
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A family of lanthanide compounds based on nitronyl nitroxide radicals: synthesis, structure, magnetic and fluorescence properties†
Xue-Hui Lva,
Shuai-Liang Yanga,
Yuan-Xia Lia,
Chen-Xi Zhang *ab and
Qing-Lun Wang*bc
*ab and
Qing-Lun Wang*bc
aCollege of Chemical Engineering and Materials Science, Tianjin University of Science and Technology, Tianjin 300457, P. R. China. E-mail: zcx@tust.edu.cn
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, P. R. China. E-mail: wangql@nankai.edu.cn
cCollege of Chemistry, Nankai University, Tianjin 300071, P. R. China
First published on 9th August 2017
Abstract
A series of lanthanide nitronyl nitroxide radical compounds, [Ln(hfac)3(NITPh-p-N(CH3)2)2]·xC7H16 (Ln = La (1), Eu (2), Gd (3), Tb (4), Dy (5), Er (6), hfac = hexafluoroacetylacetonate; NITPh-p-N(CH3)2 = 4′-dimethylamino-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide, x = 2/3 for complexes 1, 2, 3 and 6, and 0 for 4 and 5), have been synthesized and structurally characterized. X-Ray crystallographic analyses show that the structures of the six compounds are similar and all consist of isolated molecules, in which central Ln(III) ions are coordinated to six oxygen atoms from three hexafluoroacetylacetonate ligands and two oxygen atoms from nitronyl nitroxide radicals. Variable-temperature magnetic susceptibility studies reveal ferromagnetic interactions between the Gd(III) ion and nitronyl nitroxide radical in complex 3. Ac magnetic susceptibility measurements of complexes 4 and 5 exhibit frequency-dependent out-of-phase signals, indicating single-molecule magnet (SMM) behavior.
Introduction
The study of molecular nanomagnets showing slow relaxation of the magnetization is one of the key topics in the field of molecular magnetism.1 Molecular nanomagnets mainly include single-molecule magnets (SMMs)2 and single-chain magnets (SCMs).3 SMMs and SCMs not only permit the observation of fascinating quantum phenomena but also are investigated as potential candidates for future high-density data storage materials. SMMs and SCMs exhibit slow relaxation of magnetization due to a large ground-state spin quantum number (S), a significant uniaxial magneto-anisotropy (D < 0), and a good magnetic isolation of the molecule.Lanthanide (Ln) ions have become attractive candidates for constructing new SMMs and SCMs because most of them have significant magnetic anisotropy arising from the large unquenched orbital angular momentum. As a result, many lanthanide complexes with SMM behavior have become attractive synthetic targets for chemists.4
The stability of the radicals like nitronyl nitroxide at a relatively high temperature has made them potential systems in molecular magnetism.5 Recently, nitronyl nitroxides and lanthanide ions were successfully synthesized to obtain the SMMs and SCMs,6 especially heavy lanthanide ions such as terbium(III) and dysprosium(III).7 Since the terbium(III) and dysprosium(III) ions have large anisotropies,8 synthesis and characterization of terbium(III) and dysprosium(III) with the nitronyl nitroxide have already set the fashion in the field of molecular magnetism.9–11 For example, complex [Tb3(hfac)9(NIT-2thien)3]n showed the coexistence of spin canting, metamagnetism, spin dynamic relaxation and magnetic ordering.9 [Dy(hfac)3(PyNO)]2 shows a remarkable SMM behavior with complex hysteresis at 1.4 K.10 One-dimensional chain complex [Tb(hfac)3(NITPhSCH3)]n is the first example of radical–metal SMM units in the construction of an SCM system.11
It is very interesting to develop new nitronyl nitroxide complexes and better understand the nature of 2p–4f magnetic interaction. In this paper, by using a new nitronyl nitroxide radical NITPh-p-N(CH3)2 and lanthanide ions, a series of novel 2p–4f complexes were synthesized. Herein we report a detailed study of the magnetic properties of a novel family of 2p–4f complexes: [Ln(hfac)3(NITPh-p-N(CH3)2)2]·xC7H16, (Ln = La (1), Eu (2), Gd (3), Tb (4), Dy (5) and Er (6), x = 2/3 for complexes 1, 2, 3 and 6 while 0 for 4 and 5). Complexes 4 and 5 present slow relaxation of the magnetization at low temperature, suggesting single-molecule magnet behavior.
Experimental section
Materials and physical measurements
All reagents and solvents were purchased from commercial sources and used without purification. Ln(hfac)3·2H2O (La, Eu, Gd, Tb, Dy, Er)12 and NITPh-p-N(CH3)2 (ref. 13) were synthesized according to the reported methods.Infrared spectra were recorded on a Perkin-Elmer Spectrum One spectrometer using KBr pellets in the region 4000–400 cm−1. Elemental analyses for C, H, and N were measured by a Perkin-Elmer 240 elemental analyzer. The X-ray powder diffraction patterns (PXRD) have been recorded on a Shimadzu LabX XRD-6100 diffractometer, operated at 40.0 kV and 30.0 mA, using a Cu target tube and graphite monochromator. The intensity data were recorded by continuous scan in 2θ/θ mode from 5° to 50° with a step size of 0.02° and a scan speed of 5° min−1. The thermogravimetry-differential thermal analysis (TG-DTA) was studied by SDT-Q600 at a heating rate of 20 C min−1 in nitrogen atmosphere, and nitrogen gas of high purity (>99.999%) with a flow rate of 100 mL min−1 was used as carrier gas. The sample was heated from ambient temperature to 600 °C. Luminescence properties were recorded on an F-4500 FL spectrophotometer with a xenon arc lamp as the light source. The magnetic measurements were carried out with a MPMS XL-7 SQUID magnetometer. The samples are embedded in grease to avoid preferential orientation of the micro-crystallites. Diamagnetic corrections were made with Pascal's constants for all of the constituent atoms.14
Synthesis procedures
Crystal structure determination
Crystals of complexes 1–6 were mounted on glass fibers. Determination of the unit cell and data collection were performed with Mo-Kα radiation (λ = 0.71073 Å) on a Bruker SMART 1000 diffractometer and equipped with a CCD camera. The ω–φ scan technique was employed. The structures were solved primarily by direct method and second by Fourier difference techniques and refined by the full-matrix least-squares method. The computations were performed with the SHELXL-97 program.15 Non-hydrogen atoms were refined anisotropically. The hydrogen atoms were set in calculated positions and refined as riding atoms with a common fixed isotropic thermal parameter. A summary of the crystallographic data and structure refinement is given in Table 1. Selected bond distances and angles for 1–6 are listed in Table S1 (see ESI†). CCDC 1063970 (1), 1063969 (2), 1063967 (3), 1063968 (4), 957789 (5), 1063966 (6), contain the supplementary crystallographic data for this paper.†| Compound | 1 | 2 | 3 | 4 | 5 | 6 |
| Formula | C149H173La3F54N18O30 | C149H173Eu3F54N18O30 | C149H173Gd3F54N18O30 | C135H141Tb3F54N18O30 | C45H47DyF18N6O10 | C149H173Er3F54N18O30 |
| Fw | 4138.78 | 4177.93 | 4193.80 | 3998.42 | 1336.39 | 4223.83 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | C2/c | C2/c | C2/c | C2/c | P21/n | C2/c |
| a (Å) | 50.575(6) | 50.142(5) | 49.926(3) | 49.914(19) | 12.6610(12) | 49.977(11) |
| b (Å) | 12.6932(12) | 12.6433(12) | 12.6037(5) | 12.461(4) | 17.3720(15) | 12.631(3) |
| c (Å) | 28.871(3) | 28.692(2) | 28.6405(16) | 28.558(10) | 24.491(2) | 28.671(6) |
| α (°) | 90 | 90 | 90 | 90 | 90 | 90 |
| β (°) | 100.922(3) | 101.271(3) | 101.279(3) | 101.821(8) | 93.967(6) | 101.550(10) |
| γ (°) | 90 | 90 | 90 | 90 | 90 | 90 |
| V (Å3) | 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 298(3) 298(3) |
17389(3) |
17674.0(16) |
17386(11) |
5373.8(8) | 17733(7) |
| Z | 4 | 4 | 4 | 4 | 4 | 4 |
| Dcalcd (Mg m−3) | 1.511 | 1.566 | 1.576 | 1.528 | 1.652 | 1.582 |
| μ (mm−1) | 0.816 | 1.169 | 1.241 | 1.333 | 8.547 | 1.535 |
| F(0 0 0) | 8360 | 8432 | 8444 | 7992 | 2668 | 8492 |
| Crystal size (mm3) | 0.26 × 0.24 × 0.22 | 0.24 × 0.22 × 0.22 | 0.26 × 0.24 × 0.22 | 0.22 × 0.20 × 0.18 | 0.20 × 0.18 × 0.14 | 0.20 × 0.18 × 0.12 |
| θ range for data collection (°) | 1.84–25.02 | 1.45–25.02 | 1.45–25.02 | 1.46–25.02 | 3.12–72.72 | 1.45–25.02 |
| Reflections collected/unique | 68912/15954 |
68008/15737 |
70381/15574 |
59484/15238 |
42338/10500 |
68448/15647 |
| R(int) | 0.0786 | 0.0381 | 0.0423 | 0.0540 | 0.0807 | 0.0360 |
| Data/restraints/parameters | 15954/295/1244 |
15737/295/1244 |
15574/295/1244 |
15238/228/1183 |
10500/57/762 |
15647/474/1370 |
| GOF on F2 | 1.143 | 1.118 | 1.059 | 1.045 | 1.080 | 1.072 |
| R1 [I > 2δ(I)] | 0.0468 | 0.0388 | 0.0320 | 0.0433 | 0.0587 | 0.0504 |
| wR2 [I > 2δ(I)] | 0.1155 | 0.0887 | 0.0784 | 0.1110 | 0.1498 | 0.1169 |
| R1 (all data) | 0.0508 | 0.0407 | 0.0346 | 0.0511 | 0.0685 | 0.0524 |
| wR2 (all data) | 0.1180 | 0.0898 | 0.0800 | 0.1164 | 0.1682 | 0.1182 |
Results and discussion
Crystal structure
Complexes 1, 2, 3, 4 and 6 crystallize in the monoclinic space group C2/c, while 5 crystallizes in the monoclinic space group P21/n. There are solvent molecules in complexes 1, 2, 3 and 6, while no solvent molecule in complexes 4 and 5. Crystal data and details of structural determination refinement are summarized in Table 1, and selected bond distances and angles for complexes 1–6 are listed in Table S1 in the ESI.†The structural analyses show that complexes 1–6 are very similar; hence, only the crystal structure of complex 3 is described here. Complex 3 consists of the [Gd(hfac)3(NITPh-p-N(CH3)2)2]3 unit and solvent molecule of C7H16. The structure of [Gd(hfac)3(NITPh-p-N(CH3)2)2]3 unit in complex 3 is shown in Fig. 1. In complex 3, the Gd(III) ion is eight-coordinated in slightly distorted dodecahedron geometry. Two oxygen atoms of the N–O groups from nitronyl nitroxide radicals and six oxygen atoms from three different hfac anions are coordinated to the metal ions. The bond lengths of Gd(1)–O(radical) are Gd(1)–O(1) 2.3328(20) Å and Gd(1)–O(3) 2.3412(18) Å, while the bond lengths of Gd(1)–O(hfac) are in the range of 2.3481(22)–2.4155(20) Å. These bond lengths are comparable to the reported lanthanide–nitronyl nitroxide complexes.16 The angles between the N–O groups from two radical ligands and Gd(III) ion are: N(1)–O(1)–Gd(1) 137.429(16)°, N(4)–O(3)–Gd(1) 138.962(16)° and O(1)–Gd(1)–O(3) 139.416(68)°. The nitronyl nitroxide moiety O1–N1–C1–N2–O2 makes a dihedral angle of 2.020(24)° with the plane of benzene ring.
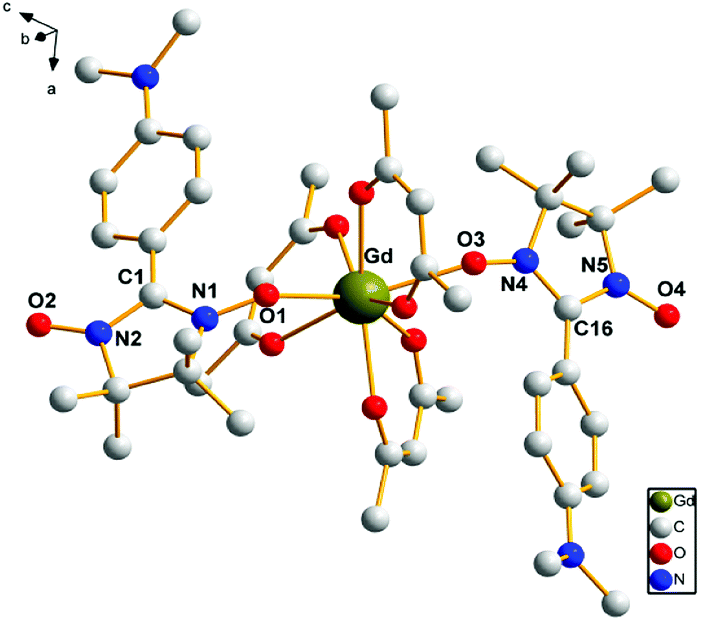 | ||
| Fig. 1 The crystal structure of complex 3. Fluorine and hydrogen atoms are not shown for the sake of clarity. | ||
In complex 3, the intermolecular hydrogen bonds occur between one carbon atom from one radical group and one fluorine atom from another hfac ion group (3.364 Å, C(6)–H6A⋯F(16)). This kind of alternate hydrogen bonds form 1D chain structure in complex 3, which is demonstrated in Fig. 2.
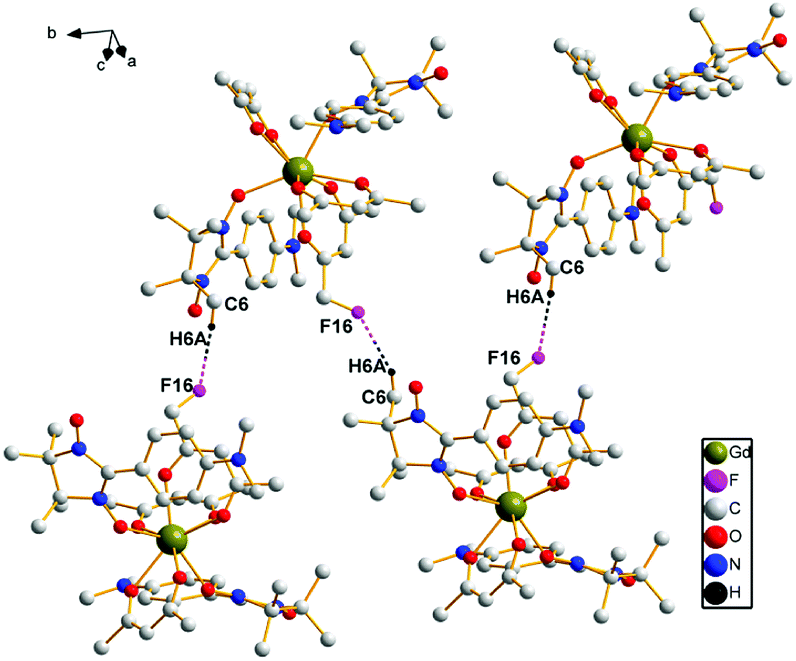 | ||
| Fig. 2 A sketch of the intermolecular hydrogen bonds of complex 3. | ||
The structure of the complexes 1, 2, 4, 5 and 6 are similar to complex 3, except for the substitution of Gd(III) with La(III), Eu(III), Tb(III), Dy(III) and Er(III) ions, which makes the bond distances and angles vary a little (Table S1†). In complex 1, the nitronyl nitroxide moiety O1–N1–C1–N2–O2 makes a dihedral angle of 1.556(27)° with the plane of benzene ring, while in complex 2 is 1.861(35)°, complex 4 is 2.493(37)°, complex 5 is 4.061(43)° and complex 6 is 3.212(57)°. The structure of complexes 1, 2, 4, 5 and 6 is shown in Fig. S1–S5 (ESI†).
The shape measure
There are many kinds of coordination geometries in eight-coordinated complexes. Three high-symmetry polyhedra are used as a method of analysis of the shape. They are: the trigonal dodecahedron (D2d-DD), the bicapped trigonal prism (C2v-TP), and the square antiprism (D4d-AP) respectively.17 There exists an intrinsic relationship between the dihedral angles (one for each pair of adjacent triangular planes) and the notion of shape, i.e., the symmetry. Therefore, the geometry of complexes was analyzed by comparing all observed dihedral angles in a given structure and the corresponding ideal values.Based on the crystal data, we carried out the calculations of the shape factor S to estimate the degree of distortion of the coordination structure in first coordination sphere. The S value is the minimal variance of dihedral angles along all edges given by eqn (1):
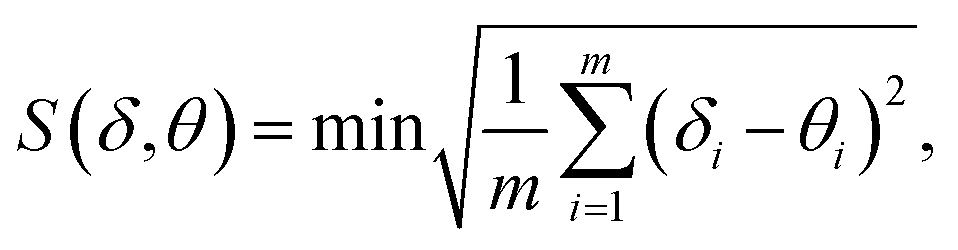 | (1) |
The following S values of shape measures calculations are showed in Table 2. By comparing the value of S, the coordination environments of complexes 1–6 are all distorted dodecahedron.
| Ln(III) | S(D2d) | S(C2v) | S(D4d) |
|---|---|---|---|
| La (1) | 6.08 | 12.06 | 9.73 |
| Eu (2) | 5.15 | 12.65 | 10.24 |
| Gd (3) | 4.95 | 12.62 | 10.43 |
| Tb (4) | 9.58 | 11.77 | 10.06 |
| Dy (5) | 5.51 | 20.98 | 18.52 |
| Er (6) | 4.83 | 20.53 | 10.58 |
Powder X-ray diffraction and TG analyses
To confirm whether the crystal structures are truly representative of the bulk materials, the X-ray powder diffraction patterns (PXRD) of complexes 1–6 have been recorded which are shown in Fig. S6–S8 (ESI†). These patterns indicate that the PXRD patterns are in good agreement with the results simulated from the single crystal date, indicating the purity of the bulk-synthesized materials.In order to investigate the thermal stability of complexes 1–6, the thermogravimetry-differential thermal analysis (TG-DTA) was studied, and the TG-DTA curve is depicted in Fig. S9–S11 (ESI†). The TG-DTA curves of 1–6 were similar. Herein, complex 1 as a representative example is selected to describe the thermal stability in details. For complex 1, the first stage takes place from 25.00 to 150 °C (the peak of DTA at 150.83 °C) with the weight loss of 11%, corresponding to losing solvent molecules. The second stage takes place from 150 to 500 °C (the peak of DTA at 302.35 °C) with the weight loss of 70%, corresponding to the skeleton of compound begins collapsed, implying the decomposition of hfac and (NITPh-p-N(CH3)2)2.
Luminescence properties
The luminescent measurements of complexes 2 and 4 were investigated at room temperature in ethanol solution. The results reveal that these compounds exhibit the characteristic emission peaks. For complex 2 (Fig. 3, top), the excitation wavelength for emission spectra is 325 nm. The emission spectra are composed of four main bands at 591 nm (5D0 → 7F1), 614 nm (5D0 → 7F2), 650 nm (5D0 → 7F3), and 700 nm (5D0 → 7F4). The electric dipole transitions emission peak at 614 nm (5D0 → 7F2) is clearly stronger than the magnetic dipole transitions peak at 591 nm (5D0 → 7F1). The fluorescence spectra show the characteristic emission peaks of europium ions.18 For complex 4 (Fig. 3, bottom), the excitation wavelength for emission spectra is 290 nm. The emission peak at 490 nm could be attributed to the 5D4 → 7F6 transition, and the other three peaks at 544, 582, and 619 nm could be assigned to the 5D4 → 7F5, 5D4 → 7F4, and 5D4 → 7F3 transitions, respectively. Among them, the 5D4 → 7F5 transition is the strongest. The fluorescence spectra show the characteristic emission peaks of terbium ions.19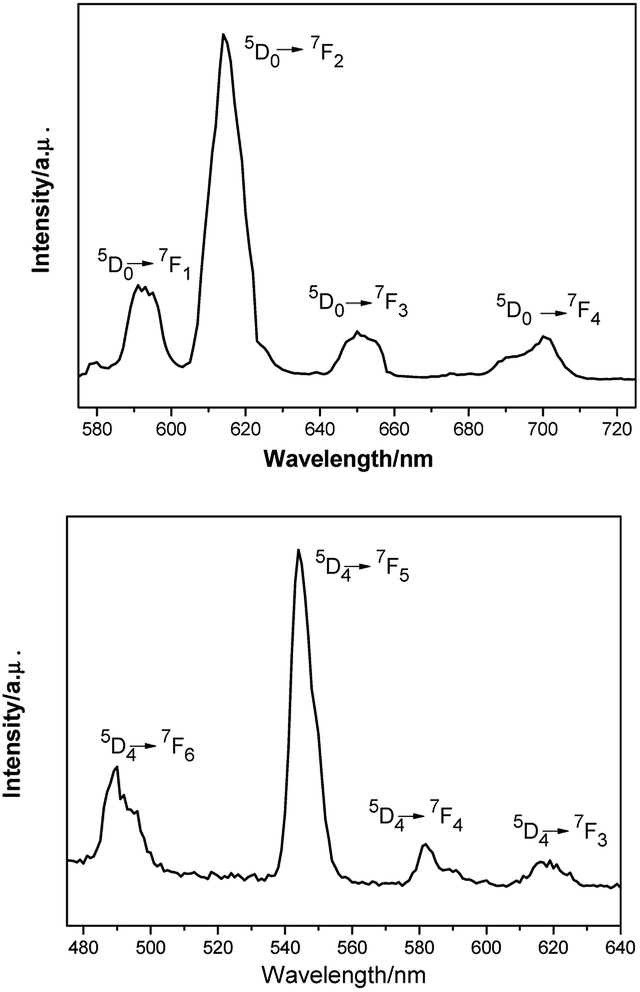 | ||
| Fig. 3 Room-temperature luminescence spectra of complex 2 (λex = 325 nm, top) and complex 4 (λex = 290 nm, bottom) in ethanol solution. | ||
Magnetic properties
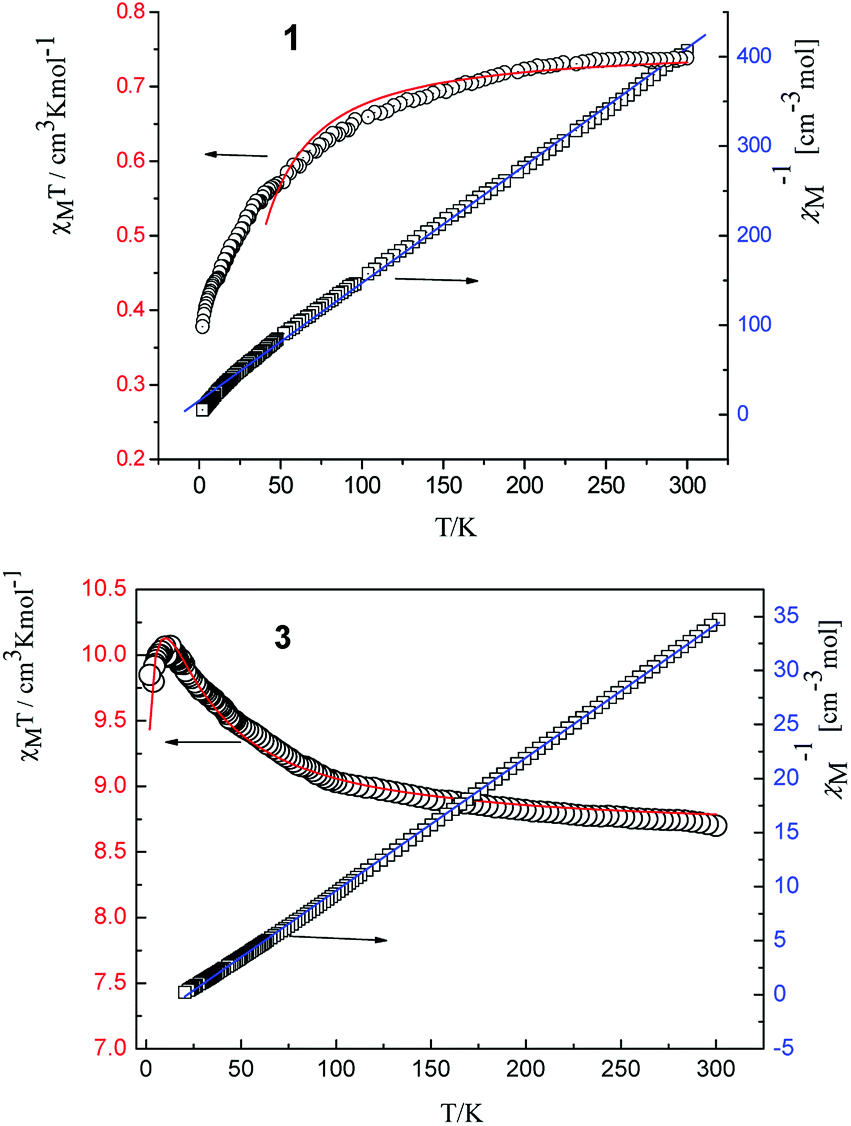 | ||
| Fig. 4 Temperature dependence of χMT (○) and plot of χM−1 vs. T of complexes 1 and 3. The solid lines represent the theoretical values based on the corresponding equations. | ||
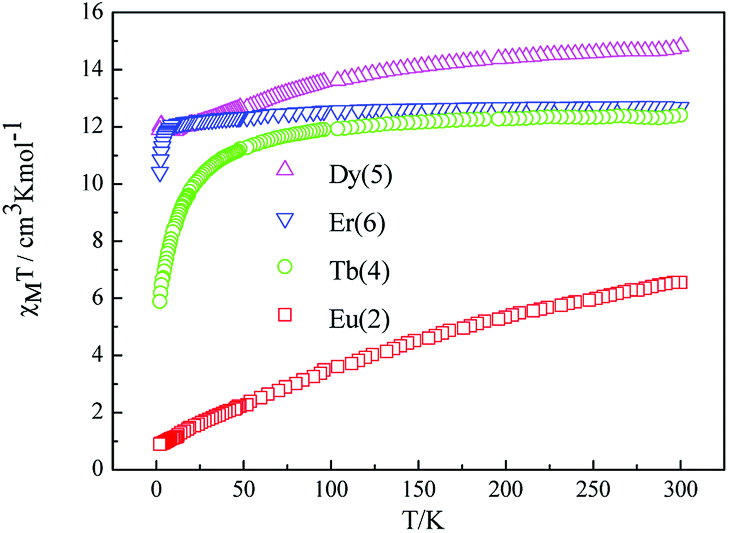 | ||
| Fig. 5 Temperature dependence of χMT for complexes 2, 4, 5, 6 at 1000 Oe. | ||
The room temperature values of χMT are 0.73, 8.64, 12.41, 14.80 and 12.69 cm3 mol−1 K for 1, 3, 4, 5, 6 respectively, in good agreement with the expected values of 0.75, 8.63, 12.23, 14.92 and 12.23 cm3 mol−1 K for one free LnIII ion (LaIII,7F0; GdIII, g = 2, 8S7/2; TbIII, g = 3/2, 7F6; DyIII, g = 4/3, 6H15/2; ErIII, g = 6/5, 4I15/2) plus two isolated radicals (g = 2, S = 1/2).19 For 2, the χMT value at room temperature is 3.09 cm3 mol−1 K is much higher than the theoretical value of 0.75 cm3 mol−1 K for EuIII ion (7F0) in the ground state and two organic radicals (S = 1/2, 0.375 cm3 mol−1 K), due to the population of the excited states of EuIII ion at room temperature. Upon cooling, the χMT versus T plots of 1–6 display different behaviors respectively. For complexes 1, 2, 4, 5 and 6, as the temperature is reduced, the χMT value continuously decreases to reach a minimum of 0.38 cm3 mol−1 K for 1, 0.419 cm3 mol−1 K for 2, 5.89 cm3 mol−1 K for 4, 10.41 cm3 mol−1 K for 5 and 11.89 cm3 mol−1 K for 6 at T = 2 K. This probably governed by the depopulation of the Stark sublevels and/or significant antiferromagnetic interaction.
For complex 1, since the La(III) ion is diamagnetic, the magnetic analysis was thus carried out by using the isotropic spin Hamiltonian Ĥ = −2J(Ŝ1Ŝ2) (eqn (2)). Also, intermolecular exchange interaction (zJ′) in the molecular field approximation was involved (eqn (3)).
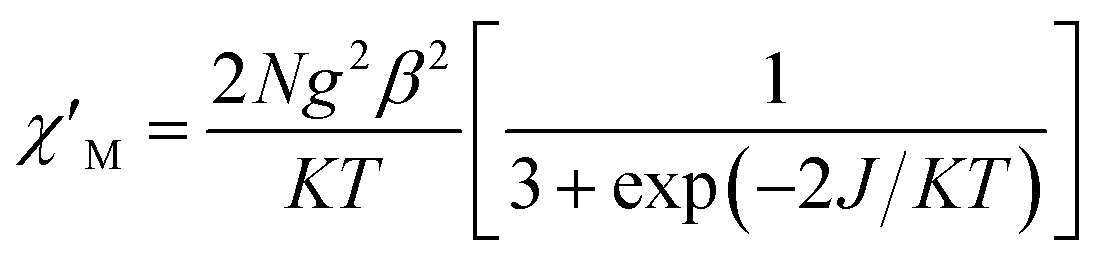 | (2) |
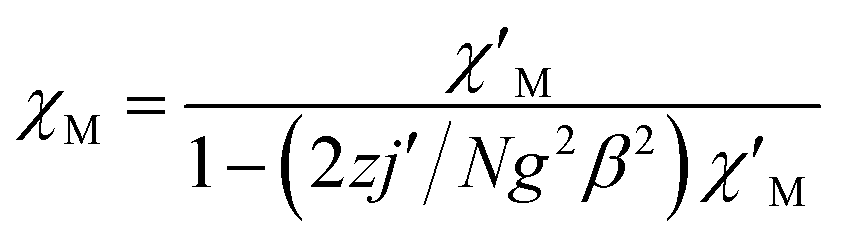 | (3) |
For complex 3, with the temperature decreases, the χMT value gradually increases to a maximum of 10.03 cm3 mol−1 K at 7.97 K, indicating the existence of ferromagnetic coupling between Gd(III) ion and nitronyl nitroxide. Below 7.97 K, χMT decreases quickly to 9.84 cm3 mol−1 K at 1.99 K. Based on the isotropic Hamiltonian Ĥ = −J1(ŜGd × Ŝrad1 + ŜGd × Ŝrad2) − J2Ŝrad1 × Ŝrad2, eqn (4) is introduced to analyze the magnetic coupling strength, where J1 and J2 represent the magnetic coupling for the Gd–radical and radical–radical, respectively.
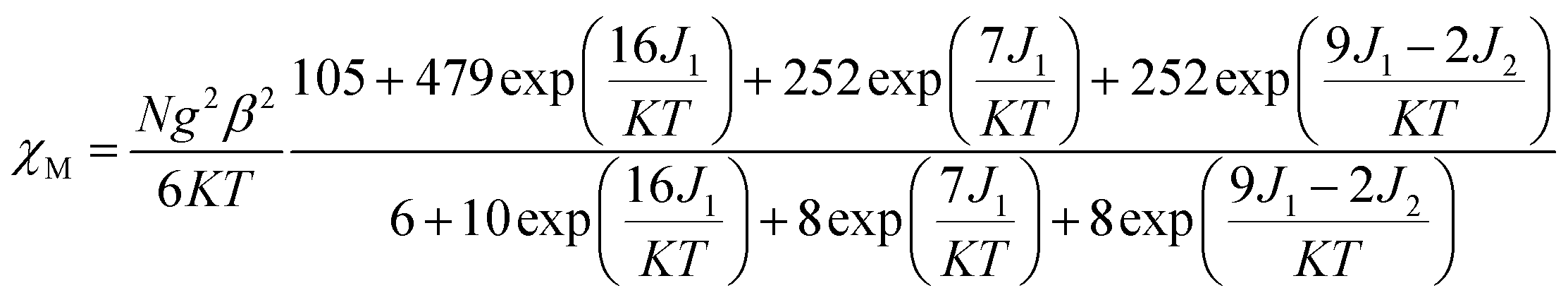 | (4) |
The best fitting results give J1 = 2.0 cm−1, J2 = −7.8 cm−1 and g = 2.00, which is in the range for other similar Gd–radical compounds.16 The positive value of J1 shows the ferromagnetic coupling between the Gd(III) ion and nitronyl nitroxide radical, while the negative J2 indicates the next nearest-neighbor (NNN) antiferromagnetic coupling between the intramolecular radicals. The plot of χM−1 vs. T obeys the Curie–Weiss law 1/χM = (T − θ)/C between 300 and 2 K, and yields C = 8.8 cm3 mol−1 K with θ = 3.2 K. The positive value of θ further confirms the existence of ferromagnetic coupling between spin carriers. The magnetization data of the field dependent magnetization at 2.0 K (Fig. S12†) climbs up quickly until 10 kOe, and then rises up gradually to 70 kOe with an effective moment of 8.38 μB, which is in smaller than the expected value of S = 9/2 under ferromagnetic interaction per GdRad2 unit (9.00 μB), indicating significant spin frustration behaviour because of the competing exchange interactions between J1 and J2.
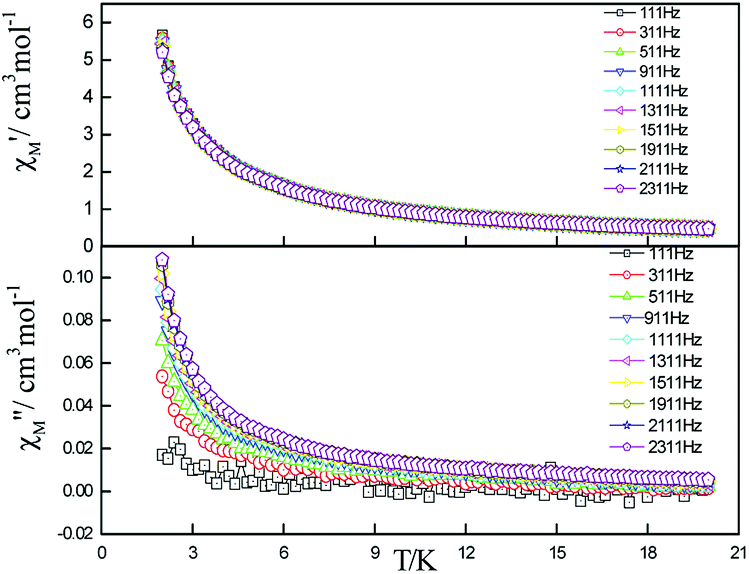 | ||
| Fig. 6 Frequency dependence of in-phase (top) and out-of-phase (down) components of the ac magnetic susceptibility for 5 under zero applied dc field. | ||
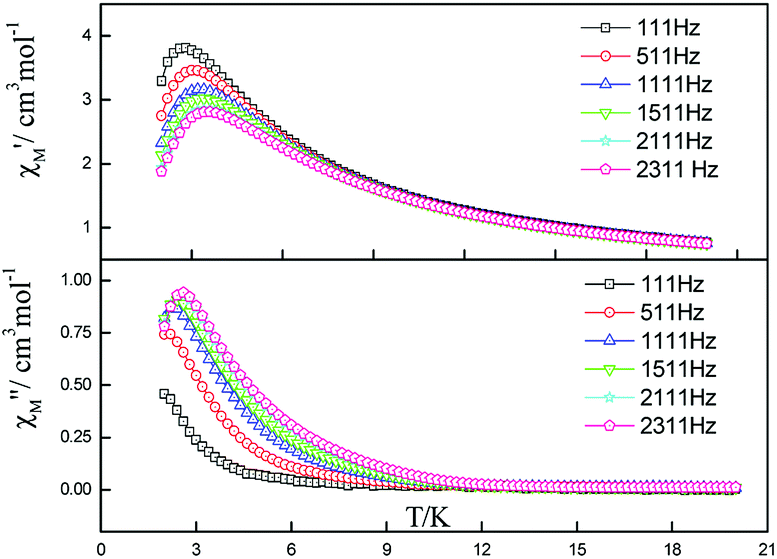 | ||
| Fig. 7 Frequency dependence of in-phase (top) and out-of-phase components of the ac magnetic susceptibility for 5 under 2000 Oe applied dc field. | ||
For complex 5, the Cole–Cole diagrams (Fig. 8), which were measured at temperatures of 2.0, 2.2 and 2.4 K, exhibit a quasi-semicircular shape that can be fitted to the generalized Debye model with α values of 0.34, 0.39 and 0.37, from 2 K to 2.4 K. The parameter φ (φ = (ΔTp/Tp)/Δ(logf)) was calculated and a value of 0.36 was obtained, which excludes the possibility of a spin-glass (0.01 < φ < 0.08).21 Plots of lnτ−1 versus T−1 display linear dependence indicating spin reversal by the thermally activated Orbach mechanism process (Fig. 9). The Arrhenius fit (τ = τ0exp(Δeff/kBT)) gives the effective energy barrier (Δeff/kB) of (23.44 ± 1.68) K and the pre-exponential τ0 of (7.15 ± 0.72) × 10−9 s (R = 0.925) by ac versus T data under a dc field of 2000 Oe, which fall in the range well for SMMs.22,23
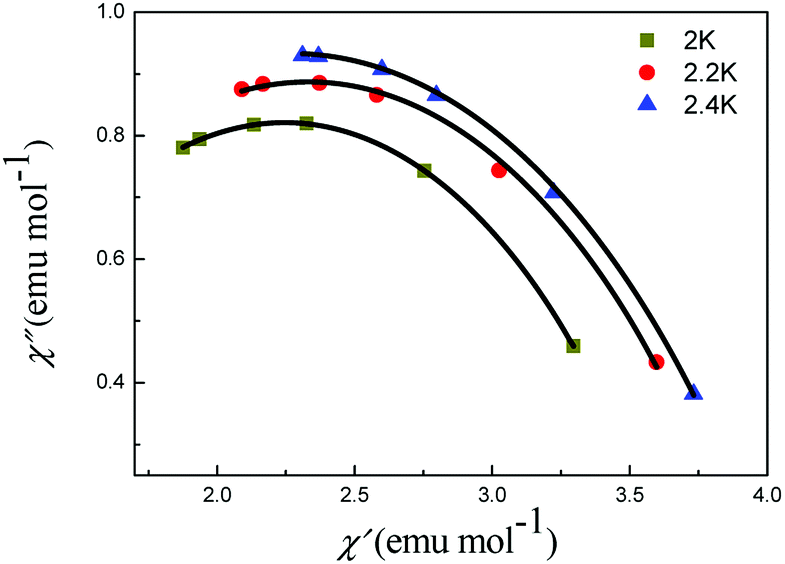 | ||
| Fig. 8 Cole–Cole plots of 5 under 2000 Oe. The black lines are results of fitting using the Debye model. | ||
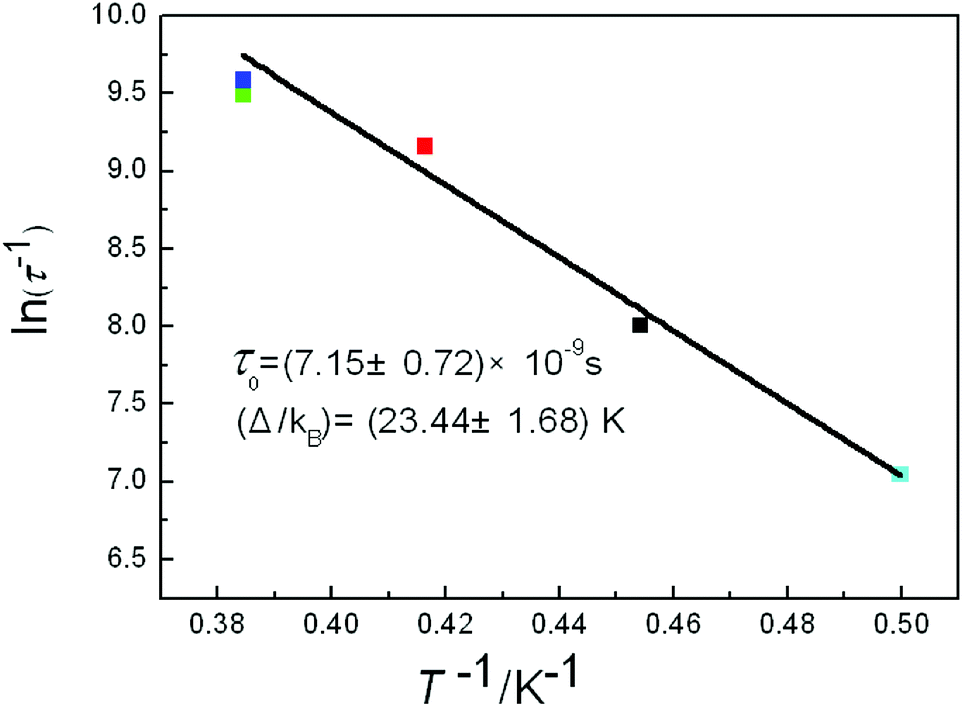 | ||
| Fig. 9 Plot of lnτ−1 vs. T−1 of 5 under 2000 Oe dc field. The black line is the fitting result using the Arrhenius law. | ||
Conclusions
In summary, six lanthanide coordination complexes have been synthesized and structurally characterized. Dc magnetic studies show that ferromagnetic metal–radical interactions take place in complex 3. Ac magnetic studies for compounds 4 and 5 show clear frequency dependence of the out-of-phase susceptibility. With the application of external field (2000 Oe), good peak shapes of the out-of-phase and in-phase signals were obtained in 5, which give an energy barrier of (23.44 ± 1.68) K. It should be noted that multiple relaxation events can occur for single lanthanide sites. Thus, a more precise result must wait for low temperature measurements (T < 1.0 K) by using a micro-SQUID or more AC curves under different magnetic fields.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21371104, 20771081, 21101096, and 21471084) and MOE Innovation Team (IRT13022) of China.Notes and references
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, 2006 Search PubMed.
- (a) R. Sessoli, H.-L. Tsai, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrichson, J. Am. Chem. Soc., 1993, 115, 1804–1806 CrossRef CAS; (b) R. Sessoli, D. Gatteschi, A. Caneschi and M. Novak, Nature, 1993, 365, 141–143 CrossRef CAS; (c) S. M. J. Aubin, M. W. Wemple, D. M. Adams, H.-L. Tsai, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1996, 118, 7746–7754 CrossRef CAS; (d) S. L. Castro, Z. Sun, C. M. Grant, J. C. Bollinger, D. N. Hendrickson and G. Christou, J. Am. Chem. Soc., 1998, 120, 2365–2375 CrossRef CAS; (e) H. Oshio, N. Hoshino and T. Ito, J. Am. Chem. Soc., 2000, 122, 12602–12603 CrossRef CAS.
- (a) A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., Int. Ed., 2001, 40, 1760–1763 CrossRef CAS; (b) R. Clérac., H. Miyasaka, M. Yamashita and C. Coulon, J. Am. Chem. Soc., 2002, 124, 12837–12844 CrossRef; (c) R. Lescouëzec, J. Vaissermann, C. Ruiz-Pérez, F. Lloret, R. Carrasco, M. Julve, M. Verdaguer, Y. Dromzee, D. Gatteschi and W. Wernsdorfer, Angew. Chem., Int. Ed., 2003, 42, 1483–1486 CrossRef PubMed; (d) T.-F. Liu, D. Fu, S. Gao, Y.-Z. Zhang, H.-L. Sun, G. Su and Y.-J. Liu, J. Am. Chem. Soc., 2003, 125, 13976–13977 CrossRef CAS PubMed.
- (a) N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed; (b) R. Sessoli and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2328–2341 CrossRef CAS; (c) L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC.
- (a) L. Bogani, C. Sangregorio, R. Sessoli and D. Gatteschi, Angew. Chem., Int. Ed., 2005, 44, 5817–5821 CrossRef CAS PubMed; (b) H.-X. Tian, R.-N. Liu, X.-L. Wang, P.-P. Yang, Z.-X. Li, L.-C. Li and D.-Z. Liao, Eur. J. Inorg. Chem., 2009, 4498–4502 CrossRef CAS; (c) F. Ortu, J. Liu, M. Burton, J. M. Fowler, A. Formanuik, M.-E. Boulon, N. F. Chilton and D. P. Mills, Inorg. Chem., 2017, 56, 2496–2505 CrossRef CAS PubMed.
- (a) G. Poneti, K. Bernot, L. Bogani, A. Caneschi, R. Sessoli, W. Wernsdorfer and D. Gatteschi, Chem. Commun., 2007, 1807–1809 RSC; (b) F. Pointillart, K. Bernot, G. Poneti and R. Sessoli, Inorg. Chem., 2012, 51, 12218–12229 CrossRef CAS PubMed; (c) R.-N. Liu, C.-X. Xiong, S.-P. Zhao, J. Wu, Q. Li and D. Fang, Inorg. Chem. Commun., 2012, 22, 104–107 CrossRef CAS.
- R.-N. Liu, L.-C. Li, X.-L. Wang, P.-P. Yang, C. Wang, D.-Z. Liao and J.-P. Sutter, Chem. Commun., 2010, 46, 2566–2568 RSC.
- S. Das, A. Dey, S. Biswas, E. Colacio and V. Chandrasekhar, Inorg. Chem., 2014, 53, 3417–3426 CrossRef CAS PubMed.
- R.-N. Liu, Y. Ma, P.-P. Yang, X.-Y. Song, G.-F. Xu, J.-K. Tang, L.-C. Li, D.-Z. Liao and S.-P. Yan, Dalton Trans., 2010, 39, 3321–3325 RSC.
- X.-H. Yi, K. Bernot, F. Pointillart, G. Poneti, G. Calvez, C. Daiguebonne, O. Guillou and R. Sessoli, Chem.–Eur. J., 2012, 18, 11379–11387 CrossRef CAS PubMed.
- X.-L. Wang, X. Bao, P.-P. Xu and L. Li, Eur. J. Inorg. Chem., 2011, 3586–3591 CrossRef CAS.
- K. Bernot, L. Bogani, A. Caneschi, D. Gatteschi and R. Sessoli, J. Am. Chem. Soc., 2006, 128, 7947–7956 CrossRef CAS PubMed.
- (a) P. Hu, M. Zhu, X.-L. Mei, H.-X. Tian, Y. Ma, L.-C. Li and D.-Z. Liao, Dalton Trans., 2012, 41, 14651–14656 RSC; (b) E. F. Ullman, J. H. Osiecki, D. G. B. Boocock and R. Darcy, J. Am. Chem. Soc., 1972, 94, 7049–7059 CrossRef CAS.
- (a) Theory and Applications of Molecular Paramagnetism, ed. E. A. Boudreaux and L. N. Mulay, Wiley-Interscience, New York, 1976 Search PubMed; (b) G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- (a) G. M. Sheldrick, SHELXS 97, University of Gottingen, Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXL 97, University of Gottingen, Göttingen, Germany, 1997 Search PubMed.
- (a) M.-Y. Song, Y.-F. Hou, L.-M. Wen, S.-P. Wang, S.-T. Yang, J.-J. Zhang, L.-N. Geng and S.-K. Shi, J. Mol. Struct., 2016, 1107, 174–181 CrossRef CAS; (b) P. Hu, H.-F. Guo, Y. Li and F.-P. Xiao, Inorg. Chem. Commun., 2015, 59, 91–94 CrossRef CAS; (c) M.-Y. Song, L.-M. Wen, S.-P. Wang, S.-T. Yang, J.-J. Zhang, L.-N. Geng and S.-K. Shi, Inorg. Chim. Acta, 2015, 430, 1–7 CrossRef CAS; (d) X.-L. Wang, Inorg. Chim. Acta, 2012, 387, 20–24 CrossRef CAS.
- (a) J. L. Hoard and J. V. Silverton, Inorg. Chem., 1963, 2, 235–242 CrossRef CAS; (b) S. J. Lippard and B. J. Russ, Inorg. Chem., 1968, 7, 1686–1688 CrossRef CAS; (c) V. V. Fomenko, L. I. Kopaneva, M. A. Porai-Koshits and T. N. Polynova, J. Struct. Chem., 1974, 15, 244–249 CrossRef; (d) D. L. Kepert, Prog. Inorg. Chem., 1978, 24, 179–183 CAS.
- (a) S. Swavey and R. Swavey, Coord. Chem. Rev., 2009, 253, 2627–2638 CrossRef CAS; (b) Z. Ahmed and K. Iftikhar, Inorg. Chim. Acta, 2012, 392, 165–176 CrossRef CAS; (c) P. Coppo, M. Duati, V. N. Kozhevnikov, J. W. Hofstraat and L. De Cola, Angew. Chem., 2005, 117, 1840–1844 CrossRef; (d) Y. Lu and B. Yan, J. Mater. Chem. C, 2014, 2, 7411–7416 RSC.
- (a) Y.-L. Wang, Y.-Y. Gao, Y. Ma, Q.-L. Wang, L.-C. Li and D.-Z. Liao, J. Solid State Chem., 2013, 202, 276–281 CrossRef CAS; (b) C. Lescop, G. Bussière, R. Beaulac, H. Bélisle, E. Belorizky, P. Rey, C. Reber and D. Luneau, J. Phys. Chem. Solids, 2004, 65, 773–779 CrossRef CAS.
- D. Visinescu, A. M. Madalan, M. Andruh, C. Duhayon, J.-P. Sutter, L. Ungur, W. V. Heuvel and L. F. Chibotaru, Chem.–Eur. J., 2009, 15, 11808–11814 CrossRef CAS PubMed.
- J. A. Mydosh, Spin glasses: an experimental introduction, Taylor & Francis, London, 1993 Search PubMed.
- (a) L. Bogani, C. Sangregorio, R. Sessoli and D. Gatteschi, Angew. Chem., Int. Ed., 2005, 117, 5967–5971 CrossRef; (b) K. Bernot, L. Bogani, R. Sessoli and D. Gatteschi, Inorg. Chim. Acta, 2007, 360, 3807–3812 CrossRef CAS.
- (a) R.-N. Liu, C.-M. Zhang, X.-L. Mei, P. Hu, H.-X. Tian, L.-C. Li, D.-Z. Liao and J.-P. Sutter, New J. Chem., 2012, 36, 2088–2093 RSC; (b) H.-X. Tian, X.-F. Wang, X.-L. Mei, R.-N. Liu, M. Zhu, C.-M. Zhang, Y. Ma, L.-C. Li and D.-Z. Liao, Eur. J. Inorg. Chem., 2013, 2013, 1320–1325 CrossRef CAS; (c) P. Hu, X.-F. Wang, Y. Ma, Q.-L. Wang, L.-C. Li and D.-Z. Liao, Dalton Trans., 2014, 43, 2234–2243 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Appendix A. CCDC 1063970 (1), 1063969 (2), 1063967 (3), 1063968 (4), 957789 (5), 1063966 (6) contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra05764d |
| This journal is © The Royal Society of Chemistry 2017 |