
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and biological evaluation of flavonoid salicylate derivatives as potential anti-tumor agents†
Xiangping Deng‡
a,
Zhe Wang‡a,
Juan Liua,
Shujuan Xionga,
Runde Xionga,
Xuan Caoa,
Yanming Chenb,
Xing Zhenga and
Guotao Tang *a
*a
aInstitute of Pharmacy and Pharmacology, Hunan Province Cooperative Innovation Center for Molecular Target New Drug Study, University of South China, Hengyang, Hunan 421001, China. E-mail: tgtzq@163.com
bMudanjiang Youbo Pharmaceutical Co. Ltd, Mudanjiang, China
First published on 3rd August 2017
Abstract
A series of flavonoid salicylate derivatives containing trimethoxybenzene and a series of chrysin salicylate derivatives were synthesized for use as anti-tumor agents, and evaluated for antiproliferative activity using three human tumor cells: MCF-7 (breast carcinoma cells), HepG2 (liver carcinoma cells), MGC-803 (gastric carcinoma cells) and the mice tumor cells MFC (forestomach carcinoma cells). A substituent group of a suitable size and the trimethoxybenzene had a certain influence on the bioactivity of the flavonoid salicylate derivatives. Compound 2 and its salicylate derivatives 7a–7g containing the trimethoxybenzene exhibited more antiproliferative activity. Among them, compound 7g displayed the most potent antiproliferative activity against MGC-803 cells and MFC cells with the concentration causing 50% inhibition of cell growth (IC50) values of 11.05 ± 1.58 μM and 13.73 ± 2.04 μM, respectively. The flow cytometry results showed that compound 7g caused the cell cycle to be arrested in the G0/G1 phase and induced apoptosis of MFC cells in a dose-dependent manner. Furthermore, compound 7g showed good anti-tumor activity in vivo. These results suggested that compound 7g could be a new, potent anti-tumor candidate which should be optimized and evaluated further.
Introduction
Tumors are the most common disease in the world and cause serious harm to human health. There is a critical need for discovering new and reliable anti-tumor drugs to improve the survival rates of tumor patients. Natural products have played an important role in the drug discovery and development because of their wide range of bioactivities, low toxicity and hardly any side effects.1–3During the last few decades, a growing number of studies have indicated that flavonoids have important effects in tumor treatment.4 Flavonoids are attractive natural plant products which widely exist in nature and have good biological activities, including anti-tumor, anti-inflammatory, anti-angiogenic, cardiovascular protective and anti-mutagenic effects.5–8 As the first discovered flavonoid vascular disrupting agent, flavonoid acetic acid (FAA) has shown great anti-tumor activity in the animal model.9 Acacetin significantly inhibited ovarian cancer, cell-induced angiogenesis and tumor growth in vivo by inhibiting the hypoxia-inducible factor 1-alpha (HIF-1α) protein level by increasing its degradation and decreasing its stability.10 Substantial studies have been conducted to identify diverse structurally modified flavonoids with biological benefits superior to their natural counterparts.11,12
Acetylsalicylic acid (ASA) and salicylic acid (SA) which are well known as anti-inflammatory drugs also show anti-tumoral properties, induction of apoptosis13,14 and changing tumor glucose utilization.15 The anti-tumor effect of ASA and SA are because of their effects on cyclooxygenase (COX)16 and 6-phosphofructo-1-kinase (PFK).15 It has been demonstrated that ASA and SA modulated PFK quaternary structure and decreased tumor cells' glucose consumption and viability, suggesting that ASA and SA could be used as an anti-tumoral agent.15
The structure of the trimethoxybenzene group, which exists in many tumor vascular disrupting agents (Fig. 1), such as CA4P and its analogues OXi4503, AVE8062,17–20 the colchicine analog ZD6126,21 BNC-105 (ref. 22) and CKD-516,23 has an important research value in tumor drug design.
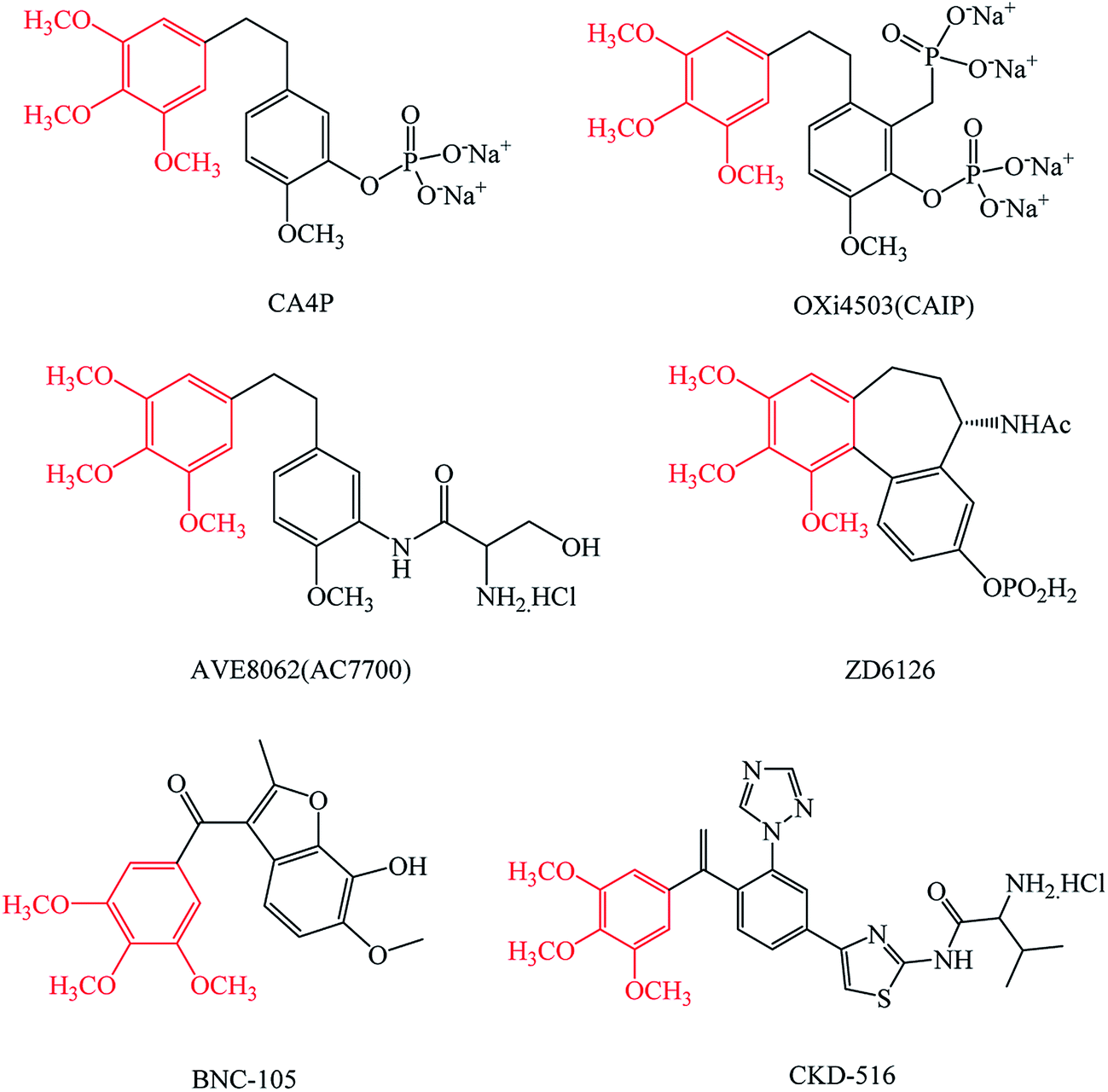 | ||
| Fig. 1 The structures of CA4P, OXi4503, AVE8062, ZD6126, BNC-105 and CKD-516. These tumor vascular disrupting agents all have a trimethoxybenzene group. | ||
Based on the previous considerations, it was proposed that introducing trimethoxybenzene groups and SA and its diverse derivatives into a flavonoid scaffold might be an effective strategy for seeking novel flavonoid derivatives with potential anti-tumor activity. Therefore, a series of flavonoid salicylate derivatives were synthesized which contained trimethoxybenzene and a series of chrysin salicylate derivatives, and these were evaluated for anti-tumor bioactivities in vitro and in vivo.
Results and discussion
Chemistry
In this study, two flavonoids and 21 flavonoid salicylate derivatives were synthesized and satisfactory analytical and spectroscopic data was obtained, which agreed well with their expected structures. The synthesis of compounds 2 and 4 are shown in Scheme 1. Commercially available 3,4,5-trimethoxyphenol or 1,3,5-trihydroxybenzene were used to produce compound 1 or 3, respectively, by the Hoesch reaction with zinc chloride (ZnCl2) as catalyst in a mixture of absolute ether and chloroform or absolute ether at 0 °C. The key intermediates 1 or 3 were reacted with an aromatic aldehyde to give flavonoid 2 or 4 with recrystallization from 95% ethanol. As shown in Scheme 2, hydroxyl of salicylic acids 5a–5h were methylation and then carboxyl were acylating chlorination to obtain 6a–6h. The three groups of the final target compounds 7a–7g, 8a–8g and 9a–9g were synthesized as shown in Scheme 3 and the values of the concentration causing 50% inhibition of cell growth (IC50) for them are listed in Table 1.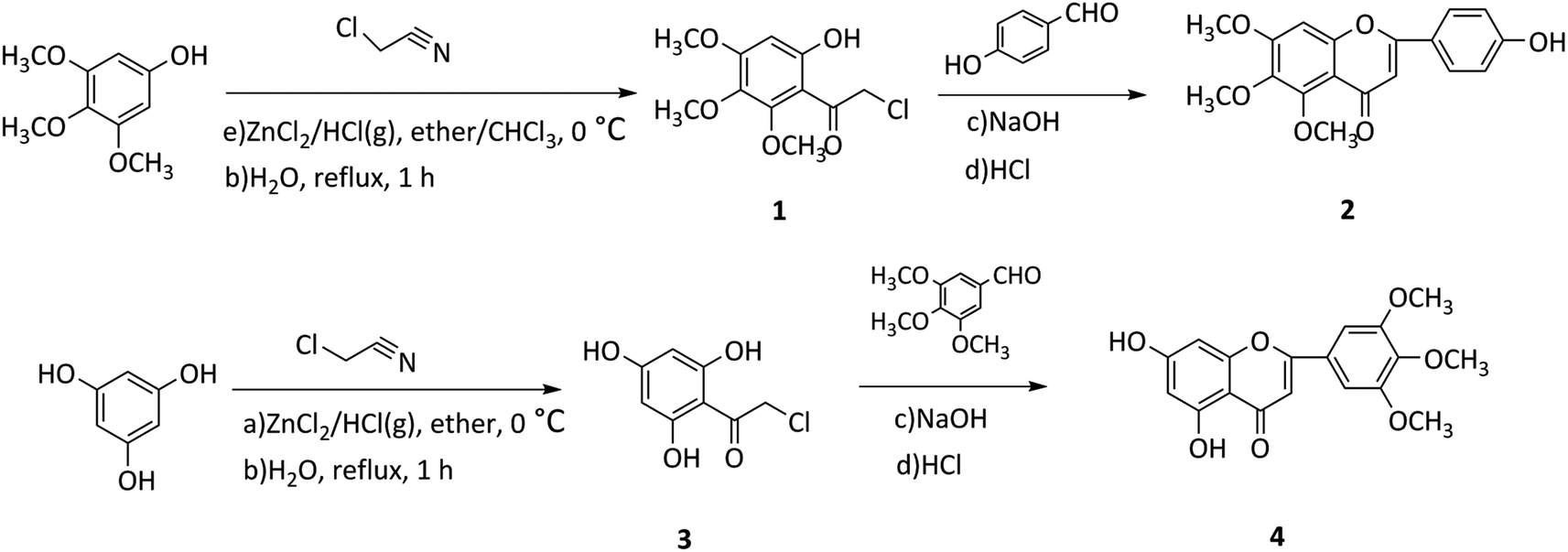 | ||
| Scheme 1 Synthetic route of flavonoids 2 and 4. | ||
 | ||
| Scheme 2 Synthetic route of compounds 6a–6h. | ||
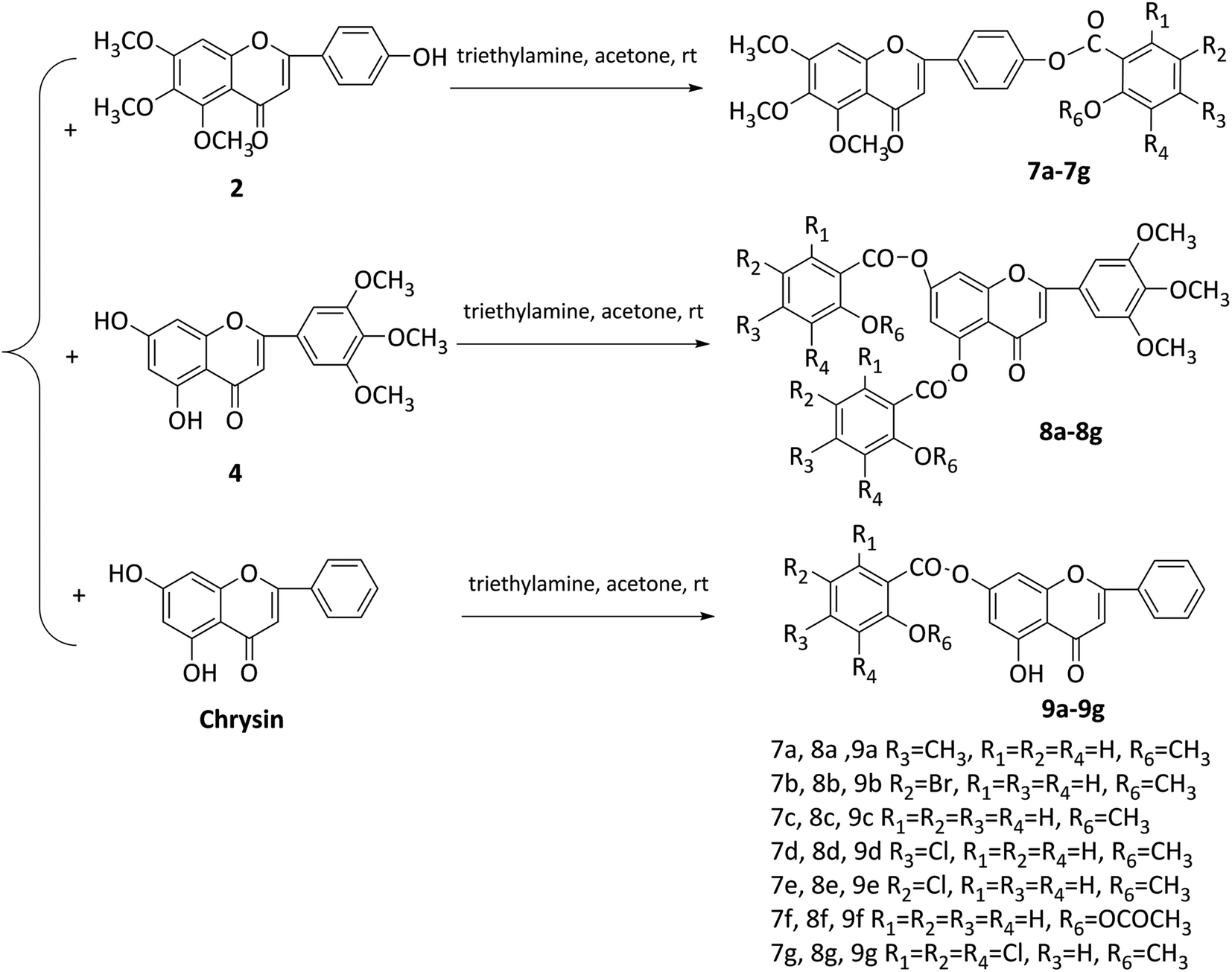 | ||
| Scheme 3 Synthetic route of compounds 7a–7h, 8a–8h and 9a–9h. | ||
| Compound | IC50 (μM) | |||
|---|---|---|---|---|
| MGC-803 | MCF-7 | HepG-2 | MFC | |
| a ND = not detected. | ||||
| 2 | 31.19 ± 6.48 | 44.23 ± 3.77 | >100 | 50.27 ± 3.71 |
| 4 | >100 | ND | 78.36 ± 5.11 | >100 |
| 7a | 27.08 ± 5.91 | >100 | >100 | 34.62 ± 3.64 |
| 7b | 52.33 ± 6.92 | >100 | ND | 64.48 ± 5.27 |
| 7c | 63.78 ± 5.01 | >100 | >100 | 89.22 ± 4.97 |
| 7d | 21.59 ± 5.75 | ND | 65.44 ± 4.63 | 25.38 ± 5.13 |
| 7e | 27.53 ± 3.93 | >100 | >100 | 40.46 ± 3.95 |
| 7f | 89.31 ± 6.05 | ND | 99.72 ± 7.41 | >100 |
| 7g | 11.05 ± 1.58 | 55.70 ± 3.75 | 60.43 ± 8.06 | 13.73 ± 2.04 |
| 8a | >100 | >100 | >100 | >100 |
| 8b | >100 | ND | ND | >100 |
| 8c | 53.35 ± 8.92 | 84.97 ± 6.98 | 76.27 ± 4.27 | 91.26 ± 6.09 |
| 8d | >100 | ND | ND | ND |
| 8e | 78.21 ± 5.98 | >100 | ND | 77.24 ± 6.72 |
| 8f | >100 | ND | >100 | >100 |
| 8g | >100 | >100 | 87.9 ± 5.61 | >100 |
| 9a | 28.28 ± 4.34 | 48.54 ± 3.29 | 62.34 ± 6.18 | 31.06 ± 2.35 |
| 9b | 84.68 ± 11.03 | >100 | >100 | 73.45 ± 5.82 |
| 9c | >100 | ND | ND | ND |
| 9d | 37.53 ± 6.81 | 53.54 ± 4.16 | 64.75 ± 3.15 | 32.37 ± 4.13 |
| 9e | 36.1 ± 7.89 | 51.49 ± 3.67 | 84.37 ± 5.02 | 44.69 ± 2.98 |
| 9f | >100 | >100 | >100 | ND |
| 9g | 21.77 ± 6.53 | ND | >100 | 25.95 ± 3.72 |
| 5-FU | 70.11 ± 3.08 | 60.43 ± 2.95 | 68.17 ± 3.29 | 79.26 ± 4.55 |
| Chyrsin | 87.21 ± 4.37 | 68.83 ± 5.14 | >100 | 91.48 ± 4.99 |
Biological activities
After comparison of the bioactivity data, compound 2 and its salicylate derivatives 7a–7g, and chrysin and its salicylate derivatives 9a–9g showed better inhibition activity than compound 4 and its salicylate derivatives or 8a–8g with the larger sized substituents, suggesting that a suitable size of substituent group for flavonoids was favorable. As a whole, compound 2 and its salicylate derivatives 7a–7g displayed higher inhibitory activity, when compared to chrysin and its salicylate derivatives 9a–9g, implying that the scaffold hopping strategy was feasible. Compounds 7f, 8f and 9f with the 5-Cl substituent showed a lower inhibitory activity. At the same time, it was noted that compounds 7a and 9a with 4-CH3 substituent on the R3 position, and compounds 7d and 9d with 4-Cl substituent on the R3 position displayed good antiproliferative activity against MGC-803 cells and MFC cells. Furthermore, compounds 7g and 9g with the 3,5,6-trichloro substituent were more active than derivatives with other substituents. Out of these compounds, compound 7g bearing the 3,5,6-trichloro substituent and the trimethoxybenzene showed the most potent antiproliferative activity against MGC-803 cells and MFC cells with IC50 values of 11.05 ± 1.58 μM and 13.73 ± 2.04 μM, respectively. Therefore, compound 7g was selected for further evaluation for anti-tumor activity.
The Kunming male mice were chosen for the anti-tumor efficacy assay in vivo. The biological activity of compound 7g against MGC-803 cells and MFC cells was found to be similar. Considering all these factors, the MFC cell lines were chosen for the cell cycle assay, the apoptosis assay, and the anti-tumor efficacy assay in vivo.
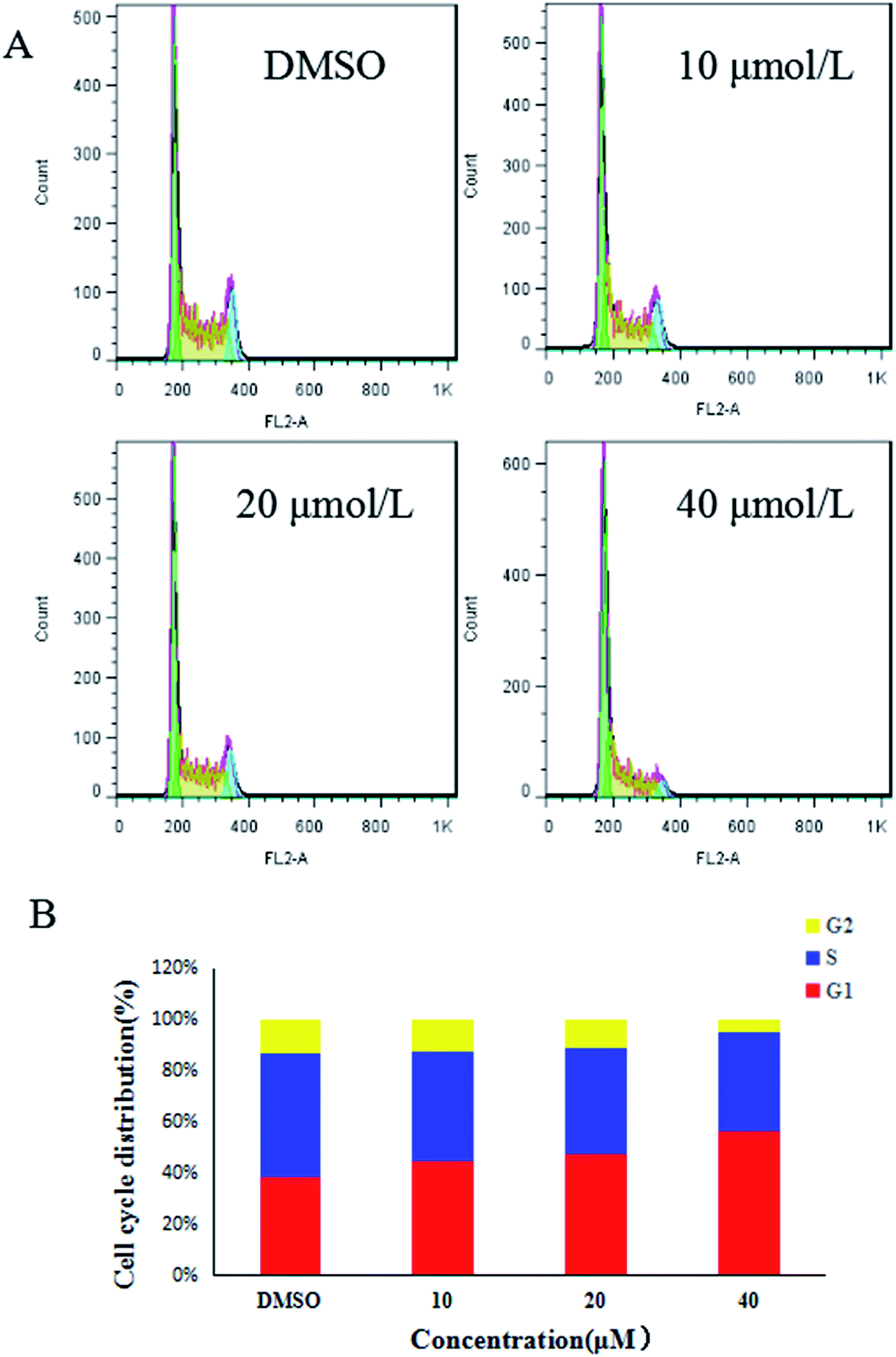 | ||
| Fig. 2 Investigation of cell cycle distribution of compound 7g using flow cytometry. Cells were treated with compound 7g at concentrations of 10, 20, or 40 μM for 24 h. Cells treated with vehicle (DMSO) were used for comparison. (A) Representative photographs from three independent experiments are shown. (B) Cell cycle proportion in (A) was quantitated. Data presented are the mean ± SD of three independent experiments. | ||
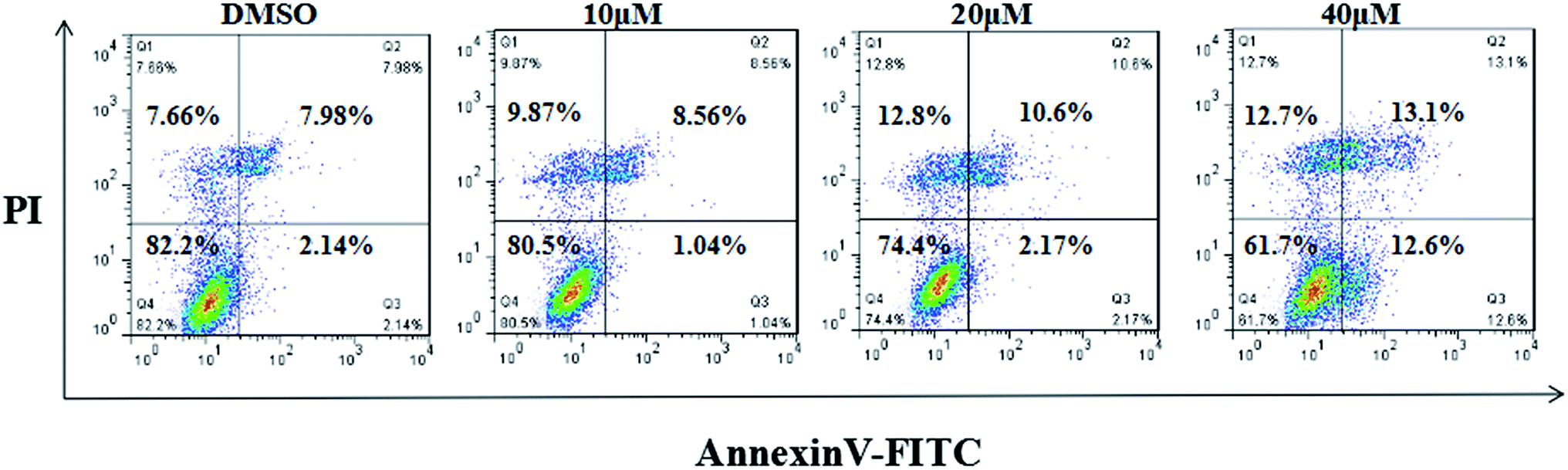 | ||
| Fig. 3 Evaluation of concentration dependent cellular apoptosis of compound 7g by flow cytometry. Cells were treated with compound 7g at 10, 20, or 40 μM for 24 h, respectively. Cells treated with vehicle (DMSO) were used for comparison. Representative photographs from three independent experiments are shown. | ||
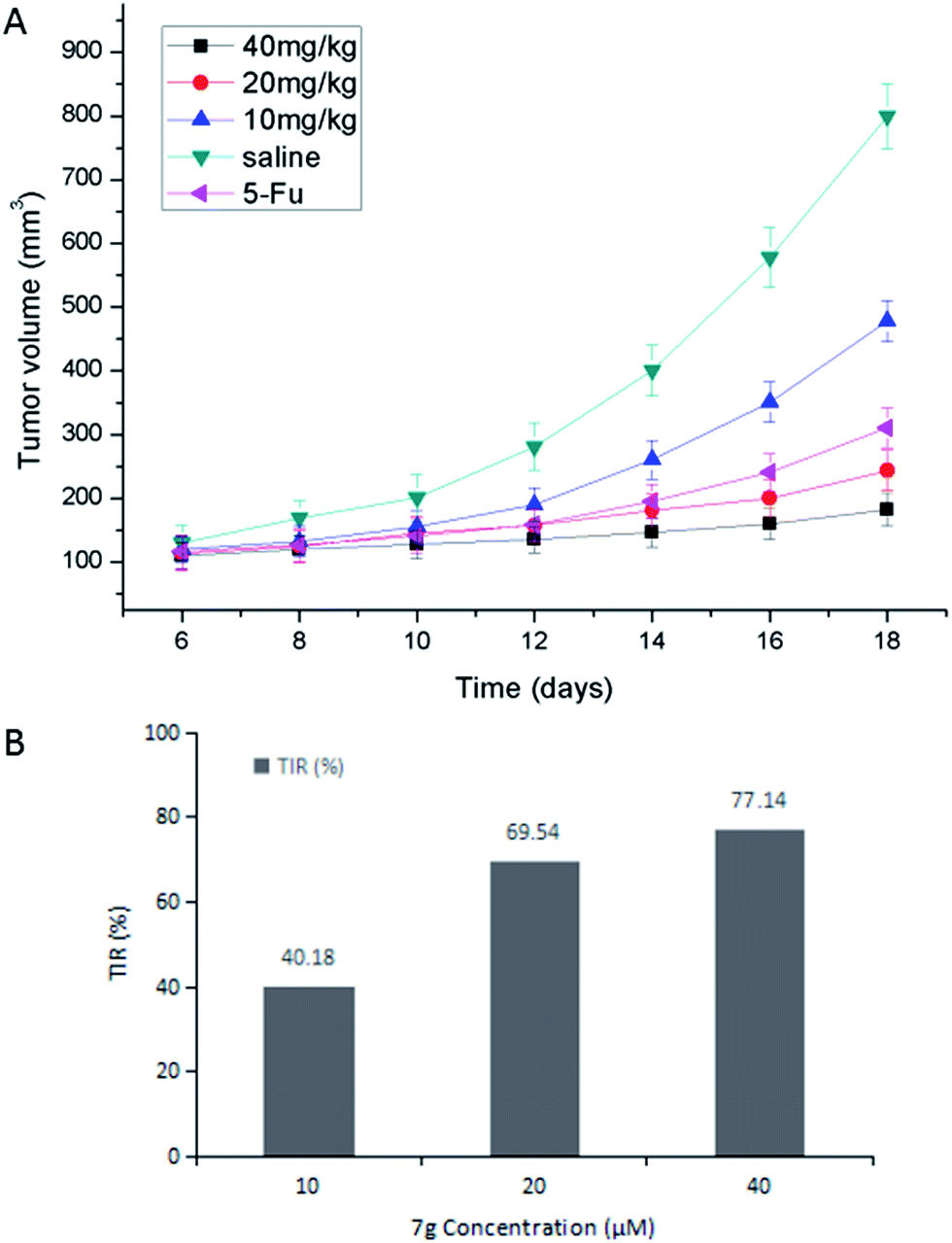 | ||
| Fig. 4 (A) Tumor volume growth curves of the mice after treatment with 5-Fu (25 mg kg−1, dissolved in saline mixed with 5% Tween 80), different doses of compound 7g (10, 20, 40 mg kg−1, dissolved in saline mixed with 5% Tween 80) and saline (mixed with 5% Tween 80). Data are shown as a mean ± SD (n = 6). The differences of the last measured tumor volume among five groups are statistically significant (P < 0.05). (B) The TIR (%) of different doses of compound 7g. | ||
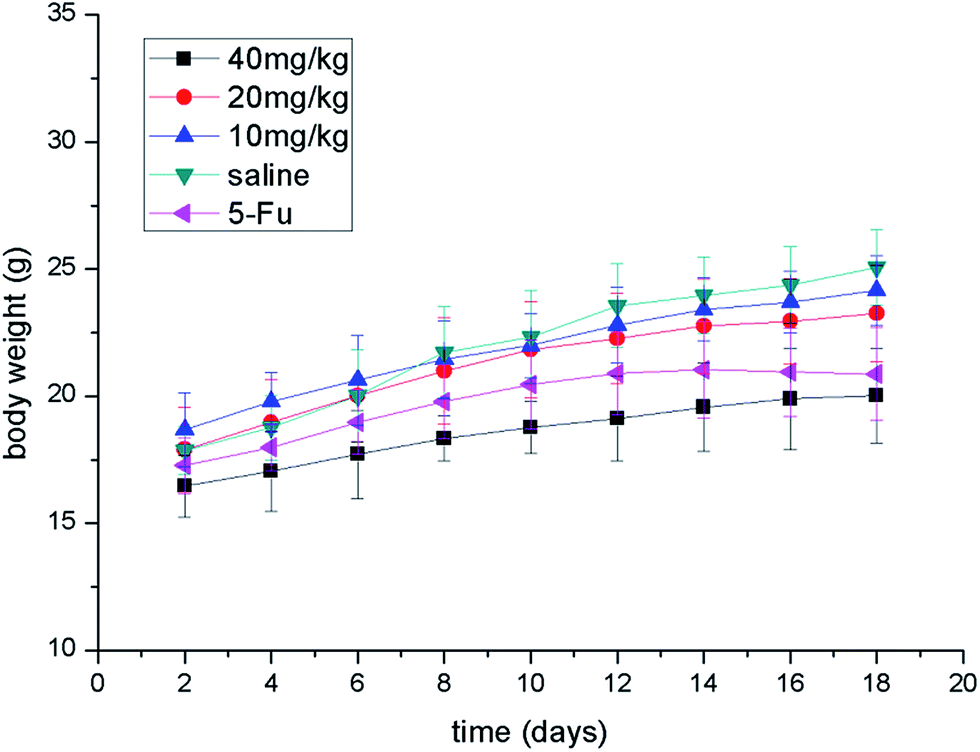 | ||
| Fig. 5 Body weight evolution curves of the mice after treatment with 5-Fu (20 mg kg−1, dissolved in saline mixed with 5% Tween 80), different doses of compound 7g (10, 20, 40 mg kg−1, dissolved in saline mixed with 5% Tween 80) and saline (mixed with 5% Tween 80). Data are shown as a mean ± SD (n = 6). | ||
Materials and methods
Chemicals
All the starting materials, solvents and reagents were reagent grade and purchased from commercial sources unless otherwise stated. Silica gel column chromatography was performed with silica gel 100–200 mesh size. Thin-layer chromatography was carried out on silica gel 60 F254 plates and visualized under (ultraviolet) UV light at 254 nm. Melting points (uncorrected) were measured using an electrothermal digital melting point apparatus (Thermo Fisher Scientific). Proton-nuclear magnetic resonance (1H-NMR) and 13C-NMR spectra were measured on a spectrometer (Bruker) at 400 MHz and 101 MHz, respectively. Mass spectra (MS) were obtained using GCT time-of-flight mass spectrometer (Waters). Chemical shifts and coupling constants are reported in ppm (δ) and Hz (J), respectively. The 1H-NMR, 13C-NMR and electrospray ionization (ESI)-MS spectral data of the compounds are supplied as ESI.†Synthesis of 5,7-dihydroxy-2-(3,4,5-trimethoxyphenyl)-4H-chromen-4-one (2)
Freshly prepared dry hydrogen chloride (HCl) gas was bubbled slowly and continuously into a reaction mixture of a solution of 3,4,5-trimethoxyphenol (0.012 mol) and 2-chloroacetonitrile (0.024 mol) in a mixed solvent of absolute ether and chloroform (30 mL) with a catalytic amount of ZnCl2 at 0 °C, for 3 h. After standing the reaction flask at 4 °C for 3 d, a yellow precipitate was prepared after filtration, washing and drying. The yellow precipitate was dissolved in hot water and heated to reflux, and a light yellow precipitate was produced. The light yellow precipitate, 2-chloro-1-(6-hydroxy-2,3,4-trimethoxyphenyl)ethan-1-one (1) was filtered off, washed with water (H2O), and dried in vacuo to give a light yellow precipitate. 4-Hydroxybenzaldehyde (0.01 mol) and 1 (0.012 mol) were added together to a stirred solution of 3 mL ethanol, then 10% sodium hydroxide [NaOH (aq)] was added dropwise into the solution. The reaction mixture was stirred at room temperature (rt) for 48 h, and acidification with 10% HCl (aq) gave a crude product, which was filtered off and purified by recrystallization from 95% ethanol to give compound 2 as golden powder.Synthesis of 2-(4-hydroxyphenyl)-5,6,7-trimethoxy-4H-chromen-4-one (4)
Freshly prepared dry HCl gas was bubbled slowly and continuously into a reaction mixture of a solution of 1,3,5-trihydroxybenzene (0.023 mol) and 2-chloroacetonitrile (0.032 mol) in absolute ether (30 mL) with a catalytic amount of ZnCl2 at 0 °C, for 3 h. After standing the reaction flask at 4 °C for 3 d, an orange precipitate was prepared after filtration, washing and drying. The orange precipitate was dissolved in hot water and heated to reflux, and a red-brown precipitate was produced. The red-brown precipitate, 2-chloro-1-(2,4,6-trihydroxyphenyl)ethan-1-one (3) was filtered off, washed with water, and dried in vacuo to give a light red-brown precipitate. 4-Hydroxybenzaldehyde (0.01 mol) and 3 (0.012 mol) were added together to a stirred solution of 3 mL ethanol, then 10% NaOH (aq) was added dropwise into the solution. The reaction mixture was stirred at rt for 48 h, and acidification with 10% HCl (aq) gave a crude product, which was filtered off and purified by recrystallization from 95% ethanol to give compound 4 as reddish brown powder.General method of synthesis for 6a–6g
A stirred solution of 4-methylsalicylic acid (0.005 mol) in distilled water (10 mL), was heated at 37 °C. The solution was adjusted to pH 10 with 30% NaOH. After stirring for 30 min, dimethyl sulfate (2 mL) was slowly added and the pH of the solution was adjusted to 7, and then 30% NaOH to adjust the pH of the solution to 10. The reaction was carried out for 2 h and the solution was hydrolyzed at 80 °C for 15 min. After cooling, the solution was adjusted to pH 5 with 10% HCl, and a white precipitate was obtained. The crude product was filtered off, washed with water, and dried in vacuo to give a white powder. The white precipitate, thionyl chloride and dichloromethane were left with stirring at 40 °C for 30 min. The methylene chloride was evaporated under reduced pressure to give a clear oily liquid (6a). Compounds 6b–6g were prepared in a similar way.General method of synthesis for 7a–7g
A solution of 6a (0.005 mol) and compound 2 (0.004 mol) in acetone (15 mL) with triethylamine (0.5 mL) at was stirred at rt for 1 h and a yellow precipitate was obtained. The yellow precipitate was filtered off, washed with acetone and dried in vacuo. The crude product was purified using column chromatography on silica gel, and eluted with dichloromethane/methanol (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give a yellow powder (7a).
:1) to give a yellow powder (7a).
Compounds 7b–7g were prepared in a similar way.
General method of synthesis for 8a–8g
A solution of 6a (0.005 mol) and compound 4 (0.004 mol) in acetone (15 mL) with triethylamine (0.5 mL) was stirred at rt for 1 h and a yellow precipitate was obtained. The yellow precipitate was filtered off, washed with acetone and dried in vacuo. The crude product was purified using column chromatography on silica gel, and eluted with dichloromethane/methanol (50:1) to give a yellow powder (8a).
Compounds 8b–8g were prepared in a similar way.
General method of synthesis for 9a–9g
A solution of 6a (0.005 mol) and chrysin (0.004 mol) in acetone (15 mL) with triethylamine (0.5 mL) was stirred at rt for 1 h and a yellow precipitate was obtained. The yellow precipitate was filtered off, washed with acetone and dried in vacuo. The crude product was purified using column chromatography on silica gel, and eluted with dichloromethane/methanol (50:1) to give a light yellow powder (9a).
Compounds 9b–9g were prepared in a similar way.
Cell culture
Various human tumor cells including MCF-7, HepG2, MGC-803 and the mice MFC carcinoma cells were purchased from the Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Gibco, NY, USA) containing 10% fetal bovine serum at 37 °C in a humidified incubator with 5% carbon dioxide (CO2).In vitro anti-tumor efficacy
In vitro anti-tumor efficacy of the compounds were evaluated using the human tumor cells MCF-7, HepG2, MGC-803 and the mice tumor cells MFC. Each tested compound was dissolved in dimethyl sulfoxide (DMSO). Cells were plated in 96-well microtiter plates at a density of 8 × 103 cells per well and incubated in a humidified atmosphere with 5% CO2 at 37 °C for 48 h. Test compounds of different concentrations (256, 128, 64, 32, 16, 8, 4, 2 μM) were added into triplicate wells and DMSO was used as the control. After 48 h, 20 μL of MTT solution (5 mg mL−1) was added to each well and incubation was continued for an additional 4 h. Formazan was dissolved in 150 μL of DMSO and added to the wells. The absorbance (optical density, OD) was monitored on a Wellscan MK-2 microplate reader (LabSystems) at 490 nm. The IC50 values was determined using the logit method. All experiments were performed three times.Cell cycle analysis
The MFC cells were plated in six-well plates (2 × 104 cells per well). Next day, the cells were incubated with different concentrations of compound 7g (0, 10, 20, 40 μM). After 24 h of culturing, the cells were washed twice with cold phosphate buffered saline (PBS) and then resuspended with cold PBS. Next, the cells were fixed and then stored in 70% cold ethanol for 12 h at 4 °C. Then 500 μL of PI stain was added to the cells in each tube, and the cells were gently vortex mixed and then incubated for 30 min at 37 °C in the dark. The cells were analyzed using flow cytometry within 24 h. All the experiments were performed in triplicate.Cell apoptosis assay
The MFC cells were plated in six-well plates (2 × 104 cells per well). Next day, the cells were incubated with different concentrations of compound 7g (0, 10, 20, 40 μM). After 24 h of culturing, the cells were washed twice with cold PBS and then resuspended in 1× binding buffer. Cells were transferred to a 5 mL culture tube, and then 5 μL of FITC Annexin V and 5 μL of PI were added to the cells. Next, the cells were gently vortex mixed and incubated for 15 min at 25 °C in the dark. Then 400 μL of 1× binding buffer was added to each tube. The cells were analyzed using flow cytometry within 1 h. All the experiments were performed in triplicate.In vivo anti-tumor efficacy
All experiments with animals were performed in compliance with the relevant laws and Chinese institutional guidelines for animal experiments, and the Institutional Animal Care and Committee of University of South China [permit number: SYXK (Xiang) 2015-0001] approved the experiments. Kunming male mice (University of South China, China) were used for the in vivo experiments. Gastric carcinoma was chosen as a model tumor to evaluate the anti-tumor efficacy of compound 7g. Gastric tumor bearing mice were established by subcutaneously injecting 1 × 107 MFC cells per 0.2 mL into the right flank region of the mice (defined as day 0). On day 1, the mice were randomly divided into five groups (n = 6). The mice were treated with different doses of compound 7g (10, 20, 40 mg kg−1, dissolved in saline mixed with 5% Tween 80) and 5-Fu (25 mg kg−1, dissolved in saline mixed with 5% Tween 80), saline (mixed with 5% Tween 80) via intraperitoneal injection on days 2, 5, 8, 11 and 14.From day 6, tumor volumes were monitored every other day by measuring two perpendicular diameters using a Vernier caliper and calculated using the formula: volume = 0.5 × length × (width)2. Body weights were recorded every other day. On day 18, the animals were sacrificed and the tumor mass was dissected, weighed, and photographed. The TIR was calculated using the formula: TIR (%) = (1 − Wt/Ws) × 100%, where Wt and Ws represent the average tumor weight of the treatment and saline groups, respectively.
Conclusion
In summary, a series of flavonoid salicylate derivatives were synthesized and these compounds were evaluated for their anti-tumor activities. These compounds exhibited moderate to good antiproliferative activity against MGC-803 cell lines and MFC cell lines. An appropriate size of substituent group and the trimethoxybenzene had a certain influence on the bioactivity of the flavonoid salicylate derivatives. Compound 2 and its salicylate derivatives 7a–7g containing the trimethoxybenzene exhibited greater antiproliferative activity. Among them, compound 7g displayed the most potent antiproliferative activity against MGC-803 cells and MFC cells with IC50 values of 11.05 ± 1.58 μM and 13.73 ± 2.04 μM, respectively. What's more, the flow cytometry results showed that compound 7g caused the cell cycle to be arrested in the G0/G1 phase and induced apoptosis of MFC cells in a dose-dependent manner. Furthermore, compound 7g also had good antitumor activity in vivo. In summary, compound 7g can be recommended as a new, potent anti-tumor candidate for further optimization and evaluation. Further study on the anti-tumor mechanism of compound 7g is in progress.Conflict of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work was supported in part by the Construct Program of the Key Discipline in Hunan Province and Hunan Province Cooperative Innovation Center for Molecular Target New Drug [Hunan Provincial Education Department document (Approval number: 2014-405)]. The authors wish to thank the Department of Laboratory Animal Science for support with the animal experiments.Notes and references
- F. E. Koehn and G. T. Carter, Nat. Rev. Drug Discovery, 2005, 4, 206–220 CrossRef CAS PubMed.
- M. S. Butler, J. Nat. Prod., 2004, 67, 2141–2153 CrossRef CAS PubMed.
- G. M. Cragg, P. G. Grothaus and D. J. Newman, Chem. Rev., 2009, 109, 3012–3043 CrossRef CAS PubMed.
- A. Triantafyllou and I. Mylonis, Free Radical Biol. Med., 2008, 44, 657–670 CrossRef CAS PubMed.
- A. Scalbert, I. T. Johnson and M. Saltmarsh, Am. J. Clin. Nutr., 2005, 81, 215S–217S CAS.
- M. Singh, M. Kaur and O. Silakari, Eur. J. Med. Chem., 2014, 84, 206–239 CrossRef CAS PubMed.
- E. Middleton Jr, C. Kandaswami and T. C. Theoharides, Pharmacol. Rev., 2000, 52, 673–751 CAS.
- B. H. Havsteen, Pharmacol. Ther., 2002, 96, 67–202 CrossRef CAS PubMed.
- H. Yoshiji, S. R. Harris and U. P. Thorgeirsson, Anticancer Res., 2000, 20, 2249–2254 Search PubMed.
- L. Z. Liu, Y. Jing, L. L. Jiang, X. E. Jiang, Y. Jiang, Y. Rojanasakul and B. H. Jiang, Biochem. Biophys. Res. Commun., 2011, 413, 299–305 CrossRef CAS PubMed.
- Z. Y. Zhu, W. X. Wang, Z. Q. Wang, L. J. Chen, J. Y. Zhang, X. C. Liu, S. P. Wu and Y. M. Zhang, Eur. J. Med. Chem., 2014, 75, 297–300 CrossRef CAS PubMed.
- W. Huang, Q. Chen, W. C. Yang and G. F. Yang, Eur. J. Med. Chem., 2013, 66, 161–170 CrossRef CAS PubMed.
- S. I. Grivennikov, F. R. Greten and M. Karin, Cell, 2010, 140, 883–899 CrossRef CAS PubMed.
- M. J. Thun, E. J. Jacobs and C. Patrono, Nat. Rev. Clin. Oncol., 2012, 9, 259–267 CrossRef CAS PubMed.
- P. J. Loll, D. Picot and R. M. Garavito, Nat. Struct. Biol., 1995, 2, 637–643 CrossRef CAS PubMed.
- G. A. Spitz, C. M. Furtado, M. Sola-Penna and P. Zancan, Biochem. Pharmacol., 2009, 77, 46–53 CrossRef CAS PubMed.
- G. R. Pettit, J. W. Lippert, D. L. Herald, E. Hamel and R. K. Pettit, J. Nat. Prod., 2000, 63, 969–974 CrossRef CAS.
- G. J. Rustin, S. M. Galbraith, H. Anderson, M. Stratford, L. K. Folkes, L. Sena, L. Gumbrell and P. M. Price, J. Clin. Oncol., 2003, 21, 2815–2822 CrossRef CAS PubMed.
- G. R. Pettit, C. Temple, V. L. Narayanan, R. Varma, M. J. Simpson, M. R. Boyd, G. A. Rener and N. B. Ansal, Anti-Cancer Drug Des., 1995, 10, 299–309 CAS.
- G. R. Pettit and J. W. Lippert III, Anti-Cancer Drug Des., 2000, 5, 203–216 Search PubMed.
- G. Micheletti, M. Poli, P. Borsotti, M. Martinelli, B. Imberti, G. Taraboletti and R. Giavazzi, Cancer Res., 2003, 63, 1534–1537 CAS.
- G. Kremmidiotis, A. F. Leske, T. C. Lavranos, D. Beaumont, J. Gasic, A. Hall, M. O'Callaghan, C. A. Matthews and B. Flynn, Mol. Cancer Ther., 2010, 9, 1562–1573 CrossRef CAS PubMed.
- J. Lee, S. J. Kim, H. Choi, Y. H. Kim, I. T. Lim, H.-m. Yang, C. S. Lee, H. R. Kang, S. K. Ahn, S. K. Moon, D.-H. Kim, S. Lee, N. S. Choi and K. J. Lee, Eur. J. Med. Chem., 2010, 53, 6337–6354 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07235j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |