DOI:
10.1039/C7RA07186H
(Paper)
RSC Adv., 2017,
7, 39325-39333
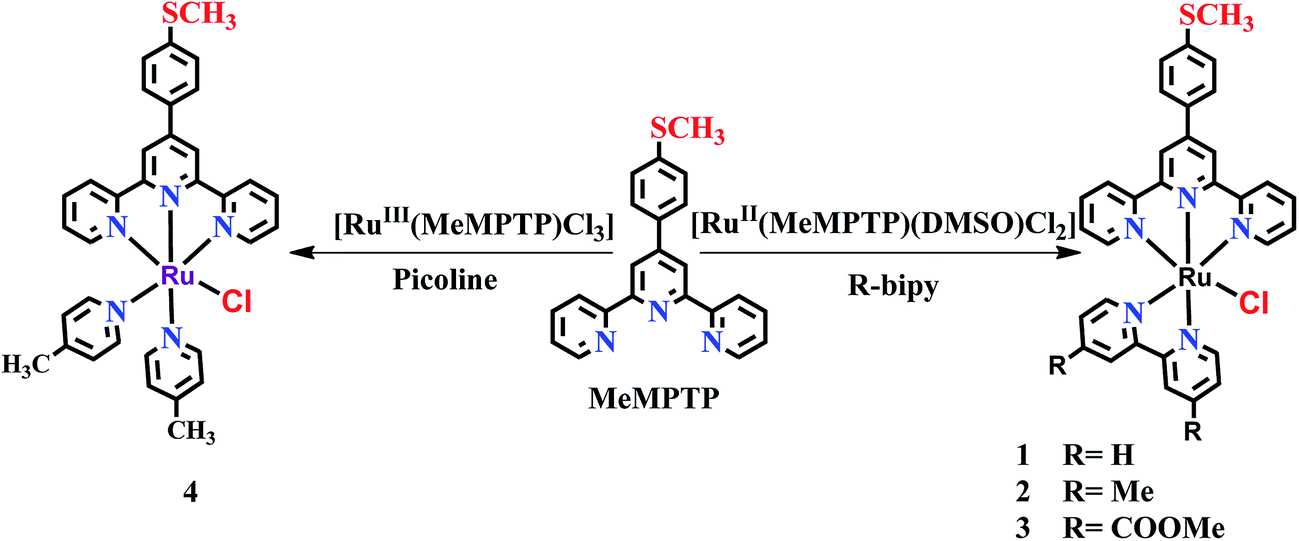
Synthesis, crystal structure and water oxidation activity of [Ru(terpy)(bipy)Cl]+ complexes: influence of ancillary ligands on O2 generation†
Received
28th June 2017
, Accepted 21st July 2017
First published on 10th August 2017
Abstract
Four new Ru(II) complexes, [RuII(MeMPTP)(bpy)Cl]PF6 (1), [RuII(MeMPTP)(dmbpy)Cl]PF6 (2), [RuII(MeMPTP)(dmcbpy)Cl]PF6 (3) and [RuII(MeMPTP)(Pic)2Cl]PF6 (4) [where, MeMPTP = 4′-(4-methylmercaptophenyl)-2,2′:6′2′′-terpyridine, bpy = 2,2′-bipyridine, dmbpy = 4,4′-dimethyl-2,2′-bipyridine, dmcbpy = 4,4′-dimethoxycarbonyl-2,2′-bipyridine and pic = 4-picoline] were synthesized and characterized via various spectroscopic techniques. The molecular structures of the complexes 1 and 2 were determined by single crystal X-ray diffraction analysis. Catalytic activity for chemical oxidation of water of the complexes 1–4 reveals that the rate of O2 evolution follows the trend 1 > 4 > 2 > 3. Except the unsubstituted complex 1, the catalytic rate for O2 generation of 2 and 4, containing electron-donating (–CH3) groups, is higher than that of 3, bearing an electron-withdrawing (–COOMe) group on the bpy, while the turn over number (TON) of the complexes follows an opposite trend. The difference in the water oxidation activity of the complexes has been correlated to the effect of the substituents on the ancillary ligands in facilitating the electron density on the Ru(II) center to achieve the higher oxidation states required for the water oxidation catalysis. Interestingly, water oxidation study of the complexes 1–4 fills the missing gap between the well-studied mononuclear ruthenium complexes based on terpy/bpy and the MeMPTP/phen ligands.
Introduction
Over the past few decades, significant advances have been made in the field of homogeneous water oxidation catalysis for the development of artificial photosynthesis by efficiently splitting water into O2 and H2.1–3 Among the immense library of water oxidation catalysts reported thus far, ruthenium complexes based on polypyridyl ligands have shown better activity owing to the low catalytic potential of Ru(II) resulting in lower kinetic barrier and higher catalytic efficiency.4–6 Particularly, the study by Thummel and coworkers has shown that mononuclear octahedral complexes of ruthenium containing a terpyridine (terpy) ligand along with bpy/py and halide co-ligands have very good catalytic activity for chemical oxidation of water to generate O2.7 Furthermore, the systematic investigation of the effect of substituents (electron-donating and -withdrawing) on polypyridyl ligands containing mononuclear ruthenium complexes, such as [Ru(terpy)(bpy)(OH2)]2+, on the water oxidation activity has been reported by Berlinguette and co-workers.8,9 Interestingly, they observed that electron-withdrawing groups (EWGs) on the bpy ligand resulted in lower catalytic activity for O2 evolution and higher catalytic turnover numbers (TONs). Conversely, the presence of electron-donating groups (EDGs) accelerated catalytic rates while decreasing TONs.8,9
On the other hand, Verani and co-workers studied water oxidation activity of mononuclear ruthenium complexes of 4-functionalised-terpyridine ligand, MeMPTP having –SMe (–R) group as an electron releasing group on terpyridine and phenanthroline coligands [RuII(terpy-R)(phen-X)Cl]+.10 Their study on the influence of the substituents (EWGs and EDGs) on phen-X ligand for catalytic water oxidation revealed an opposite trend for TON compared to those of terpy/bpy systems studied by Thummel and Berlinguette groups. In this regard, we were interested to know the reactivity pattern of mononuclear ruthenium complexes containing the missing combination of ligands (i.e. MeMPTP/bpy) and their electronic effects on the water oxidation activity of the complexes. Therefore, to fill the missing gap between terpy/bpy and MeMPTP/phen systems, we herein report the synthesis of four complexes, [RuII(MeMPTP)(bpy)Cl]PF6 (1), [RuII(MeMPTP)(dmbpy)Cl]PF6 (2), [RuII(MeMPTP)(dmcbpy)Cl]PF6 (3) and [RuII(MeMPTP)(pic)2Cl]PF6 (4) [where MeMPTP = 4′-(4-methylmercaptophenyl)-2,2′:6′2′′-terpyridine, bpy = 2,2′-bipyridine, dmbpy = 4,4′-dimethyl-2,2′-bipyridine, dmcbpy = 4,4′-dimethoxycarbonyl-2,2′-bipyridine and pic = 4-picoline] and their water oxidation properties in the presence of an aqueous solution of CeIV. Single crystal X-ray structure determination of the complexes 1 and 2 revealed octahedral coordination of RuII. Catalytic investigation of the complexes 1–4 for chemical oxidation of water in the presence of CeIV followed the trend 1 > 4 > 2 > 3 for the rate of O2 evolution. Except the unsubstituted complex 1, the catalytic rates of the complexes 2 and 4, containing EDGs (–CH3), were found to be higher than that of 3 bearing EWG (–COOMe) on the bpy, which is in accordance with the reactivity trend reported for MeMPTP/phen and terpy/bpy systems. On the contrary, the TON of the complexes followed the trend (3 > 1 > 2 > 4) similar to that of the well-known terpy/bpy systems but opposite to that of the MeMPTP/phen systems mentioned before. The difference in the water oxidation activity of the complexes 1–4 has been correlated to the effect of the substituents on the ancillary ligands in facilitating the electron density at the Ru(II) center to achieve its higher oxidation states required for water oxidation catalysis. The study reported here fills the missing gap between the mononuclear ruthenium complexes based on terpy/bpy reported by Thummel and Berlinguette groups and the MeMPTP/phen ligands reported by Verani group and brings out the differences in the electronic influence of bpy and phen ancillary ligands.
Experimental
Materials and methods
The reagents including 2,2′-bipyridine (bpy), 4,4′-dimethyl-2,2′-bipyridine (dmbpy), and 4,4′-dicarboxylic acid-2,2′-bipyridine (dcbpy) were obtained from Sigma Aldrich Chemical Company. RuCl3·xH2O was purchased from Arora Matthey Ltd. Solvents were purified by the standard literature procedures prior to use. Furthermore, 4,4′-dimethoxycarbonyl-2,2′-bipyridine (dmcbpy),11 4′-(4-methylmercaptophenyl)-2,2′:6′2′′-terpyridine (MeMPTP)12 ligands and the complexes [RuII(DMSO)4Cl2],13 [Ru(MeMPTP)Cl3]9a and [RuII(MeMPTP)(DMSO)Cl2]10 were synthesized according to the reported literature procedure.
Elemental analysis (C, H, and N) of the samples was carried out using a Thermo Fischer Flash 2000 elemental analyzer. Fourier transform infrared (FTIR) spectra of the samples were recorded with the KBr pellets on a Thermo Scientific Nicolet iS10 FT-IR spectrometer in the 4000–400 cm−1 region. UV-vis absorption spectra were recorded on a UV-2600 240V Shimadzu spectrophotometer. 1H and 13C NMR spectra were obtained on a 400 MHz JEOL JNM-ECS spectrometer. Chemical shifts (δ) are given in ppm relative to residual solvent (DMSO-d6, δ = 2.5 ppm for 1H NMR and 77.16 ppm for 13C NMR) and coupling constants (J) in Hz. LCMS-ESI (liquid chromatography mass spectrometry) spectra were recorded on a Bruker impact-HD spectrometer. Electrochemical measurements were carried out using an Autolab PGSTAT302N system. Cyclic voltammetry (CV) experiments were performed at room temperature in a one-compartment cell equipped with Ag/AgCl as a RE (reference electrode) (3 M KCl), a glassy carbon (2 mm diameter) as a WE (working electrode) and a platinum foil as a CE (counter electrode) in CH3CN containing TBAPF6 tetrabutylammonium hexafluorophosphate (0.1 M) as a supporting electrolyte at a scan rate of 100 mV s−1.
Synthesis of [RuII(MeMPTP)(bpy)Cl]PF6 (1)
[RuII(MeMPTP)(DMSO)Cl2] (100 mg, 0.165 mmol), bpy (26 mg, 0.165 mmol) and triethylamine (0.2 mL) were added in CH3OH (20 mL) and refluxed overnight under N2 atmosphere. The resulting solution was reduced to one third volume and NH4PF6 (207 mg, 1.2 mmol) was added. The dark-red precipitate formed was filtered through a frit under N2 atmosphere. The isolated solid was washed with cold CH3OH and further purified by column chromatography using neutral alumina with CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3CN (9:1). Yield: 58 mg (55%). Good quality crystals of 1 suitable for X-ray diffraction analysis were grown by slow evaporation of an ACN solution (5 mL) of 1 (2 mg) in a vial at room temperature. 1H NMR (DMSO-d6): δ = 10.11 (d, J = 4.5 Hz, 1H), 9.17 (s, 2H), 8.95 (d, J = 8.2 Hz, 2H), 8.92 (d, J = 7.3 Hz, 1H), 8.65 (d, J = 8.2 Hz, 1H), 8.36 (t, J = 7.8 Hz, 1H), 8.31 (d, J = 8.2 Hz, 2H), 8.08 (t, J = 6.8 Hz, 1H), 8.03 (t, J = 7.8 Hz, 2H), 7.78 (t, J = 7.8 Hz, 1H), 7.63 (d, J = 5.5 Hz, 2H), 7.57 (d, J = 8.2 Hz, 2H), 7.43 (d, J = 5.4 Hz, 1H), 7.39 (t, J = 6.4 Hz, 2H), 7.08 (t, J = 6.4 Hz, 1H), 2.63 (s, 3H) (Fig. S1†). 13C NMR (DMSO-d6): 14.4 (–SCH3). MS: m/z = 648.0297 [C32H25ClN5RuS]+. IR (KBr, cm−1): 1558 m, 1540 m, 1432 m, 1420 m for C
:CH3CN (9:1). Yield: 58 mg (55%). Good quality crystals of 1 suitable for X-ray diffraction analysis were grown by slow evaporation of an ACN solution (5 mL) of 1 (2 mg) in a vial at room temperature. 1H NMR (DMSO-d6): δ = 10.11 (d, J = 4.5 Hz, 1H), 9.17 (s, 2H), 8.95 (d, J = 8.2 Hz, 2H), 8.92 (d, J = 7.3 Hz, 1H), 8.65 (d, J = 8.2 Hz, 1H), 8.36 (t, J = 7.8 Hz, 1H), 8.31 (d, J = 8.2 Hz, 2H), 8.08 (t, J = 6.8 Hz, 1H), 8.03 (t, J = 7.8 Hz, 2H), 7.78 (t, J = 7.8 Hz, 1H), 7.63 (d, J = 5.5 Hz, 2H), 7.57 (d, J = 8.2 Hz, 2H), 7.43 (d, J = 5.4 Hz, 1H), 7.39 (t, J = 6.4 Hz, 2H), 7.08 (t, J = 6.4 Hz, 1H), 2.63 (s, 3H) (Fig. S1†). 13C NMR (DMSO-d6): 14.4 (–SCH3). MS: m/z = 648.0297 [C32H25ClN5RuS]+. IR (KBr, cm−1): 1558 m, 1540 m, 1432 m, 1420 m for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC stretching of pyridine rings, 836s for PF6. Anal. calcd for [C32H25N5RuClSPF6]: C, 48.46; H, 3.81; N, 8.83; S, 4.04. Found: C, 48.62; H, 3.92; N, 8.72; S, 4.19.
N and CC stretching of pyridine rings, 836s for PF6. Anal. calcd for [C32H25N5RuClSPF6]: C, 48.46; H, 3.81; N, 8.83; S, 4.04. Found: C, 48.62; H, 3.92; N, 8.72; S, 4.19.
[RuII(MeMPTP)(dmbpy)Cl]PF6 (2)
Complex 2 was prepared following a procedure similar to that used to prepare 1 except for the amount of reactants and solvents used as follows: [RuII(MeMPTP)(DMSO)Cl2] (250 mg, 0.413 mmol), dmbpy (76 mg, 0.413 mmol), triethylamine (0.5 mL), NH4PF6 (155 mg, 0.95 mmol) and CH3OH (50 mL). Yield: 133 mg (48%). Good quality crystals of 2 suitable for X-ray diffraction analysis were grown by slow evaporation of an ACN solution (5 mL) of 2 (2 mg) in a vial at room temperature. 1H NMR (DMSO-d6): δ = 9.91 (d, J = 5.5 Hz, 1H), 9.15 (s, 2H), 8.93 (d, J = 7.8 Hz, 2H), 8.79 (s, 1H), 8.51 (s, 1H), 8.31 (d, J = 8.7 Hz, 2H), 8.01 (t, J = 7.8 Hz, 2H), 7.92 (d, J = 5.4 Hz, 1H), 7.63 (d, J = 5.0 Hz, 2H), 7.55 (d, J = 8.7, 2H), 7.39 (t, J = 6.4 Hz, 2H), 7.20 (d, J = 5.9 Hz, 1H), 6.90 (d, J = 5.9 Hz, 1H), 2.74 (s, 3H), 2.62 (s, 3H), 2.31 (s, 3H) (Fig. S2†). 13C NMR (DMSO-d6): 20.9 and 20.4 (Py-CH3), 14.3 (–SCH3). MS: m/z = 676.0059, [C34H29ClN5RuS]+. IR (KBr, cm−1): 1691 m, 1611 m, 1476 m, 1405 m of pyridine rings (for CN and CC stretching), 2983w and 2881w for aliphatic CH3 group, and 841s for PF6−. Anal. calcd for [C34H29ClN5RuSPF6]: C, 49.73; H, 3.56; N, 8.53; S, 3.90. Found: C, 50.01; H, 3.71; N, 8.39; S, 3.81.
Synthesis of [RuII(MeMPTP)(dmdcbpy)Cl]PF6 (3)
Complex 3 was prepared following a procedure similar that used to prepare 1 except that dmdcbpy (44.92 mg, 0.165 mmol) was used in the place of bpy. Yield: 71.36 mg (58%). 1H NMR (DMSO-d6): δ = 10.33 (d, J = 6.1 Hz, 1H), 9.39 (s, 1H), 9.22 (s, 2H), 9.11 (s, 1H), 8.96 (d, J = 7.9 Hz, 2H), 8.48 (d, J = 6.1 Hz, 1H), 8.32 (d, J = 7.9 Hz, 2H), 8.05 (t, J = 7.3 Hz, 2H), 7.79 (d, J = 5.5 Hz, 1H), 7.62 (d, J = 5.5 Hz, 2H), 7.58 (d, J = 7.9 Hz, 2H), 7.46 (d, J = 5.5 Hz, 1H), 7.34 (t, J = 6.7 Hz, 2H), 4.10 (s, 3H), 3.85 (s, 3H), 2.63 (s, 3H) (Fig. S3†). 13C NMR (DMSO-d6): 164.8, 164.0 (–COO), 53.3, 53.1 (–OMe) and 14.3, 14.0 (–SCH3). MS: m/z = 764.0672 [C36H29ClN5O4RuS]+. IR (KBr, cm−1): 1583 m, 1532 m, 1470 m, 1429 m (for pyridine rings), 1725s (due to –COO), 838.75s. Anal. calcd for [C36H29ClN5O4RuSPF6]: C, 47.56; H, 3.21; N, 7.70; S, 3.53. Found: C, 47.71; H, 3.35; N, 7.83; S, 3.64.
Synthesis of [RuII(MeMPTP)(Pic)2Cl]PF6 (4)
Mixture of [Ru(MeMPTP)Cl3] (100 mg, 0.177 mmol), triethylamine (0.3 mL) and excess of 4-picoline (10 mL) were heated at 100 °C for 13 h. After cooling, hexane (10 mL) was added to the reaction mixture. The resultant precipitate was filtered and washed with hexane to remove unreacted 4-picoline. The residue was dissolved in water (5 mL) and aq. solution of NH4PF6 (100 mg in 3 mL) was added. The resulting solid was filtered, washed with water and dried under vacuum. Furthermore, the complex was purified by column chromatography over silica gel with DCM/acetone (1:1). Yield: 44.64 mg (37%). 1H NMR (DMSO-d6): δ = 9.17 (d, J = 5.5 Hz, 2H), 9.05 (s, 2H), 8.22 (d, J = 8.5 Hz, 2H), 7.50 (d, J = 8.5 Hz, 2H), 8.90 (d, J = 8.5, 2H), 7.86 (t, J = 6.7 Hz, 2H), 8.17 (t, J = 7.9 Hz, 2H), 7.77 (d, J = 6.1 Hz, 4H), 6.91 (d, J = 6.1 Hz, 4H), 2.59 (s, 3H), 2.08 (s, 6H) (Fig. S4†). 13C NMR (DMSO-d6): 20.0 (–PyMe), 14.3 (–SCH3). MS: m/z = 678.1032 [C34H31ClN5RuS]+. IR (KBr, cm−1): 1594 m, 1533 m, 1499 m, 1425 m for pyridine rings, 835.97s for PF6−. Anal. calcd for [C34H31ClN5RuSPF6]: C, 49.61; H, 3.80; N, 8.51; S, 3.90. Found: C, 50.03; H, 3.88; N, 8.68; S, 4.02.
X-ray structural determination
Single crystal X-ray structural data of the compounds (1 and 2) were collected on a Bruker D8 Venture PHOTON 100 CMOS diffractometer using an INCOATEC micro-focus source and graphite monochromated MoKα radiation (λ = 0.71073 Å). The program SAINT14 was employed for the integration of diffraction profiles, and absorption correction was performed with the SADABS program.15 The structures were initially solved by SIR 9216 and further refinement was done with a full matrix least square technique using SHELXL-201317 and WinGX system (Ver 2013.3).18 All of the non-hydrogen atoms were located from the difference Fourier map and refined anisotropically. All of the hydrogen atoms were fixed by HFIX at ideal positions and included in the refinement process using riding model with isotropic thermal parameters. All of the crystallographic and structure refinement data of complexes are summarized in Table 1. Selected bond lengths and angles of compounds 1–2 are summarized in Table S1.† The crystallographic information file is deposited with the CCDC numbers 1447199 and 1447200 for compounds 1 and 2, respectively.†
Table 1 Crystal data and structure refinement parameters for complexes 1 and 2
| R1 = ∑∥F0| − |Fc∥/∑|Fo|. wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. |
| Compound |
1 |
2 |
| Empirical formula |
C32H25ClF6N5PRuS |
C34H29ClF6N5PRuS |
| Formula wt |
793.12 |
821.17 |
| Crystal system |
Monoclinic |
Triclinic |
| Space group |
P21/c (no. 14) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2) (no. 2) |
| a (Å) |
13.0582(6) |
10.991(5) |
| b (Å) |
19.8028(9) |
13.475(5) |
| c (Å) |
11.9764(6) |
13.856(5) |
| α (deg) |
90 |
64.503(5) |
| β (deg) |
97.347(2) |
75.699(5) |
| γ (deg) |
90 |
66.929(5) |
| V (Å3) |
3071.5(3) |
1696.5(12) |
| Z |
4 |
2 |
| ρcalc. (g cm−3) |
1.715 |
1.608 |
| μ (mm−1) |
0.788 |
0.716 |
| F(000) |
1592 |
828 |
| Reflns collected |
54801 |
70617 |
| Indep. reflns |
7608 |
8436 |
| GOF |
1.01 |
1.05 |
| R1a/wR2b [I > 2σ(I)] |
0.0796 |
0.0395 |
| R1/wR2 (all data) |
0.2026 |
0.0989 |
Oxygen evolution measurements
The O2 evolution measurements were carried out in acidic aqueous solutions containing complex 1–4 (pro-catalyst) along with [[Ce(NO3)6](NH4)2] (550 mg, 1 mmol) and CF3SO3H (3 mL, pH = 1) in the ratio of 500:1 equiv. using Clark-type oxygen electrode (Hansatech instrument, Oxygraph Plus). Before each measurement, a fresh Teflon membrane was applied over the electrode tip, and the probe was calibrated in oxygen-free (N2 purge) followed by oxygen saturated deionized water. The CeIV solution was purged with N2 to provide an oxygen-free solution, and then the Ru(II) complex (0.1 mg) dissolved in acetonitrile (100 μL) was introduced by a syringe through a septum. The generated O2 (nmol ml−1) was measured and recorded against time by a Clark type electrode.19 Furthermore, to calculate the turnover number (TON = [maximum amount of O2 evolved]/[catalyst] in moles) for complexes 1–4, the water oxidation reactions were carried out in a 10 mL round bottom flask capped with a rubber septum under ambient conditions. (NH4)2[Ce(NO3)6] (CAN) (550 mg, 1 mmol) and CF3SO3H (3 mL, pH = 1) were stirred together and an ACN solution of the complex to be studied (100 μL, 6.3 × 10−4 M) was injected through the septum. The mixture was stirred for 24 h. The amount of O2 generated was measured via gas chromatography by injecting the head gas of reaction vessel using an air tight syringe. The GC system used was a PerkinElmer Clarus 580 GC with a thermal conductivity detector and a 5 Å molecular sieve column operating at 60 °C with helium as the carrier gas. The calibration was carried out with air as the standard (21% O2).
Results and discussion
The complexes 1–3 were synthesized by the reaction of [RuII(MeMPTP)(DMSO)Cl2] with bpy, dmbpy and dmcbpy ligands, respectively, whereas complex 4 was synthesized by the reaction of [Ru(MeMPTP)Cl3] complex with excess of 4-picoline (Scheme 1). All the complexes (1–4) were purified via column chromatography.
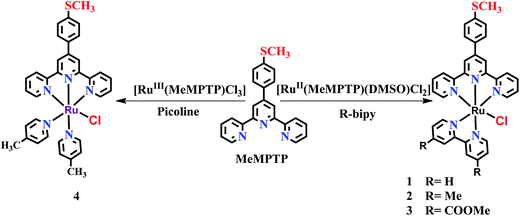 |
| | Scheme 1 Schematic pathways of the preparation of complexes 1–4. | |
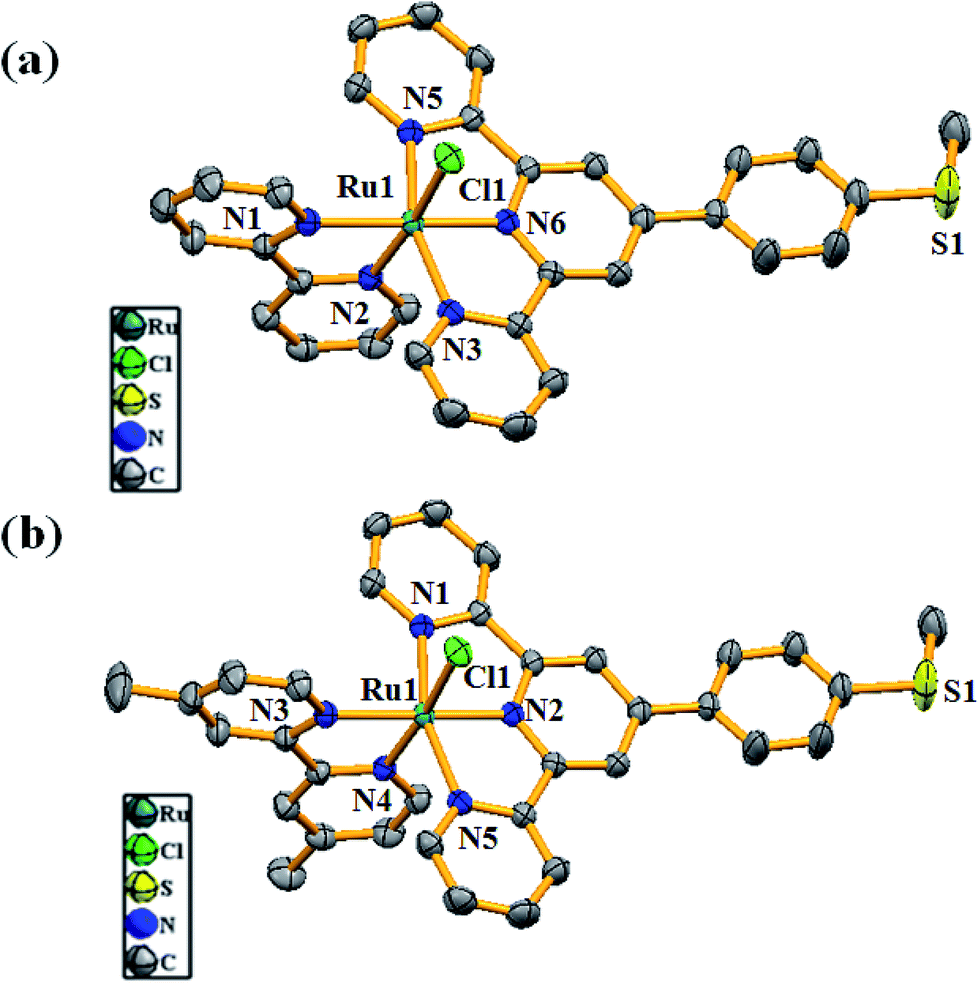
Structural characterization of complexes 1 and 2
Single crystals of the complexes (1 and 2) suitable for the X-ray diffraction study were grown by slow evaporation of the ACN solutions of the complexes. Good quality crystals were selected after careful examination with a polarized microscope and mounted on a glass fiber. Crystal structure determination reveals that both 1 and 2 are isostructural, with the Ru(II) center adopting octahedral geometry satisfied by MeMPTP and bpy/dmbpy in 1/2 along with a chloride ligand (Fig. 1a and b). The MeMPTP ligand acts as a chelating tridentate donor and coordinates to Ru(II) through two axial and one equatorial sites. The remaining equatorial sites are occupied by two N atoms from bpy/dmbpy and a chloride ligand. In the complexes, the Ru(II)–N MeMPTP bond distances fall in the range of 1.951(2)–2.070(2) Å and the Ru(II)–N1 bond distance involving central pyridine nitrogen of MeMPTP is relatively shorter than the terminal ones (Table S1†). The bond distances of Ru–N bipy vary from 2.039(2) to 2.084(2) Å. In both complexes, the bond distance of the Ru(II)–N4 trans to the chloride ligand is shorter than that of Ru(II)–N5 due to higher electro-negativity of the chloride ligand. The bond distances and angles of both complexes are found comparable with those from the similarly reported Ru-terpy based complexes (Table S1, ESI†).
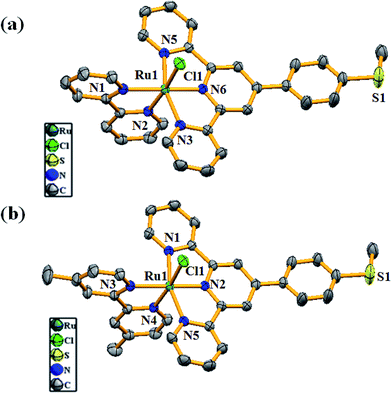 |
| | Fig. 1 ORTEP diagrams of (a) [RuII(MeMPTP)(bpy)Cl]+ (1) and (b) [RuII(MeMPTP)(dmbpy)Cl]+ (2) at 50% probability level. The hydrogen atoms, solvent molecules and counter anion are omitted for clarity. | |
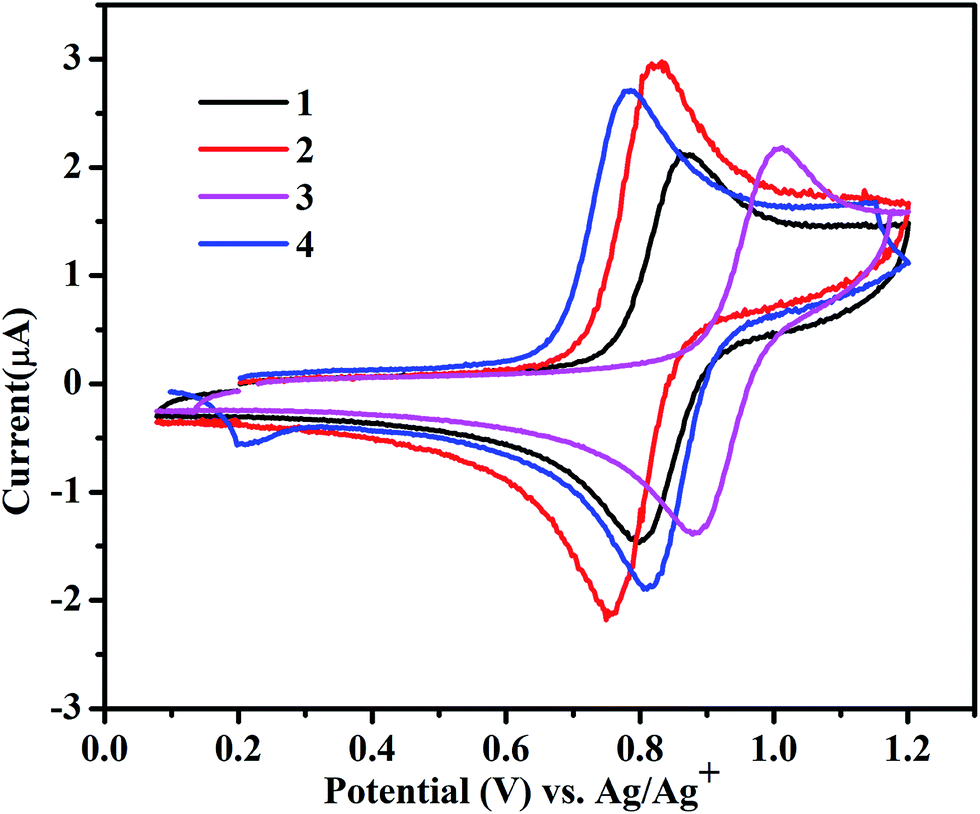
Red-ox properties of complexes 1–4
The redox behaviour of complexes 1–4 was investigated in ACN, and the results are shown in Fig. 2. The electronic environment of the Ru(II) center in these complexes varies due to the presence of electron-withdrawing and-donating functional groups and it is reflected in the RuII/RuIII redox potentials of the complexes, which are in the range of 0.77–1.01 VAg+/Ag. Among them, the un-substituted complex 1 shows RuII/RuIII couple at 0.83 VAg+/Ag, while for complex 3, with an electron-withdrawing substituent, the RuII/RuIII couple presents a higher oxidation potential at 0.94 VAg+/Ag. The high value of redox potential for 3 suggests that the electron-withdrawing COOMe group does not favor the generation of RuIII state. However, 2 and 4, with electron-donating methyl groups, show relatively lower red-ox potentials of 0.78 and 0.79 VAg+/Ag, respectively. The observed trend in the red-ox potentials is in accordance with what was described for the similar complexes reported before.10,20
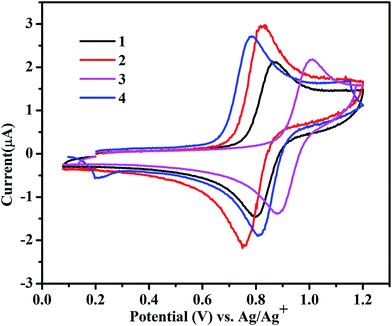 |
| | Fig. 2 Cyclic voltammograms for the RuII/RuIII couple in 4.0 × 10−4 mol L−1 ACN/TBAPF6 at 100 mV s−1. | |
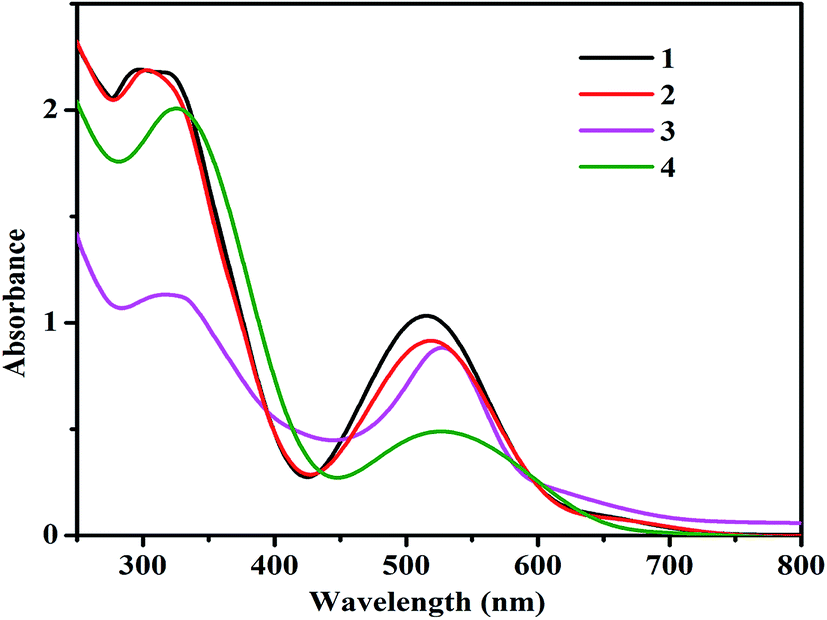
Electronic behavior of complexes 1–4
The UV-vis absorption spectra of complexes (1–4) recorded in ACN at room temperature are shown in Fig. 3. All complexes show broad absorptions with the maxima around 520 and 320 nm. The absorption maximum at 520 nm has been attributed to Ru(II) → terpy (MeMPTP) metal-to-ligand charge transfer (MLCT) transition, while the absorption band at 320 nm can be ascribed to π → π* transition. The small variance in the absorption maxima of the complexes could be arising due to the different substituent groups on the bipy in the complexes 2, 3 and the presence of pic ligands in 4.
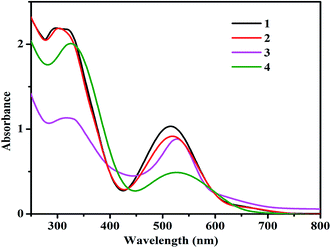 |
| | Fig. 3 UV-vis absorption spectra of the complexes (1–4) in 5 × 10−5 M ACN. | |
Catalytic activity of 1–4 for water oxidation
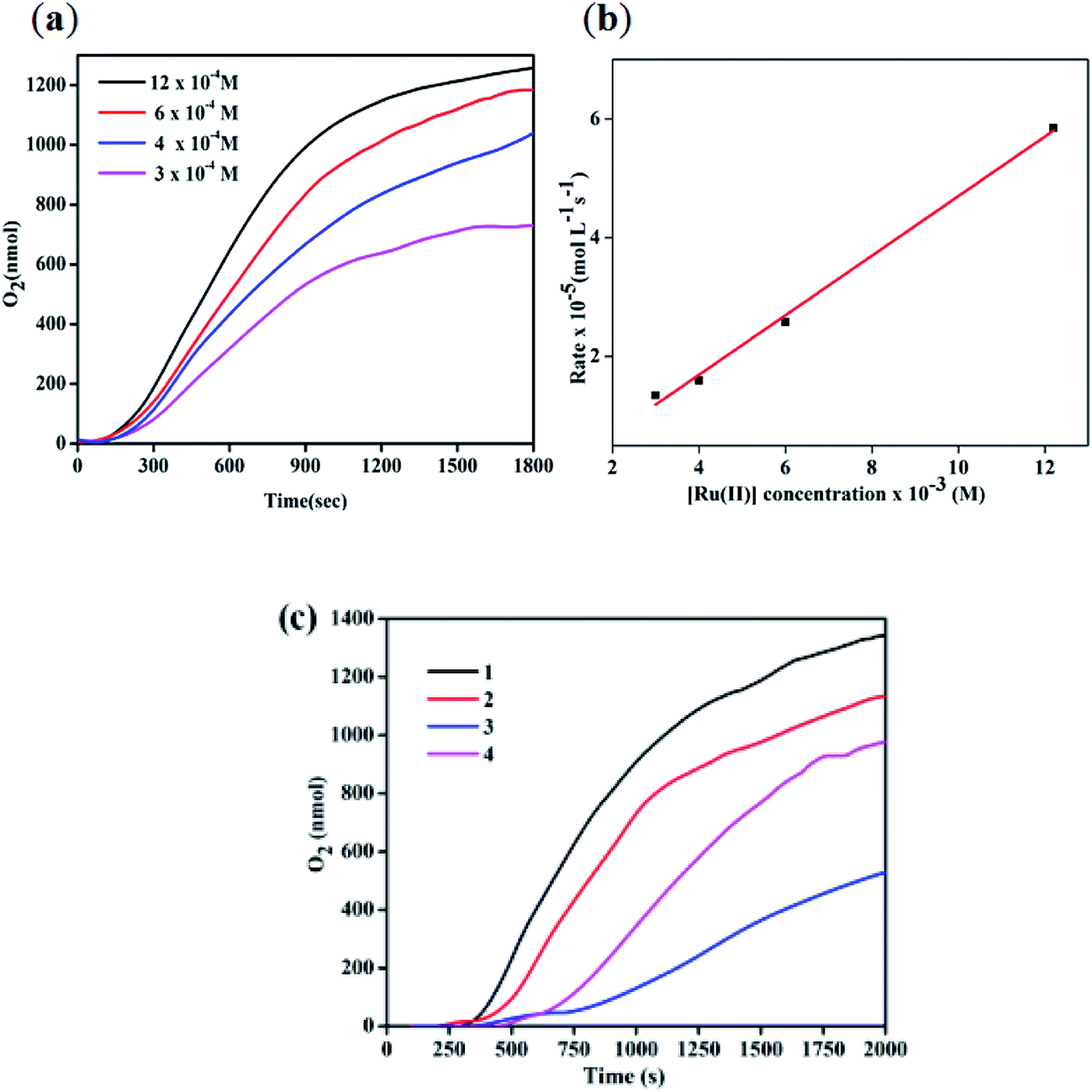
The reactivity of all four complexes for water oxidation catalysis was evaluated by measuring the rate of O2 generated in an acidic aqueous solution of (NH4)2[CeIV(NO3)6] (CAN) containing the complexes with the ratio of 500:1 equiv. using a Clark oxygen electrode of Oxygraph plus system. For the measurement, the aqueous solution of CAN was injected into the electrode chamber and degassed completely with N2 and then the complex was added to the solution. Interestingly, the O2 evolution rates measured at different concentrations of complex 2 reveal the first-order kinetic behavior and the rate of O2 generation depends on the concentration of the catalyst (Fig. 4a and b). Furthermore, all four complexes show an induction period required for the conversion of chlorido-complexes (pro-catalysts) into more active aqua-substituted counterparts. As shown in Fig. 4c, the complexes 1, 2, 3 and 4 show the induction periods of about 230, 104, 453, and 119s, respectively. Similar observations of longer induction periods for complexes containing electron-withdrawing groups and vice versa have been reported.7a,10 Furthermore, the rate of O2 generation catalyzed by the complexes was estimated by measuring the linear portion of the O2 evolution curves after the induction period (Fig. 4c) and the values are tabulated in Table 2. The highest rate was observed for complex 1, followed by 4 and 2 containing electron-donating (–Me) group, while complex 3, with an electron-withdrawing group on bipy, showed a lower rate. The observed rate of O2 generation follows similar trend observed for analogous complexes containing 1,10-phenanthroline-based auxiliary ligands reported by Verani and co-workers.10
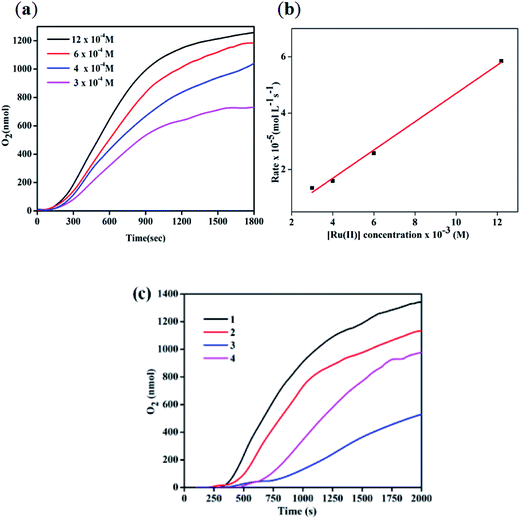 |
| | Fig. 4 (a) O2 generation as a function of time measured with the Clark electrode catalyzed by different concentrations of the complexes 2. (b) First-order plot for the rate of O2 generation of the complex 2. (c) O2 generation as a function of time measured with the Clark electrode for the complexes 1–4. | |
Table 2 Electronics, red-ox and water oxidation properties of complexes (1–4)
| Complex |
λmaxa (nm) ε, (M−1 cm−1) |
Eox1/2 (V/Ag/AgCl in ACN)b |
TONc |
Rated × 10−6 (mol L−1 s−1) |
| UV-vis spectra recorded in 5 × 10−5 M ACN. Cyclic voltammograms of (1–4) complexes recorded as 4.0 × 10−4 mol L−1 in ACN with 0.1 M TBAPF6 as the supporting electrolyte using a 100 mV s−1 scan rate at room temperature in an inert atmosphere; WE = glassy carbon, RE = Ag/AgCl, and CE = platinum foil. All potentials listed versus Ag/AgCl. (NH4)2[Ce(NO3)6] (550 mg), CF3SO3H (pH = 1), and the complex (6.3 × 10−4 M) for 24 h. CF3SO3H (pH = 1), (NH4)2[Ce(NO3)6] (550 mg) and the complex (0.1 mg in 100 μL in ACN). |
| 1 |
516, 308/20800 |
0.83 |
407 |
9.0 |
| 2 |
519, 305/18200 |
0.78 |
266 |
3.9 |
| 3 |
527, 323/17600 |
0.94 |
480 |
2.6 |
| 4 |
525, 324/10000 |
0.79 |
117 |
6.6 |
Furthermore, the TON of the complexes was measured in 10 mL septum-capped round-bottomed flask (please see Experimental part) and estimated from the amount of O2 generated after 24 h by GC. Interestingly, all four complexes show good catalytic activity with TON in the range of 100–500 (Table 2) and the trend follows a reverse relation with the rate of O2 evolution. This observation is in line with the reactivity trend reported for terpy/bipy-based mononuclear ruthenium complexes containing substituted bipy co-ligands as reported by Thummel and Berlinguette groups.7a,8
The induction period required for the generation of active catalyst has also been observed from time-dependent 1H NMR studies of complex 2 in ACN-d3/D2O mixed solvents. The 1H NMR stack plot (Fig. S5, ESI†) shows the gradual decrease in the intensity of the signal at 10.3 ppm corresponding to H1 of chlorido-complex and the appearance of a new peak at 9.8 ppm upon exchange of the chloride by an aqua ligand. As the reaction progresses, the intensity of the peak due to aqua-complex gradually increases and that of chlorido-complex decreases.
Reactivity studies: oxidative titration of complex 2 with CeIV
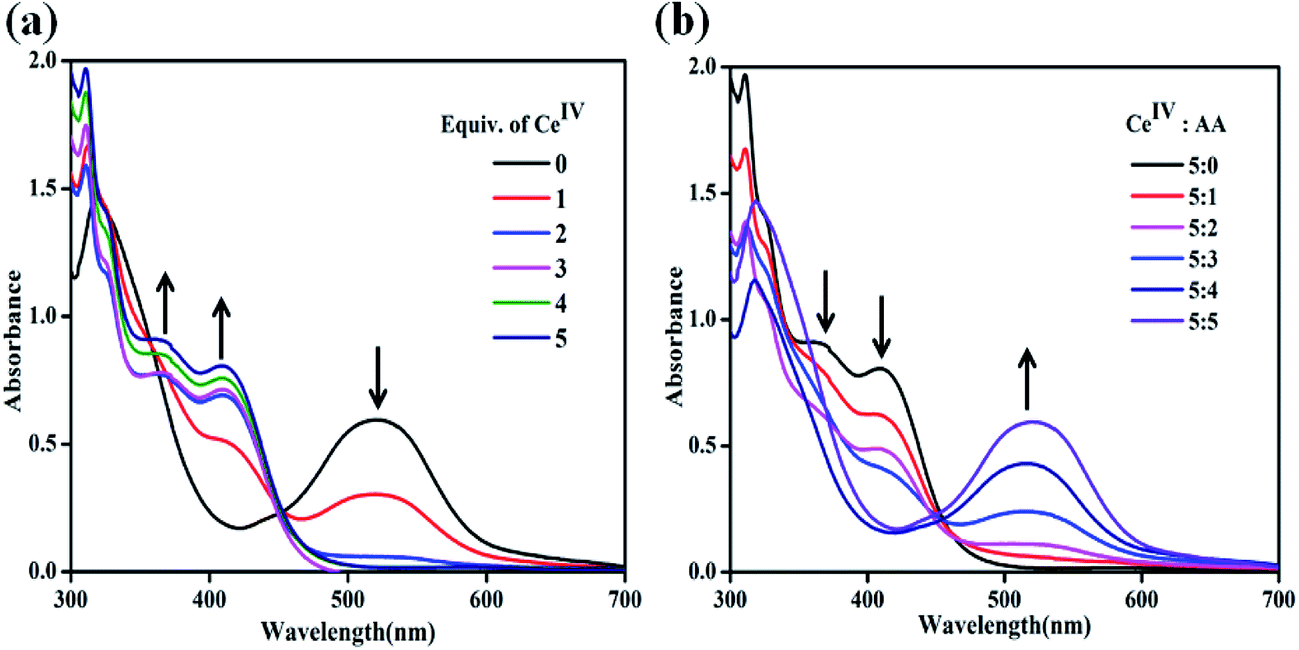
To understand the reactivity pattern of chlorido-containing pro-catalysts, UV-vis titrations have been carried out for complex 2 as a representative example against CeIV. As shown in Fig. 5a, the ACN solution of 2 shows an absorption band at 517 nm corresponding to RuII → terpy MLCT transition. The addition of 1 equiv. of CeIV results in a significant decrease in the intensity of this band and the evolution of a new band at 413 nm can be ascribed to in situ generated RuIII species (Fig. 5a). This observation indicates the partial oxidation of RuII to RuIII. A further increase in the addition of CeIV from 1 to 4 equiv. results in a continuous decrease in the intensity of RuII band at 517 nm and a rise in the intensity of the band due to RuIII species at 413 nm. Moreover, the appearance of an additional band at 364 nm, which can be attributed to in situ formations of [RuIV = O]2+ species, has been observed.10 Further addition of 5 equiv. of CeIV results in a complete disappearance of RuII band and an increase in the intensity of bands due to RuIII and [RuIV = O]2+ species. These spectroscopic changes clearly support the complete oxidation of RuII species by the addition of CeIV and the in situ formations of high valent [RuIV = O]2+ species.
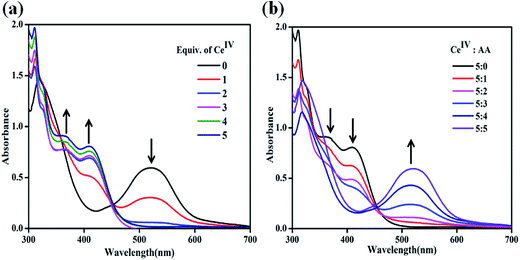 |
| | Fig. 5 (a) UV-vis spectral changes for 2 (5 × 10−5 M in ACN) upon addition of CeIV (1.25 × 10−3 M in aqueous triflic acid, 0–5 equiv., pH = 1). (b) UV-vis spectral changes for oxidized 2 upon the reductive titration with AA (1.25 × 10−3 M, 0–5 equiv.) in ACN. | |
Reductive titration of 2 with ascorbic acid (AA)
In order to further verify the oxidative conversion of RuII → RuIII by CeIV, reverse titration was performed. A solution of complex 2 containing 5 equiv. of CeIV was titrated against AA as a reducing agent. As shown in Fig. 5b, the addition of 1 equiv. of AA resulted in the reappearance of the characteristic RuII → terpy band at 517 nm and the intensity of RuIII and RuIV bands decreased, indicating the regeneration of RuII species. A subsequent addition of AA up to 5 equiv. led to the complete disappearance of the 413 nm band due to RuIII and the growth of the 517 nm band due to RuII, confirming the reductive conversion of RuIII → RuII species. In the complete redox cycle, no shift in the absorption maxima is observed, suggesting the regeneration of the original chlorido-complex, 2. This observation indicates that the redox process possibly occurs through the formation of a seven-coordinate intermediate.
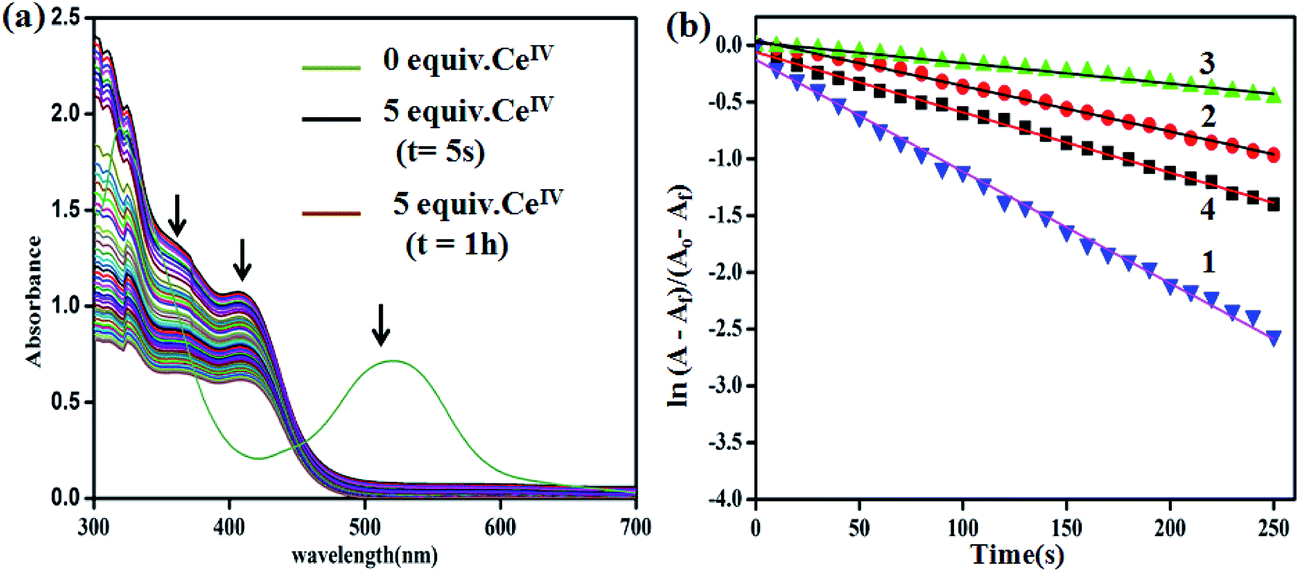
Time-dependent decay of high-valent ruthenium species
As mentioned before, the addition of 5 equiv. of CeIV to an ACN solution of 2 resulted in the appearance of two bands at 413 and 364 nm corresponding to in situ formations of RuIII and [RuIV = O]2+ species, respectively, and the complete disappearance of RuII → terpy MLCT band at 520 nm within 5 s as shown in Fig. 6a. The relative rate of decay of the 364 nm band assigned to high valent ruthenium-oxo species is calculated for the complexes 1–4 from time-dependent UV-vis spectra along with the first-order decay constants, as shown in Fig. 6b. Interestingly, the unsubstituted bipy complex 1 shows the highest rate of 9.0 × 10−3 s−1 followed by 4 (5.3 × 10−3 s−1), 2 (4.5 × 10−3 s−1) and 3 (1.8 × 10−3 s−1). This trend is in good agreement with the rate of O2 evolution discussed before. In addition, the calculated halftime lives of the complexes for the decay of Ru-oxo species are in accordance with the induction period observed for the rate of O2 evolution (Table S2†)10.
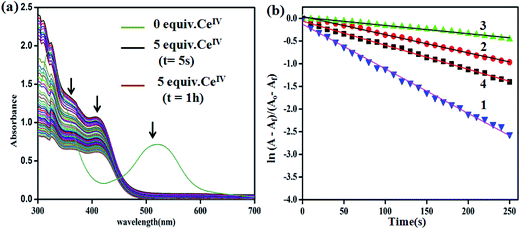 |
| | Fig. 6 (a) UV-vis spectral changes for 2 (Ru2+) over time upon addition of CeIV (1.25 × 10−3 M) in aqueous triflic acid, 5 equiv. (pH = 1). (b) First-order rate plots for 1–4 complexes. | |
Regeneration of the catalyst
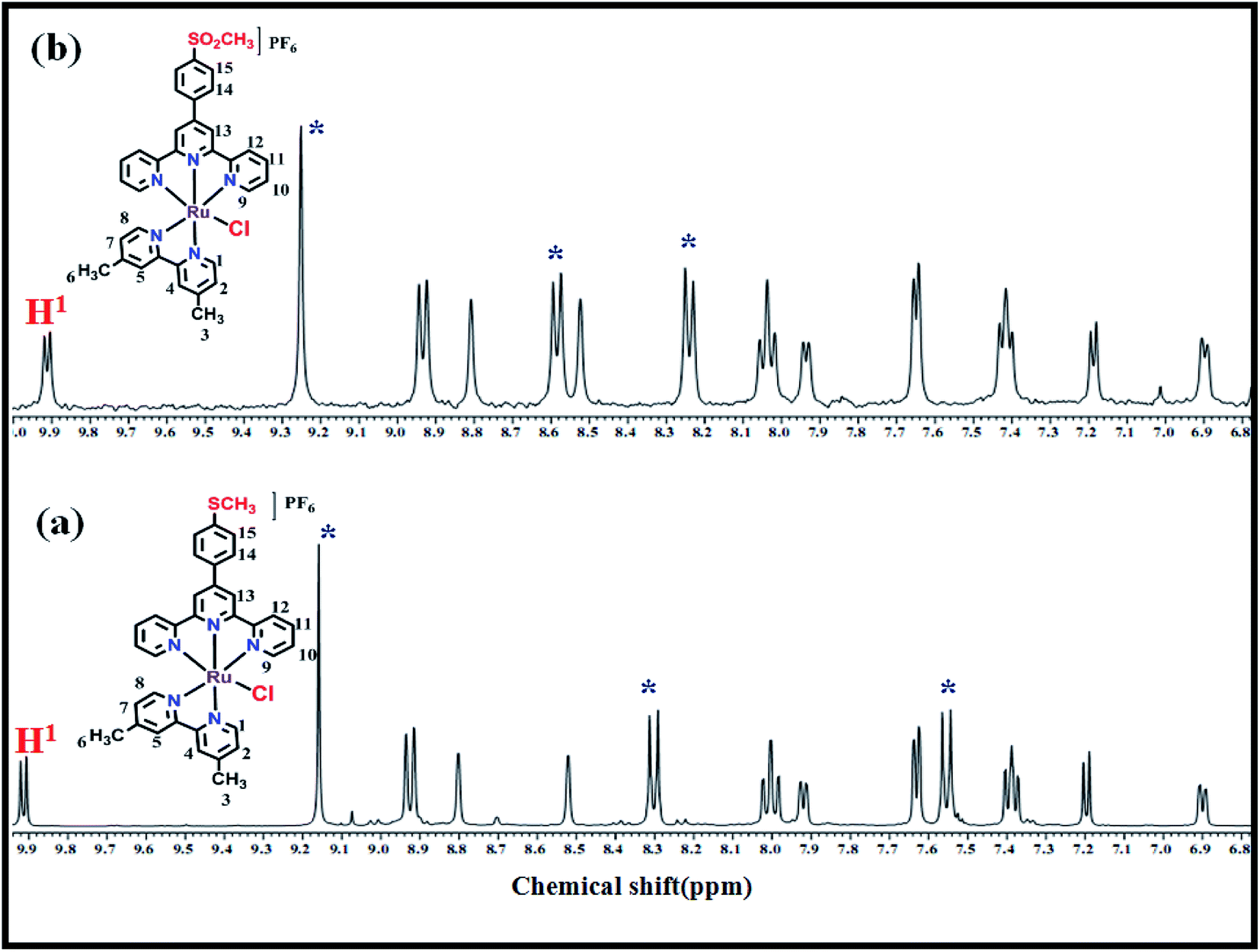
The results mentioned in the previous sections suggest that the catalytic core in the complexes is regenerated after the catalysis. For further confirmation, as a model example, we have recovered complex 2 after treating with an excess of CAN followed by addition of AA. The addition of excess CAN in aq. triflic acid solution (pH = 1) to the ACN solution of 2 resulted in the formation of a green precipitate, which was isolated by filtration. 1H NMR spectrum of the precipitate in DMSO-d6 shows the appearance of a broad peak indicating the formation of a paramagnetic high oxidation state ruthenium complex. Furthermore, the addition of AA to this green precipitate resulted in an instant conversion to a red solid that was isolated and analyzed by ESI-MS, IR and 1H NMR spectroscopic techniques. 1H NMR spectrum of the red solid obtained, shown in Fig. 7, revealed that the peak at 9.9 ppm assigned to H1 proton of dmbpy ligand did not show any shift in the regenerated complex suggesting the retention of the original coordination environment around the Ru(II). However, shifts in the peaks at 9.2, 8.3 and 7.56 ppm, corresponding to MeMPTP ligand, were observed. This indicates the possible structural change associated with MeMPTP ligand in the recovered complex. The ESI-MS analysis of the regenerated complex shows the molecular ion peak at m/z 708.0774 which is 32 mass units higher than that of complex 2 (Fig. S6†). This difference could be attributed to oxidation of the –SMe group of MeMPTP ligand to –SO2Me resulting in the shifting of the peaks in 1H NMR spectrum (Scheme S1†). Further evidence of the oxidation of the –SMe group of the ligand was obtained by treating MeMPTP ligand with CeIV. The resulting oxidized ligand shows IR peak at 1261 cm−1 corresponding to SO and ESI-MS peaks at 371.1092 and 387.1040 for SOCH3 and SO2CH3, respectively (Fig. S7†).
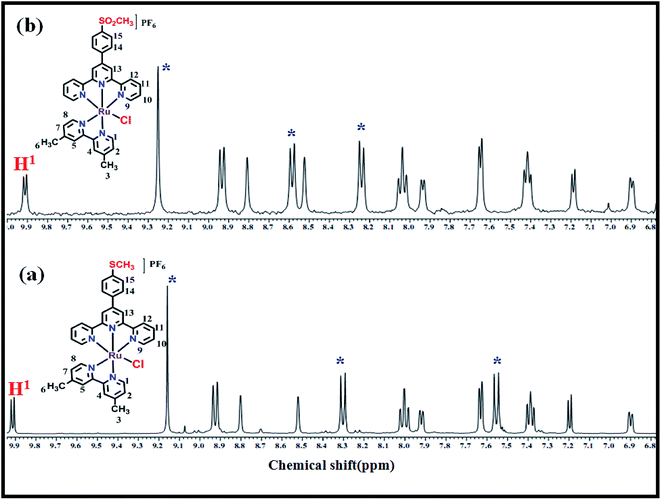 |
| | Fig. 7 1H NMR spectra of complex 2 (a) before and (b) after treating with CAN (excess) followed by reduction with AA. | |
Conclusions
In conclusion, herein we investigated the water oxidation activity of ruthenium complexes based on terpy derivative (MeMPTP) along with substituted bpy/py and picoline ligands. These systems bridge the gap between the well-known terpy/bipy and MeMPTP/phen systems studied before. Interestingly, the water oxidation measurements reveal that the catalytic rate for the O2 generation by the complexes in the presence of CeIV sacrificial oxidant follow the trend 1 > 4 > 2 > 3. Except the un-substituted complex 1, the complexes 2 and 4, with electron-donating groups on bipy/py, show higher catalytic rate than that of 3 containing electron-withdrawing substituent on bipy. This observation has been attributed to the fact that the electron-donating groups in 2 and 4 can enhance the electron density of the metal and facilitate the formation of the high-valent ruthenium species required for catalytic oxidation of water. However, the electron-withdrawing group (–COOMe) in 3 decreases the electron donation to ruthenium making it difficult to achieve the higher oxidation state species. Furthermore, all complexes (pro-catalysts) require an induction period for the catalysis and the rate of O2 generation follow the first-order relation. Importantly, the spectroscopic evidence for the in situ formations of high-valent ruthenium-oxo (RuIV = O) species, which are considered as important intermediate species in water oxidation catalysis, are observed. The TONs of the complexes follow the trend (3 > 1 > 2 > 4) and the values are comparable with those obtained for similar complexes reported before. The observed catalytic rate of O2 generation and TON of the complexes clearly demonstrate the influence of substituents on ancillary ligands on the water oxidation activity of the complexes. The MeMPTP ligand, with an electron donating –SMe group, supports the catalytic water oxidation activity of the complexes; however, in the presence of CeIV it undergoes oxidation to –SO2Me. More interesting aspects of this study are that it completes the gap between the well-known terpy/bpy systems studied by the groups of Thummel and Berlinguette and the MeMPTP/phen systems studied by Verani and brings out the differences in electronic influence of bipy and phen coligands and will be pivotal for the design of future water oxidation catalysts.
Acknowledgements
CMN gratefully acknowledges the financial support from the Council of Scientific and Industrial Research (CSIR), project ref no. 01/2691/12/EMR-II, Government of India. We would also like to thank Habib Baydoun, Ph.D scholar under supervision of Prof. Claudio N. Verani from Wayne State University, USA for the useful suggestions on calculation of TON.
References
-
(a) J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC;
(b) H. Dau and I. Zaharieva, Acc. Chem. Res., 2009, 42, 1861–1870 CrossRef CAS PubMed;
(c) M. D. Karkas, O. Verho, E. V. Johnston and B. Akermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed;
(d) G. Renger, Photosynth. Res., 2007, 92, 407–425 CrossRef CAS PubMed.
-
(a) R. Brimblecombe, G. C. Dismukes, G. F. Swiegers and L. Spiccia, Dalton Trans., 2009, 9374–9384 RSC;
(b) I. Romero, M. Rodriguez, C. Sens, J. Mola, M. R. Kollipara, L. Francàs, E. Marza, L. Escriche and A. Llobet, Inorg. Chem., 2008, 47, 1824–1834 CrossRef CAS PubMed;
(c) T. Wada, K. Tsuge and K. Tanaka, Inorg. Chem., 2001, 40, 329–337 CrossRef CAS PubMed.
-
(a) C. Sens, I. Romero, M. Rodríguez, A. Llobet, T. Parella and J. B. Buchholz, J. Am. Chem. Soc., 2004, 126, 7798–7799 CrossRef CAS PubMed;
(b) Y. Liu, S. M. Ng, S. M. Yiu, W. W. Y. Lam, X. G. Wei, K. C. Lau and T. C. Lau, Angew. Chem., Int. Ed., 2014, 53, 14468–14471 CrossRef CAS PubMed;
(c) T. A. Betley, Q. Wu, T. V. Voorhis and D. G. Nocera, Inorg. Chem., 2008, 47, 1849–1861 CrossRef CAS PubMed;
(d) Y. V. Geletii, Z. Huang, Y. Hou, D. G. Musaev, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2009, 131, 7522–7523 CrossRef CAS PubMed.
-
(a) R. Zong and R. Thummel, J. Am. Chem. Soc., 2005, 127, 12802–12803 CrossRef CAS PubMed;
(b) Z. P. Deng, H. W. Tseng, R. F. Zong, D. Wang and R. Thummel, Inorg. Chem., 2008, 47, 1835–1848 CrossRef CAS PubMed;
(c) T. J. Meyer, M. H. V. Huynh and H. H. Thorp, Angew. Chem., Int. Ed., 2007, 46, 5284–5304 CrossRef CAS PubMed;
(d) J. A. Gilbert, D. S. Eggleston, W. R. Murphy, D. A. Geselowitz, S. W. Gersten, D. J. Hodgson and T. J. Meyer, J. Am. Chem. Soc., 1985, 107, 3855–3864 CrossRef CAS.
-
(a) J. J. Concepcion, M. K. Tsai, J. T. Muckerman and T. J. Meyer, J. Am. Chem. Soc., 2010, 132, 1545–1557 CrossRef CAS PubMed;
(b) Z. Chen, J. J. Concepcion, H. Luo, J. F. Hull, A. Paul and T. J. Meyer, J. Am. Chem. Soc., 2010, 132, 17670–17673 CrossRef CAS PubMed;
(c) J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2008, 130, 16462–16463 CrossRef CAS PubMed.
-
(a) J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. M. Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954–1965 CrossRef CAS PubMed;
(b) S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS;
(c) J. W. Jurss, J. C. Concepcion, M. R. Norris, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2010, 49, 3980–3982 CrossRef CAS PubMed;
(d) L. L. Duan, A. Fischer, Y. H. Xu and L. C. Sun, J. Am. Chem. Soc., 2009, 131, 10397–10399 CrossRef CAS PubMed;
(e) S. Masaoka and K. Sakai, Chem. Lett., 2009, 38, 182–183 CrossRef CAS;
(f) L. Wang, L. Duan, Y. Wang, M. S. G. Ahlquistb and L. Sun, Chem. Commun., 2014, 50, 12947–12950 RSC;
(g) M. Yagi, S. Tajima, M. Komi and H. Yamazaki, Dalton Trans., 2011, 40, 3802–3804 RSC;
(h) M. Yagi and M. Kaneko, Chem. Rev., 2001, 101, 21–36 CrossRef CAS PubMed.
-
(a) H. W. Tseng, R. Zong, J. T. Muckerman and R. Thummel, Inorg. Chem., 2008, 47, 11763–11773 CrossRef CAS PubMed;
(b) N. Kaveevivitchai, R. Zong, H. W. Tseng, R. Chitta and R. P. Thummel, Inorg. Chem., 2012, 51, 2930–2939 CrossRef CAS PubMed;
(c) L. Tong and R. P. Thummel, Chem. Sci., 2016, 7, 6591–6603 RSC.
-
(a) D. J. Wasylenko, R. D. Palmer and C. P. Berlinguette, Chem. Commun., 2013, 49, 218–227 RSC;
(b) A. M. Asaduzzaman, D. Wasylenko, C. P. Berlinguette and G. Schreckenbach, J. Phys. Chem. C, 2015, 119, 242–250 CrossRef CAS.
-
(a) D. J. Wasylenko, C. Ganesamoorthy, B. D. Koivisto, M. A. Henderson and C. P. Berlinguette, Inorg. Chem., 2010, 49, 2202–2209 CrossRef CAS PubMed;
(b) D. J. Wasylenko, C. Ganesamoorthy, M. A. Henderson, B. D. Koivisto, H. D. Osthoff and C. P. Berlinguette, J. Am. Chem. Soc., 2010, 132, 16094–16106 CrossRef CAS PubMed.
- D. C. Wanniarachchi, M. J. Heeg and C. N. Verani, Inorg. Chem., 2014, 53, 3311–3319 CrossRef CAS PubMed.
- K. H. Chang, O. N. King, A. Tumber, E. C. Woon, T. D. Heightman, M. McDonough, C. J. Schofield and N. R. Rose, ChemMedChem, 2011, 6, 759–764 CrossRef CAS PubMed.
- N. Tuccitto, V. Torrisi, M. Cavazzini, T. Morotti, F. Puntoriero, S. Quici, S. Campagna and A. Licciardello, ChemPhysChem, 2007, 8, 227–230 CrossRef CAS PubMed.
- E. Dulière, M. Devillers and J. M. Brynaert, Organometallics, 2003, 22, 804–811 CrossRef.
- SMART(V 5.628), SAINT (V 6.45a), XPREP, SHELXTL, Bruker AXS Inc, Madison, Wisconsin, USA, 2004 Search PubMed.
- G. M. Sheldrick, Siemens Area Detector Absorption Correction Program, University of Gottingen, Gottingen, Germany, 2004 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick, SHELXL-2014 Program for Crystal Structure Solution and Refinement, University of Gottingen, Gottingen, Germany, 2014 Search PubMed.
- L. J. Farrugia, WinGX-A Windows Program for Crystal Structure Analysis, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- S. Kal, L. Mensah and P. H. Dinolfo, Inorg. Chim. Acta, 2014, 423, 201–206 CrossRef CAS.
- L. Tong, R. Zong, R. Zhou, N. Kaveevivitchai, G. Zhang and R. P. Thummel, Faraday Discuss., 2015, 185, 87–104 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1447199 and 1447200. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra07186h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*