
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-switchable nanoparticles based on amphiphilic diblock, triblock and heterograft polyoxazoline†
L. Korchiaa,
C. Bouilhac a,
A. Aubertb,
J.-J. Robina and
V. Lapinte*a
a,
A. Aubertb,
J.-J. Robina and
V. Lapinte*a
aInstitut Charles Gerhardt Montpellier UMR5253 CNRS-UM-ENSCM, Equipe Ingénierie et Architectures Macromoléculaires, Université Montpellier, France. E-mail: vincent.lapinte@umontpellier.fr
bInstitut Charles Gerhardt Montpellier UMR5253 CNRS-UM-ENSCM, Equipe Matériaux Avancés pour la Catalyse et la Santé, Université Montpellier, France
First published on 4th September 2017
Abstract
Photo-active nanoparticles (NPD, NPT, NPH) were elaborated in water from amphiphilic diblock (D), triblock (T) and heterograft (H) copolymers based on a chromatic unit, coumarin, linked to an alkyl chain and a hydrophilic polyoxazoline chain. Under UV-exposure, for graft copolymers, the coumarin moieties crosslink the nanoparticle core (NPHx) while a simple chain by chain dimerization happens with linear diblock and triblock copolymers (NPDx and NPT). The self-assembly of these copolymers was examined as well as the UV-activity and UV-reversibility of the corresponding NPs by UV spectroscopy. Their stability under light exposure and temperature was also monitored by dynamic light scattering (DLS). Finally, the loading of a lipophilic fluorescent probe (Nile red) into the NP core was evaluated by fluorescence emission. All these data illustrate the tunable size, UV-activity, stability and loading efficiency of the NPs by the starting macromolecular architecture of the amphiphilic copolymers. Triblock copolymers seem to be a promising tool for switchable NPs characterized by two opposite states: NPs alternatively with and without a photo-dimerized core.
Introduction
Amphiphilic copolymers have, for a long time, attracted much attention due to their ability to self-assemble in selective solvents.1–4 The stability of self-assembled polymeric nanoparticles is a crucial property for many applications including delivery with nano-carriers or chemistry with nano-reactors. Polymeric nano-carriers have been extensively investigated and largely used in medical applications such as drug delivery.5,6 To enhance the therapeutic effect and decrease side effects, nano-carriers should meet drastic requirement specifications such as appropriate size, stability, drug loading efficiency and controlled release kinetics. For this last points, nano-carriers often present the drawbacks of drug burst releases and instability in physiological media. To improve the stability of nanoparticles (NP)s and decrease the burst, the cross-linking has been recognized as a powerful approach.7,8 In comparison to other strategies, photo-crosslinking is an attractive way because rapid, effective and well-controlled process without by-product formation.9–13Among various photo-crosslinkable molecules, coumarin and its derivatives have attracted considerable attention.14–16 They photo-dimerize according to a [2π + 2π] cyclo-addition under UV-irradiation at λ > 310 nm. The dimer adduct can be reversibly dissociated into the two coumarin precursors at λ < 260 nm by photo-cleavage and thus reverts to the starting compound.17 The reversible photo-dimerization of coumarin has also been explored for designing photo-controllable NPs.18 For example, Luo and et al. prepared photo-crosslinked telodendrimer micelles characterized by a high drug loading efficiency, capacity and stability.19 Ji and et al. also synthesized a coumarin-based pH-responsive polymer able to form cross-linked micelles with a favorable sustained release of drug.20,21 Otherwise, He and et al. studied the photo-crosslinking of poly(ethyleneimine)s as smart drug carriers.9
From photo-controlled reversible dimerization of coumarin moieties incorporated in copolymer, NPs can be core-crosslinked and subsequently de-crosslinked under illumination at two selective wavelengths.9,22 As shown by Trenor17 or Zhao,16,23 according to the number and the position of coumarin units in the copolymer architecture, the photo-response occurring inside the core changed. For instance, Zhao and et al. described amphiphilic heterografted copolymers self-assembled into NPs where coumarin moieties were gathered in the inner part.8 Their UV-irradiation at 300 nm stabilized the NP by core-crosslinking whereas a partial reversible de-crosslinking happened. Otherwise, the simple photo-dimerization of linear polymer bearing only one coumarin per chain has already been described17 but never with amphiphilic copolymers.
In previous works, we elaborated several amphiphilic photo-responsive architectures of copolymers. Heterograft copolymers bearing numerous coumarins per chain were able, after self-assembly into aqueous media, to photo-crosslink the NP core whereas linear copolymers bearing a coumarin in terminal position of polymeric chain only photo-dimerized the core NP.24 In addition, these linear copolymers can also be first dimerized giving triblock copolymers. All of them are based on hydrophilic polyoxazoline and a hydrophobic alkyl C11 chain terminated by a coumarin moiety. Polyoxazolines have received significant attention over the last few years for various applications in technical matters25 and biomedical applications.26,27 Their potential comes from their various potential functionalizations,28–32 high hydrophilicity and biocompatibility relative to their peptidic structural analogy.33 In spite of those numerous advantages, amphiphilic polyoxazolines associated to photo-active group have been little studied.34,35
Herein, we investigated the stabilization and the UV-activity of photo-responsive NPs by core photo-dimerization or core photo-crosslinking using amphiphilic diblock, triblock and heterografted copolymers previously described (Fig. 1).24 The comparison of both core photo-responses should allow a better understanding of the core-stabilized nanoparticles in term of UV-efficiency, stability and loading ability.
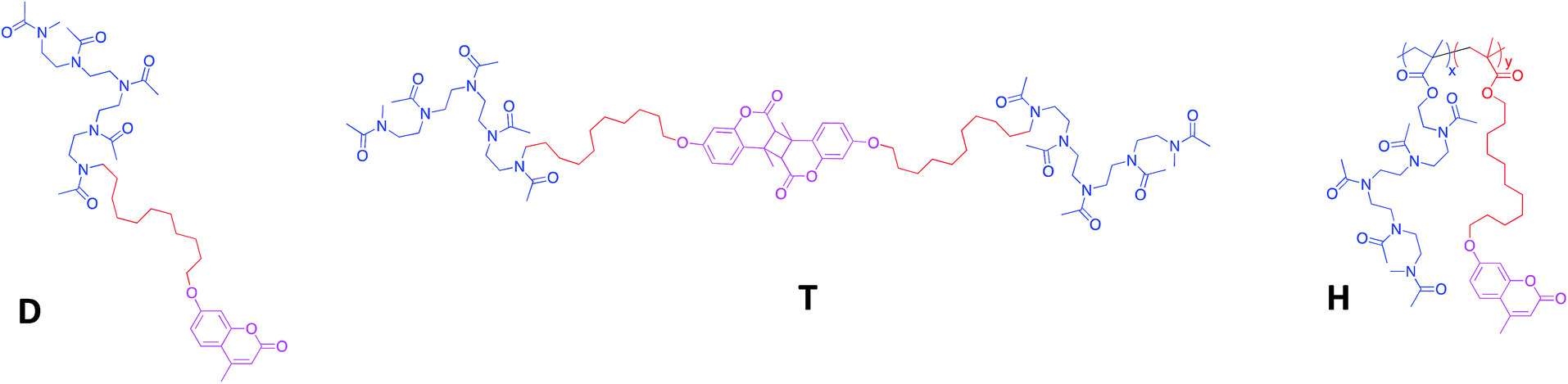 | ||
| Fig. 1 Chemical structure of the amphiphilic diblock (D), triblock (T) and heterograft (H) copolymers. | ||
Results and discussion
Comparative self-assembly of the amphiphilic diblock, triblock and heterografted photo-responsive copolymers
Three macromolecular architectures of photo-responsive copolymers (diblock, triblock and heterograft) named D, T and H, respectively, were synthesized from the same hydrophobic and hydrophilic blocks as well as the same photo-active group, 7-hydroxy-4-methylcoumarin, as shown in Fig. 1. For each copolymer, the hydrophobic alkyl chain in C11 bore a terminal coumarin moiety which constituted the core of photo-responsive NPs into aqueous media.According to the copolymer architecture and the number of coumarin units per chain, the photo-response occurring into the inner part of the NPs under UV-exposure differed. In this way, three cases were studied: the photo-dimerized NP core using diblock copolymers (NPDx), the crystallized NP core using the triblock copolymers (NPT) and the photo-crosslinked NP core using graft copolymers (NPHx). These photo-responsive nano-objects were elaborated combining the polymerization step, the aqueous self-assembly and the UV-irradiation knowing that the chronology changed in function of the route as shown in Fig. 2.
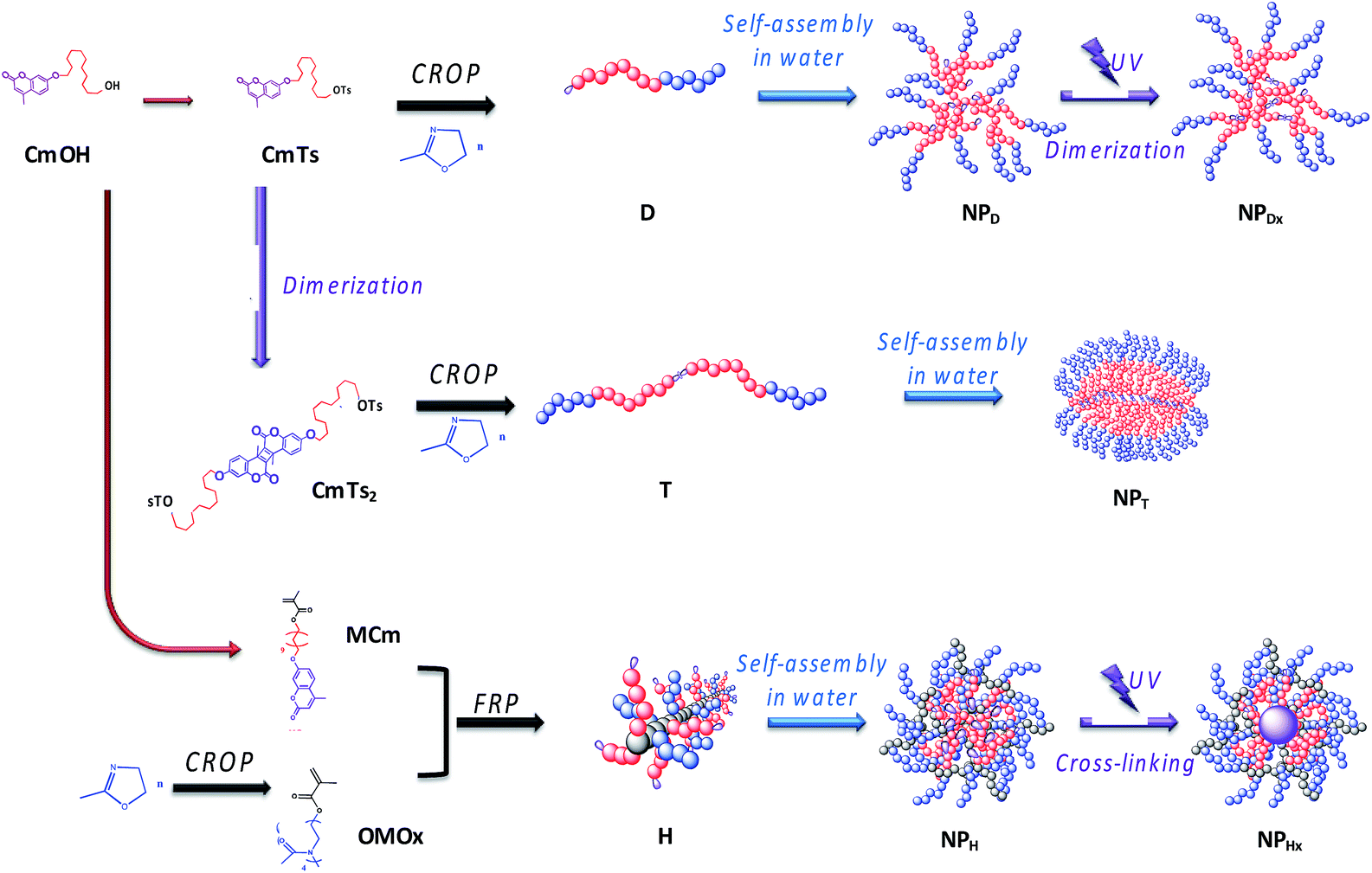 | ||
| Fig. 2 Association of polymerization step, self-assembly and UV-illumination to elaborate NPDx, NPT and NPHx nanoparticles. | ||
For NPDx, the amphiphilic photo-responsive diblock D was first synthesized by cationic ring-opening polymerization (CROP) of 2-methyl-2-oxazoline (MOx) using the hydrophobic coumarinated initiator CmTs as already described.24 Then, the diblock D self-assembled in water into photo-responsive NP (NPD) before photo-illuminating the core at 350 nm during 1 h to lead to NPDx.
Inversely, for triblock route, the photo-illumination of the NP occurred before self-assembling. The triblock copolymers T were first synthesized by photo-irradiation of the corresponding initiator CmTs into a bis-initiator CmTs2 before polymerizing MOx by CROP. They were further self-assembled in water into NP (NPT) having photo-dimerized core with a crystalline character as already demonstrated.36
For NPHx, the amphiphilic heterograft copolymers (H) were synthesized by free radical polymerization (FRP) of hydrophilic and hydrophobic methacrylate macromonomers as previously reported.37 The hydrophilic and coumarinated hydrophobic side chains were randomly distributed along the backbone. These amphiphilic copolymers were then self-assembled in water into NP (NPH) before being photo-irradiated to crosslink the core of the NP (NHPx). We noted the difference in behavior between linear di- and triblock copolymers with terminal fluorescent dye (dimerization of the NP core) and the graft copolymers bearing numerous pendent coumarin units (cross-linking of the NP core).
The amphiphilic copolymers were well-characterized by 1H NMR spectroscopy and SEC as previously reported.24,36,37 The three types of copolymers belong a hydrophilic/hydrophobic ratio, MH/Mh, around 2.5 where MH and Mh corresponded to the molecular weight of hydrophilic and hydrophobic blocks, respectively (Table 1). A copolymer H with a high molecular weight (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was selected to demonstrate the influence of numerous coumarins per chain. We noted that for a same MH/Mh ratio, the number of coumarins ranged from 1, 2 to 16 for D, T and H, respectively. The predominant hydrophilicity of these copolymers D, T and H favoured the direct dissolution in water.
000 g mol−1) was selected to demonstrate the influence of numerous coumarins per chain. We noted that for a same MH/Mh ratio, the number of coumarins ranged from 1, 2 to 16 for D, T and H, respectively. The predominant hydrophilicity of these copolymers D, T and H favoured the direct dissolution in water.
| Mna [g mol−1] | Da [—] | DPnb [—] | CMCc [mol L−1] | Nb Coumd [—] | MH/Mhd [—] | Dim.e [%] | De-dim.f [%] | |
|---|---|---|---|---|---|---|---|---|
| a Determined by SEC in DMAc using PMMA standards.b Calculated by 1H NMR.c Determined by spectrofluorimetry through coumarin emission band.d Hydrophilic/hydrophobic balance.e Monitored by UV-spectroscopy and calculated according to eqn (1) (ESI 1).f Monitored by UV-spectroscopy and calculated according to eqn (2) (ESI 1). | ||||||||
| D | 1500 | 1.2 | 13 | 2.6 × 10−6 | 1 | 2.7 | 70 | 30 |
| T | 2800 | 1.5 | 12 + 12 | — | 2 | 2.4 | 75 | 75 |
| H | 20000 |
2.6 | 36 × 4 | 2.5 × 10−6 | 16 | 2.5 | 80 | 55 |
According to DLS traces, the amphiphilic copolymers D, H and T self-assembled in water into NPs above 12, 80 and 150 nm, respectively, with dispersity index above 0.3–0.4 (Fig. 3). Regarding to TEM images, all the NPs seemed also relatively monodispersed with a lower diameter (above 10, 30 and 20 nm, respectively). The difference in size of NPs resulted from the fact that the DLS examined the NPs in suspension by contrast to TEM which studied dry NPs. We noted the difference of behaviour with more aggregations for T related to H and D. Herein, we focus on the stability of these NPs, their size or aggregation is not discussed.
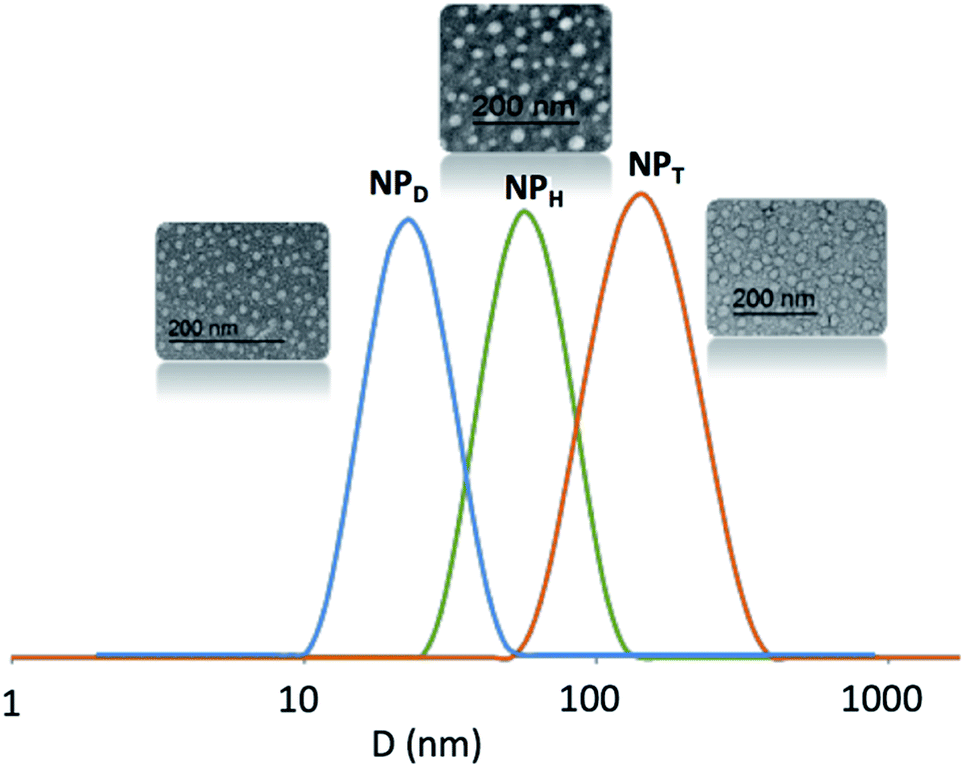 | ||
| Fig. 3 DLS results and TEM images of NPD, NPH and NPT. | ||
Photo-reversibility of the NP core stimulation
The coumarinated NPs were illuminated at 350 and 254 nm and monitored by UV-vis spectroscopy. For all the NPs, a decrease of UV absorption band of coumarin was clearly observed indicating that the [2 + 2] cycloaddition and thus the crosslinking or the dimerization of the NP core into NPHx, NPDx and NPT occurred (Fig. 4). By contrast, the reversible cycloaddition produced de-crosslinking or de-dimerization of the NP core resulting into NPH, NPD and NPD (T) respectively. This UV-activity was measured with the increase of the same UV absorption band upon the illumination at 254 nm (Fig. 4). From these data, the kinetics of dimerization and de-dimerization of coumarin units can be monitored in time. In the literature, the graft copolymers are the most commonly employed photo-responsive amphiphilic systems. Some of them, using 7-hydroxy-4-methylcoumarin, present a crosslinking rate of 60–80% for 30–90 min and a de-crosslinking close to 40% for 5–20 min.22,38,39 These crosslinking and de-crosslinking rates weren't mentioned in the literature but we calculated them using the kinetic data according the methodology of Fig. 4 According to eqn (1) and (2) (annexe ESI I†), the copolymers H were in good agreement with the literature with a crosslinking rate of 80% for 1 h and a de-crosslinking rate close to 55% in 10 min. By contrast, only one example of linear photo-responsive molecule bearing coumarin units has already been reported.40 This structure was purely hydrophilic and irradiated in bulk. In this case, 30% of dimerization and de-dimerization happened in 20 and 4 min, respectively. They explained the low efficiency and reversibility of the photo-reaction by the slow mobility of the polymer chains in bulk. In our case, the coumarin dimerization and de-dimerization rate of the nanoparticle NPD into NPDx corresponded to 70 and 30%, respectively (Table 1). To our knowledge, the copolymer D was the first example of amphiphilic photo-dimerized linear copolymer in solution. It was the fastest photo-dimerizable copolymer among D, T and H. However, its lower efficiency (70%) related to the graft copolymers H (75%) can be explained by the least number of coumarin units in the inner part of the NPs. In the same way, for H, the closeness of the coumarins justified the high crosslinking rate. By contrast, the tangle of the core polymer chains D and the low meeting probability seemed to be an explanation of the low efficiency of copolymers D in photo-dimerization. The alternative route consisting in the triblock copolymers T offered the best dimerization rate with 80%. The photo-dedimerization of coumarin was also investigated and gave the best results for the same copolymers T (75%) related to H (55%) and D (30%) (Fig. 4). The quasi-reversibility (80/75) of the triblock copolymers should be caused by preferential interactions and crystallization keeping the coumarins close to each other after de-dimerization, ensuring the next photo-dimerization in good yield. | ||
| Fig. 4 Kinetic study of a coumarin dimerization/de-dimerization cycle under UV exposure at λ = 350 and 254 nm monitored by UV-vis spectrometer for various NPs. | ||
Supplementary studies on the reversibility of the coumarin photo-dimerization were realized with successive photo-cycloaddition and photo-cleavage cycles as illustrated in Fig. 5. For the copolymers D and H, the maximum absorbance at 320 nm of each cycle slightly decreases with the number of cycles, probably due to the appearance of a small amount of asymmetric by-products of the coumarin dimer during photo-cleavage as previously described for other heterografted copolymers.41,42 The copolymers T differed from the other ones by a total photo-reversibility explained by the well-defined initial organization of the previous dimerization, also causing the crystallization of NP core.
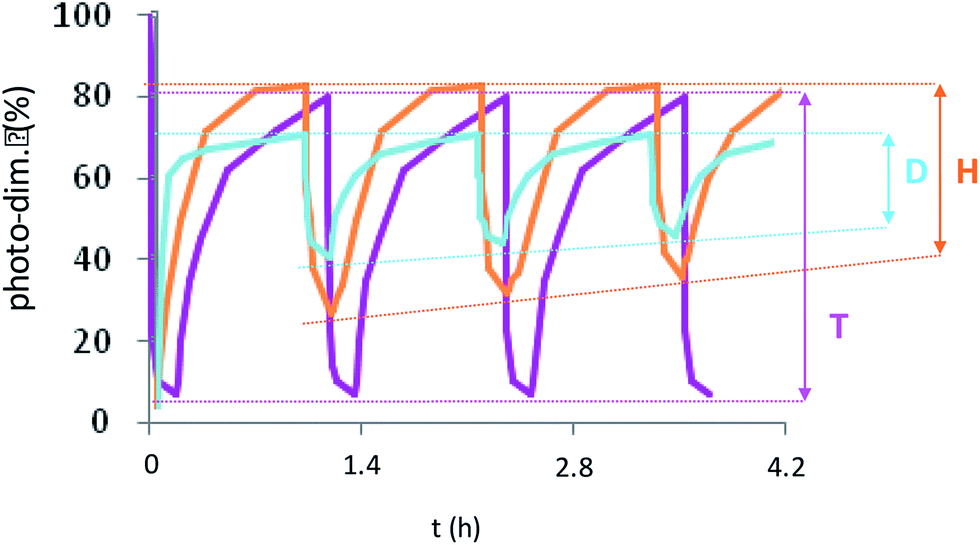 | ||
| Fig. 5 Photo-cycloaddition reversibility of NPD, NPH and NPT after repetitive irradiation cycles at 350 nm (1 h)/254 nm (10 min) (cycloaddition/cleavage) for the two first and 254 (10 min)/350 nm (1 h) (cleavage/cycloaddition) for the last. | ||
Stability of the nanoparticles
The aim of this study was to enlighten if the photo-crosslinking was preferable to the simple photo-dimerization of the coumarin units chain by chain to stabilize the nanoparticles. Several parameters including the temperature and light versus time were investigated. The formation of aggregates and the uncontrollable photo-cleavage of coumarin units were monitored by DLS and UV spectroscopy, respectively. The DLS study highlighted the appearance of aggregation of the NPs before (NPD and NPH) and after photo-crosslinking (NPHx) or photo-dimerization (NPDx and NPT) during a prolonged heating at 37 and 60 °C. Fig. 6(a), (c) and (e) correspond to the size distribution of non-irradiated NPs (NPD and NPH) in water (continuous lines) and irradiated nanoparticles NPDx and NPHx (dotted lines) and NPT. Fig. 6(b), (d) and (f) represent the same NPs after 60 days at 60 °C. The size evolutions under heating at 60 °C during 60 days for both systems were shown in Fig. SI 1.†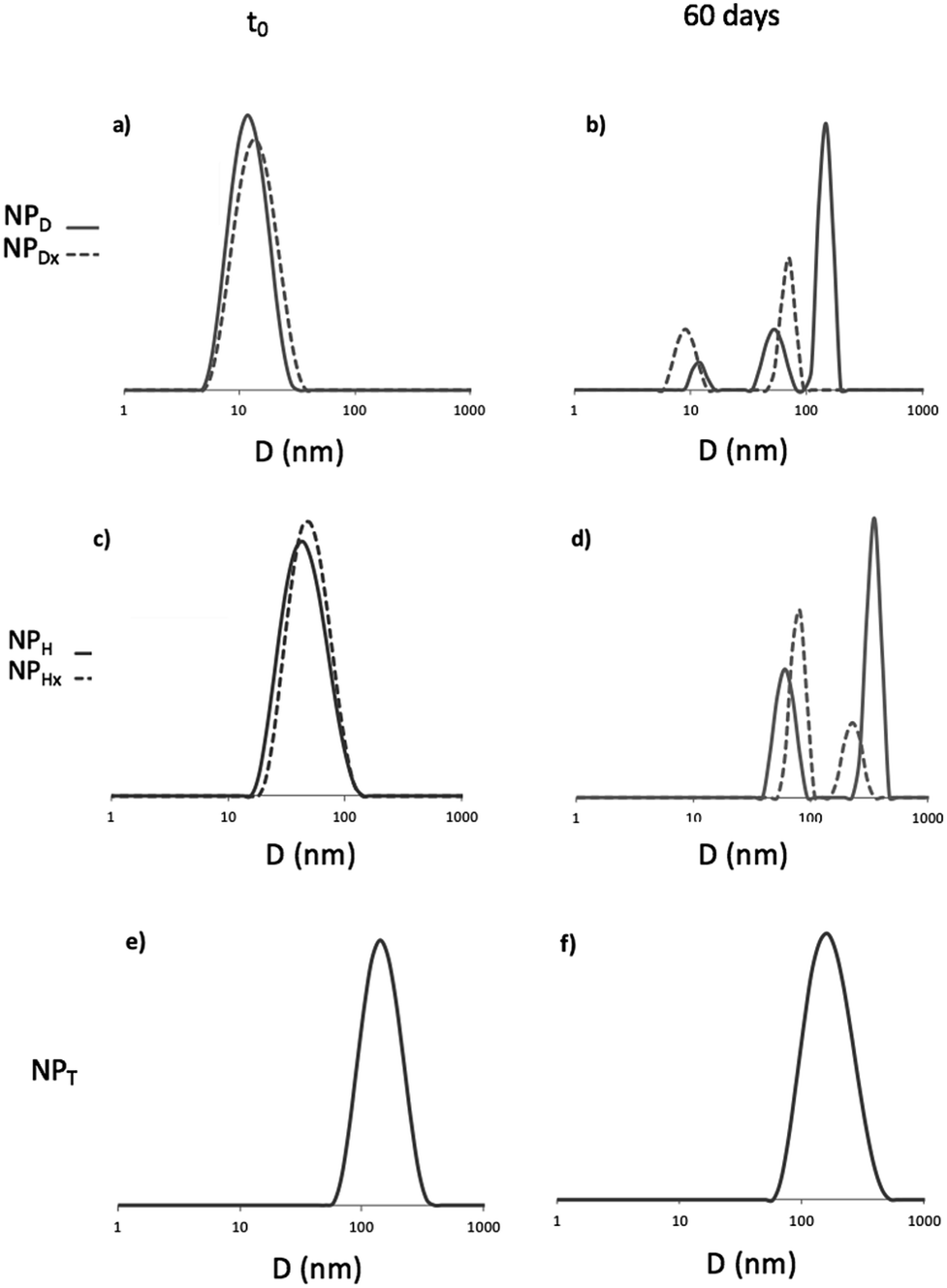 | ||
| Fig. 6 DLS traces of D, H and T before (a), (c) and (e) and after (b), (d) and (f) 60 days at 60 °C. For H and D dotted lines represented cross-linked and dimerized NPs. | ||
For D, the stability of the NPs was confirmed with the same average diameter of 12 nm before and after UV-irradiation over time and under light as well as at 37 °C (ESI II†). At higher temperature, 60 °C, after 7 days the occurrence of predominant aggregates until 150 nm of mean diameter appeared (Fig. (6b)). The dimerization delayed the instability until 15 days and limited the size of the aggregates at 60 nm (Fig. SI 1†). Initially mono-populated, the size distributions were disturbed under heating caused by aggregation and the appearance of multi-populated distributions (Fig. (6b)). Even though the dimerization of NPD into NPDx limited this aggregation in a first time, after 20 days the NPDx destabilization happened. Less impacted, NPH and NPHx presented nevertheless the same characteristics. Predominant aggregates were formed under heating at 60 °C (Fig. 6(c) and (d)) whereas they did not appear under other parameters such as light, time and the lower temperature, 37 °C (Fig. SI 2†). The stability of NPH above 80 nm of mean diameter before and after crosslinking (NPHx) was affected by heating and lead to multi-populated distribution with the main aggregates at 300 nm and above 130 nm since one month. As observed for D, the core photo-crosslinking seemed to improve the NP stability and delayed their aggregations even if the post-irradiation was not enough to preserve a long-term stability.
Concerning the triblock copolymers, the NPT were insensitive to the temperature (Fig. 6(e) and (f)) as well as light (Fig. SI 1 and SI 2†). The overlapping of the initial and the terminal DLS traces around 150 nm demonstrated the stability of the crystallized core of NPT. Thus, the crystallization of the NP core by coumarin dimerization before self-assembling in water was the more efficient way to stabilize the NPs knowing that the triblocks route also allowed the more efficient photo-dimerization of coumarin.
To pursue the investigation on the stability of NPT, NPDx and NPHx over light and temperature, the de-dimerization or de-crosslinking of coumarin was monitored by UV spectroscopy regarding the intensity of coumarin absorption band at 363 nm as shown in Fig. 7. In the dark, all the NP cores remained photo-dimerize or photo-crosslink. The light had contrasting effect on NPs with no modification of NPDx and NPT whereas 30% of de-dimerization of NPHx (eqn (2)†) occurred after 60 days. The supplementary parameter destabilizing the NP core was the temperature. For NPDx and NPHx, at 37 °C, an unwitting reversible photo-cleavage rates of 40 and 20% were attained respectively versus 65 and 80%, respectively at 60 °C. We noted a faster de-dimerization of the core for NPDx in comparison to NPHx. The crosslinking of the hydrophobic chains NPHx seemed to quite stabilize the NP core related to the simple dimerization of the polymeric chains of NPDx. As previously observed by DLS, the NP core of NPT was insensitive to light or heating. The high stability of NPT could be explained by the crystallization of the inner part of NP, that stabilized the dimerized coumarin moieties and avoided the reverse photo-reaction.
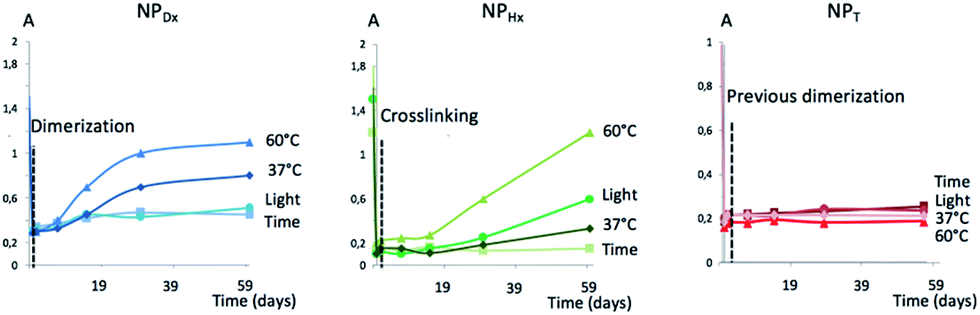 | ||
| Fig. 7 Photo-cleavage of NPDx, NPHx and NPT versus light and temperature using UV spectroscopy. | ||
Loading efficiency
To determine the effect of the coumarin dimerization on the entrapment of molecules, Nile red was loaded into NPs (NPD, NPDx, NPH, NPHx and NPT) at a standard theoretical loading capacity of 10%.9 We noted the multicyclic structural likeness between this hydrophobic fluorescent probe and the dimerized coumarin. The NP internalization of the colorimetric probe induced the red coloration of the NP suspensions from 1 h with color intensification until stagnation after 15 h. After this period, NPDx and NPHx suspensions were irradiated at 350 nm during one hour to respectively core-dimerize and core-crosslink loaded NPs. The no-overlapping of the coumarin and Nile red emission bands in fluorospectroscopy was preliminary checked. As observed on the emission spectra of both NPs and reported in Fig. SI III,† Nile red λemission was above 600 nm and coumarin λemission around 380 nm at λexcitation = 552 nm in water. The Nile red entrapment in hydrophobic environment resulted from the appearance of an emission band in water whereas the free Nile red in water was undetectable by fluorescence. To ensure that Nile red emission intensity came exclusively from loaded molecules, charged NPs were separated from the rest of the solution, meaning non-loaded Nile red and unimers. By chromatography separation, DLS analysis and fluorescence emission, large NPs, corresponding to loaded NPs and eluting faster than small molecules such as free Nile red and unimers, were separated (Fig. SI 3†). The fluorescence monitoring identified the fractions containing loaded Nile red (Fig. SI 4†) whereas DLS shown the scattering intensity at 12 nm of the NPD suspension fractions (Fig. 8). Overall loaded NPs presented same size distribution than unloaded NPs (data not shown). As observed, coumarin intensity increased with the presence of aggregates and decreased when the unimers were eluted. However, for the richer fractions in loaded NPs, Nile red radiation seemed to interfere with coumarin radiation and cover up NPs emission. This Nile red quenching effect, already proved with different molecules in others studies, explained the low coumarin emission intensity observed.43 The study was realized on NPDx, NPH, NPHx and NPT and similar profiles were observed (Fig. SI 5–8 in Fig. SI III†). We noted that core-crosslinked NPHx and core-dimerized nanoparticles, NPDx and NPT, presented lower coumarin emission intensity in comparison to NPH and NPD (above 100–400 against 800–1000). This phenomenon can be explained by the formation of cyclobutane ring during the dimerization.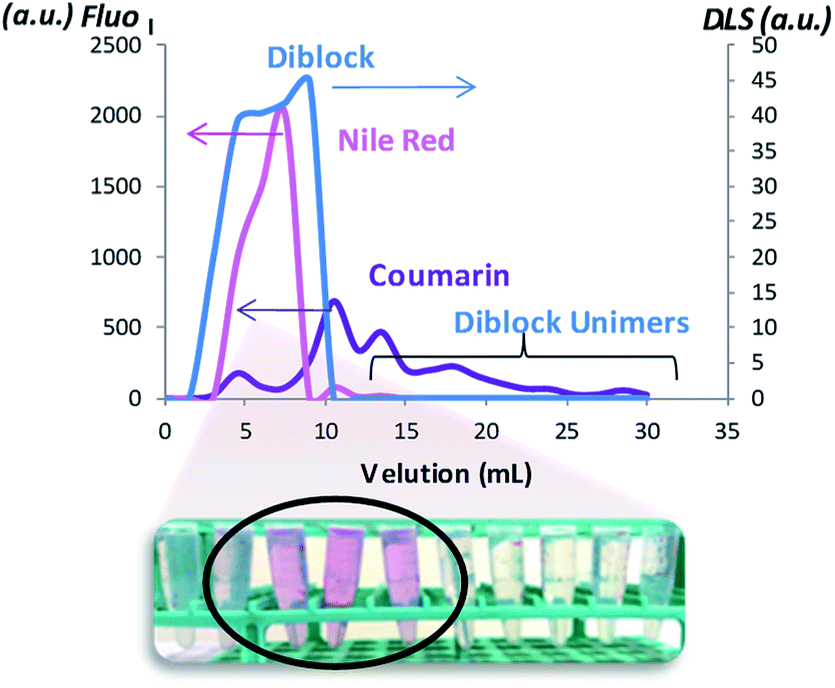 | ||
| Fig. 8 Elution of the charged NPs in Nile red. | ||
To estimate the Nile red loading rate containing in gathered fractions, the emission intensity of Nile red was measured as illustrated in Fig. 9. A significant split to high wavelength was observed for NPDx. This bathochromic effect of 20 nm probably came from interaction between dimerized coumarin and Nile red by π-stacking. Indeed, the D chain mobility already observed in UV-study made the establishment of easier interaction between coumarin moieties and Nile red than heterograft or triblock copolymers. Moreover, the structural similarity between dimerized coumarin and Nile red can accentuate the electronic resonance.
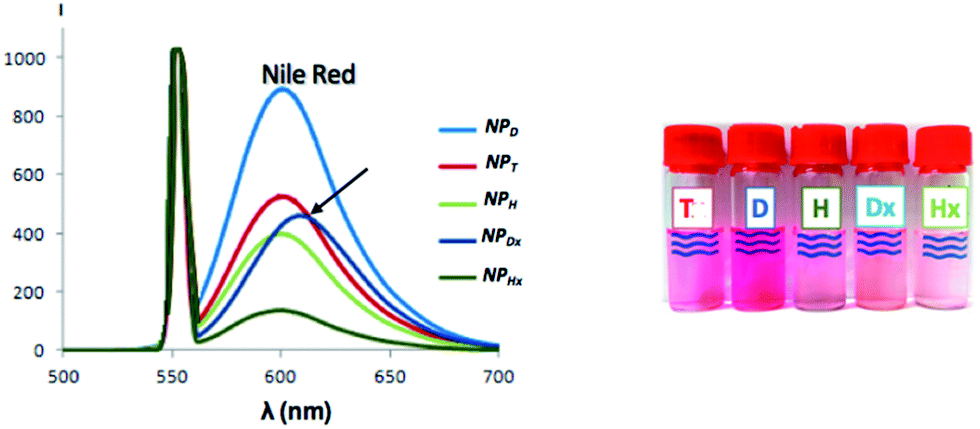 | ||
| Fig. 9 Emission spectra of Nile red entrapped into various NPs monitored by fluorescence under λexcitation above 552 nm and corresponding suspensions. | ||
For each NP, from maximal intensity around 600 nm, the loading efficiency (D.L.E.) and the loading capacity (D.L.C.) can be deduced through the previous calibration. The drug loading efficiency (D.L.E.) ranged from 11 to 80%, in relation to the amount of drug initially added (eqn (3), ESI III†) (Table 2). The experimental drug loading capacity (D.L.C.), in relation to the amount of copolymer initially introduced (eqn (4), ESI III†), ranged from 1 to 8%. In the literature, a loaded system is considered efficient for a D.L.E. above 50% and a D.L.C. above 5%. D copolymer was compared to C11POx, a similar amphiphilic copolymer with the same hydrophobic chain and length of hydrophilic chain but without terminal coumarin unit in order to evaluate the influence of the latter on the Nile red entrapment (Table 2). Slightly better results were achieved with NPD demonstrating favorable interactions between dye and the spectroscopic probe. The best D.L.E. and D.L.C. results corresponded to the NPD (80% and 8%) in contrast with the NPHx (11 and 1%) knowing that theoretical maximal D.L.E. was above 100% and D.L.C. above 10%. The NPT had an intermediate D.L.E. and D.L.C. values. Surprisingly, the crystallization of the T core did not too much hinder the entrapment of Nile red even if the impact was more perceptible with the D.L.C. (4%). Moreover, a twice decrease of loading rate after the dimerization of linear polymer NP core occurred while three times decrease by crosslinking of the NP core was observed for NPH. The photo-dimerization (44% for NPDx instead 80% for NPD) or photo-crosslinking (11% for NPHx instead 38% for NPH) seemed to expel the loaded Nile red of the hydrophobic NP core. Furthermore, the number of coumarin units inside the core, as well as the dimerization and the cross-linking, hinder the entrapment of Nile red. Thus, triblocks, from their previous dimerization, were the more efficient irradiated and stabilized approach for drug loading. In conclusion, the best amphiphilic copolymer for the entrapment of Nile red molecule was the amphiphilic diblock copolymers whereas the more studied photo-responsive systems for entrapment were based on heterografted copolymers.
| % drug loading efficiency | % drug loading capacity | |
|---|---|---|
| NPC11POx | 76 | 6 |
| NPD | 80 | 8 |
| NPDx | 44 | 5 |
| NPT | 50 | 4 |
| NPH | 38 | 3 |
| NPHx | 11 | 1 |
Experimental
Materials
CoumC11OH (CmOH), CoumC11OTs (CmTs, initiator of diblock), CmTs2 (bis-initiator of triblock), MCm (hydrophobic coumarinated monomer), MPOx (hydrophilic polyoxazoline monomer), D (diblock copolymer),24 T (triblock copolymer)36 and H (heterograft copolymers)37 were synthesized according to the procedures previously reported by our group. After photo-dimerization of the core NPD and NPH, the nanoparticles constituted of D and H copolymers were named NPDx and NPHx, respectively.Instruments
The UV-dimerization of and UV-crosslinking of ND and NH into NDx and NHx were performed in a cylindrical photochemical reactor “Rayonet RPR-200” equipped with 16 symmetrically placed lamps with emission in 350 or 254 nm. The samples were irradiated in quartz flasks placed on a circular rotating holder. UV-visible analyses were conducted with a Perkin Elmer-lambda 35 UV/Vis spectrometer equipped with PTP-1+1 Peltier System in quartz vial (l = 1 cm). Size exclusion chromatography (SEC) with N,N-dimethylacetamide (DMAc) (with 0.1 w/w% LiCl) as eluent at a flow rate of 0.8 mL min−1, calibrated with poly(methylmethacrylate) (PMMA) standards, was performed on a PL-GPC 50 Plus equipped with a Varian model 410 autosampler. The SEC apparatus comprised a refractive index detector and was filled with a 8 μm PolarGel-M pre-column (7.5 × 50 mm) and two 8 μm PolarGel-M columns (7.5 × 300 mm) thermostatted at 50 °C. For DLS analysis, the samples were concentrated at 1 mg mL−1. Solutions were filtered through 0.45 μm hydrophilic PTFE syringe filters (Millipore). Measurements were performed at 25 °C using a Zetasizer Nano ZS from Malvern Instruments equipped with a laser He–Ne source of 633 nm wave-length and an angle of 173°. The intensity-averaged values of the hydrodynamic diameter are given in this study. Sample preparation for transmission electron microscopy (TEM) analysis was done as follows: 10 μL of aqueous copolymer suspensions concentrated at 0.04 mg mL−1 were dropped onto glow discharged carbon-coated copper grids (Agar scientific, Cu-300). Then, 10 μL of a 4 w/v% aqueous uranyl acetate negative stain solution was added. TEM imaging was performed using a JEOL, JEM-2200FS microscope operating at 200 kV. Fluorimetry measurements were performed using a RF-5301 PC spectrofluorophotometer (Shimadzu) using quartz cuvettes.Methods
The nanoparticles of the copolymers D, H and T (ND, NH and NT) were prepared in water by direct copolymer dissolution in Milli-Q water, at a concentration of 1 mg mL−1. Particles were formed under 500 rpm mechanical stirring after 24 hours and then filtered through 0.45 μm hydrophilic PTFE syringe filters (Millipore) before DLS measurements.The Critical Aggregation Concentrations (CAC) were determined through the maxima intensity of coumarin emission band at 340 nm evolution according to the concentrations and monitoring by a spectrofluorometer with λex = 310 nm recorded from 300 to 700 nm. Maximal concentrations were chosen according to saturated emission limitation.
The kinetic of photo-dimerization (or cross-linking)/de-dimerization (de-crosslinking) were monitored in sealed quartz vials, to keep the concentration constant, and irradiated through the photo-chemical reactor Rayonet RPR-200 at 350 nm/254 nm, respectively. For each kinetic time, the same vial was alternately placed into the UV-vis spectrometer where the coumarin absorbance at 320 nm was recorded from 250 to 370 nm and placed under UV exposure inside the photo-reactor. Concentrations were chosen according to saturated absorbance limitation imposed by the UV-spectrometer.
The stability of the nanoparticles NPD, NPDx, NPH, NPHx and NPT were monitored by DLS following the size populations and by UV-spectrometry regarding the intensity of the coumarin band at 320 nm. The measurements were realized on sealed quartz vials during 60 days, at 20, 37 or 60 °C, either protected or exposed from natural light.
Loaded-nanoparticles were prepared with Nile red as commonly used model drug; its photo-activity permits the monitoring of entrapment efficiency of the nanoparticles throughout the calculation of the drug loading efficiency (D.L.E.) and the drug loading capacity (D.L.C.). Nile red loading by NPD, NPDx, NPH, NPHx and NPT was carried out by direct and simultaneous dissolution of Nile red probe (0.1 mg mL−1) with copolymers (1 mg mL−1) into MilliQ-water during 24 hours under mechanical stirring (500 rpm) as well as unloaded nanoparticles. Polymer concentration was chosen according to self-assembly study. NPDx and NPHx were then irradiated during 1 hour under UV-exposure at 350 nm into quartz flasks through the photo-reactor Rayonet RPR 200 and under stirring and cooling. Nile red loaded nanoparticle suspensions were subsequently eluted by size exclusion chromatography to separate unloaded molecules. A pre-packed desalting disposable PD-10 columns (GE healthcare) constituted of Sephadex® G25 medium was used as stationary phase and MilliQ-water as eluted phase. Elution flow rate was controlled by a peristaltic pump (Ismatec®, VWR), regulated at 1 mL min−1, and fractions of 1.5 mL were collected until a total elution volume above 30 mL. After DLS and spectrofluorimetry analysis, the fractions containing Nile red loaded nanoparticles were gathered for each kind of nanoparticles and then lyophilized. Free Nile red and copolymer were recuperated and dissolved into acetone to before quantification.
Quantification concentrations varying from 1 × 10−6 to 5 × 10−6 mg mL−1 in acetone were chosen according to Nile red maximal emission and monitored though a spectrofluorometer inside quartz vials under λexcitation = 552 nm and recorded from 500 to 700 nm. The loading efficiency was estimated from the Nile red emission intensity calibration according to various concentrations in acetone (cf. annexe Fig. SI 9†).
Conclusions
This study shown the manifold combinations between UV-illumination and self-assembly to yield photo-active NPs possessing dimerized or crosslinked core. The difference of behaviour between amphiphilic block and heterograft copolymers in term of photo-activity, stability of the NPs and the loading of fluorescent molecules was demonstrated. The crosslinking (NPHx) and the dimerization chain by chain (NPDx) of the NP core both slightly improved the stability of the nano-objects over temperature and light and the dimerization of coumarin units before polymerizing and self-assembling allowed a highly reversible photo-response related to post-dimerization (D into Dx) and post-crosslinking (H into Hx) of NPs. More remarkably the triblock copolymers self-organized into stable NPTs with an insensitive crystallized core over the temperature and light which seem to be a promising tool for switchable NPs.Otherwise, the gain of coumarin unit in the entrapment of aromatic molecule was demonstrated. Moreover, the drug loading efficiency and capacity in Nile red were favoured by the mobility of the core chains and disadvantaged by crystallization, dimerization and cross-linking of the core.
This study demonstrated the high opportunity of amphiphilic diblock copolymers to load active molecules while triblock copolymers offered an interesting solution to stabilize NPs. Finally, the photo-dimerization of linear copolymers in the NP core seemed to be an alternative way to the photo-crosslinking of graft copolymers for photo-active NPs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the “Ministère de l’Enseignement Supérieur et de la Recherche” and the “Centre National de la Recherche Scientifique” for financial support. They are thankful to Dr Erwan Oliviero from ICGM technical platform for TEM imaging.Notes and references
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41(18), 5969–5985 RSC.
- C. Travelet, C. Bouilhac, J. J. Robin and R. Borsali, Soft Matter, 2014, 10(38), 7545–7557 RSC.
- C. Bouilhac, C. Travelet, A. Graillot, S. Monge, R. Borsali and J. J. Robin, Polym. Chem., 2014, 5(8), 2756–2767 RSC.
- K. M. Zepon, I. Otsuka, C. Bouilhac, E. C. Muniz, V. Soldi and R. Borsali, Biomacromolecules, 2015, 16(7), 2012–2024 CrossRef CAS PubMed.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126(3), 187–204 CrossRef CAS PubMed.
- V. P. Torchilin, Pharm. Res., 2007, 24(1), 1–16 CAS.
- C. de Las Heras Alarcon, S. Pennadamand and C. Alexander, Chem. Soc. Rev., 2005, 34(3), 276–285 RSC.
- J. Jiang, B. Qi, M. Lepage and Y. Zhao, Macromolecules, 2007, 40(4), 790–792 CrossRef CAS.
- H. He, Y. Ren, Y. Dou, T. Ding, X. Fang, Y. Xu, H. Xu, W. Zhang and Z. Xie, RSC Adv., 2015, 5(128), 105880–105888 RSC.
- C. Ventura, P. Thornton, S. Giordani and A. Heise, Polym. Chem., 2014, 5(21), 6318–6324 RSC.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35(11), 1068–1083 RSC.
- T. Wang, X. Tang, J. Han, Y. Ding, W. Guo and M. Pei, Macromol. Biosci., 2016, 16(5), 774–783 CrossRef CAS PubMed.
- R. C. Lin, M. G. Mohamed, K. C. Hsu, J. Y. Wu, Y. R. Jheng and S. W. Kuo, RSC Adv., 2016, 6(13), 10683–10696 RSC.
- L. Fertier, H. Koleilat, M. Stemmelen, O. Giani, C. Joly-Duhamel, V. Lapinte and J. J. Robin, Prog. Polym. Sci., 2013, 38(6), 932–962 CrossRef CAS.
- G. Kaur, P. Johnston and K. Saito, Polym. Chem., 2014, 5(7), 2171–2186 RSC.
- Y. Zhao, Macromolecules, 2012, 45(9), 3647–3657 CrossRef CAS.
- S. R. Trenor, A. R. Shultz, B. J. Love and T. E. Long, Chem. Rev., 2004, 104(6), 3059–3078 CrossRef CAS PubMed.
- Y. Zhao, J. Mater. Chem., 2009, 19(28), 4887–4895 RSC.
- J. Liu, J. Zhou and Y. Luo, Bioconjugate Chem., 2012, 23(2), 174–183 CrossRef CAS PubMed.
- Q. Jin, X. Liu, G. Liu and J. Ji, Polymer, 2010, 51(6), 1311–1319 CrossRef CAS.
- G. Y. Liu, C. J. Chen, D.-D. Li, S. S. Wang and J. Ji, J. Mater. Chem., 2012, 22(33), 16865–16871 RSC.
- K. Iliopoulos, O. Krupka, D. Gindre and M. Sallé, J. Am. Chem. Soc., 2010, 132(41), 14343–14345 CrossRef CAS PubMed.
- J. He, L. Tremblay, S. Lacelle and Y. Zhao, Soft Matter, 2011, 7(6), 2380–2386 RSC.
- L. Korchia, C. Bouilhac, V. Lapinte, C. Travelet, R. Borsali and J. J. Robin, Polym. Chem., 2015, 6(33), 6029–6039 RSC.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48(43), 7978–7994 CrossRef CAS PubMed.
- T. X. Viegas, M. D. Bentley, J. M. Harris, Z. Fang, K. Yoon, B. Dizman, R. Weimer, A. Mero, G. Pasut and F. M. Veronese, Bioconjugate Chem., 2011, 22(5), 976–986 CrossRef CAS PubMed.
- R. Luxenhofer, Y. Han, A. Schulz, J. Tong, Z. He, A. V. Kabanov and R. Jordan, Macromol. Rapid Commun., 2012, 33, 1613 CrossRef CAS PubMed.
- C. Travelet, M. Stemmelen, V. Lapinte, J. J. Robin and R. Borsali, J. Nanopart. Res., 2013, 15(6), 1–16 CrossRef.
- B. Guillerm, S. Monge, V. Lapinte and J. J. Robin, J. Polym. Sci., Part A: Polym. Chem., 2013, 51(5), 1118–1128 CrossRef CAS.
- B. Guillerm, S. Monge, V. Lapinte and J. J. Robin, Macromol. Rapid Commun., 2012, 33(19), 1600–1612 CrossRef CAS PubMed.
- D. Rayeroux, V. Lapinte and P. Lacroix-Desmazes, J. Polym. Sci., Part A: Polym. Chem., 2012, 50(22), 4589–4593 CrossRef CAS.
- B. Guillerm, V. Darcos, V. Lapinte, S. Monge-Darcos, J. Coudane and J. J. Robin, Chem. Commun., 2012, 48(23), 2879–2881 RSC.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49(36), 6288–6308 CrossRef CAS PubMed.
- Y. Chujo, K. Sada, A. Naka, R. Nomura and T. Saegusa, Macromolecules, 1993, 26(21), 5611–5614 CrossRef CAS.
- Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1990, 23(10), 2693–2697 CrossRef CAS.
- L. Korchia, C. Bouilhac, J. J. Robin and V. Lapinte, Eur. Polym. J., 2017, 88, 636–644 CrossRef CAS.
- L. Korchia, C. Bouilhac, C. Travelet, R. Borsali, J. J. Robin and V. Lapinte, Soft Matter, 2017, 13, 4507–4519 RSC.
- M. Obi, S. Y. Morino and K. Ichimura, Chem. Mater., 1999, 11(3), 656–664 CrossRef CAS.
- J. Babin, M. Lepage and Y. Zhao, Macromolecules, 2008, 41(4), 1246–1253 CrossRef CAS.
- E. R. Draper, T. O. Mc Donald and D. J. Adams, Chem. Commun., 2015, 51(64), 12827–12830 RSC.
- J. Jiang, B. Qi, M. Lepage and Y. Zhao, Macromolecules, 2007, 40(4), 790–792 CrossRef CAS.
- J. Dai and J. C. Kim, Korean J. Chem. Eng., 2011, 29(3), 323–328 CrossRef.
- I. N. Kurniasih, H. Liang, P. C. Mohr, G. Khot, J. P. Rabe and A. Mohr, Langmuir, 2015, 31(9), 2639–2648 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07094b |
| This journal is © The Royal Society of Chemistry 2017 |