
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In silico studies of solvated F19W amyloid β (11–40) trimer†
Son Tung Ngo *ab,
Xuan-Cuong Luuc,
Minh Tung Nguyend,
Chinh N. Lec and
Van V. Vu*c
*ab,
Xuan-Cuong Luuc,
Minh Tung Nguyend,
Chinh N. Lec and
Van V. Vu*c
aComputational Chemistry Research Group, Ton Duc Thang University, Ho Chi Minh City, Vietnam. E-mail: ngosontung@tdt.edu.vn
bFaculty of Applied Sciences, Ton Duc Thang University, Ho Chi Minh City, Vietnam
cNTT Hi-Tech Institute, Nguyen Tat Thanh University, Ho Chi Minh City, Vietnam. E-mail: vanvu@ntt.edu.vn
dBinh Duong University, Thu Dau Mot City, Binh Duong Province, Vietnam
First published on 1st September 2017
Abstract
Alzheimer's disease (AD) is associated with the oligomerization and/or fibrillation of amyloid beta (Aβ) peptides, which cause damage to brain cells. Aβ oligomers and fibrils contain hydrophobic cores formed with parallel beta sheets. Mutations of F19, a residue in the hydrophobic core of Aβ peptides, slow down their aggregation process but do not alter the overall structure of the resulting fibrils. However, the effects of F19 mutations on the toxic Aβ oligomers have not been elucidated. We studied the F19W mutant of the 11–40 truncated Aβ trimer (F19W 3Aβ11–40) using replica exchange molecular dynamics (REMD) simulations. While most structural terms do not change significantly, critical polar contacts decrease by 20%, and notably, RMSD almost doubles upon F19W mutation. Six minima were found in the free energy surface of F19W 3Aβ11–40, which have lower energy barriers (by ∼1 kJ mol−1) and significantly lower total population (∼20%) compared to those of the three minima found for 3Aβ11–40 (∼60%). The binding free energy between constituting chains of the mutant trimer increases by ∼28 kcal mol−1 but fluctuates significantly (±27.1 kcal mol−1). Our results indicate that while the hydrophobic core of amyloid beta peptide is capable of adapting to structural changes, F19W mutation results in a significantly more flexible trimer. The more flexible F19W mutant oligomers would require more time to self-assemble into fibrils. Our results contribute to a better understanding of the behavior of Aβ peptides and their oligomerization/aggregation process, which is necessary to understand AD pathogenesis.
Introduction
Alzheimer's disease is highly prevalent, presenting in more than 30% of elderly people.1–5 Thus far, AD therapies are inefficient because the nature of AD has not been properly understood. There are three main proposed AD mechanisms, including the cholinergic hypothesis, tau protein hypothesis, and amyloid hypothesis. Several experimental studies implied that AD is associated with synaptic failure.6,7 Numerous studies suggest that the aggregation of tau proteins directly relates to the death of neuron cells in the brain of AD patients.8,9 The amyloid cascade hypothesis is supported by recent experiments and genetic evidence.10–15 β-Amyloid peptides have been the therapeutic targets in recent studies.16–18 Furthermore, it was shown that Aβ peptides could alter the accumulation of tau proteins.19–21Computer-aided drug design that focuses on searching potential inhibitors for the formation and/or aggregation of wild-type Aβ oligomers is of great interests.22–27 Furthermore, the trimeric oligomers were shown to be one of the most harmful Aβ peptides to the brain.15 However, the structural understanding of Aβ oligomers are limited since they exist in numerous forms.28,29 Recently, small angle neuron scattering (SANS) was used to detect Aβ1–42 pentermer/hexamer.30 SANS can provide some limited structural information but it still relied heavily on molecular dynamics (MD) simulations to gain insight into the structure of the pentamer/hexamer species. Hence, MD simulations have been instrumental in understanding the nature of Aβ oligomers. We recently obtained details of Aβ (11–40) truncated trimer (3Aβ11–40) using rigorous replica exchange MD (REMD) simulations.31–33
Several studies showed that the mutations at N-terminal region, including A2V,34 H6R,35 D7H,36 and D7N37 cause structural changes. However, the sequence 1–10 often has minimal effect on Aβ oligomers. The familial mutations in the in the middle region, including F19W,38 F20W,38 A21G,39 E22Q,40 E22K,41 E22G,42 E22Δ,43 and D23N,44 induce greater changes in Aβ oligomers. As we mentioned above, experimental studies on the conformations of Aβ oligomers are very limited, computational studies have been instrumental in this aspect.45–48
F19 is a residue in the Aβ central hydrophobic core that governs the aggregation of Aβ peptides. Experiments on F19W reveals that this residue is inserted inside the hydrophobic core of Aβ fibrils.38,49 Replacing F19 by various residues does not alter the structure of Aβ fibrils but increase the length of the lag phase and characteristic fibrillation time by about 4 folds and 1.5–4 folds, respectively.50 However, the effects of F19W mutation on Aβ trimer has not been investigated. In addition, although, both F and W residue are aromatic residues, replacing F by W significantly changes the conformations of Aβ oligomers.38,49,50 The understanding of these effects on the atomic level would thus provide the better insights into the oligomerization of Aβ peptides.
In this work, replica exchange molecular dynamics (REMD) simulations at 48 different temperatures in 400 ns were performed to gain insights into the structural change upon F19W mutation of soluble 3Aβ11–40, one of the most neurotoxic Aβ oligomers. The studies were carried out for the F19W mutant of 3Aβ11–40 (F19W 3Aβ11–40) using the same parameters that were previously used for 3Aβ11–40.31 Our results indicated that F19W mutation significantly increase the dynamic of 3Aβ11–40. Our results contribute to better understanding the aggregation of amyloid beta peptide, which would provide the basis for developing better AD therapy.
Materials and methods
Initial conformation of the mutation F19W Aβ11–40 trimer
According to previous studies,31,32 the fibril-like structure of the trimer was the repeating unit of the two-fold 12Aβ11–40 fibrils.51 The residue F19 was mutated to W19 using PyMOL tool.52 The mutant F19W trimer was then parameterized utilizing the all-atom Amber99SB-ILDN force field53 since it is the most appropriate force field representing the Aβ peptides.54 The system was solvated using the TIP3P water model.55 Three Na+ ion were added to neutralize the system. The starting conformation of the simulations includes approximately 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms totally. This conformation is displayed in Fig. 1.
000 atoms totally. This conformation is displayed in Fig. 1.
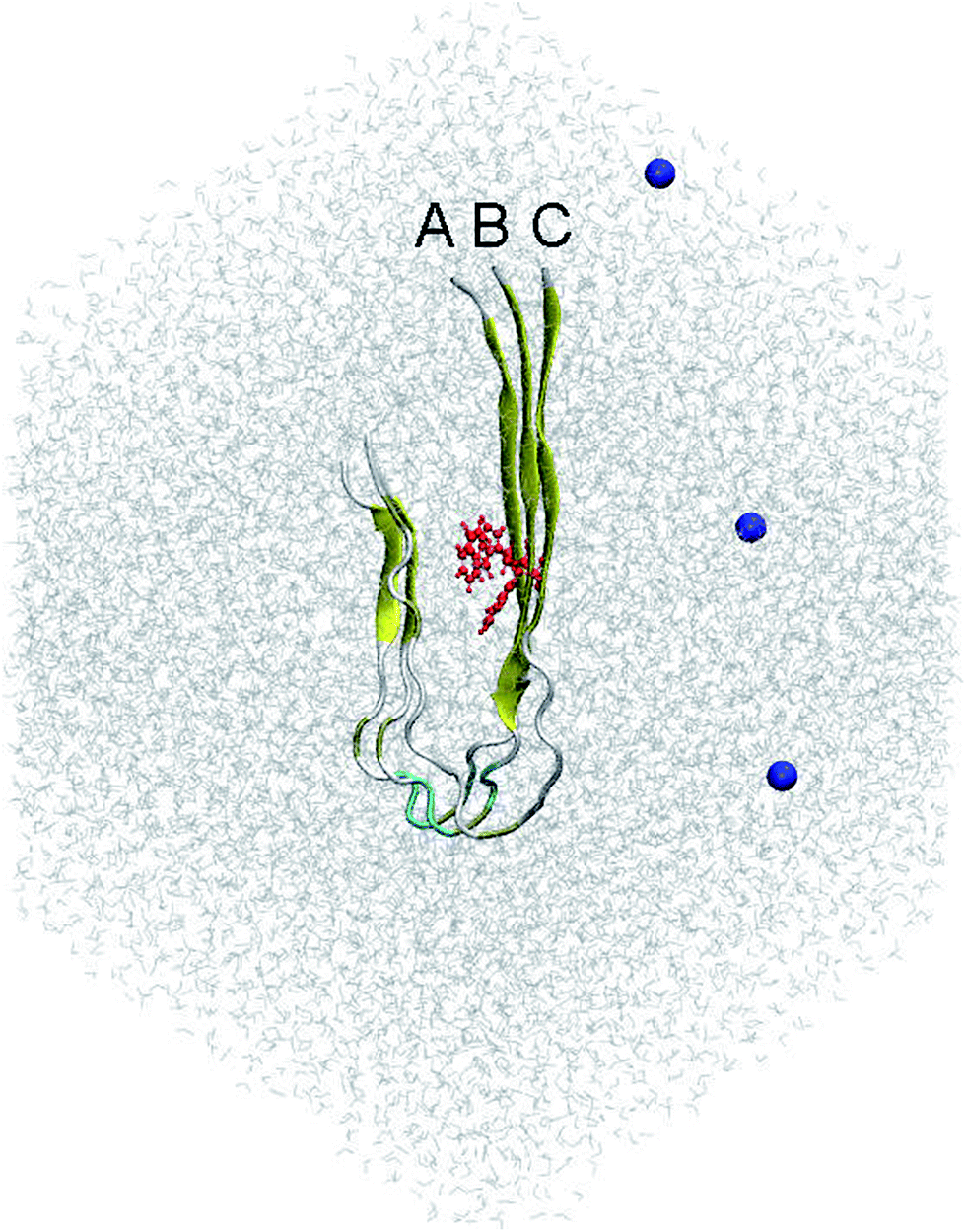 | ||
| Fig. 1 Initial conformation of the REMD simulations of the solvated F19W 3Aβ11–40. The system was minimized and relaxed following three steps of energetic minimizations and NVT simulations, respectively. In which, the red color highlighted the mutation residue F19W. Blue color noted the Na+ ions, which were used to neutralize the soluble system. | ||
Computational scheme
It is known that REMD is an extensively sampling approach to investigate the folding and aggregation of Aβ peptides.48,56–58 The simulations were carried out using GROMACS version 5.0.6.59 In particular, the structural change of the 3Aβ11–40 upon F19W mutation was investigated over 400 ns of temperature REMD simulations with 48 replicas. The parameters of the simulations were referenced to the previous study.31 F19W 3Aβ11–40 was subjected to energy minimization involving steepest descent, conjugate gradient, and L-BFGS scheme, as previously carried out for soluble 3Aβ11–40.31,60 The minimized conformation was then relaxed in NVT simulations with 500 ps length.The relaxing conformation of the solvated mutation F19W trimer was selected as the starting conformation for REMD simulations at 48 different temperatures. The replica temperatures were chosen based on the previous studies.31 In particular, there were 400000 of 1 ps exchanges over the simulations. The structural change of the F19W 3Aβ11–40 was monitored every 10 ps.
Structural analysis
A sidechain contact is counted when the distance between heavy atoms is smaller than 0.45 nm. A hydrogen bonded contact is counted when the distance between acceptor (A) and donor (D) is smaller than 0.35 nm and the angle between A–H–D is larger than 135°, where H is hydrogen atom. A salt bridge or polar contact between two residues is measured when the distance between their charged groups is smaller than 0.46 nm. RMSD, gyration radius (Rg), and free energy surface (FES) were determined using GROMACS tools. The secondary structure of the F19W peptide chains in the trimer was predicted utilizing the DSSP protocol.61The free energy difference of binding between isolated monomers to the others was determined using the MM-PBSA method.62 Details of the computational method to estimate the binding free energy between two Aβ peptides was described in the previous studies.31,63
Results and discussion
REMD simulations of soluble F19W 3Aβ11–40
Solvated F19W 3Aβ11–40 was simulated utilizing temperature REMD with 48 different replicas. The system reached the stable states after 250 ns of REMD simulations. Conformations generated during 250–400 ns were selected for analysis. All metrics were averaged over these snapshots. SASA, CCS, and secondary structure parameters of replicas at 250 ns were monitored and shown in Fig. S1.† SASA value ranges from 5929.8 to 7097.9 Å2. CCS value varies in a very large range of 1280.2–1444.7 Å2. The beta, coil, turn, and helix contents were found in the range of 17–50%, 40–70%, 0–17%, and 0–9%, respectively. The fluctuations of these parameters indicate that our simulations were not targeted to any biased conformations.All of secondary structure terms, Rg, RMSD, intermolecular sidechain contacts, and SASA were determined over different time windows. The obtained values are shown in Fig. 2, in which the red lines and black lines correspond to the metrics at 250–350 and 250–400 ns windows of REMD simulations at 300 K, respectively. The superimpositions of these two lines in each graph (less than 1% variation) in Fig. 2 indicate that our simulations converged.
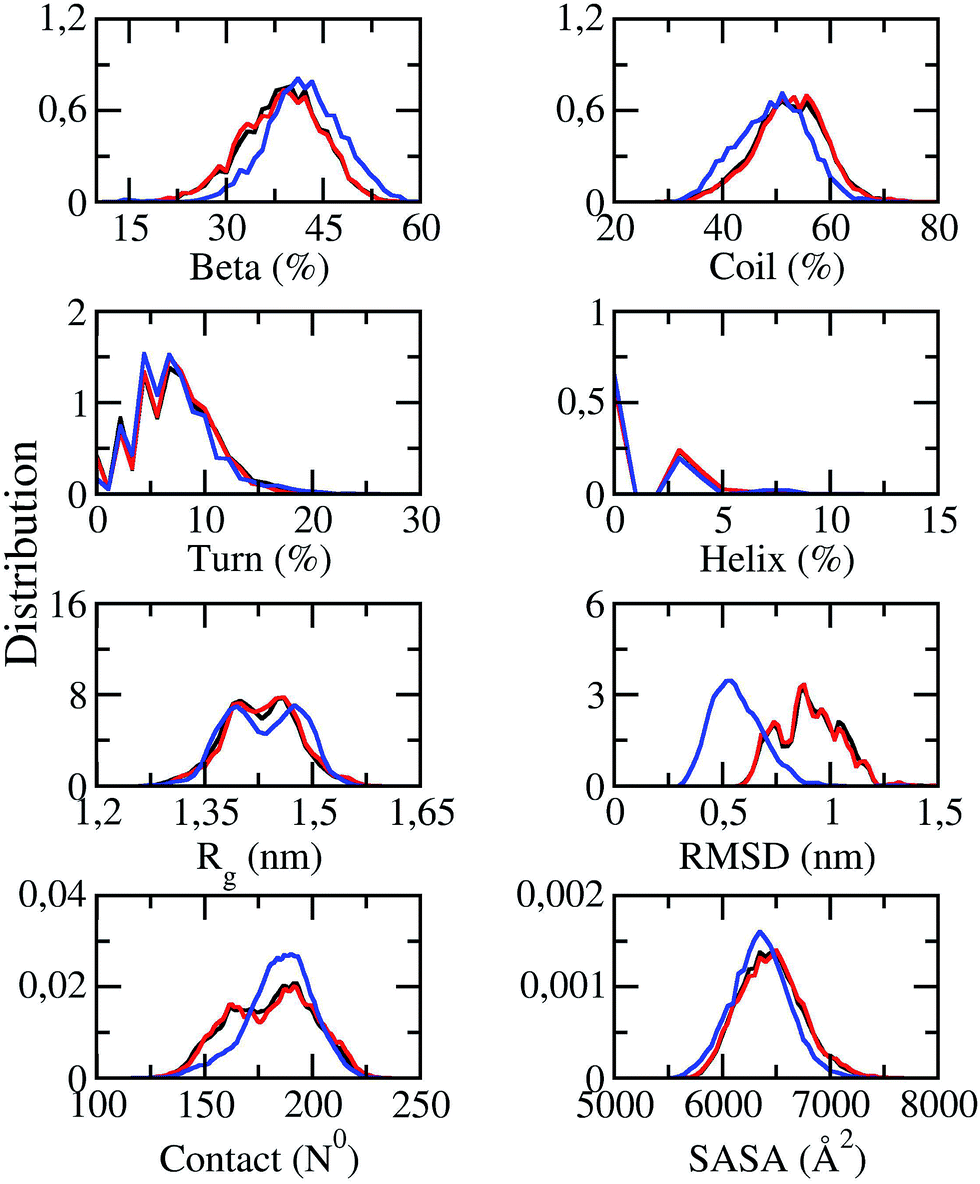 | ||
| Fig. 2 Distributions of measured values of F19W 3Aβ11–40 in REMD simulation time windows of 250–350 ns (red lines) and 250–400 ns (black lines) at 300 K. The blue lines indicate the distributions of the metrics of the wild-type Aβ11–40 trimer.31 | ||
The distributions of the secondary structure terms of F19W 3Aβ11–40 were compared to those in 3Aβ11–40 (blue lines in Fig. 2). In the last 150 ns of REMD simulations, the secondary structure parameters of 3Aβ11–40 change slightly upon F19W mutation. The mutant and wild-type truncated trimers form the same turn and helix contents at ∼7 ± 4 and ∼2 ± 2%, respectively.31 The beta content decreases by ∼3% to 39 ± 6% while the coil content increases by ∼3% to 52 ± 7% (Fig. 2).31
The average Rg value of solvated F19W 3Aβ11–40 is approximately ∼1.43 ± 0.05 nm, which is equal to that of solvated 3Aβ11–40,31 although the distribution of Rg is slightly different (Fig. 2). The SASA distribution of F19W 3Aβ11–40 is similar to that of 3Aβ11–40 with the average value slightly increases from 6351 ± 269 Å2 (ref. 31) to 6449 ± 288 Å2. In addition, the mean number of non-bonded intermolecular contacts between two heavy atoms of different chains in F19W 3Aβ11–40 is 180 ± 19, which is similar to that in 3Aβ11–40, although the distribution slightly changes. However, the mean RMSD value increases from ∼0.51 in 3Aβ11–40 to ∼0.91 ± 0.14 nm in F19W 3Aβ11–40 (Fig. 2). This large increase in RMSD while other structural terms only slightly change indicates that Aβ peptides can adapt to changes in their hydrophobic core, by shifting their backbones drastically to maintain the overall structure.
Examination of structural terms of the F19 residue in wild-type 3Aβ11–40 and W19 residue in F19W 3Aβ11–40 revealed local structural changes in the hydrophobic core of the trimer (Fig. S2†). RMSD, Rg, SASA, and CCS all increases significantly, which is expected given the larger volume and surface area of tryptophan compared to that of phenylalanine. F19W 3Aβ11–40 is likely more flexible than wild-type 3Aβ11–40. F19W mutation likely destabilizes the hydrophobic core of the 3Aβ11–40, leading to slower aggregation rate.
Effects of the mutation F19W on the distribution of secondary structure per residue
The distributions of beta, coil, turn, and helix contents per residues averaged for all three chains in soluble F19W 3Aβ11–40 are shown in Fig. 3. Distributions for individual chains are shown in Fig. S3.† Overall, these distributions are similar to those found for soluble 3Aβ11–40. The trimer contains two beta structure domains (sequences 16–20 and 31–36) at either flank of the peptide chains, intercalating three unstructured structure domains (sequences 11–15, 21–30, and 37–40). The coil content is dominant in these three unstructured domains. Turns are abundant in the middle of the peptide chains, around residues 25 and 26. Helix content is also mostly present around these two residues.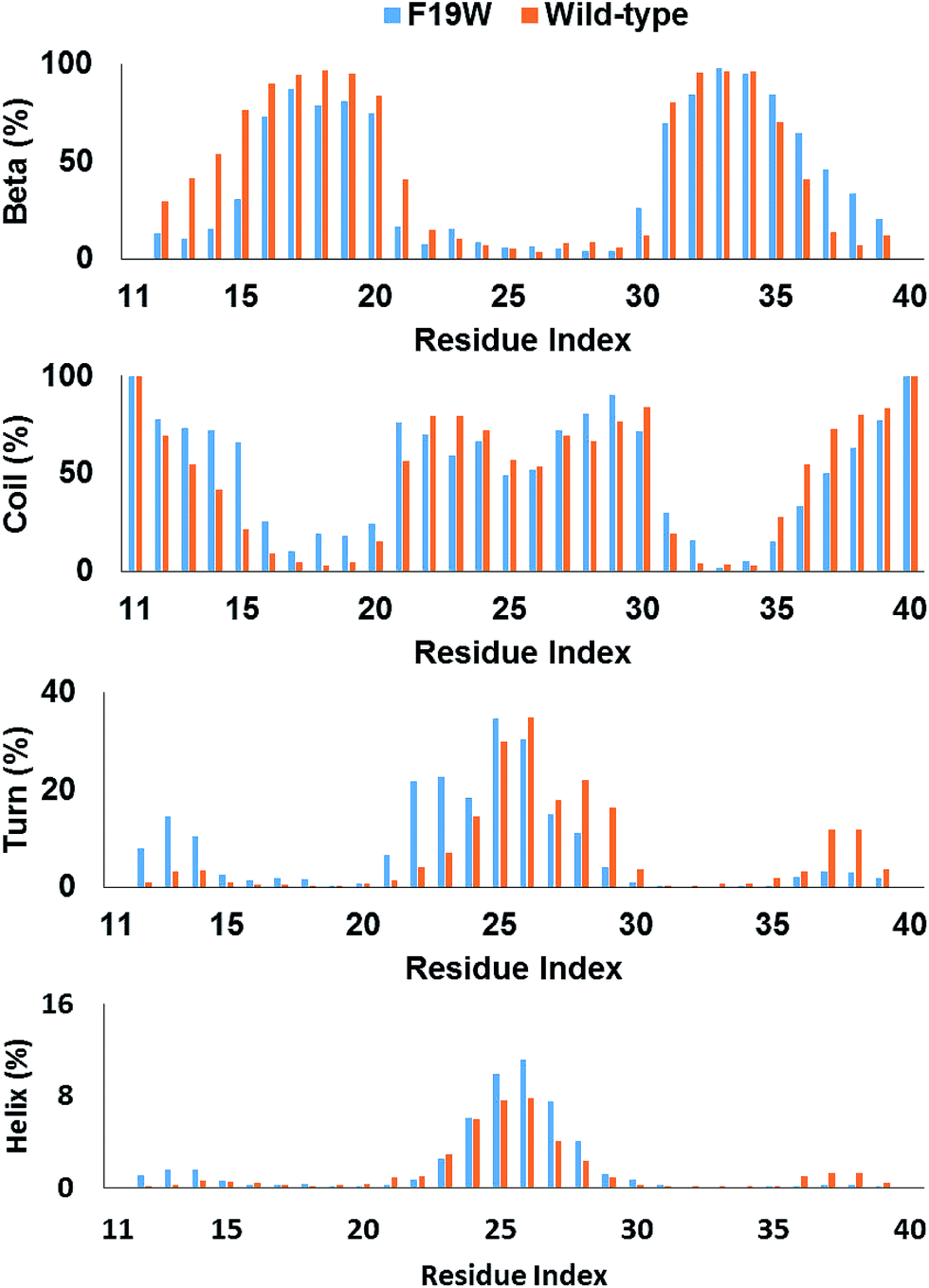 | ||
| Fig. 3 Secondary structure terms per residues averaged for all three chains of 3Aβ11–40 (ref. 31) and its F19W mutant. The data were obtained from 250 to 400 ns of REMD simulations. | ||
The secondary structure per residue shifted upon F19W mutation. Most of the 3% decrease in beta content occurs in the N-terminal beta region (sequence 12–21) whereas there is a slight increase in the beta content of the C-terminal region (sequence 35–39). Concomitantly, the decrease in coil content also occurs in these two regions. The beta content at residue 19 in 3Aβ11–40 is 95%, which decreases to 81% in F19W 3Aβ11–40.
The turn content per residue also shifted significantly. The turn content increases in the N-terminal flank of the peptide chain (residues 12–14 and 21–24) where the beta content increases. In contrast, the turn content decreases in the C-terminal flank (residues 28–30 and 37–39) with the similar magnitude. The helix content also shifted in similar fashion, with slightly increase in the residues 12–14 and slightly decrease in the residues 37–39. In addition, the helix content also slightly increases in the middle of the chains (residues 25–27).
Inter-chain contacts
The intermolecular contacts between constituting monomer of F19W 3Aβ11–40 were analyzed to explore the physical effects of the mutation on the nature of binding between isolated chains. The intermolecular contacts involve the sidechain, hydrogen bonded, and polar contacts that were measured as the criteria mentioned in the Materials & methods section. The intramolecular contact salt bridge D23–K28 was also considered. These values were probed every 10 ps over the last 150 ns of REMD simulation at 300 K.The sidechain contacts between different heavy atoms of different chains were counted and averaged over the considered snapshots. The sidechain contact maps were then constructed based on the probability of these values. The maps are shown in three graphs on the left side of Fig. 4. As observed in 3Aβ11–40,31 the most abundant interactions are observed between chain A–chain B and chain B–chain C in parallel fashion. Particularly, the sequences 11–22 and 30–36 of chain A, chain B, and chain C, where beta content is dominant, are involved in these interactions. The side chain contacts between chain A and chain C are not as abundant but observable. These results indicate that the Aβ11–40 chains form parallel structures in which chain B is in the middle of chain A and chain C. In addition, significant contacts between N-terminal of chain A and C-terminal of chain B, as well as between N-terminal of chain B and C-terminal chain C are also observed, indicating that the computational sampling was adequate, and that the parallel structure contain a turn region in the middle that allow inter-chain interactions of C-terminal and N-terminal. These results are in good agreement with previous NMR studies on the structures of Aβ10–35, Aβ1–40, and Aβ1–42 fibrils.64–67
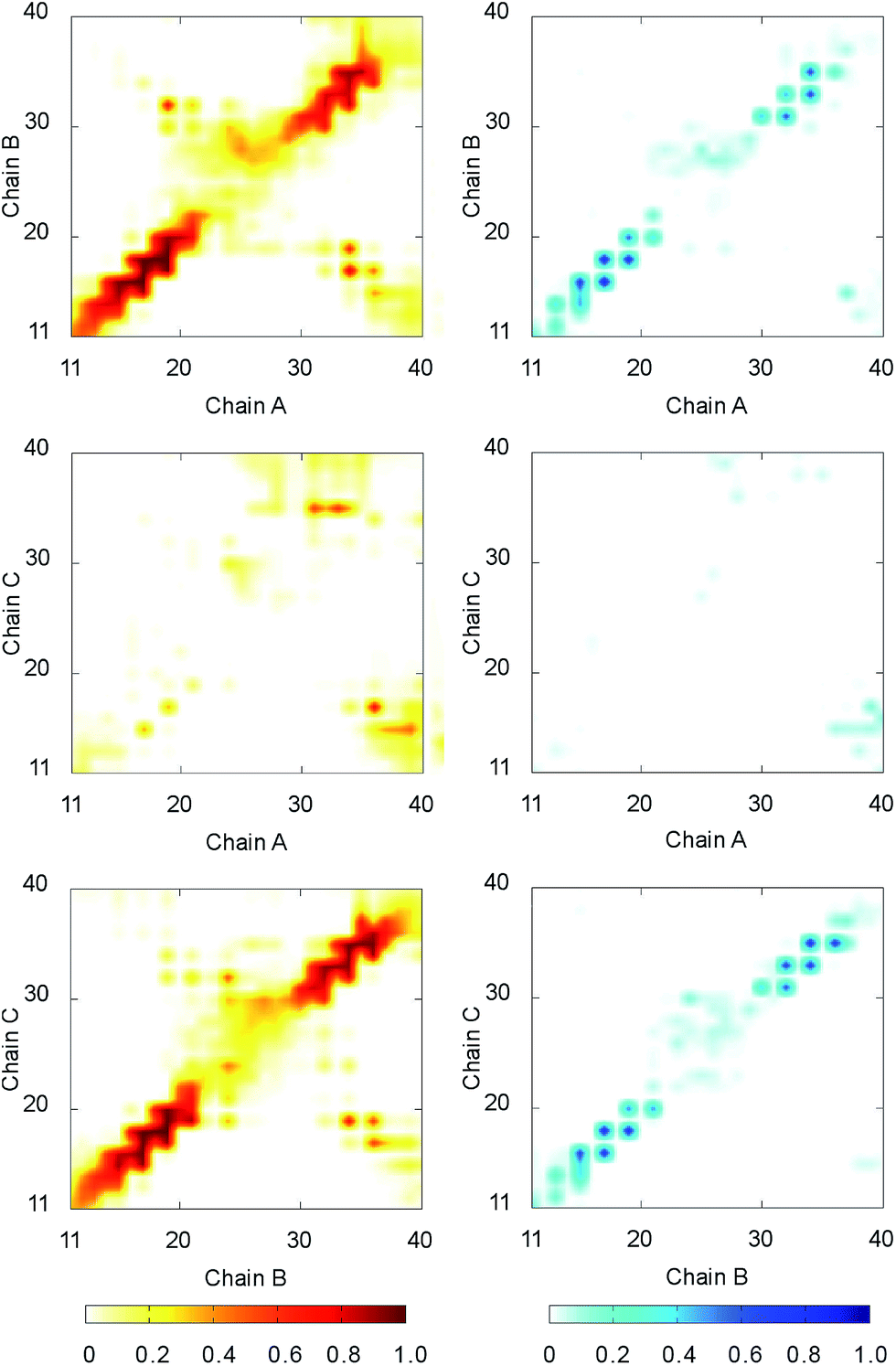 | ||
| Fig. 4 The contact maps between the constituting chains of F19W 3Aβ11–40. The graphs on the left side show sidechain contact maps. The graphs on the right side show hydrogen bonded contact maps. The metrics were estimated over the 15000 snapshots of REMD simulations from 250 to 400 ns. | ||
The hydrogen bonded contact maps are shown on three graphs on the right side of Fig. 4, which are consistent with the sidechain interaction maps. As observed for sidechain contacts, significant numbers of hydrogen bonds (H-bonds) are observed between chain A and chain B, as well chain B and chain C in parallel fashion. The sequences 11–22 and 30–36 of the chains are involved the most in hydrogen-bonded interactions. There are some sizable H-bonds between N- and C-terminal of different chains.
The salt bridge D23–K28 is known to be a critical factor maintaining the Aβ loop region.66,68,69 However, in some NMR results, this salt bridge is not observed in Aβ fibril formations.67,70 In the wild-type case,31,32 the polar contacts between D23 and V24, G25, S26, N27, and G29 were found replace the salt bridge D23–K28 securing the durability of the Aβ turn region. Therefore, in this work polar contacts between D23 and the residues in the sequence 24–29 were evaluated through measuring the distances between two charged groups of these residues.
The distribution of the distance between two charged groups of different residues was estimated and shown in Fig. S4.† Salt bridges or polar contacts were counted when the distances between two charged groups are smaller than 0.46 nm. The average population of the salt bridge D23–K28 in F19W 3Aβ11–40 is 12%, which is significantly higher than that in 3Aβ11–40 (8%). However, the numbers of polar contacts between D23 to V24, G25, S26, N27, and G29 significantly decreased upon F19W mutation (Table 1). Overall, the total populations of important polar contacts decrease by ∼20%. Moreover, the populations of intermolecular contacts H13A–Q15B and H13B–Q15C, which are associated with the stability of the N terminals, decreases from 32% to 29%. Overall, the mutation F19W significantly decreases most of the polar contacts, except for D23–K28 salt bridge. The decrease in polar contacts would lead to higher flexibility of the mutant trimer.
| Contact | WT | F19W | Contact | WT | F19W |
|---|---|---|---|---|---|
| D23–K28 | 8 | 12 | D23–S26 | 21 | 11 |
| D23–V24 | 70 | 65 | D23–N27 | 6 | 3 |
| D23–G25 | 31 | 22 | D23–G29 | 11 | 4 |
Metastable structures obtained from combination of the FES and clustering methods
The representative structures of F19W 3Aβ11–40 were detected using the combination of FES and clustering methods, which has been shown to be highly appropriate in studying the metastable structures of β-amyloid systems.31,32 The RMSD and Rg values were computed to construct the FES through a GROMACS tool “gmx sham”.71 The result is shown in Fig. 5. RMSD and Rg fall in range of 0.60–1.20 nm and 1.25–1.55 nm, respectively.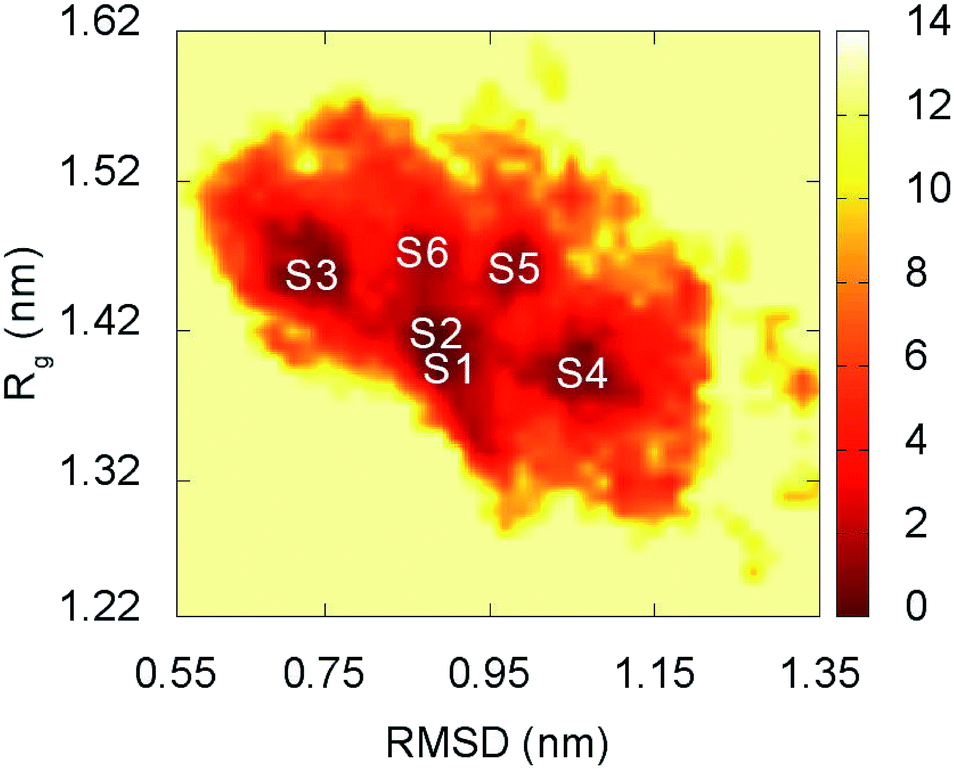 | ||
| Fig. 5 Free energy surface of F19W 3Aβ11–40 was constructed from snapshots from 250 to 400 ns of REMD simulations. S1–S6 denote the six local minima. See Table 3 and Fig. 6 for further details. | ||
There are six local minima obtained for F19W 3Aβ11–40 from clustering with Cα cutoff of 0.3 nm (Fig. 5). The minima are designated as S1–S6 with coordinates listed in Table 3. The secondary structure terms, SASA, and CCS of these structures are shown in Table 3. In average, these minima contain 39, 51, 8, and 2% of beta, coil, turn, and helix contents, respectively. The structures of S1, S4, S5, and S6 (Fig. 6) exhibit a short helix in the middle of one chain, whereas the helix content in S2 and S3 is zero. These metrics are almost the same as the mean values during the last of 150 ns of REMD simulations at 300 K. The average CCS of six structures was determined as 1347.1 ± 27.2 Å2, which is marginally larger than the value of the wild-type Aβ11–40 trimer (1330.7 Å2).31 The average SASA of these minima is 6330.2 ± 271.0 Å2, which is slightly smaller than the metric of all considered snapshots during the computations.
| Rg | RMSD | Beta | Coil | Turn | Helix | CCS | SASA | |
|---|---|---|---|---|---|---|---|---|
| a The units are (%) for the secondary structure terms, (nm) for Rg and RMSD, and (Å2) for CCS and SASA.b Average values for F19W 3Aβ11–40.c Average values for 3Aβ11–40 reported in ref. 31. | ||||||||
| S1 | 1.40 | 0.88 | 39 | 53 | 4 | 3 | 1298.4 | 6279.7 |
| S2 | 1.41 | 0.87 | 36 | 54 | 10 | 0 | 1338.7 | 6089.3 |
| S3 | 1.45 | 0.74 | 42 | 50 | 8 | 0 | 1378.8 | 6729.0 |
| S4 | 1.39 | 1.06 | 32 | 57 | 8 | 3 | 1367.6 | 6666.6 |
| S5 | 1.46 | 0.96 | 43 | 41 | 11 | 4 | 1332.9 | 6040.2 |
| S6 | 1.47 | 0.86 | 39 | 48 | 10 | 3 | 1366.0 | 6176.5 |
| Sb | 1.43 | 0.90 | 39 | 51 | 8 | 2 | 1347.1 ± 27.2 | 6330.2 ± 271.0 |
| Mc | 1.41 | 0.54 | 36 | 56 | 6 | 1 | 1330.7 | 6245.5 ± 322.8 |
| ΔEelec | ΔEvdW | ΔGsur | ΔGPB | −TΔS | ΔGbind | |
|---|---|---|---|---|---|---|
| a The unit of values is kcal mol−1.b Reported in ref. 31. | ||||||
| C to A + B | −132.0 | −136.5 | −19.8 | 176.5 | 68.5 | −43.36 |
| B to A + C | −154.7 | −219.5 | −30.2 | 234.4 | 68.0 | −102.0 |
| A to B + C | −136.7 | −142.6 | −19.3 | 186.2 | 66.4 | −46.0 |
| Average | −141.1 | −166.2 | −23.1 | 199.0 | 67.6 | −63.8 ± 27.1 |
| Average of WTb | −90.1 | −144.9 | −21.3 | 153.6 | 66.8 | −35.8 ± 7.9 |
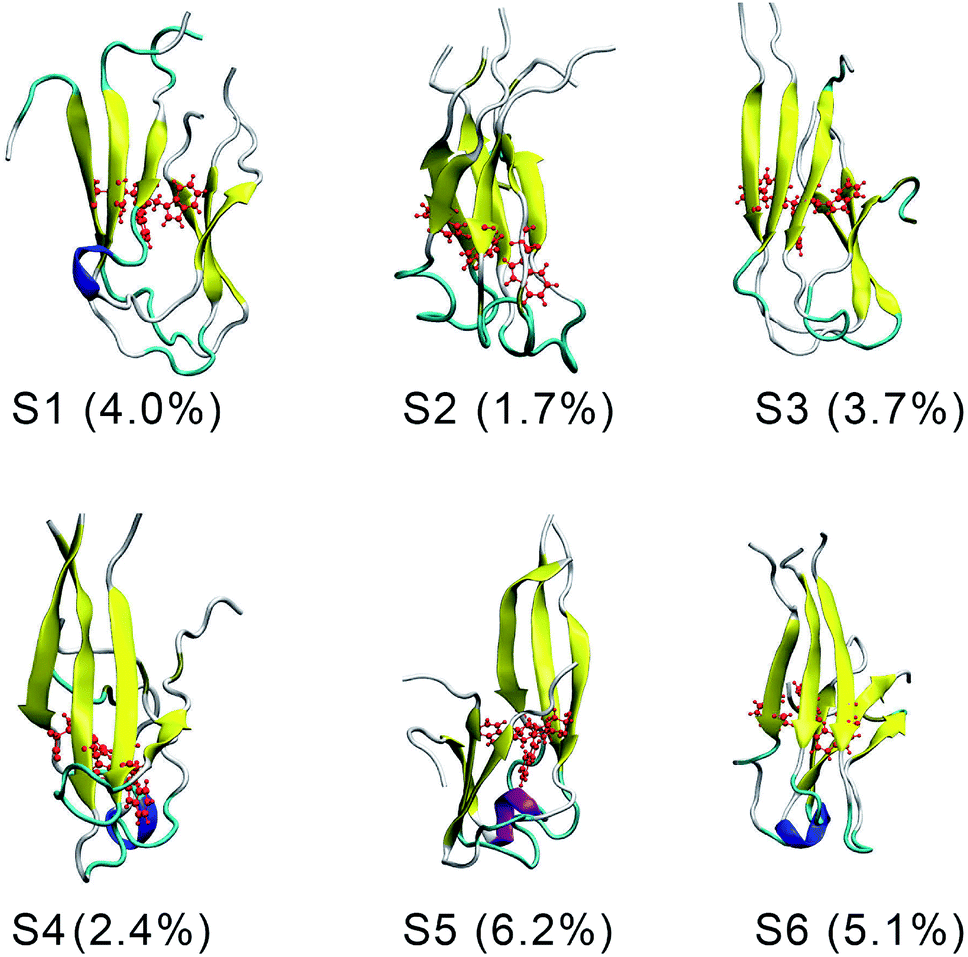 | ||
| Fig. 6 The representative conformations of the mutant trimer obtained from the snapshots from 250 to 400 ns of REMD simulations using FES and clustering methods. | ||
The structures of S1–S6 (Fig. 6) are significantly different from those of the three minima MA, MB, and MC of 3Aβ11–40,31 despite being simulated from the same initial conformation.51 The populations of S1–S6 are very small, ranging from 1.7–6.2%. In contrast, MA, MB, and MC respectively represent 47, 7, and 6% of the total population of all conformations.31 In addition, the free energy barriers of six minima S1–S6 are 12.8, 12.8, 12.3, 12.3, 11.7, and 11.7 kJ mol−1, respectively, which are about ∼1 kJ mol−1 lower than those found for the minima of 3Aβ11–40. The larger number of minima with lower energy barriers and populations indicate that the F19W 3Aβ11–40 adopts more easily interchangeable conformations. This result is consistent with the significant decrease in crucial polar contacts described above.
Binding free energy between the constituting monomers to the others
Several computational methods have been developed to probe the free energy difference of binding between two molecules in biochemical physics.72–79 Although the MM-PBSA method was shown to have some limitations, including large error,80 it has been applied widely in computational biophysics. The method was employed to determine the binding free energy between isolated peptide to the other.63 Recently, the binding free energy difference between constituting monomers in solvated 3Aβ11–40 was probed using the MM-PBSA method.31The MM-PBSA method was applied to investigate the nature of binding between the chains of soluble F19W 3Aβ11–40. The calculations were applied on S1–S6 minima (Fig. 5). The results were averaged over these 6 minima and are shown in Table 3. In this scheme, the free energy difference of binding between isolated chain to the others was evaluated. The binding free energy of chain A to the remaining chains of the trimer (−46.0 kcal mol−1) is approximately equaled to that of chain C (−43.6 kcal mol−1), with the smaller than 5%. Both of these affinities are approximately half of that of chain B to chains A and C (ΔGbind −102.0 kcal mol−1), which is consistent with parallel structures in which chain B is in the middle of chain A and chain C.
The mean vdW interaction energy ΔEvdW of F19W 3Aβ11–40 is −166.2 kcal mol−1 (Table 2), which is significantly larger than that in 3Aβ11–40 (−144.9 kcal mol−1). The combination of electrostatic ΔEelec and polar ΔGPB energies in F19W 3Aβ11–40 is 5.6 kcal mol−1 smaller than that in solvated 3Aβ11–40. The contribution of surface energy ΔGsur and conformational entropy −TΔS are approximately the same between 3Aβ11–40 and its F19W mutant. Overall, a constituting chain formed approximately −63.8 ± 27.1 kcal mol−1 of binding free energy to the other monomers in F19W 3Aβ11–40. This affinity is significantly larger than that in 3Aβ11–40 (35.8 ± 7.9 kcal mol−1). However, the error in ΔGbind of the F19W 3Aβ11–40 (27.1 kcal mol−1) is much larger than that in 3Aβ11–40 (7.9 kcal mol−1), indicating that the interactions between monomers fluctuate significantly upon F19W mutation. These results indicate that the monomers in soluble F19W 3Aβ11–40 bind to each other more tightly than in 3Aβ11–40, largely due to the increased in vdW interactions. The fluctuation in binding free energy likely results from the decrease in important polar contacts. This fluctuation is consistent with higher RMSD and lower energy barriers of the conformations of F19W 3Aβ11–40.
Conclusions
Results showed that upon F19W mutation, the secondary structure terms slightly change with 3% lest beta and 3% more coil contents. The distributions per residue of these terms shifted to significantly. In addition, the number of contacts, SASA, and Rg value only change slightly in the F19W mutant. However RMSD increases significantly from ∼0.51 in 3Aβ11–40 to ∼0.91 ± 0.14 nm in F19W 3Aβ11–40. Important polar contacts between D23 to sequence 24–29, which help stabilize of the loop region, decreases by 20%. The important intermolecular contacts H13A–Q15B and H13B–Q15C, which maintain the N-terminal region of the trimer also decreased by 3% in the mutant.Six minima were found on the free energy surface of F19W 3Aβ11–40, which account for around 20% of total conformations. In contrast, there are only 3 minima found for 3Aβ11–40, which account for 60% of all conformations. The free energy barriers of the minima in the F19W 3Aβ11–40 is ∼1 kJ mol−1 smaller than those in 3Aβ11–40. The binding free energy between the chains of F19W 3Aβ11–40, obtained using MM-PBSA method for all six minima, is ∼28 kcal mol−1 larger than that in 3Aβ11–40. However, the high error of binding free energy σΔGbind implies that the conformations of F19W 3Aβ11–40 fluctuate much more than those of 3Aβ11–40.
Altogether, our computational studies provided detailed insights into the effects of F19W mutation on soluble 3Aβ11–40. The similarity in structural terms and higher binding affinity between constituting chains indicate that the hydrophobic core of amyloid beta peptide is capable adapting to large changes. However, the decrease in critical polar contacts, higher fluctuation in binding energy, the higher number of minima with lower energy barriers and significantly lower population indicate a significant increase in the flexibility of the mutant. These results also contribute to understanding the fibrillation of Aβ peptide. The more flexible F19W mutant oligomers would require more time to self-assemble into fibrils, which is consistent the observation that mutations at F19 residues result in longer lag phase (4 folds) and characteristic fibrillation time (1.5–4 folds).50
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 103.01-2016.48 to STN, as well as by Nguyen Tat Thanh University institutional grant # 2016.03.01 to VVV.References
- D. J. Selkoe, Neuron, 1991, 6, 487–498 CrossRef CAS PubMed
.
- A. S. Henderson and A. F. Jorm, Dementia, John Wiley & Sons Ltd, 2002 Search PubMed
- J. L. Cummings, N. Engl. J. Med., 2004, 351, 56–67 CrossRef CAS PubMed
- H. W. Querfurth and F. M. LaFerla, N. Engl. J. Med., 2010, 362, 329–344 CrossRef CAS PubMed
- Alzheimer's association, Alzheimer's disease facts and figures, 2015 Search PubMed.
- D. J. Selkoe, Science, 2002, 298, 789–791 CrossRef CAS PubMed
- P. Davies and A. J. F. Maloney, Lancet, 1976, 308, 1403 CrossRef
- I. Khlistunova, J. Biernat, Y. Wang, M. Pickhardt, M. von Bergen, Z. Gazova, E. Mandelkow and E.-M. Mandelkow, J. Biol. Chem., 2006, 281, 1205–1214 CrossRef CAS PubMed
- K. SantaCruz, J. Lewis, T. Spires, J. Paulson, L. Kotilinek, M. Ingelsson, A. Guimaraes, M. DeTure, M. Ramsden and E. McGowan, et al., Science, 2005, 309, 476–481 CrossRef CAS PubMed
- T. Saito, T. Suemoto, N. Brouwers, K. Sleegers, S. Funamoto, N. Mihira, Y. Matsuba, K. Yamada, P. Nilsson and J. Takano, et al., Nat. Neurosci., 2011, 14, 1023–1032 CrossRef CAS PubMed
- J.-C. Lambert, C. A. Ibrahim-Verbaas, D. Harold, A. C. Naj, R. Sims, C. Bellenguez, G. Jun, A. L. DeStefano, J. C. Bis and G. W. Beecham, et al., Nat. Genet., 2013, 45, 1452–1458 CrossRef CAS PubMed
- D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed
- M. Bucciantini, E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson and M. Stefani, Nature, 2002, 416, 507–511 CrossRef CAS PubMed
- D. M. Walsh and D. J. Selkoe, J. Neurochem., 2007, 101, 1172–1184 CrossRef CAS PubMed
- M. K. Jana, R. Cappai, C. L. L. Pham and G. D. Ciccotosto, J. Neurochem., 2016, 136, 594–608 CrossRef CAS PubMed
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed
- M. Citron, Nat. Rev. Neurosci., 2004, 5, 677–685 CrossRef CAS PubMed
- A. Aguzzi and T. O'Connor, Nat. Rev. Drug Discovery, 2010, 9, 237–248 CrossRef CAS PubMed
- G. M. Shankar, S. Li, T. H. Mehta, A. Garcia-Munoz, N. E. Shepardson, I. Smith, F. M. Brett, M. A. Farrell, M. J. Rowan and C. A. Lemere, et al., Nat. Med., 2008, 14, 837–842 CrossRef CAS PubMed
- C. R. Muratore, H. C. Rice, P. Srikanth, D. G. Callahan, T. Shin, L. N. P. Benjamin, D. M. Walsh, D. J. Selkoe and T. L. Young-Pearse, Hum. Mol. Genet., 2014, 23, 3523–3536 CrossRef CAS PubMed
- S. Moore, L. D. B. Evans, T. Andersson, E. Portelius, J. Smith, T. B. Dias, N. Saurat, A. McGlade, P. Kirwan and K. Blennow, et al., Cell Rep., 2015, 11, 689–696 CrossRef CAS PubMed
- J. Geng, K. Qu, J. Ren and X. Qu, Mol. BioSyst., 2010, 6, 2389–2391 RSC
- Q. Nie, X.-g. Du and M.-y. Geng, Acta Pharmacol. Sin., 2011, 32, 545–551 CrossRef CAS PubMed
- J. Bieschke, M. Herbst, T. Wiglenda, R. P. Friedrich, A. Boeddrich, F. Schiele, D. Kleckers, J. M. Lopez del Amo, B. A. Grüning and Q. Wang, et al., Nat. Chem. Biol., 2012, 8, 93–101 CrossRef CAS PubMed
- S. T. Ngo and M. S. Li, Mol. Simul., 2013, 39, 279–291 CrossRef CAS
- S. T. Ngo, S.-T. Fang, S.-H. Huang, C.-L. Chou, P. D. Q. Huy, M. S. Li and Y.-C. Chen, J. Chem. Inf. Model., 2016, 56, 1344–1356 CrossRef CAS PubMed
- S. T. Ngo, D. T. Truong, N. M. Tam and M. T. Nguyen, J. Mol. Graphics Modell., 2017, 76, 1–10 CrossRef CAS PubMed
- G. Bitan, M. D. Kirkitadze, A. Lomakin, S. S. Vollers, G. B. Benedek and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 330–335 CrossRef CAS PubMed
- M. D. Kirkitadze, M. M. Condron and D. B. Teplow, J. Mol. Biol., 2001, 312, 1103–1119 CrossRef CAS PubMed
- M. Wolff, B. Zhang-Haagen, C. Decker, B. Barz, M. Schneider, R. Biehl, A. Radulescu, B. Strodel, D. Willbold and L. Nagel-Steger, Sci. Rep., 2017, 7, 2493 CrossRef PubMed
- S. T. Ngo, H. M. Hung, D. T. Truong and M. T. Nguyen, Phys. Chem. Chem. Phys., 2017, 19, 1909–1919 RSC
- S. T. Ngo, H. M. Hung, K. N. Tran and M. T. Nguyen, RSC Adv., 2017, 7, 7346–7357 RSC
- S. T. Ngo, M. T. Nguyen, N. T. Nguyen and V. V. Vu, J. Phys. Chem. B, 2017 DOI:10.1021/acs.jpcb.7b05906
- X. Zheng, D. Liu, R. Roychaudhuri, D. B. Teplow and M. T. Bowers, ACS Chem. Neurosci., 2015, 6, 1732–1740 CrossRef CAS PubMed
- J. C. Janssen, J. A. Beck, T. A. Campbell, A. Dickinson, N. C. Fox, R. J. Harvey, H. Houlden, M. N. Rossor and J. Collinge, Neurology, 2003, 60, 235–239 CrossRef CAS PubMed
- W.-T. Chen, C.-J. Hong, Y.-T. Lin, W.-H. Chang, H.-T. Huang, J.-Y. Liao, Y.-J. Chang, Y.-F. Hsieh, C.-Y. Cheng and H.-C. Liu, et al., PLoS One, 2012, 7, e35807 CAS
- Y. Wakutani, K. Watanabe, Y. Adachi, K. Wada-Isoe, K. Urakami, H. Ninomiya, T. C. Saido, T. Hashimoto, T. Iwatsubo and K. Nakashima, J. Neurol., Neurosurg. Psychiatry, 2004, 75, 1039–1042 CrossRef CAS
- R. T. McDonough, G. Paranjape, F. Gallazzi and M. R. Nichols, Arch. Biochem. Biophys., 2011, 514, 27–32 CrossRef CAS PubMed
- L. Hendriks, C. M. van Duijn, P. Cras, M. Cruts, W. Van Hul, F. van Harskamp, A. Warren, M. G. McInnis, S. E. Antonarakis and J.-J. Martin, et al., Nat. Genet., 1992, 1, 218–221 CrossRef CAS PubMed
- E. Levy, M. Carman, I. Fernandez-Madrid, M. Power, I. Lieberburg, S. van Duinen, G. Bots, W. Luyendijk and B. Frangione, Science, 1990, 248, 1124–1126 CAS
- O. Bugiani, A. Padovani, M. Magoni, G. Andora, M. Sgarzi, M. Savoiardo, A. Bizzi, G. Giaccone, G. Rossi and F. Tagliavini, Neurobiol. Aging, 1998, 19, S238 Search PubMed
- C. Nilsberth, A. Westlind-Danielsson, C. B. Eckman, M. M. Condron, K. Axelman, C. Forsell, C. Stenh, J. Luthman, D. B. Teplow and S. G. Younkin, et al., Nat. Neurosci., 2001, 4, 887–893 CrossRef CAS PubMed
- T. Tomiyama, T. Nagata, H. Shimada, R. Teraoka, A. Fukushima, H. Kanemitsu, H. Takuma, R. Kuwano, M. Imagawa and S. Ataka, et al., Ann. Neurol., 2008, 63, 377–387 CrossRef CAS PubMed
- T. J. Grabowski, H. S. Cho, J. P. G. Vonsattel, G. W. Rebeck and S. M. Greenberg, Ann. Neurol., 2001, 49, 697–705 CrossRef CAS PubMed
- O. Coskuner, O. Wise-Scira, G. Perry and T. Kitahara, ACS Chem. Neurosci., 2013, 4, 310–320 CrossRef CAS PubMed
- L. Xu, S. Shan and X. Wang, J. Phys. Chem. B, 2013, 117, 6206–6216 CrossRef CAS PubMed
- Y. Lu, G. Wei and P. Derreumaux, J. Phys. Chem. B, 2011, 115, 1282–1288 CrossRef CAS PubMed
- L. Tran, N. Basdevant, C. Prévost and T. Ha-Duong, Sci. Rep., 2016, 6, 21429 CrossRef CAS PubMed
- H. Inouye, K. A. Gleason, D. Zhang, S. M. Decatur and D. A. Kirschner, Proteins: Struct., Funct., Bioinf., 2010, 78, 2306–2321 CrossRef CAS PubMed
- J. Adler, H. A. Scheidt, M. Krüger, L. Thomas and D. Huster, Phys. Chem. Chem. Phys., 2014, 16, 7461–7471 RSC
- I. Bertini, L. Gonnelli, C. Luchinat, J. Mao and A. Nesi, J. Am. Chem. Soc., 2011, 133, 16013–16022 CrossRef CAS PubMed
- P. Schrödinger LLC, The PyMOL Molecular Graphics System, Version 1.3r1, 2010 Search PubMed
- A. E. Aliev, M. Kulke, H. S. Khaneja, V. Chudasama, T. D. Sheppard and R. M. Lanigan, Proteins: Struct., Funct., Bioinf., 2014, 82, 195–215 CrossRef CAS PubMed
- A. K. Somavarapu and K. P. Kepp, ChemPhysChem, 2015, 16, 3278–3289 CrossRef CAS PubMed
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS
- S. Jang and S. Shin, J. Phys. Chem. B, 2008, 112, 3479–3484 CrossRef CAS PubMed
- V. Knecht, J. Phys. Chem. B, 2010, 114, 12701–12707 CrossRef CAS PubMed
- L. Tran and T. Ha-Duong, Peptides, 2015, 69, 86–91 CrossRef CAS PubMed
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef
- D. F. Shanno, Math. Comput., 1970, 24, 647–656 CrossRef
- W. G. Touw, C. Baakman, J. Black, T. A. H. te Beek, E. Krieger, R. P. Joosten and G. Vriend, Nucleic Acids Res., 2015, 43, D364–D368 CrossRef CAS PubMed
- H. K. Srivastava and G. N. Sastry, J. Chem. Inf. Model., 2012, 52, 3088–3098 CrossRef CAS PubMed
- H. L. Chiang, S. T. Ngo, C. J. Chen, C. K. Hu and M. S. Li, PLoS One, 2013, 8, e65358 CAS
- T. S. Burkoth, T. L. S. Benzinger, V. Urban, D. M. Morgan, D. M. Gregory, P. Thiyagarajan, R. E. Botto, S. C. Meredith and D. G. Lynn, J. Am. Chem. Soc., 2000, 122, 7883–7889 CrossRef CAS
- R. Tycko, Biochemistry, 2003, 42, 3151–3159 CrossRef CAS PubMed
- T. Luhrs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Doeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef CAS PubMed
- Y. Xiao, B. Ma, D. McElheny, S. Parthasarathy, F. Long, M. Hoshi, R. Nussinov and Y. Ishii, Nat. Struct. Mol. Biol., 2015, 22, 499–505 CAS
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742–16747 CrossRef CAS PubMed
- A. T. Petkova, W. M. Yau and R. Tycko, Biochemistry, 2006, 45, 498–512 CrossRef CAS PubMed
- M. T. Colvin, R. Silvers, Q. Z. Ni, T. V. Can, I. Sergeyev, M. Rosay, K. J. Donovan, B. Michael, J. Wall and S. Linse, et al., J. Am. Chem. Soc., 2016, 138, 9663–9674 CrossRef CAS PubMed
- E. Papaleo, P. Mereghetti, P. Fantucci, R. Grandori and L. De Gioia, J. Mol. Graphics Modell., 2009, 27, 889–899 CrossRef CAS PubMed
- G. M. Morris, D. S. Godsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS
- J. Aqvist, C. Medina and J.-E. Samuelsson, Protein Eng., 1994, 7, 385–391 CrossRef CAS PubMed
- S. T. Ngo, B. K. Mai, D. M. Hiep and M. S. Li, Chem. Biol. Drug Des., 2015, 86, 546–558 CAS
- R. W. Zwanzig, J. Chem. Phys., 1954, 22, 1420–1426 CrossRef CAS
- S. T. Ngo, H. M. Hung and M. T. Nguyen, J. Comput. Chem., 2016, 37, 2734–2742 CrossRef CAS PubMed
- D. T. Truong, M. T. Nguyen, V. V. Vu and S. T. Ngo, Chem. Phys. Lett., 2017, 671, 142–146 CrossRef CAS
- S. T. Ngo, M. T. Nguyen and M. T. Nguyen, Chem. Phys. Lett., 2017, 676, 12–17 CrossRef CAS
- J. D. Holliday, S. S. Ranade and P. Willett, Quant. Struct.-Act. Relat., 1995, 14, 501–506 CrossRef CAS
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S4. Fig. S1 shows the secondary structural terms of 48 replicas at 250 ns of REMD simulations. Fig. S2 shows the distributions of structural terms of 19th residues in F19W 3Aβ11–40 and 3Aβ11–40. Fig. S3 shows the distributions of critical intramolecular polar contacts (D23 to V24, G25, S26, N27, K28 and G29) and intermolecular polar contacts (H13A–Q15B and H13B–Q15C) of F19W 3Aβ11–40. Fig. S4 shows the distributions of the mean secondary structure per residue of F19W 3Aβ11–40. See DOI: 10.1039/c7ra07187f |
| This journal is © The Royal Society of Chemistry 2017 |