
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dimension-dependent band alignment and excitonic effects in graphitic carbon nitride: a many-body perturbation and time-dependent density functional theory study†
W. F. Espinosa-García *ab,
J. M. Osorio-Guillénb and
C. Moyses Araujoc
*ab,
J. M. Osorio-Guillénb and
C. Moyses Araujoc
aUniversidad de San Buenaventura-Medellín, Facultad de Ingenierías, Grupo de Investigación en Modelamiento y Simulación Computacional, Carrera 56C No. 51-110, Medellín, Colombia. E-mail: william.espinosa@usbmed.edu.co
bInstituto de Física, Universidad de Antioquia UdeA, Calle 70 No. 52-21, Medellín, Colombia. E-mail: mario.osorio@udea.edu.co
cMaterials Theory Division, Department of Physics and Astronomy, Uppsala University, P.O Box 516, S75120, Uppsala, Sweden. E-mail: moyses.araujo@physics.uu.se
First published on 20th September 2017
Abstract
First-principles many-body theory and time-dependent density functional theory were used to study the dimension effects on the band alignment and optical properties of s-triazine and graphitic C3N4. The inclusion of quasiparticle corrections is very important to describe the quantum confinement and the enhancement of the electron–electron (e–e) interaction. The calculated quasiparticle gaps range from 4.0 eV (monolayer) to 3.21 eV (tetralayer). The position of the valence band maxima is found to be almost constant, whereas the conduction band minima show a strong quantum confinement effect with a variation of ∼0.7 eV respective to the bulk structure. The calculated frequency-dependent imaginary part of the dielectric function using the Bethe–Salpeter equation shows prominent excitonic effects, where there is a strong redistribution of the spectral weight to lower photon energies in the ultraviolet frequencies where the major part of the absorption occurs. On the other hand, a less intense excitonic absorption in the visible region is due to light polarization perpendicular to the surface plane. In contrast, time-dependent density functional theory also shows a redistribution of the spectral weight in the ultraviolet but it fails to describe the excitonic features in the visible region.
1 Introduction
The energy consumption projections1 around the world have motivated, in the most recent decades, widespread research to find materials and technologies with capabilities to use solar energy as a principal source of power, in order to generate energy that is environmentally friendly and at the same time is produced at a low-cost. Since the pioneering work of Fujishima and Honda,2 who demonstrated the capability of a TiO2 anode and a Pt cathode under ultraviolet irradiation with an external bias to oxidize water, many interesting approaches have been studied to synthesize liquid fuels from carbon dioxide (CO2). Nowadays some of those mechanisms have become promising alternatives to replace fossil fuels. In this context, many researchers around the world have devoted enormous efforts to find better candidates to be implemented as photocatalytic materials to reduce CO2 to liquid fuels and, consequently, alleviate the global warming impact by generating clean energy. In spite of the huge efforts, a good candidate with the desirable properties: efficient absorption of visible light, high chemical stability under typical operating conditions, band edge positions that straddle the water or CO2 redox potentials, and large hole diffusion length that is abundant in the earth’s crust3,4 has not been found yet.An active and stable photocatalyst is able to use light to drive reactions. The incoming photon energy can be used to stimulate certain desired reactions, otherwise, they are energetically unfavourable. Among the extensively studied photocatalytic materials, carbon nitrides (C3N4) have attracted attention due to their prominent properties such as high thermal stability, insolubility in most solvents, high surface area, easy processability, excellent stability, and low cost.5–8 Additionally, C3N4 has been successfully implemented in hydrogen production,9 photocatalytic water splitting,10 and oxidation reactions,11 among others. C3N4 possesses different allotropes,12,13 the graphitic structures being the most stable under ambient conditions.13,14 Among the graphitic structures, the 2D hexagonal pattern based structures (g-h-C3N4) are the most studied. G-h-triazine is made up from the polymerization and subsequent condensation of C3H3N3 rings (triazine), into a graphitic solid that consists of C3N3 rings connected by a N bridge, and g-h-heptazine is built up from tri-s-triazine (C6H3N6) ring units connected by planar amino groups. Different density functional theory studies have shown that the most stable coplanar structure at zero pressure is g-h-heptazine.8,14,15 Experimentally many different techniques can be used to synthesize the graphitic carbon nitrides, and some investigations have found many physical and chemical factors that affect the performance of the photocatalytic activity in those graphitic compounds.6,16–19 Up to today, a complete condensation of g-h-C3N4 with an ideal stoichiometric ratio of 0.75 has not been possible to obtain, and a small amount of hydrogen (≃2%) is always found.5,20
In recent years, density functional theory calculations have been employed to study mainly the electronic and optical properties of the pristine g-h-C3N4, with different exchange–correlation functionals, and including or excluding van der Waals dispersive forces.8,14,15,21 Theoretical studies have been focused on enhancing the electronic and photocatalytic properties of g-h-C3N4 by doping with different atoms.22–25 However, recombination effects could be unfavourable for the photocatalytic performance in this material. Other interesting studies have incorporated quasiparticle corrections (G0W0) to accurately calculate the band gap,8 and others to solve the more computationally expensive Bethe–Salpeter equation for monolayer structures of g-h-triazine and g-h-heptazine.26
A detailed knowledge of optical properties is always desirable to guide the photocatalytic applications. A comprehensive description of the optical properties is therefore of the utmost importance to clarify the role of the Coulomb interaction between electrons and holes, commonly called excitonic effects, which could modify the absorption properties. Additionally, the effect of quantum confinement on the optical properties of g-h-C3N4 is not totally understood, and it is well-known that the dimensionality strongly modifies the band alignment and the excitonic properties. In this paper, we applied state-of-the-art many-body Green’s function theory and Time-Dependent Density Functional Theory (TD-DFT) to investigate the quantum confinement and dimensionality effects on band gaps, band alignments and optical properties of g-h-triazine. We have chosen g-h-triazine due to its surface formation energy being higher in energy than that of g-h-heptazine, indicating that it would be easier to exfoliate g-h-heptazine than g-h-triazine. Additionally, its surfaces are almost coplanar making it easier to study the absorption process.15
2 Method of calculation
The slab models were constructed from the optimized bulk structure (for details see ref. 15) along the [001] crystallographic direction. We allowed all the ionic positions to relax without any symmetry restrictions to see any possible corrugation effects. The total energies, stress tensor and ionic forces were calculated via density functional theory using the projector augmented wave method,27,28 as has been implemented in the VASP code.28,29 The Román-Pérez and Soler30 algorithm and the van der Waals density functional (vdW-DF) implementation by Klimeš et al.31 (OptB88) were used to take into account dispersive forces. Hard PAW potentials with the valence atomic configuration 2s22p2 for C and 2s22p3 for N were used to relax the surfaces with a cut-off energy of 910 eV. All structural parameters, lattice constants, and atomic positions for each calculated system have been optimized by simultaneously minimizing all atomic forces and stress tensor components via the conjugate gradient method. Brillouin-zone integration was performed on a Monkhorst–Pack 6 × 6 × 1 k-mesh with a Gaussian broadening of 0.01 eV for full relaxation, and ionic forces were converged to 0.1 meV Å−1 and the slab cohesive energy was converged to 5 meV with respect to the vacuum thickness of the periodically separated repeated slabs and the number of layers. For the calculation of the total energies a Γ-centred 6 × 6 × 1 k-mesh was used in conjunction with the tetrahedron method including Blöchl corrections.32The quasiparticle energies were calculated by a nonself-consistent GW approximation (G0W0),33 where the Green function G0 and the screened Coulomb W0 are calculated using the OptB88 Kohn–Sham eigenvalues and eigenfunctions of the ground state structures. We kept the same input parameters as above, however, we used PAW potentials optimized specifically to yield accurate high-energy scattering properties up to 10 Ry above the vacuum level. This was achieved by placing one projector in the valence band and an additional projector typically 5–8 Ry above the vacuum level.
Then, we calculated the imaginary part of the dielectric function including local field effects by means of TDDFT with Bootstrap (BO) kernel34 and the Bethe–Salpeter equation (BSE) beyond the Tamm–Dankoff approximation using the full-potential augmented-plane wave method with local orbitals.35 The muffin-tin radii of C and N are set to RMT = 1.225 a.u. The parameter RMT|G + k|max setting the number of plane waves was converged to 7. The irreducible wedge of the Brillouin zone was sampled with a uniformly spaced k-point grid of 12 × 12 × 9 and 6 × 6 × 4 for the RBO and BSE calculations, respectively. We used 128 empty bands for RBO, whereas the BSE Hamiltonian was set with 10 valence and 16 conduction states. Finally, the energy dispersion relations were adjusted by a scissor correction of the conduction bands to follow the QP energies. For consistency, the scissor corrections were done using the G0W0 band gap at the Γ-point.
3 Results and discussion
3.1 Electronic properties
In previous work in ref. 15, we calculated the band structure and band alignments incorporating quasiparticle corrections of bulk g-h-triazine and g-h-heptazine. In that work, we showed the vital importance of including van der Waals dispersive forces, in order to give an appropriate physical description of the surface properties of this two-dimensional material. From the structurally optimized parameters used in ref. 15, we have built different surfaces varying the number of layers. Fig. 1 shows the calculated band gaps as a function of the number of layers using standard density functional theory (OptB88) and quasiparticle (QP) corrections on top of the OptB88 exchange–correlation functional (OptB88+G0W0). The calculated quasiparticle OptB88+G0W0 (OptB88) gaps range from 4.0 (1.56) eV for the monolayer to 3.21 (1.47) eV for the tetralayer. The large QP corrections (from 2.44 eV for the monolayer to 1.74 eV for the tetralayer) can be explained by the role of quantum confinement. This quantum effect plays an important role in those low-dimensional systems, and additionally the enhancement of the e–e interaction due to the less effective screening increases the QP corrections as the number of layers decrease.36 All the calculated band gaps decrease monotonically towards the expected bulk band gap, being almost linear in the case of OptB88. This behaviour in OptB88 indicates the poor physical contribution to the quantum and quasiparticle effects. The difference in band gaps for the monolayer and the bulk g-h-triazine with OptB88 is only 0.09 eV, indicating the importance of including the QP corrections to describe the quantum confinement and enhancement of the e–e interaction. The monolayer band gap agrees well with a previous result in ref. 26, where the used starting point for the G0W0 calculation was the LDA eigenvalues and eigenfunctions. Recently, it has become well-known that the starting point of G0W0 may affect the results,37 and so we attribute the discrepancy in the monolayer band gap to the different starting points in the G0W0 calculation.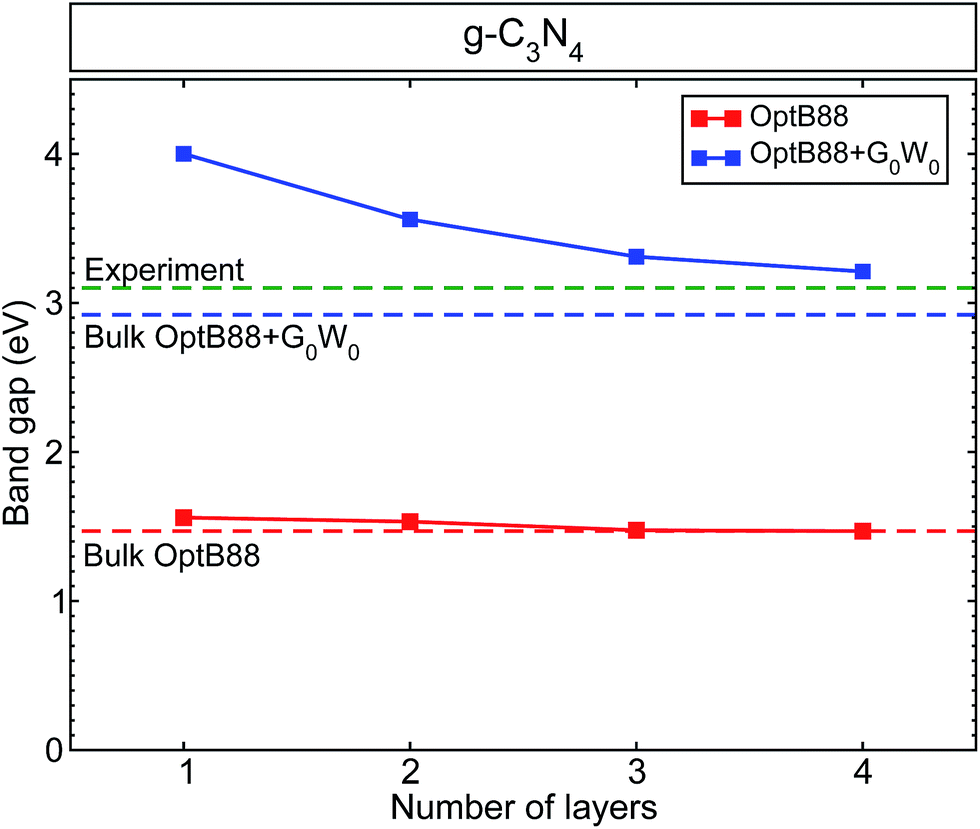 | ||
| Fig. 1 The calculated band gap for graphitic-like C3N4 as a function of the number of layers. We have included as a reference the calculated band gap for bulk g-h-triazine with OptB88 (red dashed line), OptB88+G0W0 (blue dashed line), and the experimental value40 (green dashed line). | ||
All the surfaces with both approximations (OptB88 and OptB88+G0W0) present a direct band gap at the Γ-point, where the top of the valence band and bottom of the conduction band are doubly degenerated. Similar to the bulk structure, the top of the valence band is mainly built up by N 3px,y-states with a small contribution from the C 2px,y-states and N 2s-states, whereas the bottom of the conduction band is built up by the hybridization of C 2pz-states and N 3pz-states (see Fig. S1, in the ESI†). An important factor in determining the photocatalytic activity of a material is the position of the band edges. To calculate accurately the band alignment in this system, we have used the slab model implemented in ref. 38. Thermodynamically, a redox reaction is favourable if the redox free energy lies within the band edges of the material. Fig. 2 shows the schematic representation of the calculated band edges as a function of the number of layers, with some selected important redox energies (at pH = 7) to produce fuels like methane, methanol, and formic acid using CO2 as a reactant. The QP corrections with respect to OptB88 in the valence band maxima are rather small, whereas the important corrections in the positions of the conduction band minima show a strong enhancement of the e–e interaction. The variation of the QP values in those positions from the monolayer to the bulk is ∼0.7 eV. This enhancement of the e–e interaction is due principally to the fact that most of the conduction band minima are built from pz orbitals, which extend into the vacuum region, and particularly in the monolayer the screening effects in those orbitals are rather small, indicating an intensification of the e–e interaction and causing the augmentation of the QP values. In contrast, in the in-plane bonds which are built mainly of px,y-states, the screening effects are determinant, and therefore the corrections in the QP values are small.
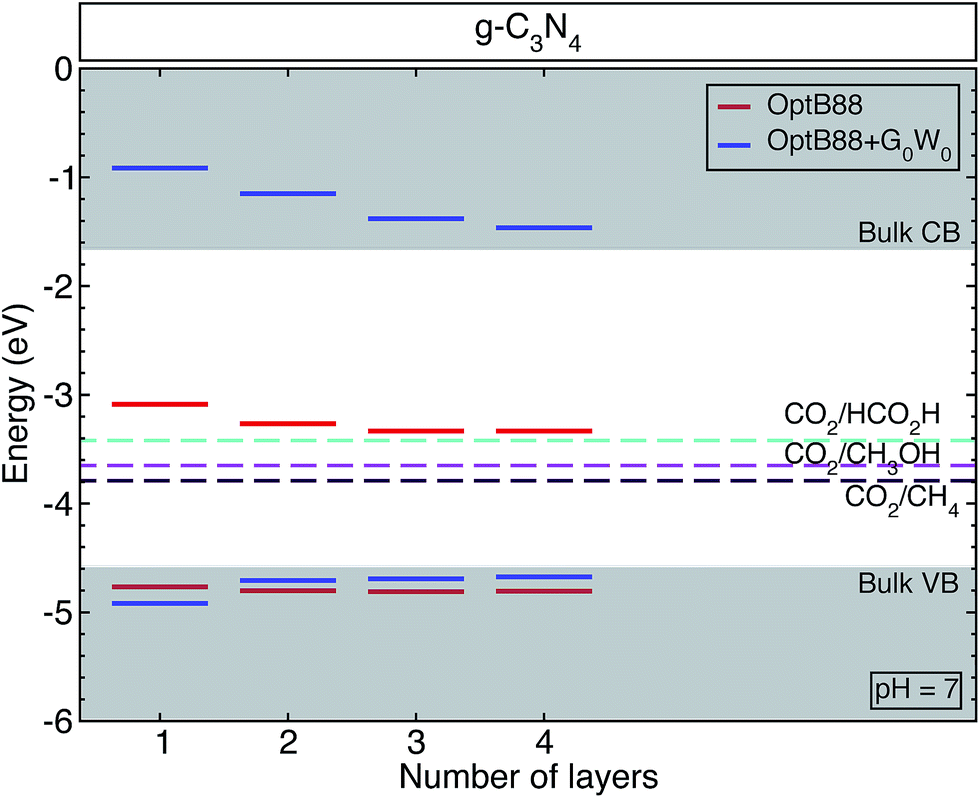 | ||
| Fig. 2 The calculated band edges for graphitic-like C3N4 as a function of the number of layers. The gray shaded areas show the valence and conduction bands for bulk g-h-triazine calculated with OptB88+G0W0. We have also added, for comparison, the measured reduction reactions producing methane CH4, methanol CH3OH, and formic acid HCO2H at pH = 7. | ||
For a semiconductor to be implemented as a photocatalyst, the VBM (CBM) has to be more negative (positive) than the reduction (oxidation) potential. This requirement ensures that the reaction is favourable without a bias voltage. The free energies of some possible pathways to reduce CO2 are located inside the band edges of the studied surfaces and bulk structure, indicating that g-h-triazine could be implemented as a photocatalytic material to reduce CO2. However, the QP corrections move towards too high positive values for the CBM in those structures, thus for a better efficiency g-h-triazine must be functionalized to obtain a better performance of this material.
3.2 Optical properties
In Fig. 3, we present the average of the imaginary part of the dielectric function for the building block of g-h-triazine, commonly known as s-triazine (C3H3N3) (left), and for the monolayer of g-h-triazine (right). The inclusion of the electron–hole (e–h) interaction generates important modifications in the dielectric function, where strongly bound exciton states appear below the onset of the IPA spectra. From the figure we can observe two contrasting effects, the influence of the quasiparticle corrections opening the band gap and the excitons causing a redshift in the absorption onset. In the left panel, the IPA onset starts around 6.6 eV with two other well-defined peaks around 8.8 eV and 9.4 eV. These main peaks describe the transition between the s-states to the p-states. The ALDA and BSE results are similar to the IPA, where the important differences are the spectral weight redistribution and the oscillator strength. The former has the same three peaks with a tiny redshift, with a considerable increment (decrease) in the first (other two) peak(s). In the latter the first (second) peak is moved towards higher (lower) energies (closer to the first), and the third peak presents a prominent shoulder around 8.2 eV. The right side of Fig. 3 shows the monolayer dielectric function of g-h-triazine. There is clear evidence of the fact that the e–h pair is poorly described by the ALDA kernel, which does not improve the results in the IPA. The main difference is in the spectral weight and in an energy enhancement with respect to the IPA peak. | ||
| Fig. 3 The calculated average of the imaginary part of the dielectric function for s-triazine and a monolayer of g-h-triazine by independent particle approximation (IPA), TD-ALDA,41 TD-BO34 and BSE. The grey shaded area highlights the energies where the monolayer shows a significant response to perpendicular light polarization and it does not present any response to parallel light polarization. A Lorentzian function with FWHM = 0.1 eV was used to broaden the calculated spectra. | ||
On the other hand, TD-BO and BSE present a strong exciton peak located in the ultraviolet region at 6.0 eV and 4.9 eV respectively. Both are redshifted with respect to the onset in the IPA (or ALDA) spectra, as one would expect. There are two other well-defined excitonic peaks located in the visible region at 2.75 eV and in the ultraviolet region at 3.6 eV. Those excitonic peaks are described only by the BSE and are absent from the TD-BO. The inset (see also the inset in the left panel) of this figure highlights the perpendicular light polarization of the monolayer, indicating a significant response to this polarization and that these peaks come from this component of the dielectric function. In this energy range, there is no response in the monolayer to the parallel light polarization. Those peaks are generated due to the strong excitonic effect and the less efficient screening (which commands the e–h interaction) in the monolayer of g-h-triazine. Additionally those peaks are not properly described by the other methods. Such excitonic peaks could make g-h-triazine surfaces exhibit some absorption of light in the visible region, and increase the photocatalytic activity of this material. Due to the failure of the TD-DFT exchange–correlation functionals used, we shall only use the BSE results for the remainder of the discussion.
A previous BSE study26 was performed for the monolayer and bulk g-h-triazine. It was concluded that the monolayer surface does not respond for the light polarization perpendicular to the surface, in contradiction with the BSE results obtained here. As it was shown above, the optical response of s-triazine reveals an excitonic resonance in the near ultraviolet region (see Fig. 3), and because of the dimensionality changes of the system it becomes more prominent and situated in the visible region for the g-h-triazine monolayer. Finally, this feature is revealed by perpendicular light polarization on the monolayer. Additionally, the calculation for the bulk was done with too large an interlayer distance (4.05 Å), in comparison to the ideal value for these graphitic compounds (3.27 Å).20,39 This huge interlayer distance modifies the electronic structure and, therefore, the optical properties of the bulk structure.
Fig. 4 shows the changes in the average of the imaginary part of the dielectric function for s-triazine and the g-h-triazine monolayer, bilayer, and bulk using BSE. In this figure, we can appreciate the changes in the excitonic peaks as we increase the dimensionality of the system. Increasing the number of layers generates a redshift in the excitonic peaks and an appreciable oscillator strength increment. The main absorption peaks in the middle of the ultraviolet region are located at 4.9 eV, 4.8 eV and 4.7 eV for the monolayer, bilayer and bulk, respectively. These peaks are generated by the response of the system to the parallel light polarization. In the inset, we observe an excitonic peak located at 3.2 eV in the near ultraviolet due to the s-triazine molecule. After increasing the dimensionality and dehydrogenating the surfaces, the excitonic peak is redshifted to the visible region relative to the s-triazine peak, and it is moved to 2.8 eV, 2.7 eV, and 2.5 eV for the monolayer, bilayer and bulk of g-h-triazine, respectively. These peaks are generated by the response of the surfaces to perpendicular light polarization. The role of the van der Waals interaction is apparent in this two-dimensional material, where this dispersive force does not generate an important screening effect, and then this weaker screening leads to an important excitonic effect in the bulk structure. The low dimensional structures are characterized by narrower peaks below the band gap, as in the bulk structure. The excitonic peaks in the visible spectrum are characterized by a small oscillator strength, and explain the poor absorption of g-h-C3N4 in the visible region. In order to quantify the absorption as the dimensionality changes in g-h-triazine, the average absorption coefficient for the monolayer, bilayer, and bulk is shown in Fig. 5 using BSE. It is noted that a redshift occurs in the absorption as the number of layers is increased. The excitonic absorption starts to be appreciable around 2.7 eV, 2.6 eV, and 2.2 eV for the monolayer, bilayer, and bulk g-h-triazine, respectively. Particularly in the case of the bulk structure, the absorption starts around 2.2 eV (α ∼ 0.8 × 105), and then for energies around 2.8 eV it becomes almost constant (α ∼ 0.5 × 105) until 4.5 eV, where it grows considerably to reach its maximum at 4.8 eV in the ultraviolet region. The quantum yield of g-h-C3N4 as a photocatalytic material in water splitting is reported to be around 0.1% at λ = 420–460 nm, even with the assistance of Pt,10 therefore those peaks located in the visible region are shown in Fig. 5 and their respective low absorption values explain the capability of g-h-C3N4, and particularly g-h-triazine, as a photocatalytic material to activate oxidative H2O/O2 and reductive H+/H2 reactions for water splitting under visible irradiation, and also demonstrate its low efficiency.
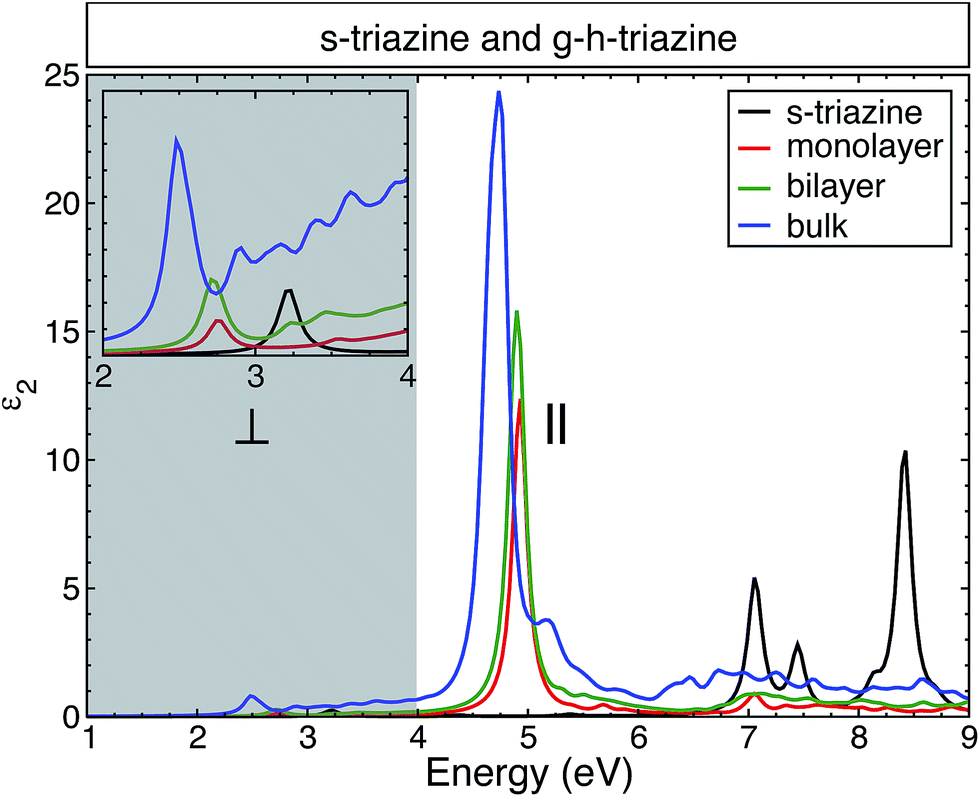 | ||
| Fig. 4 The calculated average of the imaginary part of the dielectric function for s-triazine and the g-h-triazine monolayer, bilayer and bulk using BSE. The grey shaded area highlights the energies where the extended structures show a significant response to perpendicular light polarization and they do not present any response to parallel light polarization. A Lorentzian function with FWHM = 0.1 eV was used to broaden the calculated spectra. | ||
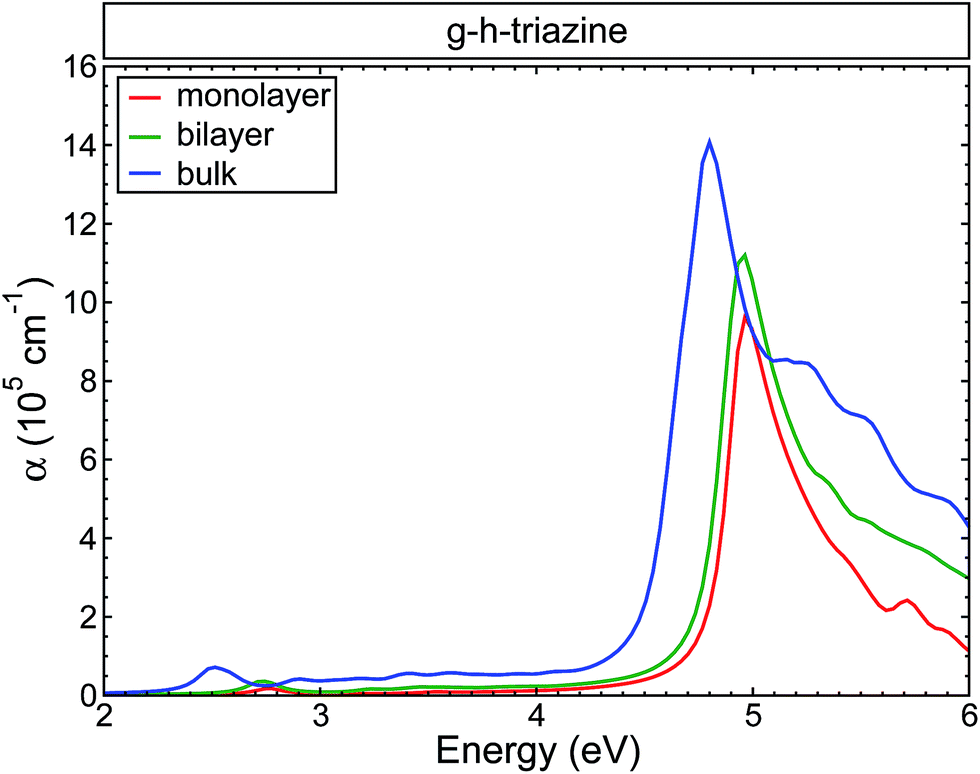 | ||
| Fig. 5 The calculated absorption coefficient for the g-h-triazine monolayer, bilayer and bulk using BSE. A Lorentzian function with FWHM = 0.1 eV was used to broaden the data. | ||
4 Conclusions
In summary, we have studied the dimension effects on the band alignments and optical properties of s-triazine and graphitic C3N4 based on DFT, TD-DFT, the GW approximation, and the Bethe–Salpeter equation also taking into account van der Waals dispersive forces. In order to properly describe the quantum confinement and the enhancement of the electron–electron interaction, and consequently the less effective screening, quasiparticle corrections have to be included in all the calculations of g-h-C3N4. It is precisely the less effective screening that is responsible for the strong quantum confinement effect in the conduction band minima with a variation of ∼0.7 eV from the monolayer to the bulk structure, whereas the valence band maxima are found to be almost constant. The optical properties show a strong redistribution of the spectral weight to lower photon energies, the presence of excitonic peaks in the visible region is described only with the BSE equation, and is totally absent with TD-ALDA, TD-BO. The excitonic absorption in the visible region is due to light polarization perpendicular to the surface plane. Quantum confinement (dimensionality) effects are not the best route to improve the performance of g-h-C3N4 as a photocatalytic material in the visible region.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Universidad de San Buenaventura-Medellín (project code M4829) and by the Vatenskapsrådet (VR), Swedish Energy Agency, and STandUP for energy. The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Center at Linköping University and the Instituto Tecnológico Metropolitano ITM. J. M. O. G. would like to thank CODI – Vicerrectoría de Investigación – Universidad de Antioquia (Estrategia de Sostenibilidad 2016–2017).References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- C. Bohn, A. Agrawal, E. Walter, M. Vaudin, A. Herzing, P. Haney, A. Talin and V. Szalai, J. Phys. Chem. C, 2012, 116, 15290–15296 CAS.
- Y. Ping, D. Rocca and G. Galli, Chem. Soc. Rev., 2013, 42, 2437 RSC.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- T. Sano, S. Tsutsui, K. Koike, T. Hirakawa, Y. Teramoto, N. Negishi and K. Takeuchi, J. Mater. Chem. A, 2013, 1, 6489–6496 CAS.
- Y. Xu and S.-P. Gao, Int. J. Hydrogen Energy, 2012, 37, 11072–11080 CrossRef CAS.
- Y. Wang, J. Yao, H. Li and D. Su, J. Am. Chem. Soc., 2011, 133, 2362–2365 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2008, 8, 76–80 CrossRef PubMed.
- C. E. Chan Thaw, A. Villa, G. M. Veith, K. Kailasam, L. A. Adamczyk, R. R. Unocic, L. Prati and A. Thomas, Chem.–Asian J., 2012, 7, 387–393 CrossRef CAS PubMed.
- A. Y. Liu and M. L. Cohen, Science, 1989, 245, 841–842 CAS.
- D. Teter and R. Hemley, Science, 1996, 271, 53–55 CAS.
- J. Gracia and P. Kroll, J. Mater. Chem., 2009, 19, 3013–3019 RSC.
- J. M. Osorio-Guillén, W. F. Espinosa-García and C. M. Araujo, J. Chem. Phys., 2015, 143, 094705 CrossRef PubMed.
- I. Papailias, T. Giannakopoulou, N. Todorova, D. Demotikali, T. Vaimakis and C. Trapalis, Appl. Surf. Sci., 2015, 358, 278–286 CrossRef CAS.
- J. Lin, Z. Pan and X. Wang, ACS Sustainable Chem. Eng., 2014, 2, 353–358 CrossRef CAS.
- B. Lotsch and W. Schnick, Chem.–Eur. J., 2007, 13, 4956–4968 CrossRef CAS PubMed.
- S. Lyth, Y. Nabae, S. Moriya, S. Kuroki, M.-A. Kakimoto, J.-I. Ozaki and S. Miyata, J. Phys. Chem. C, 2009, 113, 20148–20151 CAS.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- J. Cui, S. Liang, X. Wang and J.-M. Zhang, Phys. Chem. Chem. Phys., 2015, 17, 23613–23618 RSC.
- J. Liu, J. Alloys Compd., 2016, 672, 271–276 CrossRef CAS.
- Q. Guo, Y. Zhang, J. Qiu and G. Dong, J. Mater. Chem. C, 2016, 4, 6839–6847 RSC.
- K. Ding, L. Wen, M. Huang, Y. Zhang, Y. Lu and Z. Chen, Phys. Chem. Chem. Phys., 2016, 18, 19217–19226 RSC.
- X. Ma, Y. Lv, J. Xu, Y. Liu, R. Zhang and Y. Zhu, J. Phys. Chem. C, 2012, 116, 23485–23493 CAS.
- W. Wei and T. Jacob, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 085202 CrossRef.
- P. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Román-Pérez and J. Soler, Phys. Rev. Lett., 2009, 103, 096102 CrossRef PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef PubMed.
- P. Blöchl, O. Jepsen and O. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef.
- M. Shishkin and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 035101 CrossRef.
- S. Sharma, J. K. Dewhurst, A. Sanna and E. Gross, Phys. Rev. Lett., 2011 DOI:10.1103/PhysRevLett.107.186401.
- K. Dewhurst, S. Sharma, L. Nordström, F. Cricchio, F. Bultmark and H. Gross, http://elk.sourceforge.net.
- L. Yang, M. L. Cohen and S. G. Louie, Nano Lett., 2007, 7, 3112–3115 CrossRef CAS PubMed.
- X. Leng, F. Jin, M. Wei and Y. Ma, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 532–550 CrossRef CAS.
- H. Jiang, J. Phys. Chem. C, 2012, 116, 7664–7671 CAS.
- A. Schwarzer, T. Saplinova and E. Kroke, Coord. Chem. Rev., 2013, 257, 2032–2062 CrossRef CAS.
- V. Khabashesku, J. Zimmerman and J. Margrave, Chem. Mater., 2000, 12, 3264–3270 CrossRef CAS.
- Time-Dependent Density Functional Theory, ed. M. A. L. Marques, C. A. Ullrich, F. Nogueira, A. Rubio, K. Burke and E. K. U. Gross, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, vol. 706 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07134e |
| This journal is © The Royal Society of Chemistry 2017 |