid chromatography on a HighLoad 10/30 Superdex-200 column (GE Healthcare), injecting 100 μL of protein suspension (7 μM homogeneous recombinant protein) at 0.4 mL min−1 flow rate and isocratic elution with 1 CV of 50 mM Tris HCl pH 7.5 containing 200 mM NaCl. Protein elution was monitored at 215, 254 and 280 nm. The LMW and HMW Gel Filtration Calibration Kits (GE Healthcare) were used to prepare a calibration curve. The values of elution volumes (Ve) of protein standards (ferritin, aldolase, conalbumin, ovalbumin, ribonuclease A and carbonic anhydrase) were used to calculate their corresponding partition coefficient (Kav). The latter values were plotted against the logarithm of the molecular mass of standards, and the resulting linear function employed to obtain an estimate for LbIU-NH molecular mass in solution. The Kav values were determined from eqn (1). Blue dextran 2000 (GE Healthcare) was used to determine the void volume (V0). Vt is the total bed volume of the column.
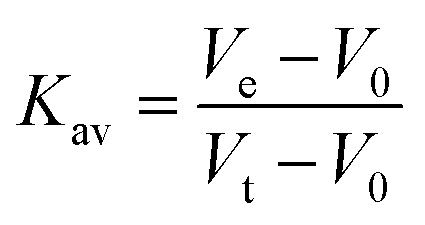 | (1) |
To evaluate if disulfide bonds play a role in maintaining the quaternary structure of LbIU-NH, recombinant protein (7 μM) was incubated in either the presence or absence of 100 mM dithiothreitol (DTT) in Tris HCl 50 mM pH 7.5 for 2 hours at room temperature. Volume elution profiles were analyzed and the molecular mass in solution was determined using a mobile phase containing 5 mM DTT as described elsewhere.26
Steady-state kinetics parameters
Recombinant LbIU-NH enzyme activity was measured by a continuous spectrophotometric assay in quartz cuvettes (1 cm) using a UV-visible Shimadzu spectrophotometer UV2550 equipped with a temperature-controlled cuvette holder. Kinetic properties of LbIU-NH for inosine and p-nitrophenyl β-D-ribofuranoside (p-NPR) were spectrophotometrically determined using the difference in absorption between the nucleoside and the purine base. Enzyme activity was measured in the presence of varying concentrations of inosine (0.2–1.5 mM) and p-NPR (0.1–1.0 mM) in 50 mM Tris HCl pH 7.5 at 25 °C. The reactions were started with addition of 5 μL of recombinant LbIU-NH (80 nM final concentration) that resulted in decreasing linear absorbance time courses for the conversion of nucleoside substrate into products; while hydrolysis of p-NPR followed the release of p-nitrophenylate anion at 400 nm in reaction solutions containing 20 nM of recombinant enzyme (final concentration). All assays were performed at least in duplicate. The Δε value employed was 0.92 mM−1 cm−1 at 280 nm and 14600 M−1 cm−1 at 400 nm.16,17 The experimental data were fitted to Michaelis–Menten equation (eqn (2)) for a hyperbolic saturation curve,27,28 in which v is the initial velocity, V is the apparent maximum initial velocity, A is the varying substrate concentration, and KM represents the apparent Michaelis–Menten constant.
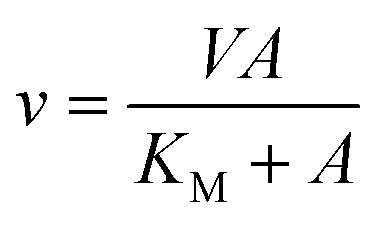 | (2) |
The kcat values were calculated from eqn (3), in which [E]t corresponds to the total concentration of LbIU-NH enzyme subunits.
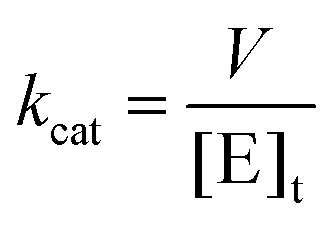 | (3) |
As no saturation for the uridine reaction could be detected by the continuous spectrophotometric assay, reverse-phase HPLC using a Dionex Ultimate 3000 with UV/VIS detector was employed to monitor the conversion of uridine into uracil. Briefly, assay mixtures containing Tris HCl 50 mM pH 7.5, 40 nM of LbIU-NH and various uridine concentrations (0.5–7.0 mM) were incubated at 25 °C. At seven time intervals (1, 3, 5, 7.5, 10 12.5 and 15 min), solutions were boiled for 3 min to stop the reaction and centrifuged at 10600 × g for 3 min. As no enzyme activity could be detected for adenosine, guanosine and cytidine by the continuous spectrophotometric assay, the HPLC discontinuous assay was employed to detect LbIU-NH enzyme activity, if any, for these substrates with some minor modifications. Assay mixtures contained Tris HCl 50 mM pH 7.5, 136 nM of LbIU-NH, and either adenosine (0.25–4.0 mM), guanosine (0.2–2.0 mM), or cytidine (0.5–4.5 mM) were incubated at 25 °C at 6 time intervals (5, 10, 15, 20, 30 and 40 min) of reaction, after which solutions were boiled for 3 min and centrifuged at 10600 × g for 3 min.
The supernatant (10 μL) for each reaction was injected onto a reverse-phase Nucleodur 100-5 C-18 HPLC column (250 × 4.6 mm, Macherey-Nagel). The mobile phase was ammonium acetate 10 mM (Merck®, Darmstadt, Germany) with a flow rate of 0.5 mL min−1. Elution of substrate and product was monitored at 254 nm, and the integrated peak area of product was compared to standard solutions to calculate the concentration of product formed at a specific time interval, yielding initial velocity values. The results were fitted either to Michaelis–Menten (eqn (2)) for hyperbolic curves or to the Hill equation (eqn (4)) for sigmoidal curves.27,28 For eqn (4), v represents the initial velocity, V is the apparent maximum initial velocity, A is the varying substrate concentration, K0.5 is the substrate concentration in which the velocity is half of the maximum velocity and n is the Hill coefficient.
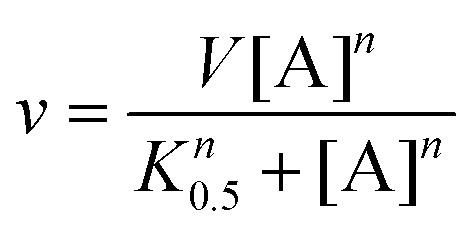 | (4) |
One unit of enzyme activity (U) was defined as the amount of enzyme catalyzing the conversion of 1 μmol of substrate into product per minute at 25 °C.
Isothermal titration calorimetry
ITC experiments were carried out using an iTC200 Microcalorimeter (Microcal, Inc., Pittsburgh, USA). Reference cell (200 μL) was loaded with Milli-Q water during all experiments and sample cell (200 μL) was filled with 100 μM of LbIU-NH recombinant enzyme in Tris HCl 50 mM pH 7.5. The injection syringe (39.7 μL) was filled with the products at different concentrations: D-ribose at 10 mM, hypoxanthine and uracil at 5 mM using the same buffer to prepare all ligand solutions. Ligand binding isotherms were measured by direct titration (ligand into macromolecule). The stirring speed was 500 rpm at 25 °C and 37 °C with constant pressure for all ITC experiments. The binding reaction started with one injection of 0.5 μL followed by 19 injections of 2.0 μL each at 300 s intervals. Control titrations (ligand into buffer) were performed in order to subtract the heats of dilution and mixing for each experiment prior to data analysis. ITC data were fitted to eqn (5), in which ΔH is the enthalpy of binding, ΔG is the Gibbs free energy change, ΔS is the entropy change, T is the absolute temperature in Kelvin, R is the gas constant (1.987 cal K−1 mol−1) and Ka is the equilibrium association constant. The dissociation constant, Kd, was calculated as the inverse of Ka (eqn (6)). All data were evaluated using the Origin 7 SR4 software (Microcal, Inc.)|
ΔG = ΔH − TΔS = −RTlnKa
| (5) |
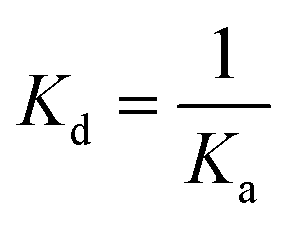 | (6) |
Energy of activation
The energy of activation (Ea) was assessed by measuring kcat values of LbIU-NH as a function of increasing temperature. Initial velocities were measured in the presence of fixed-saturating concentration of inosine (1.4 mM) at temperatures ranging from 15 to 35 °C (from 288.15 to 308.15 K). Prior to data collection, LbIU-NH was incubated for several minutes in all tested temperatures and assayed under standard conditions to ascertain enzyme stability. All assays were performed in duplicates. Ea was calculated from the slope (Ea/R) of the Arrhenius plot fitting the data to eqn (7), in which R is the gas constant (8.314 J mol−1 K−1) and the constant A represents the product of the collision frequency (Z), and a steric factor (p) based on the collision theory of enzyme kinetics.29 A simplistic approach was adopted to explain a complex phenomenon and that A is independent of temperature.
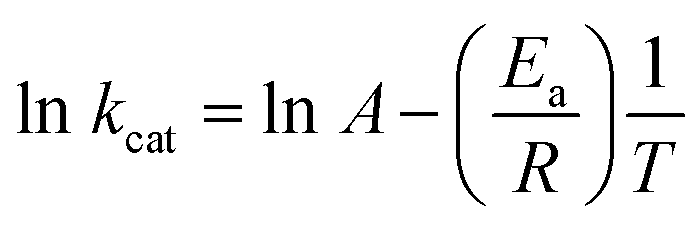 | (7) |
The enthalpy (ΔH#), Gibbs free energy (ΔG#) and entropy (ΔS#) of activation were estimated using the following equations (eqn (8)–(10)) derived from the transition state theory of enzymatic reactions.29
| ΔH# = Ea − RT | (8) |
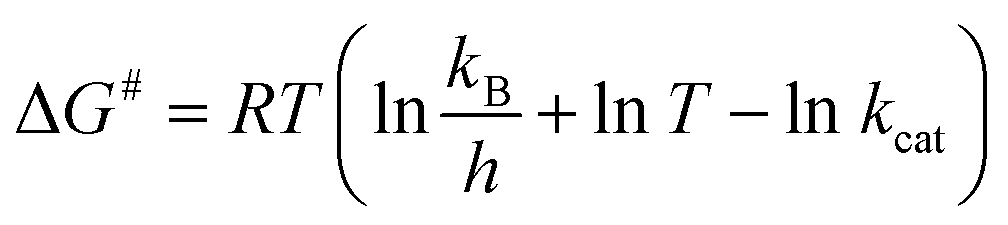 | (9) |
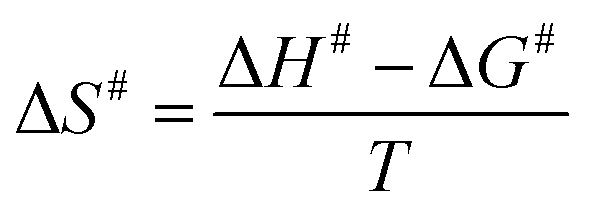 | (10) |
Energy values are in kJ mol−1, with kcat in s−1, to conform to the units of the Boltzmann (kB) (1.3805 × 10−23 J K−1) and Planck (h) (6.6256 × 10−34 J s−1) constants, and R for eqn (8) and (9) is as for eqn (7). Errors on ΔG# were calculated using eqn (11).29
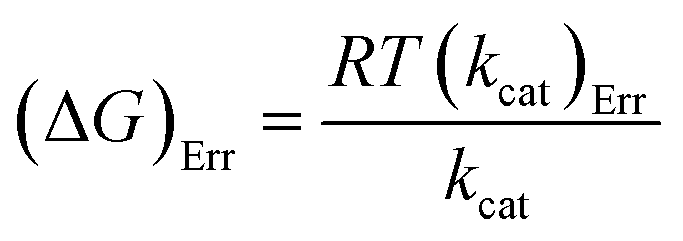 | (11) |
Solvent kinetic isotope effects (SKIE) and proton inventory
Solvent kinetic isotope effects were determined by measuring initial velocities of LbIU-NH in presence of varying concentrations of inosine (0.2 mM to 1.4 mM) in either H2O or 90% D2O in 50 mM Tris HCl pH 7.5 buffer. The proton inventory was assessed at fixed-saturating concentration of inosine (1.4 mM) with various mole fractions of D2O (0–90%) in 50 mM Tris HCl pH 7.5 buffer. Data were fitted to eqn (12), which assumes isotope effects on both V/K and V. In this equation, EV/K and EV are the isotope effects minus 1 on V/K and V, respectively, and Fi is the fraction of isotopic label in substrate A.30
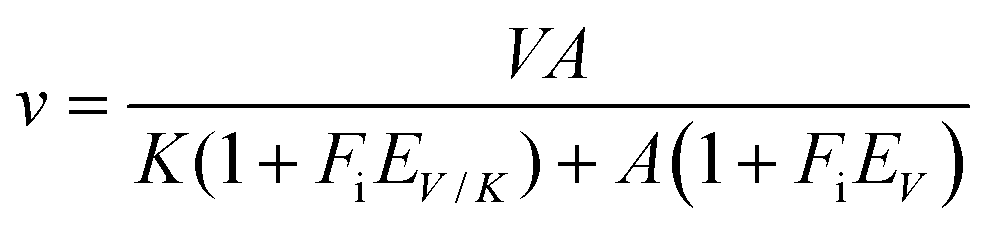 | (12) |
pH-rate profiles
The dependence of kinetic parameters on pH was determined by measuring initial velocities in the presence of varying inosine concentrations (0.2–1.4 mM) in 100 mM 2-(N-morpholino)ethanesulfonic acid (MES)/N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid (HEPES)/2-(N-cyclohexylamino)ethanesulfonic acid (CHES) buffer mixture over the following pH values: 5.0, 6.0, 6.5, 7.0, 7.5, 8.0 and 8.5.29 All measurements were carried out at least in duplicates. The pH-rate profile was generated by plotting logarithm value of kcat or kcat/KM of the substrate versus pH values. The data were fitted to either eqn (13) or (14), in which y is the apparent kinetic parameter, C is the pH-independent plateau value of y, H is the hydrogen ion concentration, and Ka and Kb are, respectively, the apparent acid and base dissociation constant for the ionizing group. Prior to performing the pH-rate profiles, LbIU-NH was incubated for 2 min at 25 °C over a broader pH range in either 100 mM citrate or CHES buffer and enzyme activity was measured in assay mixtures containing inosine (1.4 mM) in Tris HCl 50 mM pH 7.5 buffer. These measurements were carried out to show whether changes in enzyme activity were due to changes in proton concentration or to protein denaturation.Eqn (13) describes a bell-shaped pH profile for a group that must be protonated for binding/catalysis and another group that must be unprotonated for binding/catalysis, and participation of a single ionizing group for the acidic limb (slope value of +1) and participation of a single ionizing group for the basic limb (slope value of −1).
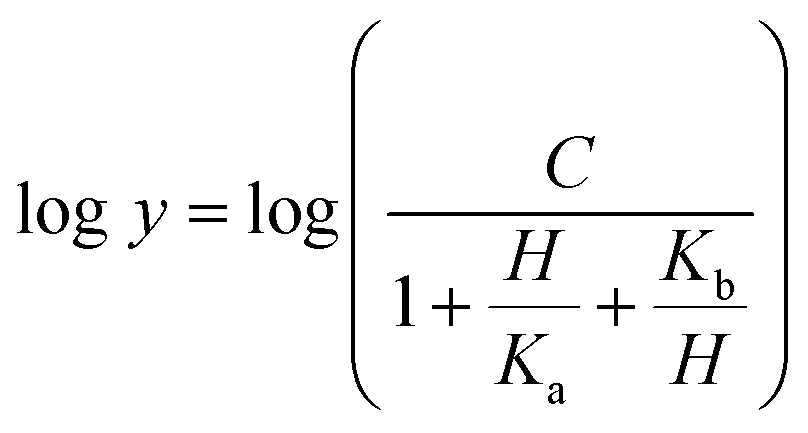 | (13) |
Eqn (14) describes a pH profile for a group that must be unprotonated for binding/catalysis and participation of a single ionizing group for the acidic limb (slope value of +1).
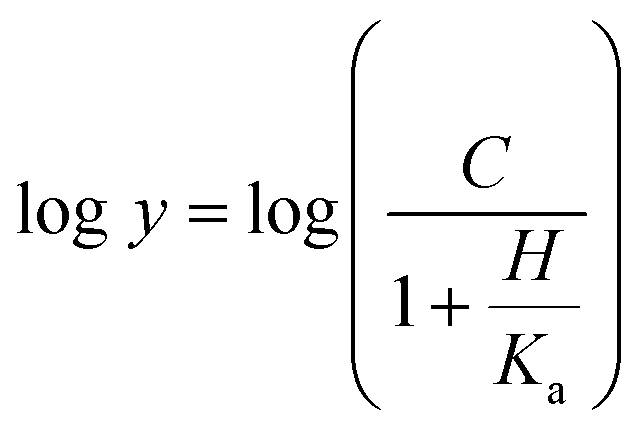 | (14) |
Pre-steady-state kinetics
Pre-steady-state kinetic parameters of the reaction catalyzed by LbIU-NH were determined using an Applied Photophysics SX.18MV-R stopped-flow spectrofluorimeter on absorbance mode to assess whether product release is part of the rate-limiting step. The decrease in absorbance was monitored at 280 nm for 0.5 s collecting 400 points (1 mm slit width = 4.65 nm spectral band) and an optical path of 2 mm. The experimental conditions were 10 μM of LbIU-NH, 2 mM of inosine in Tris HCl 50 mM pH 7.5 (mixing chamber concentrations). The control experiments were performed as the experimental conditions above in the absence of enzyme. The dead time of the equipment is 1.37 ms.The pre-steady-state time course of the reaction was fitted to eqn (15) for a linear decay, in which A is the absorbance at time t, A0 is the absorbance at time zero, and k is the apparent first-order rate constant for product formation.31
| A = A0 + kt | (15) |
Crystal structure determination
Crystals were obtained by sitting-drop vapor diffusion at 291 K in a solution containing 0.2 M ammonium acetate, 0.1 M HEPES pH 7.5 and 45% 2-methyl-2,4-pentanediol (MPD). The addition of 10% ethylene glycol to the original crystal growth conditions was used for cryoprotection. Diffraction data from crystals of LbIU-NH were collected on beamline MX2 at LNLS equipped with a Pilatus 2M detector using a wavelength of 1.459 Å, an oscillation range of 0.3° and a 3 s exposure time per image at 100 K. The data were indexed, integrated and scaled using the programs iMOSFLM32 and SCALA33 from the CCP4 suite.32,34 The asymmetric unit was estimated with respective Matthews' coefficients.35,36 The phase problem was solved by molecular replacement using MOLREP program.33 The search model was the structure of IU-NH from Leishmania major (PDB access code 1EZR, subunit A). The structure was refined using Phenix.refine37,38 (10% of the reflections for the calculation of Rfree parameter) and Coot for model building39 using σa-weighted 2Fo − Fc and Fo − Fc electron-density maps. The parameters R and Rfree were used as the principal criterion for validating the refinement protocol, and the stereochemical quality of the model was evaluated with PROCHECK40 and MolProbity.41 The final protein concentration of LbIU-NH for crystallization was 2.7 mg mL−1. The solution also contained 10 mM of D-(−)-ribose (linear), whereas a β-D-ribose (ribofuranosidic form) was found in the LbIU-NH structure. We have employed this crystallization strategy to take advantage of mutarotation processes. Mutarotation is known to occur with monosaccharides as there is an equilibrium between open, and α and β anomeric forms. The simple linear form is assumed to be the intermediate of four species of D-ribose: α-furan (7.1%), β-furan (12.3%), α-pyran (22.4%), and β-pyran (58.2%).42 The spontaneous α-to-β exchange rate constants of furanosidic forms are at least one-order of magnitude higher than the pyranosidic forms.42 It is thus likely that LbIU-NH bound the β-furanosidic form is due to its large percentage in solution and/or due to its larger affinity constant. The atomic coordinates were deposited in the Protein Data Bank, PDB ID code 5TSQ.Results and discussion
Cloning, expression, purification and mass spectrometry
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis of Escherichia coli Rosetta (DE3) host cells with recombinant pET23a(+):LbrM.18.1610 plasmid showed expression in the soluble fraction of a recombinant protein with an apparent subunit molecular mass of ∼34 kDa. This result is in agreement with the expected molecular mass of LbIU-NH (34340 Da). A three-step purification protocol (anionic exchange followed by hydrophobic interaction and size exclusion) was developed that yielded 10 mg of homogenous recombinant LbIU-NH protein (Fig. S1, ESI† data) per gram of wet cell paste (frozen cells). The protein band in SDS-PAGE of approximately 34 kDa was excised, submitted to trypsin digestion protocol and the peptides analyzed by LC-MS/MS. From the analysis in duplicate, LbIU-NH (A4H9Q9) identity was confirmed, with the identification of 43 unique peptides, 400 spectral counts and sequence coverage of 74.8%.
Oligomeric state determination
A value of 136019 for the apparent molecular mass of homogeneous recombinant LbIU-NH was estimated by gel filtration chromatography, fitting the elution volume of the single peak to eqn (1) (Fig. S2†). The gel filtration result suggests that LbIU-NH is a tetramer in solution.
In order to investigate the possibility that disulfide bonds play a role in maintaining the quaternary structure of LbIU-NH, profiles for recombinant protein elution from a gel filtration column following incubation with 100 mM DTT were analyzed (Fig. S3†). No difference was observed, suggesting that disulfide bonds play no role in maintaining the quaternary structure of LbIU-NH. Oxidation of DTT can be observed as there is an increase in absorbance at 280 nm (23 mL elution volume), which implies that protein disulfide bonds were reduced (Fig. S3†). No related peak was observed in the absence of DTT (Fig. S3,† black line). Specific enzyme activity measurements of elution peaks (23 mL) of LbIU-NH in the presence (Fig. S3;† red line) and absence of DTT (Fig. S3;† blue line) yielded fairly similar values (error within 32%). These results suggest that disulfide bonds play a negligible, if any, role in LbIU-NH enzyme activity.
Initial velocity and substrate specificity
Initial velocity experiments were carried out to determine the steady-state kinetic constants and substrate specificity (inosine, adenosine, cytidine, uridine, guanosine and p-NPR) of tetrameric LbIU-NH. Saturation curves for specific activity of LbIU-NH against increasing concentration of inosine (Fig. 1A), adenosine (Fig. 1B), cytidine (Fig. 1C), uridine (Fig. 1D), guanosine (Fig. 1E) and p-NPR (Fig. 1F) were evaluated. Steady-state kinetic parameters are given in Table 1. Owing to limited solubility, the steady-state parameters for guanosine could not be determined as no saturation was achieved. Nevertheless, a linear increase in activity could be detected (Fig. 1E) suggesting that the initial phase of the hyperbolic curve was being observed, which only allows an estimate for kcat/Km to be obtained for guanosine (Table 1).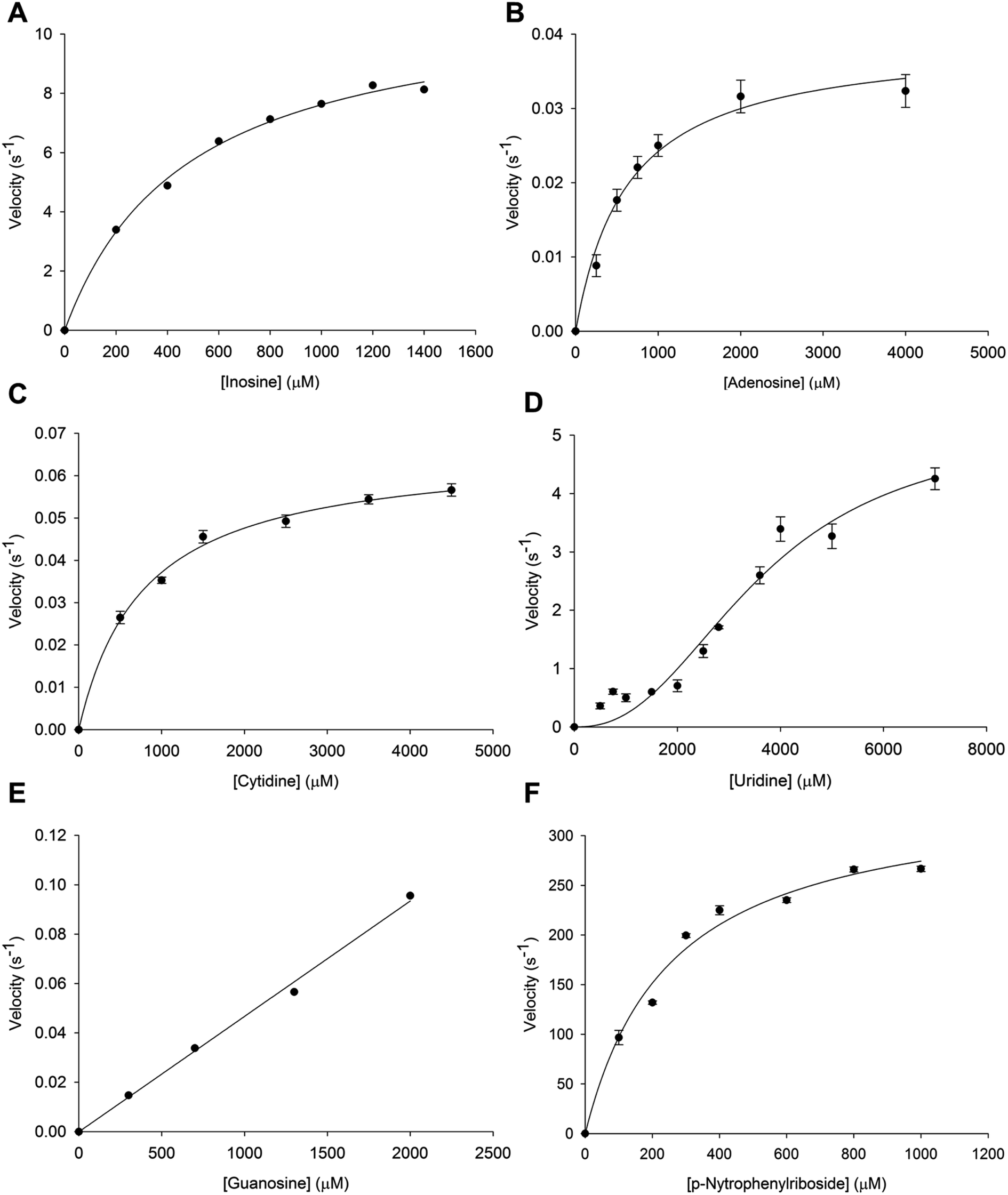 | ||
| Fig. 1 Determination of kinetic parameters for LbIU-NH. Specific activity/velocity plotted against increasing concentrations of (A) inosine, (B) adenosine and (C) cytidine obeying the Michaelis–Menten kinetics, (D) uridine showing a sigmoidal profile, (E) guanosine and (F) p-nitrophenyl β-D-ribofuranoside. | ||
| Substrate | KM/K0.5 (mM) | Vmax (U mg−1) | kcat (s−1) | kcat/KM (M−1 s −1) |
|---|---|---|---|---|
| Inosine | 0.50 ± 0.04 | 19.6 ± 0.7 | 11.2 ± 0.4 | 2.2 (±0.2) × 104 |
| Uridine | 3.6 ± 0.4 | 9 ± 1 | 5.1 ± 0.6 | — |
| Adenosine | 0.6 ± 0.1 | 0.069 ± 0.004 | 0.039 ± 0.002 | 65 ± 10 |
| Cytidine | 0.7 ± 0.07 | 0.115 ± 0.003 | 0.066 ± 0.002 | 94 ± 9 |
| Guanosine | 47 ± 2 | |||
| p-Nitrophenyl β-D-ribofuranoside | 0.25 ± 0.04 | 10 ± 0.6 | 344 ± 20 | 1.3 (±0.2) × 106 |
Steady-state kinetics results showed that KM value for inosine (Table 1) was similar to other Leishmania species such as L. major (KM = 445 ± 209 μM) and L. donovani (KM = 329 ± 143 μM).5,17 The KM value for p-NPR (Table 1) is also similar to L. major (KM = 185 ± 31 μM) and L. donovani (KM = 178 ± 39 μM).5,17 These results were expected due the sequence similarity among these species. Interestingly, the LbIU-NH kcat value for inosine is 10.6-fold lower as compared to L. major (119 s−1),17 and similar to L. donovani (7.6 s−1).5 On the other hand, LbIU-NH kcat for p-NPR is similar to L. major (185 s−1)17 and 12.5-fold larger as compared to L. donovani (26.7 s−1).5 The apparent second-order rate constant (kcat/KM) for inosine conversion into products for L. donovani (2.3 × 104 M−1 s−1)5 and L. braziliensis (2.2 × 104 M−1 s−1) are fairly similar whereas 10-fold lower as compared to L. major (27 × 104 M−1 s−1).17 The LbIU-NH kcat value for uridine substrate is lower than both L. major (32 s−1)17 and L. donovani (9.5 s−1).5 However, uridine substrate data were fitted to the Hill equation due to its sigmoidal profile suggesting positive homotropic cooperativity for this substrate, with a Hill coefficient (n) value of 2.4. To the best of our knowledge, the sigmoidal profile has never been reported for NH enzymes of parasitic protozoa. However the nucleoside hydrolase from Mycobacterium tuberculosis has been shown to display positive cooperativity for inosine and adenosine.43
Even though recombinant LbIU-NH catalyzed the hydrolysis of purine and pyrimidine nucleosides, fairly low activities were observed for adenosine, cytidine and guanosine (Table 1). These results indicate that L. braziliensis has apparently evolved to preferentially catalyze hydrolysis of inosine and, to a lesser extent, uridine. Hypoxanthine is considered to be the major precursor for purine salvage in L. braziliensis. The LbIU-NH enzyme displays the three features that characterize the L. major enzyme as a nonspecific hydrolase, which are the recognition of inosine and uridine as the favorable substrates, efficient utilization of p-NPR as substrate and activity with all naturally occurring purine and pyrimidine nucleosides (adenosine, guanosine and cytidine).17 However, the apparent second-order rate constant values of LbIU-NH for the latter substrates are considerably lower than L. major enzyme values.17 Trypanosoma vivax nucleoside hydrolase shows preference for inosine, adenosine and guanosine (IAG-NH) as suggested by the apparent second-order constant values, kcat/KM.44 Although the T. vivax IAG-NH kcat value (5.19 s−1) for inosine is similar to LbIU-NH kcat one (11.2 s−1), the overall dissociation constant (KM) is 96-fold lower for T. vivax IAG-NH as compared to LbIU-NH, resulting in a significant lower kcat/KM value for the latter enzyme. For adenosine, the kcat/KM value is approximately 2600-fold larger for T. vivax IAG-NH44 than for LbIU-NH (Table 1). Owing to solubility issues of guanosine, only kcat/KM could be estimated (Table 1), suggesting a value approximately 10500-fold lower than for T. vivax IAG-NH enzyme.44 The LbIU-NH kcat value for uridine (Table 1) is approximately 220-fold larger than T. vivax IAG-NH.44 However, LbIU-NH displayed positive cooperativity for uridine with a Hill coefficient (n) value of 2.4, whereas T. vivax IAG-NH displayed Michaelis–Menten saturation curve for this substrate.44 Although the overall dissociation constant values for cytidine are fairly similar for both enzymes, the T. vivax IAG-NH kcat value44 is at least 5-fold larger than LbIU-NH. The steady-state kinetic data suggest that LbIU-NH is a non-specific hydrolase, catalyzing the hydrolysis of naturally occurring purine and pyrimidine nucleosides. However, a preference for inosine is observed (Table 1).
Thermodynamic parameters and order of release of products
Isothermal titration calorimetry (ITC) allows monitoring of binding reactions with direct measurements of heat taken or released upon binding of a ligand, providing the binding enthalpy of the process (ΔH), an estimate for the stoichiometry of the interaction (n), and the equilibrium constant (Ka). These results allow the dissociation constant (Kd), Gibbs free energy (ΔG) and the entropy (ΔS) to be calculated. ITC binding assays showed that D-ribose can bind to free LbIU-NH enzyme (Fig. 2). The ITC data at 25 °C (Fig. 2A) yielded the following thermodynamic parameters: ΔH = −1.3 ± 0.4 kcal mol−1, ΔG = −5.0 ± 1.3 kcal mol−1 and ΔS = 12.5 ± 3.4 cal mol−1 K−1, n = 1.6 ± 0.4 sites, and Kd = 192 ± 48 μM. The ITC data at 37 °C (Fig. 2B) yielded the following parameters: ΔH = −5.1 ± 2.8 kcal mol−1, ΔG = −4.8 ± 0.6 kcal mol−1 and ΔS = 0.92 ± 0.51 cal mol−1 K−1, n = 0.8 ± 0.4 sites, and Kd = 385 ± 44 μM. No binding of free bases (hypoxanthine, and uracil) could be detected by ITC.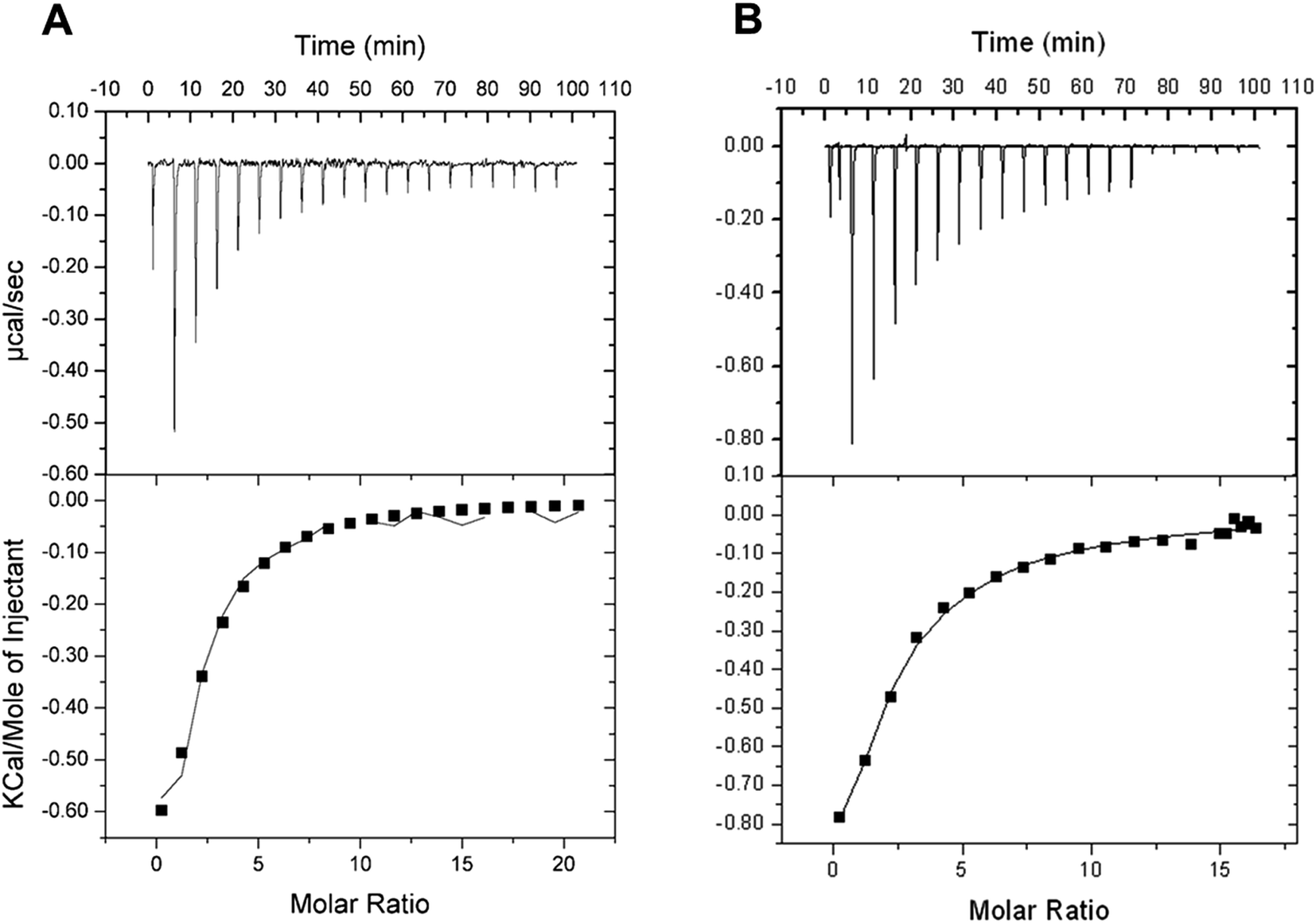 | ||
| Fig. 2 Ligand binding assay for D-ribose. The top panel represents raw data of the heat pulses and the bottom panel shows the integrated heat pulses (mol of injectant as a function of the molar ratio). (A) at 25 °C and (B) at 37 °C. | ||
The binding of D-ribose to LbIU-NH detected by ITC measurements at 25 °C showed that binary complex formation is an exothermic process as heat was released to the system (Fig. 2A). No heat changes were detected for hypoxanthine and uracil binding to LbIU-NH (data not shown), suggesting that these ligands cannot bind to free enzyme. Binding of adenine, guanine and cytosine could not be evaluated due to low solubility of these ligands in aqueous solutions. ITC data for D-ribose were best fitted to single set of sites model yielding an n (stoichiometry) value of 1.6 site per monomer of LbIU-NH, indicating that one molecule of D-ribose binds to each enzyme active site with equal affinity. The thermodynamic profile at, for instance, 25 °C suggests that D-ribose binding is a spontaneous process (ΔG = −5.0 kcal mol−1) with favorable enthalpic (−1.3 kcal mol−1) and entropic (12.5 cal mol−1 K−1) contributions. The negative enthalpy can arise from favorable redistribution of the network of interactions (hydrogen bonds and/or van der Waals) between the reacting species (including solvent).45 Hydrophobic interactions are related to the relative degrees of disorder in the free and bound system and thus these interactions are reflected in the entropy change. The release of water molecules from the reacting species to bulk solvent usually results in favorable positive entropic contribution.45 The ITC data for D-ribose binding at 37 °C (Fig. 2B) allowed to calculate a value of −317 cal mol−1 K−1 for the constant pressure heat capacity (ΔCp = ∂ΔH/∂ΔT). The negative value for ΔCp may be correlated with burial of surface area upon D-ribose binding to LbIU-NH.45 However, it should be pointed out that to give a more accurate ΔCp value, ITC data collection at various temperatures should be pursued. The favorable enthalpic and entropic contributions resulted in a favorable negative ΔG value. The fairly common enthalpy–entropy compensation phenomenon45 was observed for D-ribose binding to LbIU-NH as similar ΔG values were observed at 25 °C (−5.0 kcal mol−1) and 37 °C (−4.8 kcal mol−1) despite changes in both ΔH and ΔS values as a function of increasing temperature.
The ITC data indicate that the free base is the first product to be released followed by D-ribose dissociation to yield free LbIU-NH enzyme for the next round of catalysis. Product inhibition studies for C. fasciculata nucleoside hydrolase have revealed a rapid-equilibrium random mechanism, in which both base (hypoxanthine) and D-ribose can bind to free enzyme.16 However the dissociation constant value for D-ribose (700 μM) is approximately 10-fold lower than for hypoxanthine (6.2 mM).16 The ITC data analysis for α-D-ribose binding to LbIU-NH yielded a value of 192 μM, which is lower than for C. fasciculata nucleoside hydrolase,16 and lower than for M. tuberculosis nucleoside hydrolase (10 mM).43 As no heat change could be detected upon binding of hypoxanthine (5 mM) to LbIU-NH, there appears to be no binding of this base to free enzyme. Product inhibition studies for C. fasciculata nucleoside hydrolase indicate weak hypoxanthine affinity for free enzyme.16 Owing to limited solubility, larger concentrations of hypoxanthine could no be employed in the ITC experiments here described.
Energy of activation
The energy of activation for the enzyme catalyzed chemical reaction was assessed by measuring the dependence of kcat on temperature for inosine. Thermodynamic activation parameters for LbIU-NH-catalyzed chemical reaction were derived from data fitting to eqn (7)–(10), yielding the following values: Ea = 3.8 ± 0.1 kcal mol−1, ΔH# = 3.3 ± 0.2 kcal mol−1, ΔS# = −42.75 ± 1.3 cal mol−1 K−1, and ΔG# = 16.1 ± 0.02 kcal mol−1.The Ea value of 3.8 kcal mol−1 for inosine represents the minimum amount of energy necessary to initiate the LbIU-NH-catalyzed chemical reaction. The linearity of the Arrhenius plot (Fig. 3) indicates that there is no change in the rate-limiting step over the temperature range employed (15–35 °C). The values of free activation energy (ΔG#) represent the energy barrier required for reactions to occur. The ΔG# values can also be regarded as the variation of the Gibbs energy between the enzyme:substrate(s) activated complex and reactants (enzyme and substrates) in the ground state. The negative value for the entropy of activation (ΔS# = −42.75 cal mol−1 K−1) suggests that there is loss of degrees of freedom on going from the ground state to activated state. The enthalpy of activation (ΔH# = 3.3 kcal mol−1) is related to the ease of bond breaking and making in the generation of the activated complex (transition-state complex) from reactants in the ground state. The lower ΔH# value, the faster the rate. The enthalpy of activation for LbIU-NH (ΔH# = 3.3 kcal mol−1) suggests that more interatomic interactions (hydrogen bonds and/or van der Waals) are formed in the transition-state complex as compared to reactants in the ground state in bulk solvent.
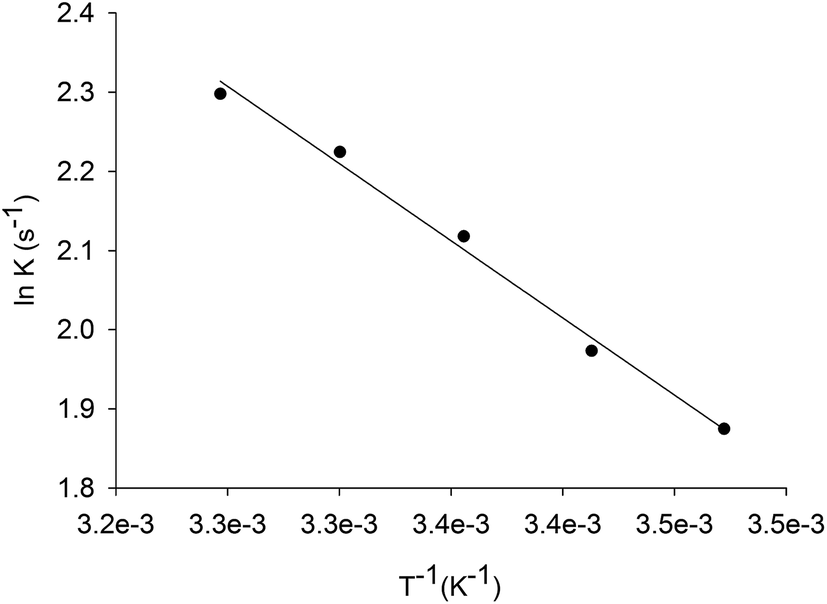 | ||
| Fig. 3 Arrhenius plot for inosine. Values for the maximum velocity of LbIU-NH-catalyzed chemical reaction were determined as a function of increasing temperatures (15 to 35 °C) at fixed-saturating concentration of inosine (1.4 mM). | ||
Solvent kinetic isotope effects (SKIE) and proton inventory
Solvent kinetic deuterium isotope effects were determined to evaluate the contribution, if any, of proton transfer from solvent to a rate-limiting step in the LbIU-NH-catalyzed chemical reaction. Data fitting to eqn (12) yielded the following SKIE values: D2OVinosine = 1.4 ± 0.1 and D2OV/Kinosine = 1.2 ± 0.1 (Fig. 4). Solvent isotope effects on V are related to solvent-exchangeable protons being transferred during events following the ternary complex formation capable of undergoing catalysis (fully loaded enzyme), which include the chemical steps, possible enzyme conformational changes and product release (leading to regeneration of free enzyme). Solvent isotope effects on V/K arise from solvent-exchangeable protons being transferred during steps in the reaction mechanism from the binding of substrates to the first irreversible step, usually considered the release of the first product.46 The value of 1.4 ± 0.1 for the solvent kinetic deuterium isotope effect on V indicates that solvent proton transfer partially limits hydrolysis of inosine in a step that occurs after substrate binding. The value of 1.2 ± 0.1 for the solvent kinetic deuterium isotope effect on V/K indicates a modest, if any, contribution to proton transfer step(s) between the free enzyme and substrate up to the first irreversible step. Similar solvent kinetic isotope effects were observed for C. fasciculata nucleoside hydrolase: 1.3 for V and 0.99 for V/K.22 To assess the number of protons transferred during the solvent isotope-sensitive step, a proton inventory experiment was carried out. A linear relationship between V and the mole fraction of D2O was obtained for LbIU-NH (Fig. 4 – inset), suggesting that a single proton is transferred in the step that exhibits the solvent isotope effect. The mode of action of nonspecific nucleoside hydrolases involves a transition state with advanced cleavage of the N9–C1′ ribosidic bond (0.22 bond order remaining), and attacking water nucleophile lagging behind N-ribosidic bond breaking with approximately 0.03 bond order from the attacking oxygen to C1′, leaving the pentose as an oxocarbenium cation with C1′ rehybridized nearly completely to sp2.17,19 The hypoxanthine leaving group is protonated at N7 prior to reaching the transition state, creating a neutral, planar and hydrophobic leaving group.17,19 The ribooxocarbenium ion is assisted by conformational distortion of the sugar using protein contacts and a catalytic site Ca2+ to permit both the ring oxygen and the unshared electrons of 5′-hydroxymethyl oxygen to participate in the cleavage of the sugar-base bond. Solvent isotope effects are global and isotope exchange can occur at several protic positions of the enzyme. Accordingly, assigning the solvent isotope effects to a particular chemical step is not straightforward. Initial rate conditions for nucleoside hydrolases imply that cleavage of the ribosidic bond must be considered the first irreversible step.22 It has thus been proposed that proton transfer is not a major part of the transition state, with the proton transfer having been completed prior to transition-state formation.22 However, it is tempting to suggest that enzyme-activated water molecule may account for the, albeit modest, SKIE results for LbIU-NH.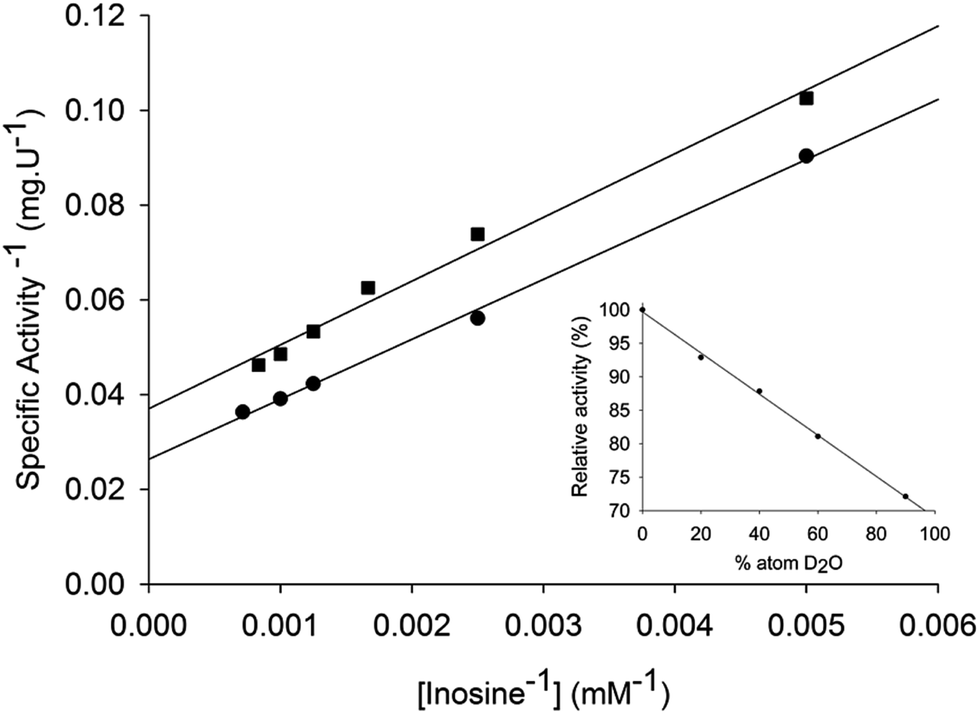 | ||
| Fig. 4 Solvent kinetic isotopic effects for LbIU-NH. Enzyme activity was measured at increasing concentrations of inosine (0.2–1.4 mM) in assay mixtures containing either 0 (●) or 90 (■) atom % D2O. The inset represents the proton inventory (0, 20, 40, 60 or 90 atom % D2O) with fixed-saturating concentration of inosine (1.4 mM). | ||
pH-rate profiles
The pH dependence of kcat and kcat/KM for inosine was determined to probe acid–base catalysis and to evaluate the apparent acid dissociation constant for ionizing groups involved in the mode of action of LbIU-NH. The bell-shaped pH-rate data for kcat (Fig. 5A) were fitted to eqn (13), yielding apparent pKa and pKb values of, respectively, 5.2 ± 1.1 and 7.9 ± 1.9. The data of pH-rate profile for kcat/Kinosine were fitted to eqn (14), yielding a value of 5.7 ± 0.5 for pKa (Fig. 5B).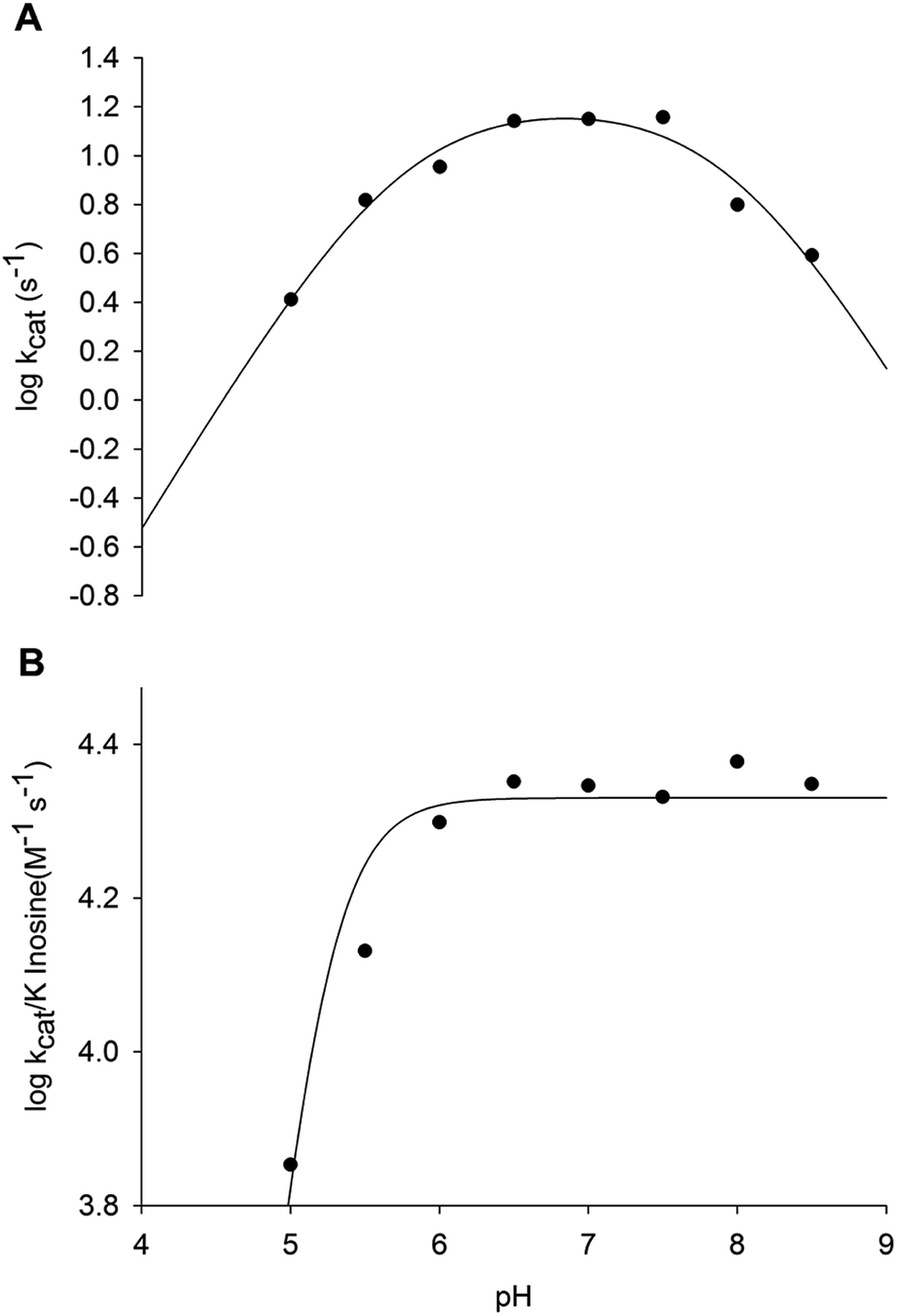 | ||
| Fig. 5 Dependence of kinetic parameters on pH. (A) pH dependence of logkcat data were fitted to eqn (10); (B) pH dependence of logkcat/Kinosine data were fitted to eqn (11). | ||
The bell-shaped pH-rate profile for kcat suggests participation of an ionizing group with apparent pKa value of 5.2 that must be unprotonated and another group with pKb value of 7.9 that must be protonated for catalysis to occur (Fig. 5A). The imidazole side chain of His240 residue is a plausible candidate for the group with pKb value of 7.9, which is likely involved in protonation of hypoxanthine leaving group. The carboxyl group of Asp241 may be the unprotonated group involved in stabilization of the developing charge on the ribosyl oxocarbenium ion. Alternatively, the carboxyl groups of Asp241 or Asp10 may be involved in proton abstraction of an enzyme-bound water molecule for nucleophilic attack on C1′ of nucleosides. The Ca2+ ion may lower the pKa of this water molecule prior to proton transfer to the carboxyl group of Asp241 or Asp10. The His240 amino acid is conserved in L. braziliensis and its analogue has been proposed to be involved in leaving group activation of nucleoside hydrolases from C. fasciculata and L. major.17,47 It has been shown that the His241Ala C. fasciculata IU-NH mutant showed a 2100-fold decrease in kcat for inosine whereas there was a 2.8-fold increase in kcat with p-nitrophenyl β-D-ribofuranoside.47 These results showed that His241 of C. fasciculata IU-NH (His240 of LbIU-NH) is the proton donor for leaving group activation as the p-nitrophenyl β-D-ribofuranoside substrate requires no stabilization of the nitrophenylate ion as leaving group.47 The carboxylate group of Asp10 of T. vivax IAG-NH is involved in proton abstraction from a water molecule, while Asp40 donates a proton to N9 of the purine ring.44 However, this mechanism for leaving group activation cannot be extended to LbIU-NH because the equivalent amino acid at this position is an asparagine residue (Asn39), as found for C. fasciculata and L. major IU-NHs (Fig. S4†).
The pH-rate data for kcat/Kinosine (Fig. 5B) indicates that the deprotonation of a group with pKa of 5.7 is required for inosine binding. Four aspartic acid residues were found to interact with the α-D-ribose moiety of nucleosides in the crystal structure of C. fasciculata IU-NH.20 These residues are conserved in LbIU-NH. The residues Asp10, Asp15, Asp241 and Thr126 are involved in interactions with Ca2+ ion in LbIU-NH crystal structure (see below). The mechanistic role of Ca2+ is to orient substrate in the active site and to position the solvent for attack at C1′ following the formation of the ribooxocarbenium ion.48 If any of these residues is in the protonated form, they will not be able to interact with Ca2+, precluding the orientation of the substrate and therefore abolishing substrate binding. Nevertheless, site-directed mutagenesis efforts will have to be pursued to ascertain whether or not His240 and Asp241 (or Asp10) play any role in catalysis, and which aspartate (Asp10, Asp15, Asp241) plays a role in substrate binding.
Pre-steady-state kinetics
Stopped-flow measurements of product formation were carried out to assess whether or not product release contributes to the rate limiting step. The pre-steady-state time course showed a linear decay (Fig. 6), and the data were thus fitted to eqn (15), yielding a value of 0.0208 (±0.0002) s−1 for the apparent first-order rate constant of product formation (k) and 2.3049 (±0.0001) for A0. The value of 0.0208 for k (ΔA280 nm/t) corresponds to a catalytic rate constant value of 11.3 s−1, which is in good agreement with the results for steady-state kinetics (Table 1, 11.2 s−1). There appears to be no burst of product formation (Fig. 6) at large subunit concentrations of LbIU-NH (10 μM) and fixed-saturating concentration of inosine (2.0 mM). Apparently, no signal was lost in the dead time of the equipment (1.37 ms). It was puzzling that the absorbance values for assay mixtures containing LbIU-NH and inosine were larger than the values for the control experiment (Fig. 6), suggesting a hyperchromic effect. As NHs expel water molecules en route to product formation, the dielectric constant of the enzyme's active site is likely lower (polarity characteristic of an organic solvent). Estimates for the molar absorptivity values at 280 nm (ε280) for inosine in solvents with different dielectric constants (D) were evaluated (Fig. S5†). The values for ε280 were: 1.7 (±0.3) × 103 M−1 cm −1 for Tris HCl 50 mM (Dwater = 78.74), 2.4 (±0.1) × 103 M−1 cm −1 for MeOH (DMeOH = 32.6) and 3.7 (±0.1) × 103 M−1 cm−1 for DMSO (DDMSO = 49). It is thus likely that the larger ε280 for the solution containing enzyme and inosine can account for the stopped-flow results. For electrostatic catalysis a reduction in the dielectric constant would result in an increase in the force between two charged molecules. Interestingly, charged molecules are present in the mode of action of nonspecific nucleoside hydrolases.17,19 An alternative possibility is the difference of ε280 for the keto (lower value for the 6-keto) and the enol forms of inosine.49 The stopped-flow measurements were repeated with a fresh enzyme preparation following three dialyses with buffer A after the purification protocol (to evaluate possible ionic effect). In addition, to rule out any interference coming from the buffer system used, the protein sample was concentrated in an Amicon ultrafiltration cell (molecular weight cutoff of 30 kDa) and the flow through was used as a blank for the experiment (Fig. S6†). As similar data were obtained, the stopped-flow results suggest that changes in dielectric constants in going from bulk solvent to enzyme active site can account for larger absorbance values solutions containing LbIU-NH and inosine as compared to control (Fig. 6).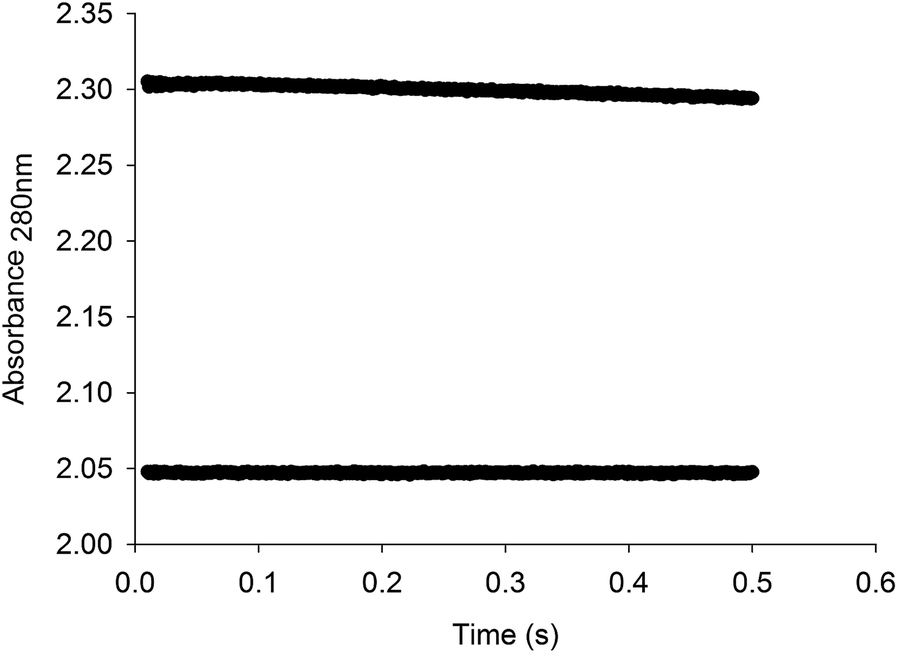 | ||
| Fig. 6 Stopped-flow trace for product formation. Measurements of the decrease in absorbance at 280 nm due to the conversion of inosine to hypoxanthine. The data were fitted to eqn (15) for a linear decay, yielding a value of 0.0208 s−1 for the apparent first-order rate constant of product formation, which corresponds to a catalytic rate constant value of 11.3 s−1. The bottom stopped-flow trace represents the control experiment (absence of LbIU-NH). | ||
Crystal structure determination
Crystals of LbIU-NH were obtained in orthorhombic space group (I222) and diffracted to a minimum d-spacing of 1.53 Å. The content of the asymmetric unity was determined by Matthews coefficient (CMatthews = 2.32), which corresponds to one monomer and solvent content of 47%. The initial electron density maps (2Fo − Fc and Fo − Fc) clearly showed evidence of a metal ion at the expected position as well as the presence of a β-D-ribose in the active site, confirming the correct phase attribution by molecular replacement. The refined model consists of one polypeptide chain, one calcium ion, one molecule of β-D-ribose and 278 water molecules (Table 2 shows data collection and refinement statistics). The electron density map (2Fo − Fc) is continuous for the main chain. The biological unity is sitting on spatial position so that the biological tetramer is generated from the symmetry operations of the space group I222. The overall structure is similar to those reported for L. major IU-NH (LmIU-NH, PDB ID 1EZR),17 C. fasciculata (CfIU-NH, PDB ID 1MAS),20 and Escherichia coli (EcIU-NH, PDB ID 1Q8F).50 The structure consists of one domain chain containing all the structural elements of these homologous enzymes (Fig. 7A). As observed for EcIU-NH, the LbIU-NH structure shows cis-bonds between residues Pro11-Gly12 and Tyr280-Pro281. All charged/polar residues in the active site are conserved (Fig. S4†). The calcium ion is anchored by residues Asp10, Asp15, Asp241 and Thr126, forming a binding site.51,52 The metal ion has eight coordination sites, five from the protein, in which Asp15 is involved in two interactions, two from the β-D-ribose molecule, and one water molecule (Fig. 7B). The β-D-ribose binding site is comprised of twelve residues, in which nine are involved in hydrogen bonds (Asp14, Asp15, Asn39, His82, Thr126, Asn160, Glu166, Asn168, and Asp241) and three (Met152, Phe167, and His240) are making hydrophobic contacts. With regard to the conformational changes upon ligand binding, the loop between β3–α3 is found in a closed form (Fig. S7†), which allows the residue His82 to maintain contact to the β-D-ribose. This loop is also found either in a closed form in the crystal structures of other holo nucleoside hydrolases or in an open form in the apo crystal structure (Fig. S8†).18,50,53 An sp2-hybridized C1′ predicts a ribose with C3′-exo conformation (below the plane of the ring), whereas the C3′-endo puckering (above the plane of the ring) is favored for free inosine.48 The β-D-ribose residue adopts a C2′-endo conformation similar to E. coli pyrimidine nucleoside hydrolase (PDB ID 1YOE; to be published), and in contrast to structures in complex with nucleosides that show the ribose having O4′-endo conformation.44 As described in the Experimental section, the crystallization solution contained D-(−)-ribose to take advantage of mutarotation of linear pentose. The LbIU-NH crystal structure thus provides a snapshot of interactions between enzyme and a moiety of nucleoside substrates.| Beamline | MX2 (LNLS) | |
| Wavelength (Å) | 1.45 | |
| Resolution range (Å) | 21.65 (1.53) | |
| Space group | I222 | |
| Unit cell (a and b, c; Å) | 74.66, 94.08, 123.12 | |
| Unique reflections | 64501 (8569) |
|
| Multiplicity | 4.3 (2.3) | |
| Completeness (%) | 98.9 (90.8) | |
| Mean I/sigma(I) | 10.0 (3.3) | |
| Rmerge (%) | 0.053 (0.431) | |
| Bfactor Wilson plot (Å2) | 23.7 | |
| CC1/2 | 0.991 (0.705) | |
| Rwork | 15.34 (18.84) | |
| Rfree | 16.68 (20.25) | |
| Number of atoms | ligands | 1 |
| Ions | 1 | |
| Waters | 278 | |
| Protein residues | 1023 | |
| RMS (bonds) (Å) | 0.011 | |
| RMS (angles) (°) | 0.972 | |
| Ramachandran favored (%) | 98.6 | |
| Ramachandran outliers (%) | 0 | |
| Clashscore | 2.64 | |
| Average B-factors (Å2) | Macromolecule | 20.946 |
| Ligands | 20.053 | |
| Solvent | 38.510 | |
| PDB code | 5TSQ | |
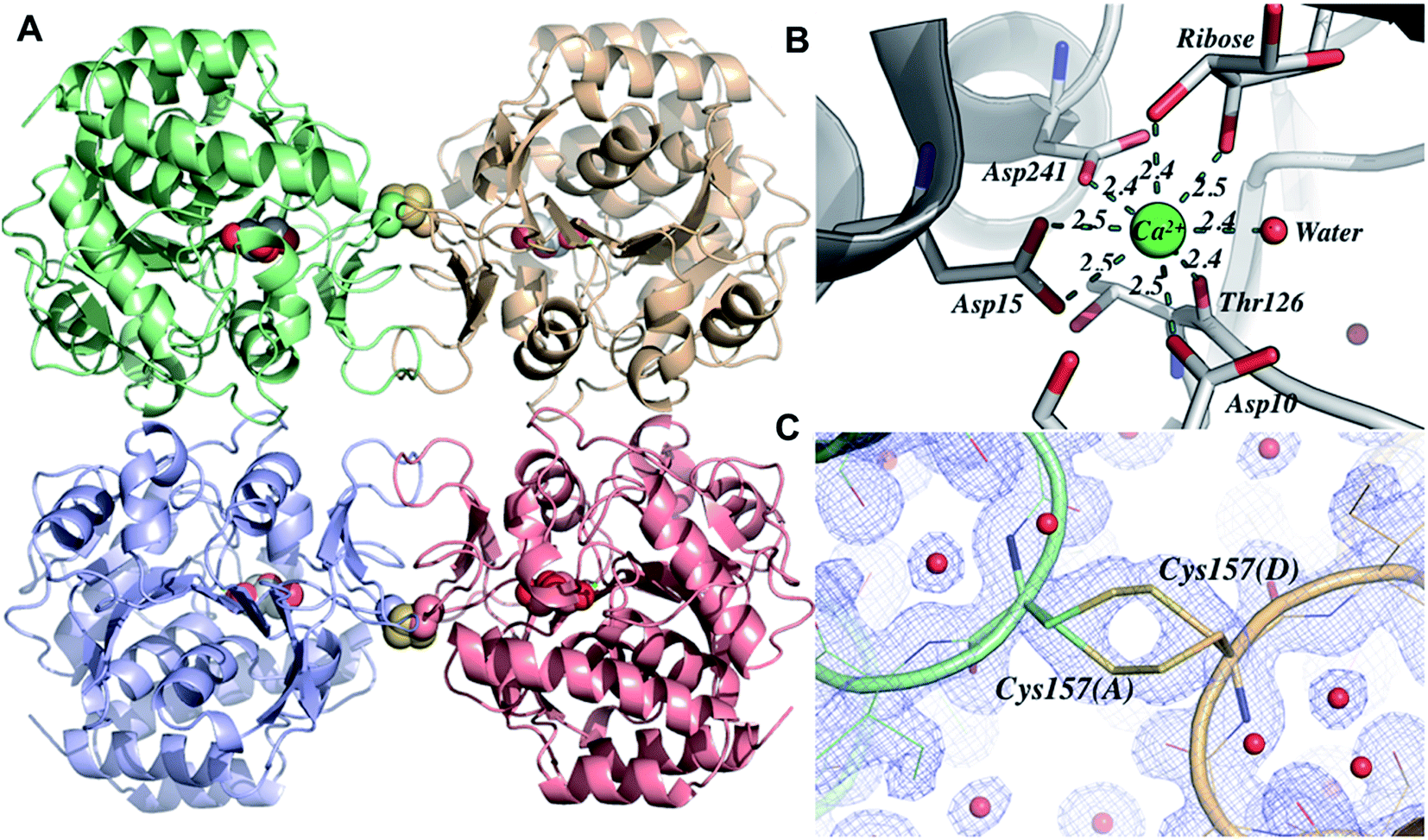 | ||
| Fig. 7 Overall structure of LbIU-NH. (A) The quaternary structure of LbIU-NH obtained from the symmetry operations of the spatial group I222. The main chains are represented as cartoon and colored by subunit: A is pale green, B is light blue, C is light red, and D is wheat. The cysteine residues involved in disulfide bonds are represented by their van der Waals raddi. (B) Calcium binding site, where the amino acids are represented as sticks and colored by CPK except by the carbon atom that is colored in green. The distances are shown in angstroms (Å). (C) Region where the disulfide bond was found, revealing a clear density map. Image generated using PyMOL. | ||
The surface contacts of the quaternary structure are very similar when compared to others IU-NHs, the dimer A–B interface is formed by mostly hydrophobic interactions involving helices α4, α7, the loop connecting β3–α3 and the loop connecting β9–β10 (contact area: 846.2 Å2). The interface A–C is formed by interactions involving the loop connecting β5–α6 and the sheet formed by β9–β10 (contact area: 1068.8 Å2). Interestingly, the LbIU-NH structure shows an intermolecular disulfide bond between the interface of subunits A–D (and B–C), which has not been reported for other known structures of IU-NHs (Fig. 7C). The link occurs between the Cys157 and its symmetrical at the neighbor subunit, and the bond occurs coincidently with one 2-fold axis of the space group I222.
Conclusion
Experimental efforts to determine the mode of action of LbIU-NH are presented, showing that this enzyme is a nonspecific hydrolase which catalyzes the hydrolysis of inosine and uridine as favorable substrates and that it catalyzes the hydrolysis of all naturally occurring purine and pyrimidine nucleosides (adenosine, guanosine and cytidine) as well as and p-nitrophenyl β-D-ribofuranoside. An interesting functional feature of LbIU-NH is the positive homotropic cooperativity for uridine, with a Hill coefficient (n) value of 2.4. To the best of our knowledge, the sigmoidal profile has never been reported for NH enzymes of parasitic protozoa. The ITC data suggested that binding of D-ribose to LbIU-NH free enzyme is a spontaneous process (ΔG less than 0 kcal mol−1) with favorable enthalpic (ΔH less than 0 kcal mol−1) and entropic (ΔS greater than 0 kcal mol−1) contributions. These data suggest favorable redistribution of the network of interactions (hydrogen bonds and/or van der Waals) between the reacting species (including solvent) and likely release of water molecules from the reacting species to bulk solvent. The ITC data indicate an ordered mechanism, in which the free base is the first product to be released followed by D-ribose dissociation to yield free enzyme. The thermodynamics for the energy of activation data indicate that the negative value for the entropy of activation suggests loss of degrees of freedom on going from the ground state to activated state, and the positive value for the enthalpy of activation indicates that more interatomic interactions (hydrogen bonds and/or van der Waals) are formed in the transition-state complex as compared to reactants in the ground state in bulk solvent. Solvent kinetic deuterium isotope effect (SKIE) on V indicates that solvent proton transfer partially limits hydrolysis of inosine in a step that occurs after substrate binding. The SKIE on V/K indicates a modest contribution to proton transfer step(s) between the free enzyme and substrate up to the first irreversible step. The linear proton inventory data suggest that a single proton is transferred in the step that exhibits the solvent isotope effect. The pH-rate profile for kcat suggests participation of an ionizing group with apparent pKa value of 5.2 that must be unprotonated and another group with pKb value of 7.9 that must be protonated for catalysis to occur. Analysis of the results obtained with the pH profile experiment, sequence alignment and structure data provided pieces of evidence that the amino acids His240 (protonated) and Aps241 (unprotonated) are involved in catalysis. In addition, pH data for kcat/Kinosine indicates that a group with pKa of 5.7 is required for inosine binding, and possibly three residues (Asp10, Asp15 and Asp241) could be involved in this process. However, these data cannot distinguish which one is involved and site-directed mutagenesis should be pursued to provide a solid experimental basis to confirm which aspartate plays a role in substrate binding. Pre-steady-state kinetics data suggest that product release does not contribute to the rate limiting step. The X-ray structure of LbIU-NH in complex with β-D-ribose and Ca2+ solved at 1.53 Å resolution (PDB ID code 5TSQ) is also reported. Interestingly, the LbIU-NH structure shows an intermolecular disulfide bond between the interface of subunits A–D (and B–C), which has not been reported for other known structures of IU-NHs. The structure allowed identification of the amino acid side chains involved in binding of substrates and catalysis. The results presented here, can be useful for the rational design of LbIU-NH enzyme inhibitors.Author contributions
PFD and LKBM designed, performed and analyzed all biochemical experiments, and drafted the paper. JFRB, LFSMT and ONS crystallized and solved the X-ray structure of LbIU-NH. CVB, LG, FTS and ADV designed vectors, and performed cloning and expression. AFPM and CVB designed, performed and analyzed the mass spectrometry experiments. ASD and GOP designed and performed HPLC method. KP and PM carried out the spectroscopic experiments. FTS also contributed to writing and formatting the manuscript. EMCF, LAB and DSS designed experiments and revised critically the manuscript.Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.Acknowledgements
This work was supported by funds awarded by Decit/SCTIE/MSMCT-CNPq-FNDCT-CAPES to National Institute of Science and Technology on Tuberculosis (INCT-TB) to D. S. S. and L. A. B. L. A. B. (CNPq, 520182/99-5), D. S. S. (CNPq, 304051/1975-06), O. N. S. (CNPq, 305984/2012-8), and E. M. C. F. (CNPq, 306706/2014-8) are Research Career Awardees of the National Research Council of Brazil (CNPq). P. F. D. acknowledges a scholarship awarded by CAPES. L. K. B. M. is a Post-Doctoral Fellow of FAPERGS/CAPES.References
- J. J. Shaw, Mem. Inst. Oswaldo Cruz, 2006, 101, 577–579 CrossRef PubMed.
- D. E. Teixeira, M. Benchimol, J. C. F. Rodrigues, P. H. Crepaldi, P. F. P. Pimenta and W. de Souza, Atlas Didático Ciclo de vida da Leishmania, Fundação CECIERJ, Rio de Janeiro, Brazil, 1st edn, 2013 Search PubMed.
- World Health Organization, Control of the leishmaniases, WHO, Geneva, 2010 Search PubMed.
- J. L. Martin, P. A. Yates, R. Soysa, J. F. Alfaro, F. Yang, K. E. Burnum-Johnson, V. A. Petyuk, K. K. Weitz, D. G. Camp, R. D. Smith, P. A. Wilmarth, L. L. David, G. Ramasamy, P. J. Myler and N. S. Carter, PLoS Pathog., 2014, 10, e1003938 Search PubMed.
- L. Cui, G. R. Rajasekariah and S. K. Martin, Gene, 2001, 280, 153–162 CrossRef CAS PubMed.
- J. Alvar, I. D. Vélez, C. Bern, M. Herrero, P. Desjeux, J. Cano, J. Jannin and M. den Boer, PLoS One, 2012, 7, e35671 CAS.
- P. D. Marsden, Rev. Soc. Bras. Med. Trop., 1985, 18, 187–198 CrossRef.
- F. Frézard, C. Demicheli and R. R. Ribeiro, Molecules, 2009, 14, 2317–2336 CrossRef PubMed.
- J. D. Berman, Clin. Infect. Dis., 1997, 24, 684–703 CrossRef CAS PubMed.
- A. Meyerhoff, Clin. Infect. Dis., 1998, 28, 42–48 Search PubMed.
- S. Sundar, T. K. Jha, C. P. Thakur, J. Engel, H. Sindermann, C. Fischer, K. Junge, A. Bryceson and J. Berman, N. Engl. J. Med., 2002, 347, 1739–1746 CrossRef CAS PubMed.
- S. W. LaFon, D. J. Nelson, R. L. Berens and J. J. Marr, Biochem. Pharmacol., 1982, 31, 231–238 CrossRef CAS PubMed.
- J. J. Marr, R. L. Berens and D. J. Nelson, Biochim. Biophys. Acta, 1978, 544, 360–371 CrossRef CAS.
- J. M. Boitz, B. Ullman, A. Jardim and N. S. Carter, Trends Parasitol., 2012, 28, 345–352 CrossRef CAS PubMed.
- W. Versées, E. Van Holsbeke, S. De Vos, K. Decanniere, I. Zegers and J. Steyaert, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, 59, 1087–1089 CrossRef.
- D. W. Parkin, B. A. Horenstein, D. R. Abdulah, B. Estupiñán and V. L. Schramm, J. Biol. Chem., 1991, 266, 20658–20665 CAS.
- W. Shi, V. L. Schramm and S. C. Almo, J. Biol. Chem., 1999, 274, 21114–21120 CrossRef CAS PubMed.
- E. Iovane, B. Giabbai, L. Muzzolini, V. Matafora, A. Fornili, C. Minici, F. Giannese and M. Degano, Biochemistry, 2008, 47, 4418–4426 CrossRef CAS PubMed.
- B. A. Horenstein and V. L. Schramm, Biochemistry, 1993, 32, 7089–7097 CrossRef CAS PubMed.
- M. Degano, D. N. Gopaul, G. Scapin, V. L. Schramm and J. C. Sacchettini, Biochemistry, 1996, 35, 5971–5981 CrossRef CAS PubMed.
- R. L. Miller, C. L. Sabourin, T. A. Krenitsky, R. L. Berens and J. J. Marr, J. Biol. Chem., 1984, 259, 5073–5077 CAS.
- B. A. Horenstein, D. W. Parkin, B. Estupiñán and V. L. Schramm, Biochemistry, 1991, 30, 10788–10795 CrossRef CAS PubMed.
- A. Shevchenko, H. Tomas, J. Havlis, J. V. Olsen and M. Mann, Nat. Protoc., 2006, 1, 2856–2860 CrossRef CAS PubMed.
- P. C. Carvalho, D. B. Lima, F. V. Leprevost, M. D. Santos, J. S. Fischer, P. F. Aquino, J. J. Moresco, J. R. Yates and V. C. Barbosa, Nat. Protoc., 2016, 11, 102–117 CrossRef CAS PubMed.
- J. K. Eng, T. A. Jahan and M. R. Hoopmann, Proteomics, 2013, 13, 22–24 CrossRef CAS PubMed.
- T. S. Tsao, E. Tomas, H. E. Murrey, C. Hug, D. H. Lee, N. B. Ruderman, J. E. Heuser and H. F. Lodish, J. Biol. Chem., 2003, 278, 50810–50817 CrossRef CAS PubMed.
- R. A. Copeland, Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists, John Wiley, N.J Hoboken, Chichester, 2005 Search PubMed.
- I. H. Segel, Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems, Wiley-Interscience, New York, 1975 Search PubMed.
- T. Lonhienne, E. Baise, G. Feller, V. Bouriotisand and C. Gerday, Biochim. Biophys. Acta, 2001, 1545, 349–356 CrossRef CAS.
- P. F. Cook and W. W. Cleland, Enzyme kinetics and mechanism, Garland Science, London and New York, 2007 Search PubMed.
- K. Hiromi, Kinetics of Fast Enzyme Reactions: Theory and Practice, Kodansha Ltd., Tokyo, 1979 Search PubMed.
- T. G. Battye, L. Kontogiannis, O. Johnson, H. R. Powell and A. G. Leslie, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 271–281 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- A. G. Leslie, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 48–57 CrossRef PubMed.
- B. W. Matthews, J. Mol. Biol., 1968, 33, 491–497 CrossRef CAS PubMed.
- K. A. Matthews and B. Rupp, Protein Sci., 2003, 12, 1865–1871 CrossRef PubMed.
- P. D. Adams, R. W. Grosse-Kunstleve, L. W. Hung, T. R. Ioerger, A. J. McCoy, N. W. Moriarty, R. J. Read, J. C. Sacchettini, N. K. Sauter and T. C. Terwilliger, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 1948–1954 CrossRef.
- P. V. Afonine, R. W. Grosse-Kunstleve, N. Echols, J. J. Headd, N. W. Moriarty, M. Mustyakimov, T. C. Terwilliger, A. Urzhumtsev, P. H. Zwart and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 352–367 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- R. A. Laskowski, M. W. MacArthur and J. M. Thornton, PROCHECK: validation of protein structure coordinates, Kluwer Academic Publishers, The Netherlands, 2001 Search PubMed.
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21 CrossRef CAS PubMed.
- K.-S. Ryu, C. Kim, C. Park and B.-S. Choi, J. Am. Chem. Soc., 2004, 126, 9180–9181 CrossRef CAS PubMed.
- P. L. Wink, Z. A. Sanchez Quitian, V. S. Rodrigues Jr, L. A. Rosado, G. O. Petersen, D. M. Lorenzini, T. Lipinski-Paes, L. F. S. M. Timmers, O. N. de Souza, L. A. Basso and D. S. Santos, Arch. Biochem. Biophys., 2013, 538, 80–94 CrossRef CAS PubMed.
- W. Versées, K. Decanniere, R. Pellé, J. Depoorter, E. Brosens, D. W. Parkin and J. Steyaert, J. Mol. Biol., 2001, 307, 1363–1379 CrossRef PubMed.
- J. E. Ladbury, M. L. Doyle and M. L. B. Doyle, Biocalorimetry 2: applications of calorimetry in the biological sciences, John Wiley, Chichester, 2004 Search PubMed.
- D. B. Northrop, Biochemistry, 1975, 14, 2644–2651 CrossRef CAS PubMed.
- D. N. Gopaul, S. L. Meyer, M. Degano, J. C. Sacchettini and V. L. Schramm, Biochemistry, 1996, 35, 5963–5970 CrossRef CAS PubMed.
- M. Degano, S. C. Almo, J. C. Sacchettini and V. L. Schramm, Biochemistry, 1998, 37, 6277–6285 CrossRef CAS PubMed.
- A. Psoda and D. Shugar, Biochim. Biophys. Acta, 1971, 247, 507–513 CrossRef CAS.
- B. Giabbai and M. Degano, Structure, 2004, 12, 739–749 CrossRef CAS PubMed.
- W. Yang, H.-W. Lee, H. Hellinga and J. J. Yank, Proteins: Struct., Funct., Genet., 2002, 47, 344–356 CrossRef CAS PubMed.
- E. Pidcock and G. R. Moore, J. Biol. Inorg Chem., 2001, 6, 479–489 CrossRef CAS PubMed.
- A. Fornili, B. Giabbai, G. Garau and M. Degano, J. Am. Chem. Soc., 2010, 132, 17570–17577 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07268f |
| This journal is © The Royal Society of Chemistry 2017 |