
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible-light-induced degradation of polybrominated diphenyl ethers with AgI–TiO2†
Ying-Ying Shaoa,
Wei-Dong Yea,
Chun-Yan Sun *a,
Chu-Lin Liua and
Qi Wangb
*a,
Chu-Lin Liua and
Qi Wangb
aDepartment of Chemistry, Shaoxing University, Shaoxing 312000, Zhejiang, PR China
bSchool of Environment Sciences and Engineering, Zhejiang Gongshang University, Hangzhou, PR China
First published on 10th August 2017
Abstract
As typical persistent organic pollutants, polybrominated diphenyl ethers (PBDEs) have aroused high environmental concern because of their toxicity and recalcitrant degradation. Here we report the visible-light-induced degradation of PBDEs with AgI–TiO2 (>420 nm). A series of nano-structured AgI–TiO2 particles were synthesized by a deposition–precipitation method and characterized by SEM, TEM, XRD, XPS, BET and UV-Vis-DRS. The AgI–TiO2 hybrids showed high efficiency for degradation of PBDEs and excellent photostability in cyclic run experiments. The absorbance intensity and absorption ability of AgI–TiO2 hybrids and the properties of the solvents were found to greatly influence the efficiency of degradation of PBDEs. The 0.2-AgI–TiO2 (the mass ratio of Ag to P25 was about 20%) in CH3OH solution presented the highest reaction rate with 0.29 ± 0.02 h−1. Most interestingly, mechanistic pathways in the degradation of PBDEs were different under UV and visible light. Unlike the common single-electron transfer in the UV-TiO2 system, the debromination mechanism in the visible light/AgI–TiO2 system showed multi-electron transfer. The analysis of products showed that many different lower brominated congeners simultaneously appeared under visible light rather than being stepwise formed under UV. This implies that the special structure of AgI–TiO2 performs remarkably enhanced electron transfer, multi-electron transfer, and results in high photocatalytic activity. This research may provide a green and efficient method to remove halogenated pollutants by using visible light.
1. Introduction
Persistent organic pollutants (POPs) have aroused significant concern because they show persistent and bioaccumulative effects in the environment.1,2 Polybrominated diphenyl ethers (PBDEs) have attracted particular attention in this field due to their extensive use as flame retardants and their global distribution.3 During the past few years, researches on PBDEs have greatly spread and mighty endeavors have been devoted to the transformation of PBDEs in the environment.3–5 However, compared with extensive studies on the environmental behavior of PBDEs, there has been a total neglect of exploring a feasible way to eliminate PBDEs contamination. Only a few studies have been made on degradation of PBDEs.6–10 PBDEs are strongly resistant to oxidative degradation. The reported treatments of PBDEs mainly focused on reductive degradation. For example, zerovalent iron reduction was an effective method for debromination of PBDEs. Li and Keum studied the reductive debromination of PBDEs by zerovalent iron powder.6 Jiang et al. reported that resin-bound zerovalent iron nanoparticles exhibited much higher activity.7 Hydrothermal treatment was also introduced as a potential method for the removal of PBDEs.11 Recently, we found that PBDEs could undergo efficient debromination under visible light irradiation in the presence of various carboxylate anions.12 Nevertheless, the efficiency of degradation still needs to be improved.Photocatalysis has been proven to be a more effective way to degrade PBEDs.8,13 In our previous studies,8 PBDEs underwent rapid debromination by TiO2 under UV irradiation, but UV accounts for only 5% of the solar spectrum. Therefore, in order to effectively utilize solar energy, it is important to realize the photocatalytic degradation of PBDEs under visible light, which is 50% of the solar spectrum. Silver halides (AgX, X: Br, I) are famous photosensitive materials activated by visible light, and have been widely used as source materials in photochemical reactions.14 But one obstacle to their application is instability under irradiation.15 When AgI particles are loaded onto the surface of TiO2, the photoelectrons from AgI can be transferred to the conduction band of TiO2 which inhibits the formation of AgI. Thus AgI–TiO2 hybrids perform with high catalytic activity and stability under visible light irradiation for the degradation of organic pollutants.15–24 For example, Hu et al. found that AgI–TiO2 appeared to enhance the efficiency of the degradation of dyes under visible light irradiation.16 Nanostructured17,19,21 and three dimensional22 AgI–TiO2 also showed improved photocatalytic activity. Compared with extensive researches on the photocatalytic oxidation ability of AgI–TiO2, the photocatalytic reduction performance of AgI–TiO2,23 especially on the degradation of halogen organic pollutants, is quite rare, which may reveal another important application for AgI–TiO2. Considering that PBDEs can undergo reduction with TiO2 by UV light irradiation, we suppose that electrons generated from AgI–TiO2 under visible light might produce an effective reduction of PBDEs under appropriate conditions, which makes the visible-light-driven photocatalytic degradation of PBDEs possible.
In this work, a series of AgI–TiO2 photocatalysts were synthesized by deposition–precipitation method. BDE209 (Fig. S1†), the major product of PBDEs,1 was selected as a target of PBDEs. All prepared AgI–TiO2 hybrids exhibited high photocatalytic reduction ability for degradation of BDE209 under visible light irradiation. The superior stability of AgI–TiO2 was observed in cyclic run experiments. Most interestingly, mechanistic pathways of degradation of PBDEs in a visible light/AgI–TiO2 system showed multi-electron transfer, which was totally different from the common single-electron transfer in the UV/TiO2 system. To the best of our knowledge, this is the first report on the photocatalytic degradation of PBDEs under visible light irradiation.
2. Materials and methods
2.1 Materials
BDE209 was obtained from Aldrich Chemical Company (USA). P25 (49 m2 g−1) was supplied by Degussa. Al2O3 and SiO2 were purchased from Aladdin Chemistry Co., Ltd. (Shanghai, China). AgNO3 was analytical grade and obtained from Beijing Chemical Reagent Company. All other chemicals were of analytical grade, purchased from Shanghai Chemical Reagent Company and used without further purification. Double distilled water was used throughout the experiments.2.2 Synthesis and characterization of photocatalysts
AgI–TiO2 hybrids were prepared as follows:16 the suspension was prepared by adding 1 g of P25 in 100 mL of H2O. After ultrasonic treatment of the suspension for 10 min, a certain amount of KI was then dispersed into the suspension, followed by another 30 min of ultrasonic treatment. Then, a certain amount of AgNO3 in NH4OH (25 wt%) solution was added to the suspension and continued under stirring for 5 h. The yellow powder (AgI–TiO2) was filtered, washed with H2O, and dried in a vacuum oven at 55 °C. The initial mass ratio of Ag to P25 was about 10%. Thus the as-prepared AgI–TiO2 was denoted as 0.1-AgI–TiO2. A series of X-Ag–TiO2 (X: 0, 0.05, 0.1, 0.15, 0.2, and 0.3) were prepared by adjusting the initial mass ratio of AgNO3 to P25. AgI–SiO2 and AgI–Al2O3 hybrids were synthesized by a process similar to AgI–TiO2, except that TiO2 was replaced by SiO2 or Al2O3. In all cases, the molar ratio of initial AgNO3 to KI was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
The morphologies of AgI–TiO2 were obtained by SEM (Hitachi S4300) and TEM (Philips CM200 FEG TEM at 200 kV). The XRD patterns of the catalysts were measured on a Rigaku D/Max-2500 diffractometer with Cu Kα radiation (1.5406 Å). The XPS spectra of AgI–TiO2 were performed by using an ESCA lab 220i-XL spectrometer with an Al Kα (1486.6 eV) X-ray source and a charge neutralizer. N2 adsorption–desorption isotherms of AgI–TiO2 were obtained by a Quantachrome Autosorb-IQ instrument at 77 K. The specific surface areas of the AgI–TiO2 samples were obtained by the BET method.
2.3 BDE209 dehalogenation experiments
A stock solution of BDE209 (1 × 10−3 mol L−1) in THF was diluted to 1 × 10−5 mol L−1 with solvents. 10 mg of AgI–TiO2 was added to 9.95 mL of BDE209 solution in a Pyrex vessel and 0.05 mL of isopropanol was added as the hole trapping agent. The total volume of the solution was 10 mL. The reaction solution continued under magnetic stirring during the irradiation experiment. The Pyrex vessel was purged with N2 for 15 min to remove O2 and protected under N2 during the irradiation. The suspensions were stirred for 30 min in the dark before irradiation to reach adsorption equilibrium. A PLS-SXE300 Xe lamp (Beijing Trusttech Co. Ltd.) equipped with a cutoff filter to cut the irradiation below 420 nm was used as the light source (Fig. S2†). At given time intervals, 1 mL aliquots were used for analysis.BDE209 was quantified with a SHIMADZU HPLC system (LC-20AT pump and UV/VIS SPD-20A detector) with a DIKMA Platisil ODS C-18 column (250 × 4.6 mm, 5 μm film thickness). The mobile phase was 2% water in acetonitrile at 1 mL min−1 and the detector wavelength was 240 nm. The quantification of BDE209 was obtained from a calibration cure with a BDE-209 standard. Product analysis was performed by GC-μECD (Agilent 7890A, Agilent Technologies Co. USA), with a programmable pressure on-column injection port and a DB-5 capillary column (30 m × 50 μm, i.d. × 0.1 μm film thickness). Splitless 10 μL injection was performed manually at 300 °C. The carrier gas was helium with a constant flow rate of 1.0 mL min−1. The oven temperature was kept at 100 °C for 2 min, increased to 15 °C min−1 up to 230 °C, then increased to 5 °C min−1 of 10 min. The standard samples of BDE203, BDE204 and PBDEs (EO5113) were used to identify the degradation products. At given times, 1 mL aliquots were collected and centrifuged, and filtered to remove the solid catalyst particles before analysis with HPLC and GC-μECD.
In all cases, the degradation of BDE209 was assumed to follow pseudo-first order kinetics, and the rate constant (k) was calculated with the following equation:
| ln(C0/C) = kt | (1) |
3. Results and discussion
3.1 Characterization of photocatalysts
As can be seen from Fig. 1 and S3,† the morphology and microstructure of AgI–TiO2 were obtained by SEM and TEM. The SEM image showed that the shapes of AgI–TiO2 particles were irregular spheres. The particle sizes were 20–40 nm (Fig. 1a). The TEM image displayed the inner structure of the catalyst. AgI displayed nanoparticles with sizes of 10–30 nm dispersed on the surface of TiO2 (Fig. 1b). There was a firm connection between TiO2 and AgI, which helped the transfer of photo-excited electrons between AgI and TiO2. | ||
| Fig. 1 Images of 0.2-AgI–TiO2: (a) SEM, (b) TEM. | ||
The XRD patterns of the TiO2 and X-AgI–TiO2 (X: 0.05, 0.1, 0.15, 0.2, and 0.3) are shown in Fig. 2. Without modification of AgI, TiO2 showed the common anatase and rutile phases of P25. With the addition of AgI, the diffraction peaks attributed to AgI were observed and gradually became stronger. Peaks at 2θ values of 22.38°, 23.70°, 25.30°, 39.15°, 42.70° and 46.31° were indexed as the (100), (002), (101), (110), (103) and (112) planes of β-AgI (JCPDS 09-0374).19 In addition, it could be deduced that all the peaks of γ-AgI overlapped with the peaks of (002), (110), (112) of β-AgI. According to reported papers, hexagonal β-AgI and cubic γ-AgI always coexist at ambient temperature.15 No diffraction peaks assigned to Ag0 were observed in any of the samples. Therefore this indicated that the as-prepared AgI–TiO2 hybrids were a mixture of β-AgI and γ-AgI dispersed on the surface of TiO2 particles.
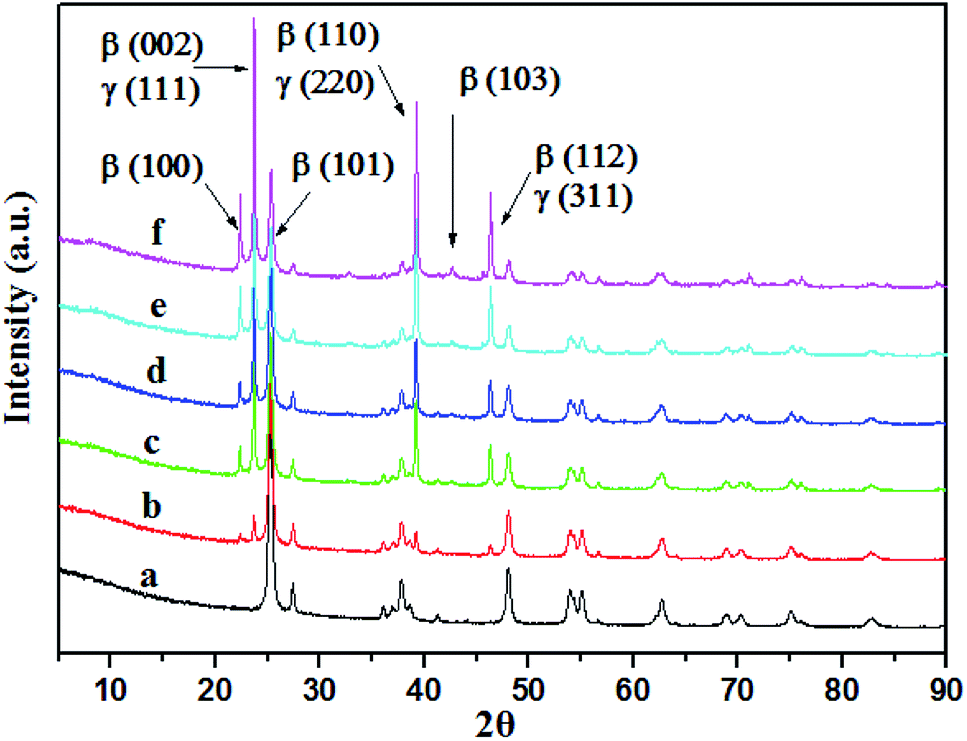 | ||
| Fig. 2 XRD patterns of TiO2 and AgI–TiO2. (a) TiO2, (b) 0.05-AgI–TiO2, (c) 0.1-AgI–TiO2, (d) 0.15-AgI–TiO2, (e) 0.2-AgI–TiO2, (f) 0.3-AgI–TiO2. | ||
XPS measurements were performed to determine the valence state of Ag and I in 0.2-AgI–TiO2. As shown in Fig. 3, the binding energy of Ag3d5/2 was 368.5 eV, which showed that there was Ag+ in the 0.2-AgI–TiO2 and no Ag0 was formed. Different from previous reports, the value of Ag3d5/2 in AgI–TiO2 (368.5 eV) was larger than that reported in AgI/TiO2 (367.5 eV),16 but close to the value in nanostructured AgI–TiO2 (368.8 eV),18 because the binding energies of Ag3d5/2 are commonly changed according to the size of the Ag nanostructure.18 The XPS peak of I3d5/2 appeared at 619.7 eV, which was assigned to I−. The peak corresponding to I2 (620.1 eV) was not observed.18
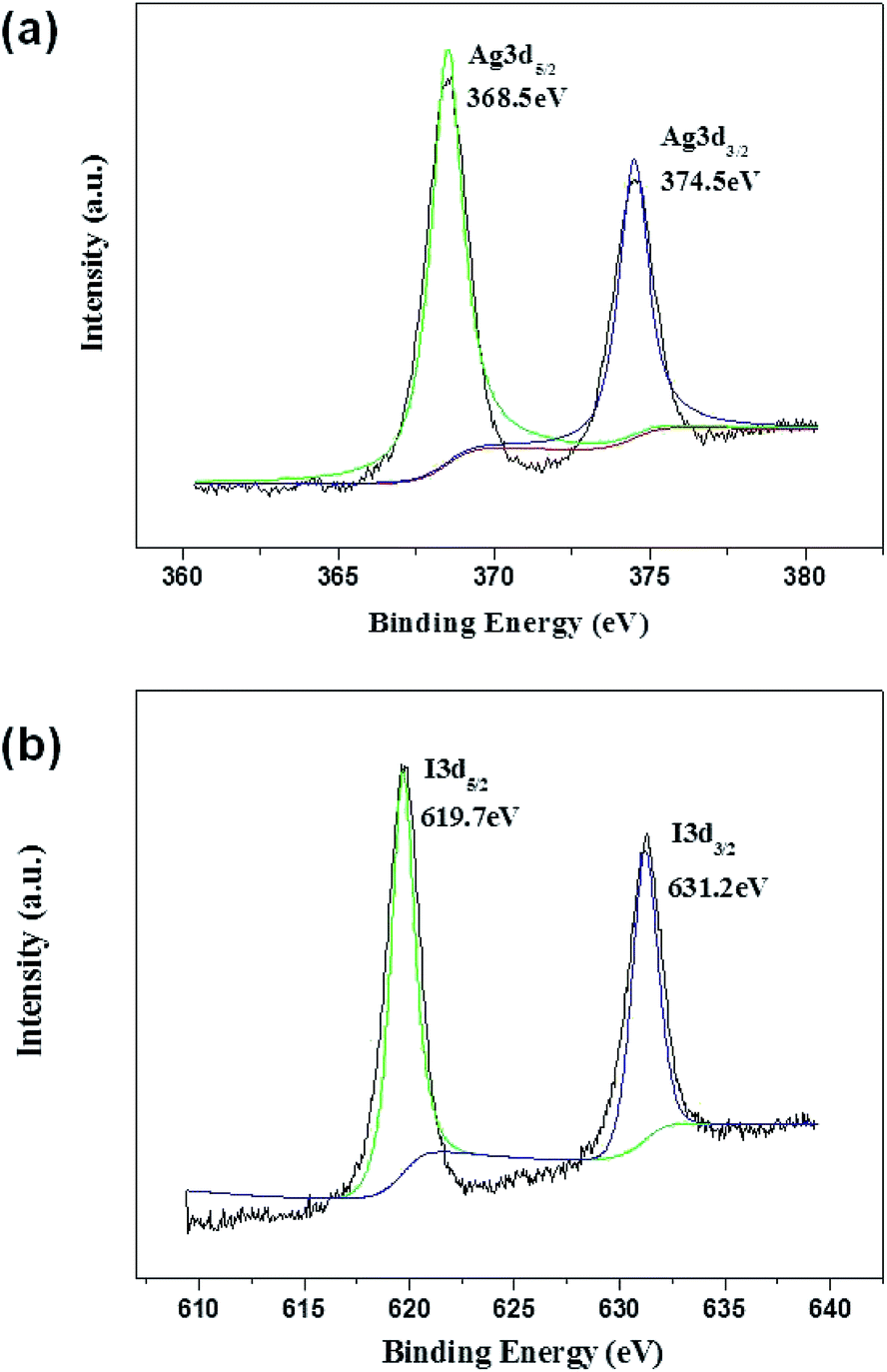 | ||
| Fig. 3 XPS spectra of Ag3d (a) and I3d (b) for 0.2-AgI–TiO2. | ||
The optical properties of the AgI–TiO2 samples were investigated by UV-Vis-DRS. As shown in Fig. 4, without modified AgI particles, TiO2 showed absorption only in the UV region, and the maximum absorbance was at 382 nm. When AgI was combined with TiO2, all the AgI–TiO2 samples with different AgI loading amounts exhibited new absorption in the visible light region (λ > 400 nm). Moreover, the enhanced absorption was observed with an increasing amount of AgI content and the absorption edges were red shifted to the maximum extent at 450 nm, with the maximum absorbance at 425 nm. Therefore, the absorption of AgI–TiO2 in the visible light region was attributed to the β-AgI and γ-AgI in the hybrids, which have a strong and wide absorption band in the visible light region.
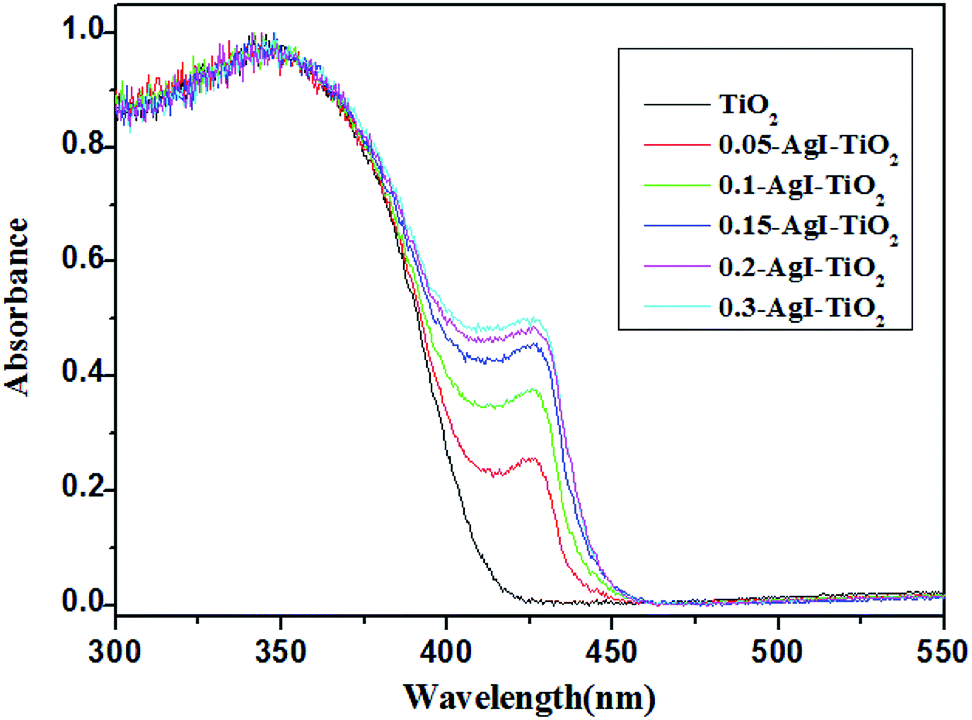 | ||
| Fig. 4 UV-Vis-DRS of TiO2 and AgI–TiO2. | ||
3.2 Degradation of BDE209 with AgI–TiO2 under visible light irradiation
As can be seen from Fig. 5, the direct photolysis of BDE209 did not occur under visible light irradiation. The degradation of BDE209 was scarcely observed in the presence of TiO2 under visible light irradiation. BDE209 also exhibited little degradation when the reactions were carried out with AgI–TiO2 under dark conditions. However, rapid degradation of BDE209 occurred in the presence of 0.2-AgI–TiO2 under visible light irradiation, and about 70% of the BDE209 disappeared after 3 h of irradiation. The kinetics was fitted by a pseudo-first-order process, giving a rate constant of 0.29 ± 0.02 h−1. This indicated that the BDE209 could undergo efficient photocatalytic degradation by AgI–TiO2 under visible irradiation.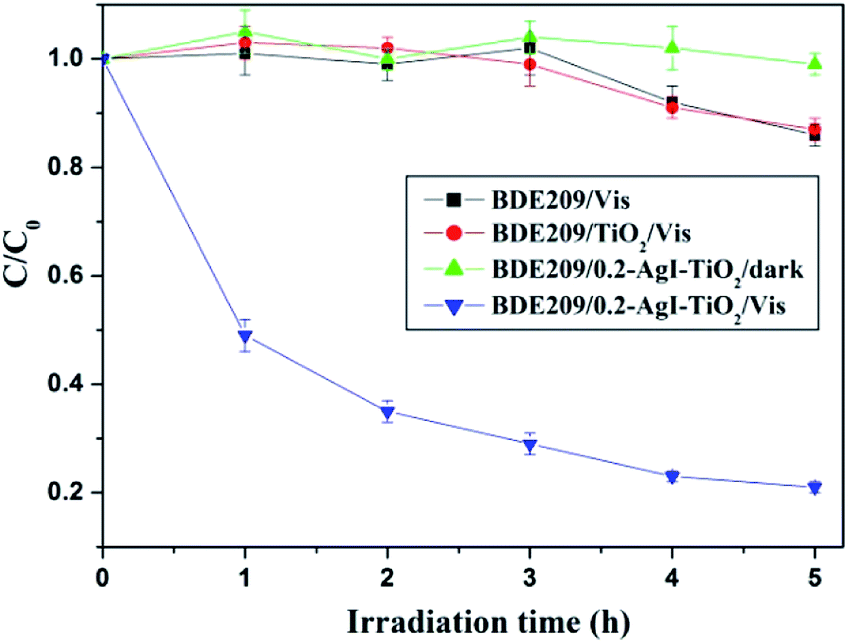 | ||
| Fig. 5 Temporal curves of the photodegradation of BDE209 under different conditions. BDE209: 1.0 × 10−5 mol L−1, catalyst: 1 mg mL−1, solvent: CH3OH, wavelength > 420 nm, anoxic condition. | ||
3.3 Effects of various parameters
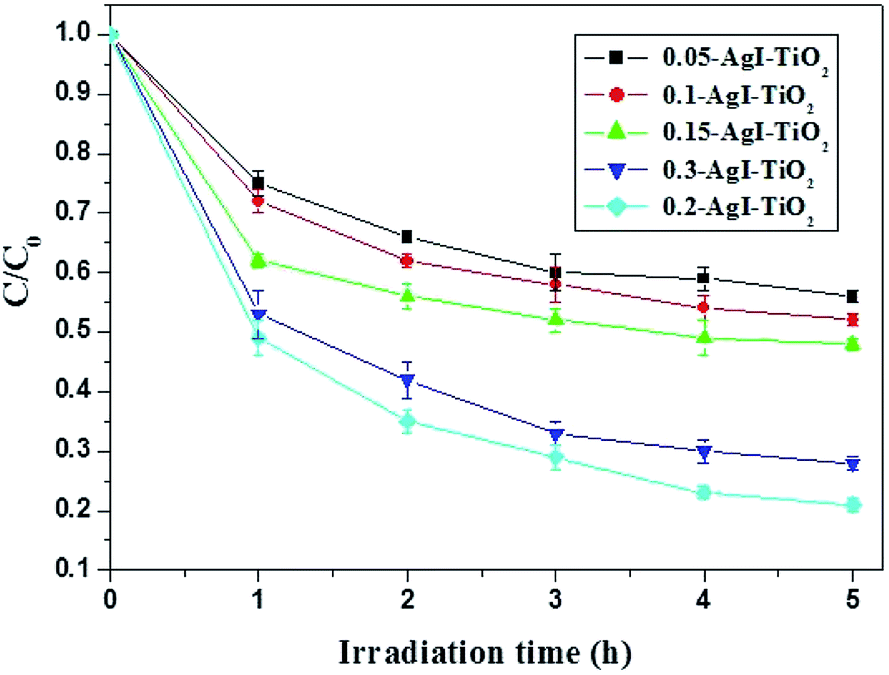 | ||
| Fig. 6 Temporal curves of the photodegradation of BDE209 under different mass ratios of AgI/TiO2 under visible irradiation. BDE209: 1.0 × 10−5 mol L−1, AgI–TiO2: 1 mg mL−1, solvent: CH3OH, wavelength > 420 nm, anoxic condition. | ||
In order to reveal the nature of the effects of the photocatalyst, the surfaces of the AgI–TiO2 samples were obtained with N2 adsorption and desorption isotherms and estimated by the BET method. As presented in Table 1, after addition of AgI, the specific surface areas of the catalysts showed a slight decrease. But the loaded amount of AgI had little effect on the specific surface areas of the AgI–TiO2 samples, which changed from 37.96 to 40.16 (m2 g−1). As a heterogeneous catalytic reaction, the photocatalytic reaction occurs primarily on the surface of the catalyst, so the substrate's adsorption ability may have a large effect on the reaction rate. Meanwhile, in the photocatalytic reaction, the absorbance intensity of the catalyst under irradiation is another key factor for the degradation rate. As can be seen from the UV-Vis-DRS (Fig. 4), the absorption intensity of 0.2-AgI–TiO2 was similar to that of 0.3-AgI–TiO2. But the amount of BDE209 absorbed on the surface of 0.2-AgI–TiO2 was more than that of 0.3-AgI–TiO2. Therefore, among all the hybrids, the degradation of BDE209 with 0.2-AgI–TiO2 was fastest, at nearly 3 times that of 0.05-AgI–TiO2.
| Catalyst | AgI/TiO2 mass ratio | SBET (m2 g−1) | Absorption amount (%) | Kinetic constant k (h−1) |
|---|---|---|---|---|
| TiO2 | 42.2 | 31.0 | ||
| 0.05-AgI–TiO2 | 0.05 | 38.4 | 28.8 | 0.106 ± 0.01 |
| 0.1-AgI–TiO2 | 0.10 | 39.2 | 29.0 | 0.120 ± 0.02 |
| 0.15-AgI–TiO2 | 0.15 | 39.9 | 29.8 | 0.127 ± 0.03 |
| 0.2-AgI–TiO2 | 0.20 | 40.2 | 30.2 | 0.293 ± 0.02 |
| 0.3-AgI–TiO2 | 0.30 | 38.1 | 28.4 | 0.238 ± 0.02 |
Although AgI can be easily photo-decomposed into Ag0, AgI–TiO2 displayed high photostability in the run experiments. As shown in Fig. 7, after four cycles, AgI–TiO2 still showed high photoreduction ability for the degradation of BDE209. The reaction rates were not obviously decreased in the run experiments. The photo-generated electrons from AgI could be transformed to the conduction bands of TiO2 not Ag+, so the stability of AgI was greatly enhanced in the AgI–TiO2 hybrids.16
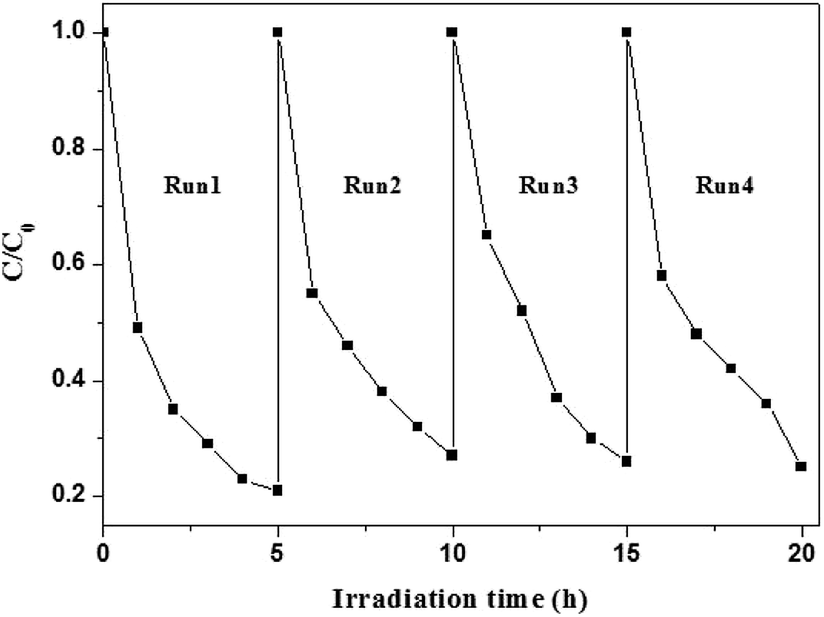 | ||
| Fig. 7 Cycling runs in the degradation of BDE209 with AgI–TiO2 under visible irradiation. BDE209: 1.0 × 10−5 mol L−1, 0.2-AgI–TiO2: 1 mg mL−1, solvent: CH3OH, wavelength > 420 nm, anoxic condition. | ||
3.4 New degradation pattern under visible light irradiation
In general, the degradation of PBDEs is caused by the loss of bromine atoms by reduction. It is known that the bioaccumulation and toxicity of PBDEs depend on the number of bromine atoms and their substituted positions. So it is very important to reveal the debromination pattern by product analysis. The less brominated products are more resistant to reduction than the more brominated congeners, so BDE209 commonly prefers to undergo debromination in a stepwise way. For example, in our previous study of the TiO2/UV system,8 the degradation of BDE209 showed a stepwise manner via single-electron transfer by TiO2. At the beginning of the irradiation, only 9Br-BDEs were observed and the amount of 9Br-BDEs increased. Then 9Br-BDEs disappeared and 8Br-BDEs were formed. In this stepwise method of degrading, the lower bromine products formed only after the higher bromine parent congeners had disappeared. However, it is most interesting that the debromination pattern in the degradation of PBDEs in visible light is totally different to that in UV.The degradation products were identified and quantified by HPLC and GC-μECD with the relative retention times of chemical standards and the orders of GC elution obtained from the literature (8, 12 and 13) (Fig. 8 and S5†). After visible light irradiation, BDE209 was reduced to its lower bromine congeners by AgI–TiO2. Before the irradiation, only the dominant GC peak of BDE209 (10Br) was observed. However, it was interesting that 6–9 Br congeners appeared simultaneously after 5 h of irradiation. With further prolonged irradiation time to 24 h, the lower bromine congeners were observed and their amounts gradually increased. The product analysis showed that a large amount of the different lower bromine congeners were formed simultaneously (Fig. 8). This implied that the degradation of BDE209 by AgI–TiO2 did not follow the stepwise mechanism via single electron transfer. The simultaneous generation of different brominated congeners required more electrons to be transferred at the same time from AgI–TiO2 hybrids to BDE209. Therefore, a distinctive multi-electron transfer mechanism occurred in the AgI–TiO2/visible system. A similar debromination mechanism with multi-electron transfer was also reported in the degradation of BDE209 by graphene/TiO2.25 This special debromination pattern via multi-electron transfer may skip some reduction steps, avoiding the formation of some more toxic, more refractory intermediates.
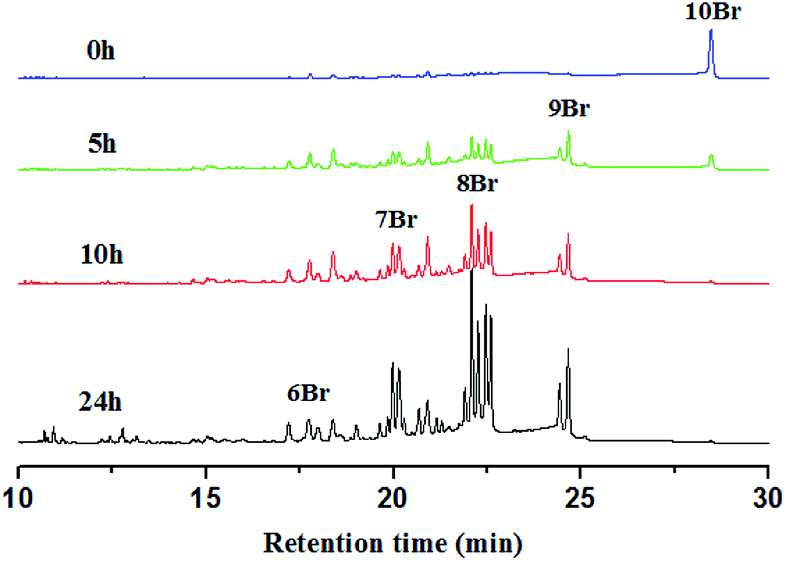 | ||
| Fig. 8 GC-μECD chromatograms of degradation products of BDE209 with AgI–TiO2 with different irradiation times. Reaction conditions: BDE209: 1.0 × 10−5 mol L−1; 0.2-AgI–TiO2: 1 mg mL−1, solvent: CH3OH, wavelength > 420 nm, anoxic condition. | ||
3.5 The mechanism of reaction
UV-Vis-DRS showed that photocatalytic activity under visible light irradiation was attributed to AgI. With loading of AgI particles, a new absorption in the visible light region was observed and the absorption intensity became stronger with an increase in the amount of AgI. This was also proved by control experiments with AgI–SiO2 and Ag–Al2O3. As shown in Fig. S6,† a certain extent of degradation of BDE209 was observed in the presence of AgI–SiO2 or Ag–Al2O3 under visible light irradiation, but the reaction was almost stopped when about 40% BDE209 had degraded. However, about 80% of BDE209 was degraded with AgI–TiO2 under the same experimental conditions. This indicated that in the AgI–SiO2 or Ag–Al2O3 system, AgI was excited by visible light and photoelectrons from AgI could transfer to BDE209, which resulted in the degradation of BDE209. But AgI was soon decomposed and the reaction stopped due to the instability of AgI under the irradiation. However, when the AgI particles were loaded onto the surface of TiO2, the photoelectrons from AgI could be transferred via the conduction band of TiO2 and inhibited the decomposition of AgI. Therefore AgI–TiO2 hybrids showed a higher catalytic efficiency than AgI–SiO2 and Ag–Al2O3. Unlike the instability of AgI, AgI–TiO2 hybrids performed with good stability for the degradation of BDE209 under visible light irradiation, which was proved by the cyclic run experiments. An oxygen control experiment showed that the degradation of BDE209 was largely prohibited in presence of O2 (Fig. S6†). This indicates that BDE209 prefers to be reduced by the electrons in the conduction bands of AgI–TiO2 and is resistant to oxidization by VB holes or reactive oxygen species in the presence of O2.Based on the above experiments, a possible reaction mechanism is proposed in Fig. 9. Under visible light irradiation, AgI was excited and then photogenerated e–h+ pairs from AgI were separated. In the anaerobic condition with N2, the VB holes were scavenged by CH3OH and the photogenerated electrons of AgI accumulated and preferred to transfer to the conduction bands of TiO2, which resulted in the inhibition of the recombination of e–h+ pairs and the more efficient separation of e–h+ pairs. Subsequently, the enhanced electron transfer, multi-electron transfer, occurred between the BDE209 and AgI–TiO2. BDE209 received more than one electron and was degraded to different lower bromo congeners at same time. Because the photo-generated electrons from AgI were transformed to the conduction bands of TiO2 not Ag+, AgI–TiO2 showed excellent photostability in the run experiments.
 | ||
| Fig. 9 The mechanism of degradation of BDE209 in CH3OH with AgI–TiO2 under visible light irradiation.23 | ||
4. Conclusions
A series of nanostructured AgI–TiO2 particles were synthesized by a deposition–precipitation method and characterized by SEM, TEM, XRD, XPS, BET and UV-Vis-DRS. The AgI–TiO2 hybrids showed high photo-reduction capability and good stability for degradation of BDE209 under visible light irradiation. The absorbance intensity and absorption ability of the AgI–TiO2 hybrids and the properties of solvents greatly influenced the efficiency of the degradation of PBDEs. The 0.2-AgI–TiO2 hybrid in CH3OH solution presented the highest reaction rate at 0.29 ± 0.02 h−1. Mechanistic pathways in degradation of PBDEs were different under UV and visible light. The debromination in the visible-light-induced AgI–TiO2 system showed a multi-electron transfer mechanism, which is different from the single-electron transfer in the UV-TiO2 system. The AgI–TiO2 hybrid showed an enhanced electron transfer in the photocatalytic process and resulted in high activity in photocatalytic reduction. This research may provide a green and efficient method of removing halogenated pollutants.Acknowledgements
The generous financial support by the National Science Foundation of China (No. 21107073 and 21477080) and the China Scholarship Council is gratefully acknowledged.References
- L. S. Brinbaum and D. F. Staskal, Environ. Health Perspect., 2004, 112, 9–17 CrossRef.
- P. P. He, C. S. He, Q. Liu and Y. Mu, RSC Adv., 2017, 7, 27214–27223 RSC.
- B. X. Mai, S. J. Chen, X. J. Luo, L. G. Chen, Q. S. Yang, G. Sheng, P. Peng, J. Fu and E. Zeng, Environ. Sci. Technol., 2005, 39, 3521–3527 CrossRef CAS PubMed.
- H. M. Stapleton, B. Brazil, R. D. Holbrook, C. L. Mitchelmore, R. Benedict, A. Konstantinov and D. Potter, Environ. Sci. Technol., 2006, 40, 4653–4658 CrossRef CAS PubMed.
- J. Bezares-Cruz, C. T. Jafvert and I. Hua, Environ. Sci. Technol., 2004, 38, 4149–4156 CrossRef CAS PubMed.
- Y. S. Keum and Q. X. Li, Environ. Sci. Technol., 2005, 39, 2280–2286 CrossRef CAS PubMed.
- A. Li, C. Tai, Z. S. Zhao, Y. W. Wang, Q. H. Zhang, G. B. Jiang and J. T. Hu, Environ. Sci. Technol., 2007, 41, 6841–6846 CrossRef CAS PubMed.
- C. Y. Sun, D. Zhao, C. C. Chen, W. H. Ma and J. C. Zhao, Environ. Sci. Technol., 2009, 43, 157–162 CrossRef CAS PubMed.
- X. Li, J. Huang, G. Yu and S. B. Deng, Chemosphere, 2010, 78, 752–759 CrossRef CAS PubMed.
- A. Z. Huang, N. Wang, M. Lei, L. H. Zhu, Y. Y. Zhang, Z. F. Lin, D. Q. Yin and H. Q. Tang, Environ. Sci. Technol., 2013, 47, 518–525 CrossRef CAS PubMed.
- K. S. Nose, S. J. Hashimoto, S. Takahashi, Y. K. Noma and S. I. Sakai, Chemosphere, 2007, 68, 120–125 CrossRef CAS PubMed.
- C. Y. Sun, W. Chang, W. H. Ma, C. C. Chen and J. C. Zhao, Environ. Sci. Technol., 2013, 47, 2370–2377 CrossRef CAS PubMed.
- C. Y. Sun, J. C. Zhao, H. W. Ji, W. H. Ma and C. C. Chen, Chemosphere, 2012, 89, 420–425 CrossRef CAS PubMed.
- C. Hu, T. W. Peng, X. X. Hu, Y. L. Nie, X. F. Zhou, J. H. Qu and H. He, J. Am. Chem. Soc., 2010, 132, 857–862 CrossRef CAS PubMed.
- A. C. Zhang, L. X. Zhang, X. Z. Chen, Q. F. Zhu, Z. C. Liu and J. Xiang, Appl. Surf. Sci., 2017, 392, 1107–1116 CrossRef CAS.
- C. Hu, X. Hu, L. Wang, J. H. Qu and A. Wang, Environ. Sci. Technol., 2006, 40, 7903–7907 CrossRef CAS PubMed.
- D. D. Yu, J. Bai, H. O. Liang, J. Z. Wang and C. P. Li, Appl. Surf. Sci., 2015, 349, 241–250 CrossRef CAS.
- D. Wu and M. Long, ACS Appl. Mater. Interfaces, 2011, 3, 4770–4774 CAS.
- Y. Li, H. Zhang, Z. Guo, J. Han, X. Zhao, Q. Zhao and S. Kim, Langmuir, 2008, 24, 8351–8357 CrossRef CAS PubMed.
- S. Song, F. Hong, Z. He, Q. Cai and J. Chen, J. Colloid Interface Sci., 2012, 378, 159–166 CrossRef CAS PubMed.
- J. G. Yu, G. P. Dai and B. B. Huang, J. Phys. Chem. C, 2009, 113, 16394–16401 CAS.
- C. H. An, W. Jiang, J. Z. Wang, S. T. Wang, Z. H. Ma and Y. P. Li, Dalton Trans., 2013, 42, 8796–8801 RSC.
- Q. Wang, X. D. Shi, J. J. Xu, J. C. Crittenden, E. Q. Liu, Y. Zhang and Y. Q. Cong, J. Hazard. Mater., 2016, 307, 213–220 CrossRef CAS PubMed.
- L. Yang, M. G. Gao, B. Dai, X. H. Guo, Z. Y. Liu and B. H. Peng, Appl. Surf. Sci., 2016, 386, 337–344 CrossRef CAS.
- M. Lei, N. Wang, L. H. Zhu, C. S. Xie and H. Q. Tang, Chem. Eng. J., 2014, 241, 207–215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07106j |
| This journal is © The Royal Society of Chemistry 2017 |