
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew and bioactive natural products from an endophyte of Panax notoginseng†
Jun Xie‡
a,
Ying-Ying Wu‡a,
Tian-Yuan Zhanga,
Meng-Yue Zhanga,
Wei-Wei Zhua,
Elizabeth A. Gullenb,
Zhao-Jie Wangc,
Yung-Chi Chengb and
Yi-Xuan Zhang *a
*a
aSchool of Life Science and Biopharmaceutics, Shenyang Pharmaceutical University, Shenyang, 110016, People's Republic of China. E-mail: zhangyxzsh@163.com; Fax: +86-24-23986576; Tel: +86-24-23986576
bDepartment of Pharmacology, Yale University School of Medicine, New Haven, CT 06520, USA
cYunnan Provincial Academy of Science and Technology, Kunming, 650051, People's Republic of China
First published on 2nd August 2017
Abstract
Five new derivatives of macrolide antibiotic Brefeldin A (BFA, 6), named as Brefeldin E1–E5 (1–5), along with Brefeldin A 7-O-acetate (7), mycotoxins (8–9) and mangrovamides A (10) were produced by an endophytic fungus, Penicillium sp., which was isolated from the healthy root of Panax notoginseng. The structures of 1–5 were established on the basis of their spectroscopic data, while the absolute configurations were assigned using a modified Mosher's method. All compounds were evaluated for their cytotoxic, antiviral and antimicrobial activities. Compounds 1–5 and 8–10 displayed low or moderate cytotoxicity against a panel of cancer cell lines. Compounds 1, 2, 4, 5, and 8–10 showed moderate antimicrobial activity. Compound 6 showed strong anticancer and antiviral properties. Additionally, it demonstrated broad-spectrum activity against human pathogenic bacteria and fungal pathogens that can cause root-rot disease in Panax notoginseng, including Escherichia coli, Staphylococcus aureus, Bacillus cereus, Klebsiella pneumonia, Candida albicans, Fusarium solani, Cylindrocarpon didynum and Alternaria panax. Compound 7, which could be mediated by 6 through the acetylation at the 7-hydroxyl, showed similar bioactivities to compound 6. Further studies of the cellular mechanism of compounds 6 and 7 showed that they arrested HepG2 cells at the S phase. Due to the similarities in the basic carbon skeleton and the chemical construction correlations between compounds 1–7, the plausible biosynthetic pathway of the BFA series of compounds has been proposed and their structure–activity relationships are also discussed.
Introduction
Panax notoginseng F. H. Chen (Araliaceae) is a valued traditional Chinese medicinal herb.1 In traditional herbal prescriptions, it has been widely used for thousands of years as a highly important medicinal plant for its antioxidant,2 antidiabetic3 and antitumor4 pharmacological effects. As a perennial plant, P. notoginseng should be grown in the field for at least three years to obtain high-quality raw roots.5 Because of the required long-term continuous cultivation,6–8 P. notoginseng is vulnerable to attacks by soil-borne pathogens including fungi, bacteria, and nematodes. Endophytic fungi play a key role in plant defense and could be used as a promising source for biocontrol agents.9A total of 89 fungi were obtained from the roots, stems, leaves, and seeds of P. notoginseng by Youkun Zheng et al.,10 63.4% exhibited activity against at least one of the pathogens tested. Specifically endophytic Trichoderma gamsii secreted volatile organic compounds with antagonistic effects on the pathogenic root-rot fungi of P. notoginseng.11 Furthermore, many endophytic fungi are capable of synthesizing various bioactive compounds that are used today as therapeutic agents against numerous diseases.12 The endophytic fungus Taxomyces andreanae provided a novel and promising approach to produce the compound paclitaxel.13 The endophytic Alternaria sp. obtained from Sabina vulgaris could produce podophyllotoxin14 which is a well-known aryltetralin lignan with potent anticancer, antiviral, antioxidant, antibacterial, immunostimulation and anti-rheumatic properties. Entrophospora infrequens, obtained from plant Nothapodytes foetida, has the ability to produce camptothecin (CPT).15 Two famous CPT derivatives, hycamtin (topotecan) and camptostar (irinotecan), have already been in clinical use to overcome ovarian, small lung refractory ovarian and other cancers all over the world.16
Known as one of the most versatile “body defense factories”, Penicillum janthinellum, can produce gibberellins17 that are known for its plant protection effects. It also produces emodin,18 fumitremorgin C,19 hadacidin,20 and epipolythiodioxopiperazine (ETP) alkaloids21 that are known to have good antitumor activities. Here we report the investigation of metabolites produced by an endophytic strain, Penicillium sp. SYP-ZL1031, that was isolated from the root of a healthy three-year-old P. notoginseng. Using NMR and MS data analysis, along with Mosher's method based on MTPA esters, five new compounds 1–5 named brefeldin E1–E5 and brefeldin A (BFA, 6), brefeldin A 7-O-acetate (7), mycotoxins (8–9), mangrovamides A (10) were identified. BFA is a 16-membered ring macrolide antibiotic initially isolated from Penicillium decumbens.22 It has been successively separated from P. simplicissimum,23 P. cyaneum24 and Eupenicillium brefeldianum.25 BFA has been used as a leading anticancer agent,26 which interferes with the traffic endoplasmic reticulum (ER) to the Golgi membrane, causing the disassembly and redistribution of Golgi apparatus proteins in eukaryotic cells.27,28 In addition, BFA showed activity against neuroendocrine tumor and prostatic cancer cells via a p53-independent mechanism.26,29,30 Here we introduce and describe an endophytic fungus, Penicillium sp., that produces BFA series compounds. This study aims to screen new or known active compounds with biological cytotoxic, antiviral, antimicrobial activities.
Results and discussion
The investigation of metabolites produced by an endophytic strain, Penicillium sp. SYP-ZL1031. SYP-ZL1031 was cultured at 28 °C in rice. From the ethyl acetate extracts of the fermented broths, ten compounds including five new compounds (1–5) and five known compounds, BFA (6), brefeldinA 7-O-acetate (7), mycotoxins (8–9) and mangrovamides A (10) (Fig. 1) were isolated. They were all obtained from endophytic strain for the first time.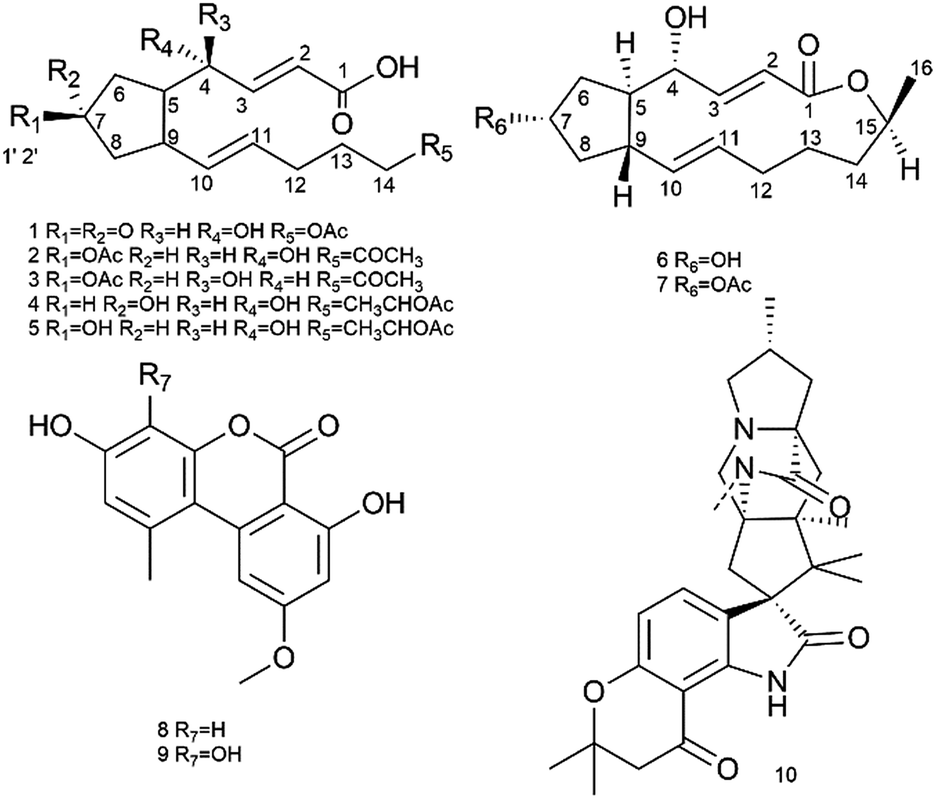 | ||
| Fig. 1 Chemical structures of compounds 1–10. | ||
Structural elucidation of the new compounds (1–5)
Using a combination of HR-ESI-MS and NMR spectroscopy, a colorless oil, brefeldin E1 (1), was analyzed for C16H22O6. The UV λmax at 203 nm indicated the presence of an α,β-unsaturated double bond. The absorption bands at 3420 (strong wide wave), 1737 and 1657 cm−1 in IR spectrum suggested the presence of hydroxyl carboxylic acid carbonyl, and α,β-unsaturated ketone carbonyl functionalities (S46†). The circular dichroism (CD) curve (S51-A†) of 1 showed a positive Cotton Effect (CE) at Δε208 4.64, a negative CE at Δε294 − 2.03. The 1H and 13C-NMR spectra (S1–S2† and Table 1) together with HSQC data (S4†) of 1 showed the presence of one methyl [δH 2.03 s and δC 21.0], five methylenes, seven methines, including one oxygenated [δH 4.38 brs δC 69.7] and four olefins [δH 6.61 dd (J = 16.1, 5.0 Hz, H-3), 6.01 d (J = 16.1, Hz, H-2), 5.60 dt (J = 15.2, 6.8 Hz, H-11), 5.46 dd (J = 15.2, 8.3 Hz, H-10); δC 146.3, 127.1, 132.6, 133.8], and three carbonyls (δC 220.2, 174.9, 173.1). The planar structure of 1 (Fig. 2A) was established by the 2D NMR data. A five-membered ring was deduced by the COSY (S3†) correlations (H-5/H-6, H-8/H-9) and the HMBC (S5†) correlations from H-6 to C-7 and H-8 to C-7. The additional COSY correlations (H-5/H-4/H-3/H-2) and HMBC correlations from H-4 to C-9, C-3, C-2 and H-3 to C-1 constructed fragment A. Fragment B from C-9 to C-16 was constructed by the COSY correlations from H-9 to H-14 and HMBC correlations from H-14 and H-16 to C-15. These two fragments were further connected to the five-membered ring via C-5 and C-9, as evidenced by the HMBC correlations from H-9 to C-11, and from H-4 to C-9. The double bonds in Δ2,3 and Δ10,11 were assigned to be E geometry on the basis of the coupling constants (J2–3 = 16.1, J10–11 = 15.2 Hz). The relative configurations of the H-5 and H-9 to H-4 were determined based on the coupling constants and NOESY (S6†) correlations. The method of homonuclear decoupling31,32 is applied to obtain the coupling constants H-4/H-5 (J4–5 = 1.4 Hz). We can infer from the coupling constant H-4/H-5 (J4–5 = 1.4 Hz) that the angle between H-4 and H-5 is about 90 degree and H-4/H-5 were on trans-orientation of the molecule.33,34 On the other hand, based on the NOESY (S6†) correlations, H-9 did not have a correlation with H-5, but instead with H-4, demonstrating that H-9/H-5 were on trans-orientation of the molecule, while H-9/H-4 were on cis-orientation.35 The absolute configuration of compound 1 was determined through Mosher's method.36 The ΔδSR values between 1a and 1b (R- and S-MTPA esters of 1 on OH-4, respectively) were positive for H-2/3 and negative for H-5/6/8 (Fig. 3 and S53a†), which indicated the 4S configuration.| Pos | 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 174.9 C | 175.4 C | 171.4 C | 172.9 C | 172.9 C | |||||
| 2 | 6.01, d (16.1) | 127.1 CH | 5.99, d (15.5) | 126.7 CH | 5.99, d (15.5) | 122.2 CH | 5.98, d (15.6) | 124.9 CH | 5.99, d (15.5) | 128.0 CH |
| 3 | 6.61, dd (5.0, 16.1) | 146.3 CH | 6.67, dd (5.1, 15.5) | 147.6 CH | 6.89, dd (4.3, 15.5) | 152.3 CH | 6.75, d (15.6) | 149.5 CH | 6.58, dd (5.3, 15.5) | 146.6 CH |
| 4 | 4.38, brs | 69.7 CH | 4.22, brs | 70.9 CH | 4.24, brs | 71.2 CH | 4.20, m | 71.4 CH | 4.19, m | 71.5 CH |
| 5 | 2.12, m | 49.2 CH | 1.92, m | 50.8 CH | 1.94, m | 50.6 CH | 1.95, m | 50.5 CH | 1.95, m | 50.6 CH |
| 6 | 2.41, dd (10.4, 18.1) | 46.6 CH2 | 1.91, m | 33.4 CH2 | 1.90, m | 33.9 CH2 | 1.84, m | 35.9 CH2 | 1.86, m | 35.8 CH2 |
| 2.13, m | 1.67, m | 1.68, m | 1.50, m | 1.58, m | ||||||
| 7 | 220.2 C | 5.05, m | 76.9 CH | 5.05, m | 76.8 CH | 4.19, m | 73.0 CH | 4.17, m | 73.1 CH | |
| 8 | 2.27, dd (11.2, 18.7) | 38.7 CH2 | 2.28, m | 40.8 CH2 | 2.29, m | 40.9 CH2 | 2.14, m | 43.8 CH2 | 2.13, m | 43.7 CH2 |
| 2.20, dd (7.6, 18.7) | 1.51, m | 1.52, m | 1.43, m | 1.42, m | ||||||
| 9 | 2.87, m | 43.0 CH | 2.49, m | 44.5 CH | 2.49, m | 44.6 CH | 2.45, m | 44.7 CH | 2.45, m | 44.6 CH |
| 10 | 5.46, dd (8.3, 15.2) | 133.8 CH | 5.37, dd (8.4, 15.2) | 135.5 CH | 5.36, dd (8.5, 15.3) | 135.4 CH | 5.39, dd (7.9, 15.2) | 135.8 CH | 5.39, dd (7.5, 15.2) | 135.9 CH |
| 11 | 5.60, dt (6.8, 15.2) | 132.6 CH | 5.45, dt (6.7, 15.2) | 131.7 CH | 5.45, dd (6.7, 15.3) | 131.9 CH | 5.44, dt (6.3, 15.2) | 131.5 CH | 5.43, dt (6.2, 15.2) | 131.4 CH |
| 12 | 2.15, m | 30.0 CH2 | 1.62, m | 24.8 CH2 | 1.62, m | 24.7 CH2 | 2.00, m | 33.4 CH2 | 2.02, m | 33.4 CH2 |
| 2.15, m | 1.62, m | 1.62, m | 2.00, m | 2.02, m | ||||||
| 13 | 1.72, m | 29.6 CH2 | 2.02, m | 33.0 CH2 | 2.01, m | 33.0 CH2 | 1.38, m | 26.6 CH2 | 1.37, m | 26.6 CH2 |
| 1.72, m | 2.02, m | 2.01, m | 1.38, m | 1.37, m | ||||||
| 14 | 4.08, t (6.6) | 65.1 CH2 | 2.48, t (7.3) | 43.7 CH2 | 2.48, t (7.4) | 43.7 CH2 | 1.57, m | 36.5 CH2 | 1.59, m | 36.6 CH2 |
| 4.08, t (6.6) | 2.48, t (7.3) | 2.48, t (7.4) | 1.57, m | 1.50, m | ||||||
| 15 | 173.1 C | 212.2 C | 212.1 C | 4.87, m | 72.5 CH | 4.87, m | 72.5 CH | |||
| 16 | 2.03, s | 21.0 CH3 | 2.13, s | 30.1 CH3 | 2.13, s | 30.0 CH3 | 1.20, d (6.2) | 20.4 CH3 | 1.20, d (6.2) | 20.4 CH3 |
| 1′/17 | 172.9 C | 172.8 C | 171.2 C | 173.0 C | ||||||
| 2′/18 | 1.99, s | 21.3 CH3 | 2.00, s | 21.3 CH3 | 2.00, s | 21.4 CH3 | 2.00, s | 21.4 CH3 | ||
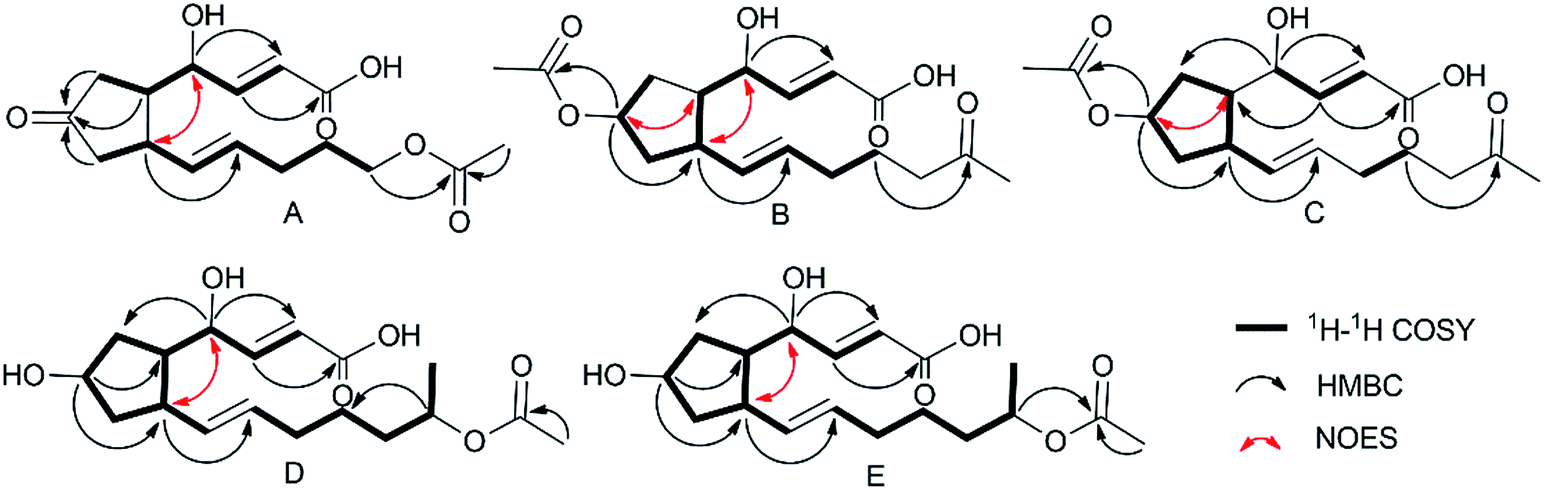 | ||
| Fig. 2 Key 2D NMR correlations of 1 (A), 2 (B), 3 (C), 4 (D), 5 (E). | ||
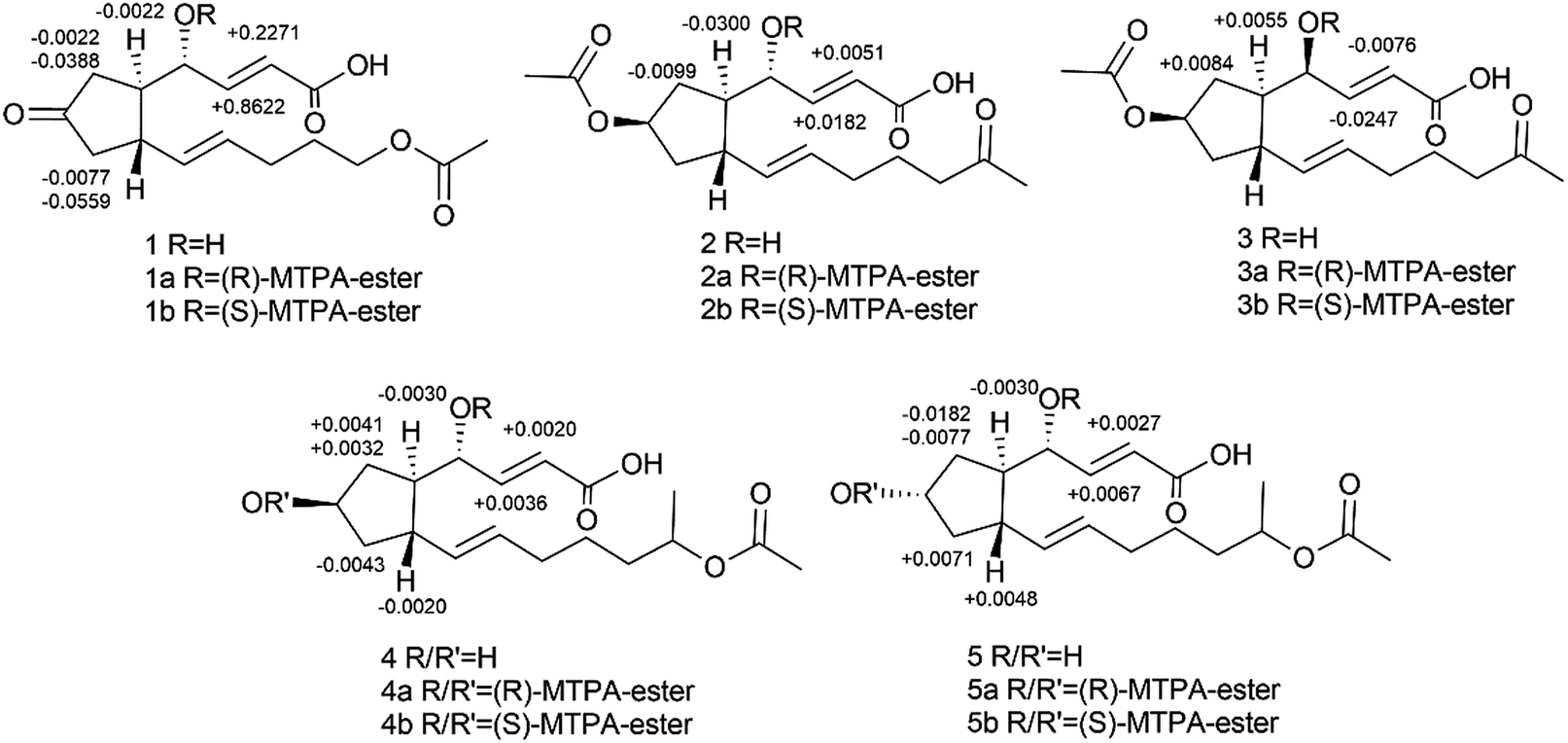 | ||
| Fig. 3 Δδ values (δb–δa in ppm) obtained for the MTPA esters of compounds 1–5. | ||
Brefeldin E2 (2) and E3 (3) were extracted as colorless oils. Using HR-ESI-MS and NMR data, it was deduced that both had formula of C18H26O6. The CD curve (S51-B†) of 2 showed a positive CE at Δε208 5.57, a negative CE at Δε195 − 3.32. The CD curve (S51-C†) of 3 showed two positive CE at Δε197 0.50 and Δε223 1.05. The 1H and 13C-NMR data of 2 and 3 (S8–S9, S15–S16† and Table 1) were similar to those of 1, and the molecular weight differed by 28 Dalton from 1. The major differences in NMR data between compounds 2, 3 and compound 1 were found to be the presence of the O-acetyl group in C-7 (δC 172.9/172.8 and 21.3/21.3, which were assigned to C-1′ and C-2′ in 2 and 3). In addition, the O-acetyl group in C-14 of compound 1 was replaced by a ketone group in compound 2 and 3. The HMBC (S12 and S19†) correlation from H-7 and H-13 (δH 5.05/5.05m, 2.02/2.01m) to C-1′ and C-15 (δC 172.9/172.8/212.2/212.1) confirmed the planar structure of 2/3. The relative configurations of the H-5, H-9 and H-4 in 2 were using the same method as compound 1. The coupling constants H-4/H-5 (J4–5 = 0.9 Hz) deduced the angle between H-4/H-5 was about to 90 degree and H-4/H-5 were on trans-orientation of the molecule.33,34 The NOESY (S13†) correlations revealed that H-9/H-5 were on the trans-orientation of the molecule, and H-9/H-4 were on the cis-orientation. The other key NOESY (S13†) of compound 2 were observed between H-7, H-5 and H-9. H-7 did not have a correlation with H-9, but with H-5, demonstrated that H-7/H-9 were on a trans-orientation of the molecule while H-7/H-5 were on the cis-orientation. The relative configurations (S20†) of the H-5, H-9 and H-4 in 3 were different from compound 1. The coupling constants between H-4 and H-5 (J4–5 = 3.8 Hz) were calculated using homonuclear decoupling31,32 method. The coupling constant J4–5 = 3.8 Hz indicated that the angle between H-4 and H-5 is about 0 or 180 degrees and H-4/H-5 were on cis-orientation of the molecule.33,34 Based on the NOESY correlations, H-9 had no correlation with H-5 and H-4, which demonstrated H-9/H-5 were on the trans-orientation of the molecule, and H-5/H-4 were on the cis-orientation. The relative configurations of H-7, H-5 and H-9 in 3 were the same as compound 2. The absolute configurations of compounds 2 and 3 were also determined based on Mosher's method (Fig. 3 and S53b,c†). The ΔδSR values between 2a and 2b (R- and S-MTPA esters of 2 on OH-4, respectively) were negative for H-5/H-6 and positive for H-2/H-3 (Fig. 3 and S53b†), which indicated the 4S configuration. The opposite result in compound 3 where 3a and 3b (R- and S-MTPA esters of 3 on OH-4, respectively) were positive for H-5/H-6 and negative for H-2/H-3 (Fig. 3 and S53c†), indicated the 4R configuration.
Brefeldin E4 (4) and E5 (5) were extracted as a colorless oil and assigned the same molecular formulas C18H28O6 using HR-ESI-MS analysis. The CD curve (S51-D†) of 4 showed a positive CE at Δε210 3.74. The CD curve (S51-E†) of 5 showed a positive CE at Δε207 4.76, a negative CE at Δε194 − 1.59. Detailed analysis of 1D NMR data (S22–S23, S29–S30† and Table 1) indicated that compounds 4 and 5 showed a striking resemblance to compounds 1, respectively, except for the presence of C-15 (δC 72.5/72.5 in 4/5), C-16 (δC 20.4/20.4 in 4/5), and the C-7 ketone of compound 1 was replaced to be a hydroxyl group in compounds 4 and 5. The HMBC (S26†) correlation from H-15 (δH 4.87m) to C-13 (δC 26.6) confirmed the planar structure of 4. And the HMBC (S33†) correlation from H-15 (δH 4.87m) to C-17 (δC 173.0) confirmed the planar structure of 5. The relative configuration (S27 and S34†) of the H-5, H-9 and H-4 in compound 4 and 5 were the same to compound 1 based on the NOESY correlations and coupling constants H-4/H-5 (J4–5 = 0.4/0.8 Hz in 4/5).33,34 We can infer from the coupling constant H-4/H-5 (J4–5 = 0.4/0.8 Hz in 4/5) that the angle between H-4 and H-5 is about 90 degree and H-4/H-5 were on trans-orientation of the molecule.33,34 On the other hand, based on the NOESY (S27 and S34†) correlations, H-9 did not have a correlation with H-5, but instead with H-4, demonstrating that H-9/H-5 were on a trans-orientation of the molecule, and H-9/H-4 were on the cis-orientation. The absolute stereochemistry of positions C-4 and C-7 of brefeldin E4 (4) and E5 (5) were determined by the application of Mosher's method.37 Which suggests 4S,7R for compound 4 (Fig. 3 and S53d†) and 4S,7S for compound 5 (Fig. 3 and S53e†).
The structures of compounds 6–10 were determined by HR-ESI-MS and NMR spectroscopy (1H and 13C-NMR). BFA (6) and brefeldin A 7-O-acetate (7) were deduced to be very compatible with spectroscopic data found in the literature.38,39 The 1H and 13C NMR of compound 8–10 were in good agreement with alternariol-5-O-methyl ether,40 3′-hydroxyalternariol-5-O-methyl ether40 and mangrovamides A41 respectively. The similar CD profiles and same sign of optical rotations of compound 10 suggested that the absolute configuration of 10 was the same as mangrovamides A.
In vitro cytotoxic activity and cell cycle analysis
The isolated metabolites on cytotoxic and antiviral were evaluated. As shown in the Table 2, it was found that brefeldin E1–E5 (1–5), with the open-ring of macrolide of BFA, and compound 8–10 displayed low cytotoxicities and antiviral activity respectively. In contrast, the compounds 6–7 were identified as being excellent activity compounds when evaluated against cell lines (293, HepG2, Huh7 and KB), showing an ID50 value from 0.024 μM to 0.62 μM and antiviral (HCV, HBV) with ID50 value from 0.013 μM to 0.022 μM, respectively. Both BFA (6) and brefeldin A 7-O-acetate (7) demonstrate the same inhibitory behavior of protein translocation from endoplasmic reticulum to the Golgi.27 The life cycle of viral replication also requires this process. Thus, it is not surprising that there is an impact on HCV and HBV. In cancer cell lines, cell cycle arrest is one of the most important strategies used to stop or limit cancer proliferation.42 As shown in Table 2, compounds 6–7 markedly suppressed the proliferation of HepG2 cells compared with positive drug LODDC with ID50 values of 0.024 μM. To explore whether the cytotoxicity of compounds 6–7 was due to cell cycle arrest, we first examined the effect on cell cycle distribution using propidiumiodide (PI) staining with flow cytometry analysis. Compounds 6–7 definitely increased the percentage of HepG2 cells at the S phase in a concentration of 0.072 μM and 0.1 μM at 48 h (Fig. 4 and 5). This demonstrated that compounds 6–7 inhibited cell growth via inducing S phase arrest in HepG2 cells.| Compound | ID50 (μM) | |||||
|---|---|---|---|---|---|---|
| 293 | HepG2 | Huh7 | KB | HCV | HBV | |
| a Results are expressed as ID50 values ± standard deviation in μM. LODDC, interferon, 3TC and DMSO were used as positive and negative controls.b Key: 293 = human renal epithelial cell line; HepG2, Huh7; = human hepatocellular carcinoma cell line; KB = human oral epidermoid carcinoma cell line; HCV = hepatitis C virus; HBV = hepatitis B virus. | ||||||
| 1 | >40 | >40 | >40 | >30 | >20 | >20 |
| 2 | >40 | >40 | >40 | >30 | >20 | >20 |
| 3 | >40 | >40 | >40 | >15 | >20 | >20 |
| 4 | >40 | >40 | >40 | >30 | >20 | >20 |
| 5 | >40 | >40 | >40 | >30 | >20 | >20 |
| 6 | 0.45 ± 0.008 | 0.024 ± 0.005 | 0.028 ± 0.006 | 0.0625 ± 0.0075 | 0.022 | >0.008 |
| 7 | 0.62 ± 0.1 | 0.024 ± 0.002 | 0.035 ± 0.005 | 0.06 ± 0.009 | 0.0132 ± 0.0068 | >0.004 |
| 8 | 16.9 ± 1.1 | >20 | >20 | >30 | >20 | >10 |
| 9 | 15.5 ± 0.9 | >20 | >20 | >30 | >20 | >10 |
| 10 | >50 | 3.7 ± 0.6 | 2.8 ± 0.6 | 11.6 ± 1.2 | 1.2 ± 0.13 | >1.8 |
| LODDC | 0.36 ± 0.00 | 0.071 ± 0.008 | 0.047 ± 0.008 | 0.058 ± 0.003 | Nd | Nd |
| Interferon | Nd | Nd | Nd | Nd | 0.013 ± 0.002 | Nd |
| 3TC | Nd | Nd | Nd | Nd | Nd | 0.04 |
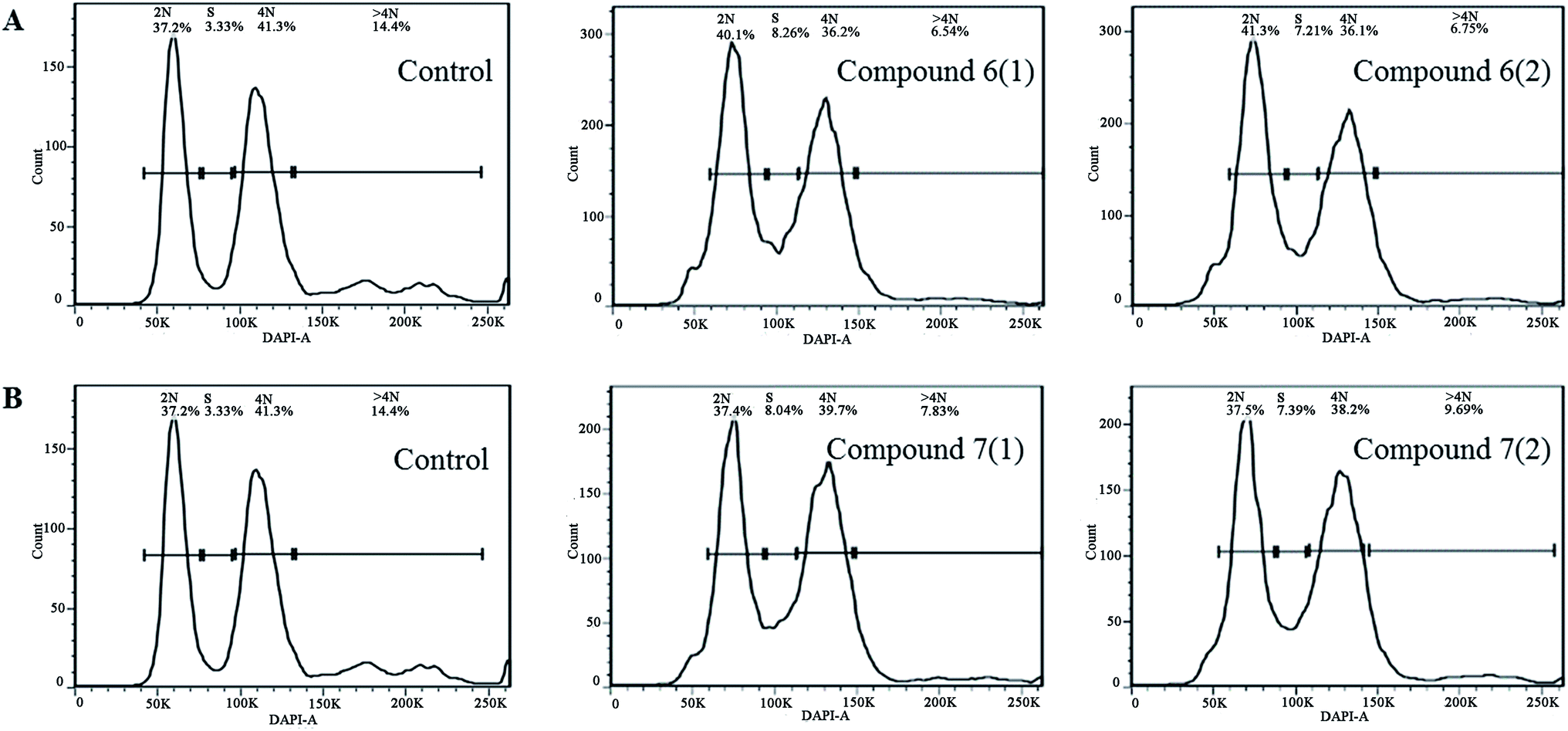 | ||
| Fig. 4 The flow cytometry histograms of HepG2 cancer cells after treatment with compound 6 (A) and 7 (B) for 30 h. The untreated group was used for negative control. | ||
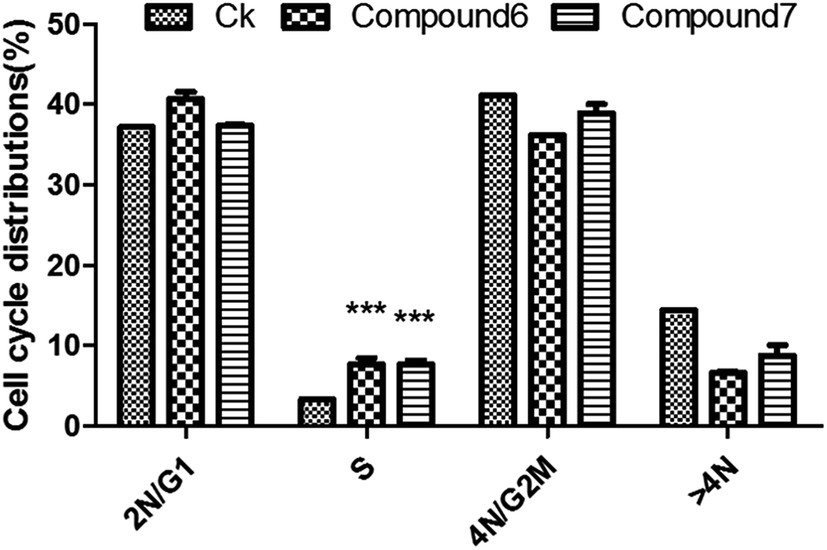 | ||
| Fig. 5 Cell cycle distribution in HepG2 cells treated with compounds 6–7. Values are % events. *p < 0.05 between drug-treated and control group. Data are mean ± SD of two independent experiments. | ||
Antimicrobial assay
The 96-well flat-bottomed assay43 was used to study the antimicrobial activity. To better understand the antibacterial activity of the compound 1–10, log-phase bacterial suspensions of Escherichia coli, Staphylococcus aureus, Bacillus cereus, Klebsiella pneumonia and Candida albicans were incubated together with test samples, and the OD600 values were measured after 24 hours. The results showed that the OD600 value of the brefeldin E1 (1), E4–E5 (4–5), brefeldin A (6), brefeldin A 7-O-acetate (7), 3′-hydroxyalternariol-5-O-methyl ether (9) and mangrovamides A (10) groups were significantly lower than those of CK, indicating that compounds 1, 4–7, 9–10 had satisfactory antibacterial activity on at least one of the pathogens tested (Fig. 4A–E). Of those compounds, compound 5 showed antibacterial activity to E. coli and S. aureus. The results indicated that the compound 5 had inhibitory effect on both Gram-negative and Gram-positive bacteria. Moreover, compound 6–7 had strong and broad-spectrum activities against human pathogenic bacteria (Fig. 6A–E).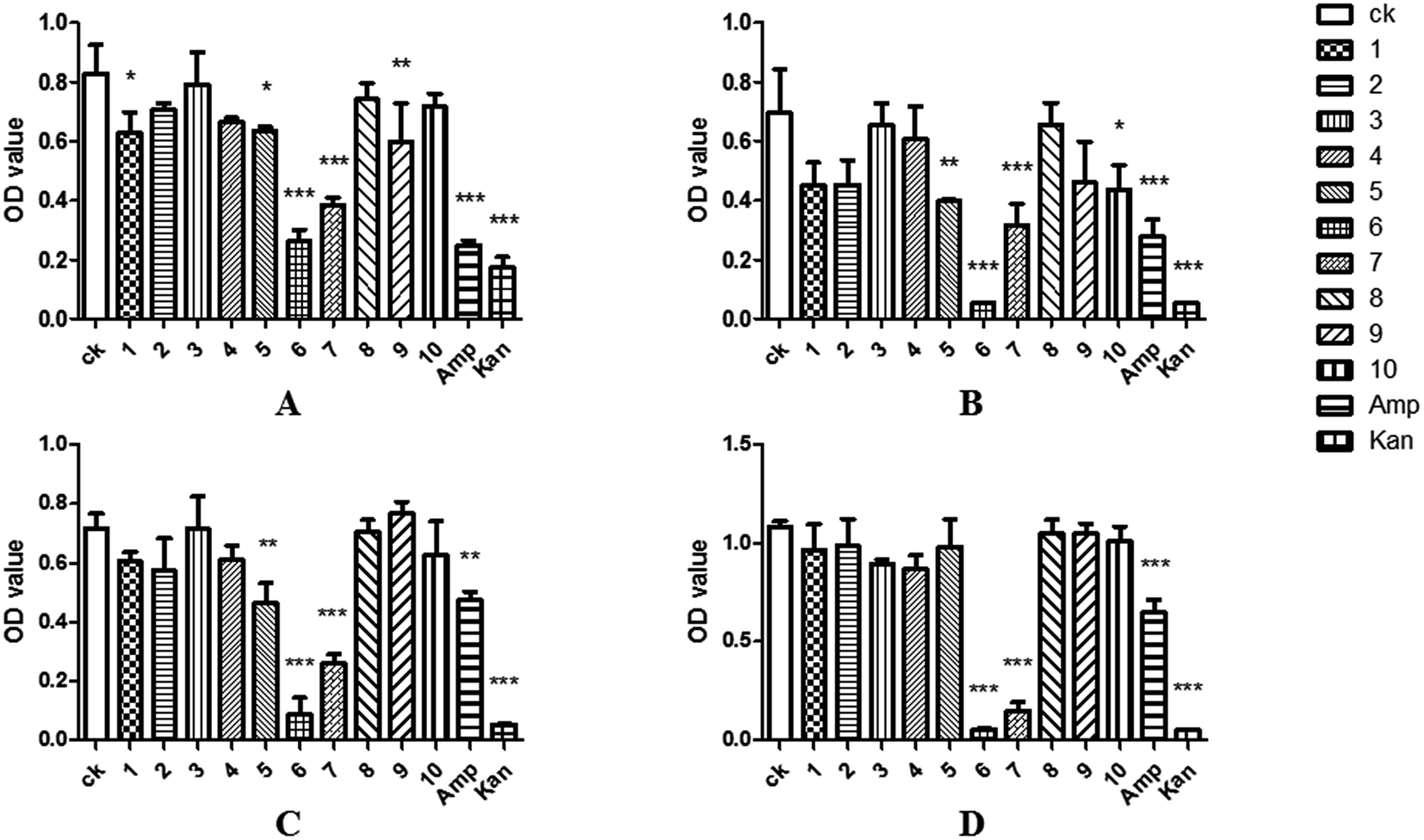 | ||
| Fig. 6 Antibacterial activities of compounds 1–10 against E. coli (A), S. aureus (B), B. cereus (C), K. pneumonia (D); ampicillin (Amp) and kanamycin (Kan), were used as positive controls. Data are mean ± SD of three independent experiments, *(P < 0.05) indicate statistically significant differences between ck and compounds 1–10. | ||
As endophytic fungi, the metabolic effects of SYP-ZL1031 on pathogenic fungi of P. notoginseng were also evaluated. The experimental method is similar to the previously mentioned43 before. Compounds 1–2, 4–10 had the ability to inhibit the growth of Cylindrosporium sp. on different levels. Meanwhile, compound 6 and 7 exhibited strong antagonistic activities against three fungal pathogens (Cylindrocarpon didynum, Alternaria panax, Fusarium solani) that can cause root-rot disease in Panax notoginseng (Fig. 7F–H). The results suggest that SYP-ZL1031 endophytic fungi may be a potential candidate for the metabolites of bioactive compounds, and has the potential to be used as a biological control agent against root-rot disease of P. notoginseng.
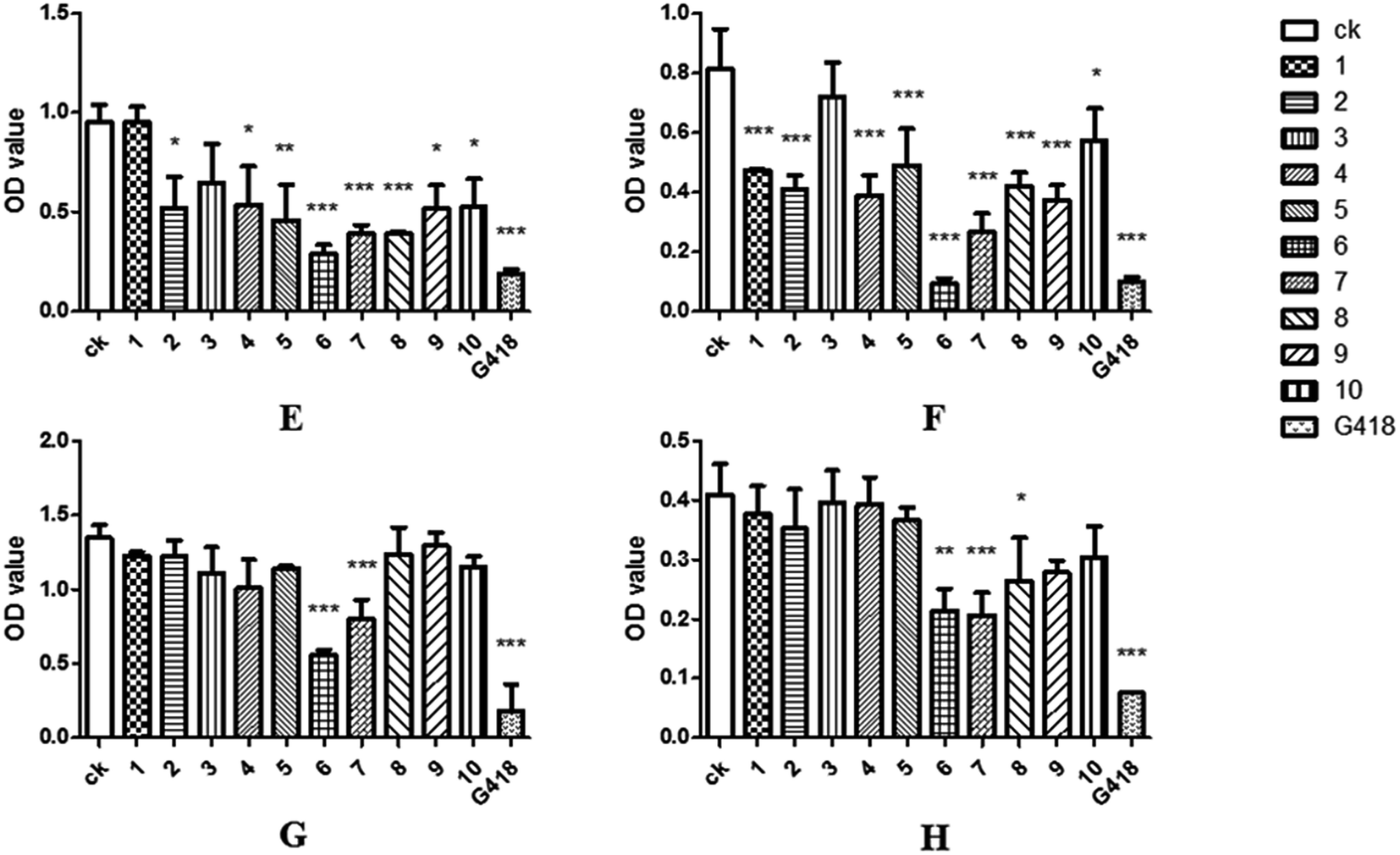 | ||
| Fig. 7 Antifungal activities of compounds 1–10 against C. albicans (E) Cylindrocarpon didynum, (F), Alternaria panax (G), Fusarium solani (H); G418 was used as positive control. Data are mean ± SD of three independent experiments, *(P < 0.05) indicate statistically significant differences between ck and compounds 1–10. | ||
Structure–activity relationship
Structure–activity relationship was also discussed between compounds 1–7. BFA series new compounds 1–5, are the ring-opened BFA seco-acid. By comparing compounds 1–5 and compounds 6–7, the presence of ring-opened reduced the biological activities against all tested cancer cell lines, virus and pathogenic bacteria/fungi (Table 2, Fig. 6 and 7), which revealed that the conformational rigidity of 13-membered lactone in compounds 6–7 had a great influence on their antitumor, antiviral and antimicrobial potency. Furthermore, C-7 hydroxyl group (compound 6) may be a preferred substituent groups than that of acetyl group (compound 7), for its stronger biological activities according to the ID50 values of cancer cell lines. Compounds 1–5 had the same skeleton apart from the type of substituent groups at C-4 and C-7. A comparison of the antimicrobial data between 4 and 5 indicated that the C-7S configuration in compound 5 showed significant inhibition of pathogenic bacteria/fungi growth. The results revealed that difference in configuration at stereocenter C7 showed different antimicrobial activities.Plausible biosynthetic pathways
Our isolation of compounds 1–7 raises interesting questions about their biosynthesis. BFA (6), known as a 16-membered ring macrolide antibiotic,22 belongs to the polyketides group which constitute a large class of natural products with similar biosynthetic grounds.44 The basic carbon skeleton of BFA series compounds might be derived from an acetate starter group, with malonate acting as the chain extender.43 The molecules have then, in some cases, been made by decarboxylation/hydroxylation/aldol cyclization/enolization reactions, which were likely to be the precursors of BFA series compounds. The plausible biosynthetic pathways of 1–7 are shown in (S52†).The structures of compounds 1–7 could be explained as being derived from poly-β-keto chains, formed by coupling of acetic acid (C2) units via condensation reactions.44 The formation of the poly-β-keto chain could be envisaged as a series of Claisen reactions. Two molecules of acetyl-CoA could participate in a Claisen condensation giving acetoacetyl-CoA, and this reaction could be repeated to generate a poly-β-keto ester of appropriate chain length. During this period, the conversion of acetyl-CoA into malonyl-CoA increased the acidity of the α-hydrogens, and thus provided a better nucleophile for the Claisen condensation. Further cyclization and other modifications can create a wide range of BFA series compounds. The stereochemistry in the chain is controlled by the condensation and reduction steps during chain extension, such as compounds 1–5. And a reassuring feature is that there appears to be a considerable degree of stereochemical uniformity throughout the known macrolide antibiotics 6 and 7.44
Conclusions
In conclusion, the endophyte metabolites study described the isolation and structural identification of five new BFA analogues, along with five known compounds from Penicillium sp. Their cytotoxic activities were evaluated against four kinds of human cancer cell lines. Compounds 6–7 had higher cytotoxicity towards a panel of cancer cells. Among them, compounds 6–7 exhibited higher cytotoxic activities than the positive control LODDC for HepG2 and Huh7 cells. By means of cell cycle detection using flow cytometry, we discovered that compounds 6–7 inhibited HepG2 cell proliferation by arresting at the S phase. The antimicrobial results showed that compound 1, 4–7, 9–10 had a satisfactory antibacterial activity at least on one of the pathogens tested. Compoud 1–2 and 4–10 had the ability to inhibit the growth of Cylindrocarpon didynum. Besides, the structure–activity relationships were discussed based on cytotoxicity and antimicrobial results, and the plausible biosynthetic pathways of compounds 1–7 were also proposed. The investigation indicated that endophytic fungi Penicillium sp. SYP-ZL1031 produced potential candidate of bioactive metabolites for antitumor, antiviral, antibacterial and have the potential as biological control agents of root-rot disease of P. notoginseng.Experimental section
General experimental procedures
Optical rotations were measured with a P-2000 Digital Polarimeter (JASCO, United Kingdom). UV spectra were recorded on an Inesa L5S spectrophotometer. IR spectra were obtained with an Equinox55 spectrophotometer in KBr discs. Circular dichroism (CD) spectrum was recorded on a MOS-450 spectrometer (Bio-Logic Science, France). The NMR spectra were run on Bruker AVANCE-125 or AVANCE-600 NMR spectrometers (Rheinstetten, Germany). HR-ESI-MS data were obtained on a Bruker Customer micr OTOF-Q 125 mass spectrometer (MA, Germany).Isolation and identification of fungal material
The three-year-old healthy P. notoginseng was collected from Wenshan, Yunnan province of China. After surface sterilization with 75% EtOH for 60 s, the root of P. notoginseng was rinsed in sterile water. Then the root was aseptically cut into small pieces and pressed onto potato dextrose agar plates. The DNA sequence of the 18S-ITS1-5.8S-ITS2-28S, which derived from the fungal strain has been submitted and deposited at GenBank with accession number KU360251. BLAST search result revealed that the isolate belongs to the genus Penicillium, and showed high identity (100%) to the species of Penicillium sp., so we named it Penicillium sp. SYP-ZL1031.Fermentation and extraction
Fermentation was carried out in 50 Erlenmeyer flasks (250 mL) each containing 75 g rice. Sterile water (100 mL) was added to each flask, and the contents were autoclaved at 121 °C for 30 min. After cooling to room temperature, each flask was inoculated with 2.0 mL of the spore and incubated at 28 °C for 30 days. The fermented material was extracted thoroughly with ethyl acetate (20 L) and all the organic solvent was concentrated under reduced pressure to afford the crude extract (75 g), which was dissolve again using 90% MeOH–H2O. Then, it was extracted by the same volume hexane to obtain the whole crude extract (50 g).Purification
The extract chromatographed over silica gel column chromatography (CC) (25 × 10 cm) using CH2Cl2–MeOH gradient elution (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1) to give four fractions (A–D). The fraction B (27.5 g) eluted with MeOH–H2O (1:9 to 10:0) was further separated by ODS column to obtain fifteen subfractions (B1–B15). Fractions B4–B12 showed antimicrobial activity by 96-well flat-bottomed plate assay.
:1 to 1:1) to give four fractions (A–D). The fraction B (27.5 g) eluted with MeOH–H2O (1:9 to 10:0) was further separated by ODS column to obtain fifteen subfractions (B1–B15). Fractions B4–B12 showed antimicrobial activity by 96-well flat-bottomed plate assay.
Then the subfraction B4 was purified by Sephadex LH-20, giving five subfractions (fraction B4-1 to fraction B4-5). Fraction B4-2 was further purified by semi-preparative HPLC (CH3CN–H2O, v/v 25:75, 3.0 mL min−1) to yield compound 1 (9.8 mg). Compounds 2 (9.3 mg) and 3 (10.4 mg) were obtained from B4-4 and B4-5 separately by semi-preparative HPLC (CH3CN–H2O, v/v 22:78, 3.0 mL min−1). Fraction B4-1 was applied on semi-preparative HPLC (CH3CN–H2O, v/v 20:80, 3.0 mL min−1) to afford the compound 4 (9.2 mg) and 5 (14.8 mg). A colorless needle crystal was crystallization precipitation in MeOH of subfraction B7, which was compound 6 (38.9 mg). Subfraction B12 was further passed over a Sephadex LH-20 column and then purified by semipreparative HPLC (CH3CN–H2O, v/v 35:65, 3.0 mL min−1) to produce compound 7 (9.7 mg) and compound 10 (10.3 mg). Subfraction B10 was further purified by semi-preparative HPLC (CH3CN–H2O, v/v 30:70, 3.0 mL min−1) to yield compound 8 (9.0 mg) and 9 (8.1 mg).
ε) 203 nm; IR (KBr) νmax 3420, 2923, 2856, 1737, 1657, 1566, 1385, 1245, 1156, 1109, 1052, 1032, 1018, 606 cm−1; 1H and 13C-NMR data, see Table 1; HR-ESI-MS m/z 333.1312 [M + Na]+ (calcd for C16H22NaO6).ε) 203 nm; IR (KBr) νmax 3423, 2924, 1711, 1656, 1381, 1248, 1113, 1051, 1031, 603 cm−1; 1H and 13C-NMR data, see Table 1; HR-ESI-MS m/z 361.1623 [M + Na]+ (calcd for C18H26NaO6).ε) 203 nm; IR (KBr) νmax 3442, 2169, 1711, 1632, 1402, 1384, 1267, 1117, 1015, 823, 703 cm−1; 1H and 13C-NMR data, see Table 1; HR-ESI-MS m/z 361.1616 [M + Na]+ (calcd for C18H26NaO6).ε) 200 nm; IR (KBr) νmax 3391, 2931, 2857, 1732, 1712, 1658, 1556, 1418, 1379, 1251, 1125, 1082, 1025, 981, 883, cm−1; 1H and 13C-NMR data, see Table 1; HR-ESI-MS m/z 363.1787 [M + Na]+ (calcd for C18H28NaO6).ε) 200 nm; IR (KBr) νmax 3398, 2929, 2857, 1736, 1659, 1563, 1406, 1247, 1125, 1083, 1023, 979, 885, 803, 725, 610 cm−1; 1H and 13C-NMR data, see Table 1; HR-ESI-MS m/z 363.1783 [M + Na]+ (calcd for C18H28NaO6).Cytotoxic activity screening45,46
Human renal epithelial cell (293), hepatocellular carcinoma cell (HepG2, Huh7) and oral epidermoid carcinoma cell (KB) were obtained from the American Type Culture Collection (USA). Cytotoxicity of 1–10 were evaluated against 293 (human renal epithelial cell line) cells. Incubated cells at 2500 cells per well onto 96 wells overnight. Added drugs and incubated for three days. Added 10 μL dye and incubated for one hour at 37 °C. Read results at 450 nm on microplate reader (Thermo Scientific, USA). And HepG2, Huh7 (human hepatocellular carcinoma cell), KB (human oral epidermoid carcinoma cell) were also screened as follow. Seeded cells at 5000 cells per well onto 48 wells and incubated overnight. Added drugs and incubated for 3 doubling times. Removed media and added 0.5% methylene blue in 50% ethanol. Incubated plates at room temp for one hour. Washed plates gently with water and air dried for overnight. Added 0.2 mL 1% sarkosyl and rotated at room temp for 3 hours. Read results at 595 nm on microplate reader (Thermo Scientific, USA).HCV screening47
The HCV genotype 1b (Con1 isolate) subgenomic replicon cell line with a luciferase reporter (Huh-luc/neo-ET) was kindly provided by Ralf Bartenschlager from the University of Heidelberg.48 Seeded cells at 20000 cells per well onto 48 wells. Incubated them overnight then added drugs. Incubated 3 days, then removed media and washed with PBS. Added 50 μL lysis buffer and rotated 15 min at room temp. Frozen cells then thawed and transfered the lysate to a white plate. Added 50 μL luciferase buffer and read results on Farcyte luminometer (Thermo Scientific, USA).
HBV screening49
HBV intracellular replicative intermediates were isolated from transfected cells as described by Zhong et al.50 Southern Blot method was used to analyze HBV. DNA isolated from 2.2.15 (HepG2 transfected with HBV) cells after being treated with drugs for 9 days and incubated a total of 12 days. Interferon was used as the positive control.Cell cycle assay51
The cell cycle analyzed by flow cytometry. Upon CPT treatment at 1000 nM for 24 h, HepG2 cells with or without added compounds 6–7 (0.072 μM and 0.1 μM) were harvested, centrifuged at 300 ×g for 5 min, washed twice with PBS, and fixed in 100% methanol on ice for at least 1 h. The cells were then washed once with PBS and resuspended in 1 mL PBS containing 250 μg mL−1 RNase A (type I-A; Sigma-Aldrich) and propidium iodide (50 μg mL−1). Flow cytometry was carried out on BD FACSCalibur and data was analyzed using Flow Jo software.Antimicrobial activity screening52
Escherichia coli ATCC25922, Staphylococcus aureus ATCC6538, Bacillus cereus ATCC14579, Candida albicans ATCC 10231, Klebsiella pneumoniae ATCC 11296 were cultivated at 37 °C for 24 h in LB media using glass tubes. Cylindrocarpon didynum, Alternaria panax, Fusarium solani were cultivated at 28 °C for 7 days in PDA media using culture dishes. The LB/PDB bacterial/spore suspension was diluted to the predetermined starting concentration (optical density at 600 nm; OD600 = 0.2), and 1 mL of bacterial/spore solution was added into 24 mL media. The bacterial/spore solution was transferred into a 96-well plate (198 μL per well) and the 2 μL of compounds 1–10 (20 g L−1) reached the final reaction volume of 200 μL per well. Here, untreated bacterial solution served as the negative control group, and bacterial solutions treated with ampicillin (0.02 g L−1), kanamycin (0.02 g L−1), geneticin (G418) (0.02 g L−1) served as positive control groups, respectively. The growth was measured after 24 hours of incubation at 37/28 °C using a multiplate photometric reader. Every experiment was performed in three biological replicates in triplicate to validate the screening method.Statistical analysis
The statistical analysis was performed using OriginPro 9.0.0 software. All the data were present as means ± SD for triplicate experiments. One-way analysis of variance (ANOVA) followed by Turkey's test was used to express the statistical differences between groups. The value of P < 0.05 was considered statistically significant.Conflict of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the Institute of Nuclear Magnetic and Mass Spectrometry Testing Center, Shenyang Pharmaceutical University. This work is supported by grants from the Key Project of Yunnan Provincial Natural Science Foundation (2016ZF001-001, 2017IB038), Yung-Chi Cheng academician workstation of Yunnan provincial academy of science and technology (2015IC017) and National Science and Technology Major Project (2017ZX09305001-002).References
- T. T. X. Dong, X. M. Cui, Z. H. Song, K. J. Zhao, Z. N. Ji, C. K. Lo and K. W. K. Tsim, J. Agric. Food Chem., 2013, 51, 4617–4623 CrossRef PubMed.
- N. Zhou, Y. Tang, R. F. Keep, X. Ma and J. Xiang, Phytomedicine, 2014, 21, 1189–1195 CrossRef CAS PubMed.
- R. Uzayisenga, P. A. Ayeka and Y. Wang, Phytother. Res., 2014, 28, 510–516 CrossRef PubMed.
- P. Wang, J. Cui, X. Du, Q. Yang, C. Jia, M. Xiong, X. Yu, L. Li, W. Wang, Y. Chen and T. Zhang, J. Ethnopharmacol., 2014, 154, 663–671 CrossRef CAS PubMed.
- H. B. Guo, X. M. Cui, N. An and G. P. Cai, Genet. Resour. Crop Evol., 2010, 57, 453–460 CrossRef.
- S. G. Lee, Res. Plant Dis., 2004, 10, 248–259 CrossRef.
- J. H. Lee, Y. H. Yu, Y. H. Kim, S. H. Ohh and W. M. Park, Plant Pathol. J., 1990, 6, 13–20 Search PubMed.
- L. Ma, Y. H. Cao, M. H. Cheng, Y. Huang, M. H. Mo, Y. Wang, J. Z. Yang and F. X. Yang, Antonie van Leeuwenhoek, 2013, 103, 299–312 CrossRef PubMed.
- Z. Y. Fan, C. P. Miao, X. G. Qiao, Y. K. Zheng, H. H.Chen, Y. W. Chen, L. H. Xu, L. X. Zhao and H. L. Guan, J. Ginseng Res., 2016, 40, 97–104 CrossRef PubMed.
- Y. K. Zheng, C. P. Miao, H. H. Chen, F. F. Huang, Y. M. Xia, Y. W. Chen and L. X. Zhao, J. Ginseng Res., 2016, 40, 1–8 CrossRef PubMed.
- J. L. Chen, S. Z. Sun, C. P. Miao, K. Wu, Y. W. Chen, L. H. Xu, H. L. Guan and L. X. Zhao, J. Ginseng Res., 2016, 40, 315–324 CrossRef PubMed.
- S. K. Deshmukh, S. A. Verekar and S. V. Bhave, Front. Microbiol., 2014, 5, 715–725 Search PubMed.
- G. Strobel, Science, 1993, 260, 214–216 Search PubMed.
- L. Liang, H. E. Jun and Y. U. X. Ping, Acta Agric. Boreali-Occident. Sin., 2006, 15, 85–89 Search PubMed.
- S. C. Puri, V. Verma, T. Amna, G. N. Qazi and M. Spiteller, J. Nat. Prod., 2005, 68, 1717–1719 CrossRef CAS PubMed.
- S. Sirikantaramas, T. Asano, H. Sudo, M. Yamazaki and K. Saito, Curr. Pharm. Biotechnol., 2007, 8, 196–202 CAS.
- A. L. Leitãoa and F. J. Enguita, Microbiol. Res., 2016, 183, 8–18 CrossRef PubMed.
- M. Marinho, A. M. D. R. Rodrigues-Filho, E. Moitinho, M. D. L. R. Santos and S. Lourivaldo, J. Braz. Chem. Soc., 2005, 16, 280–283 CrossRef.
- A. Svendsen and J. C. Firsvad, Mycol. Res., 1994, 98, 1317–1328 CrossRef CAS.
- R. Nicoletti, M. L. Ciavatta, E. Buommino and M. A. Tufano, Int. J. Biomed. Pharmaceut. Sci., 2008, 2, 1–23 Search PubMed.
- M. Zhu, X. Zhang, H. Feng, J. Dai, J. Li, Q. Che, Q. Gu, T. Zhu and D. Li, J. Nat. Prod., 2017, 80, 71–75 CrossRef CAS PubMed.
- V. L. Singleton, N. Bohonos and A. J. Ullstrup, Nature, 1958, 181, 1072–1073 CrossRef CAS PubMed.
- V. Betina, J. Fuska, A. Kjaer, P. Nemec and R. H. Shapiro, J. Antibiot., 1966, 19, 115–117 CAS.
- V. Betina, P. Nemec, J. Dobias and Z. Barath, Folia Microbiol., 1962, 7, 353–357 CrossRef CAS.
- Y. J. Wang, Y. F. Wu, F. Xue, Z. X. Wu, Y. P. Xue, Y. G. Zheng and Y. C. Shen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 895, 146–153 CrossRef PubMed.
- J. W. Zhu, H. Nagasawa, F. Nagura, S. B. Mohamad, Y. Uto, K. Ohkura and H. Hori, Bioorg. Med. Chem., 2000, 8, 455–463 CrossRef CAS PubMed.
- Z. Kossaczka, J. Drgonova, B. Podobova, V. Betina and V. Farkas, Can. J. Microbiol., 1995, 41, 971–977 CrossRef CAS PubMed.
- B. S. Jeunemaitre and C. Hawes, Biol. Cell., 1992, 74, 325–328 CrossRef.
- D. E. Larsson, H. Lövborg, L. Rickardson, R. Larsson, K. Oberg and D. Granberg, Anticancer Res., 2006, 26, 4125–4129 CAS.
- W. Eric, R. G. Sellers and D. M. Peehl, J. Urol., 2000, 164, 836–841 CrossRef.
- N. H. Meyer and K. Zangger, ChemPhysChem, 2014, 15, 49–55 CrossRef CAS PubMed.
- A. Lupulescu, G. L. Olsen and L. Frydman, J. Magn. Reson., 2012, 218, 141–146 CrossRef CAS PubMed.
- H. Furukawa, T. Hamada, M. K. Hayashi, T. Haga, Y. Muto, H. Hirota, S. Yokoyama, K. Nagasawa and M. Ishiguro, Mol. Pharmacol., 2002, 62, 778–787 CrossRef CAS PubMed.
- T. Kimura, N. Matubayasi and M. Nakahara, Biophys. J., 2004, 86, 1124–1137 CrossRef CAS PubMed.
- B. Yu, N. Zhu and Z. Du, Helv. Chim. Acta, 2010, 93, 324–328 CrossRef CAS.
- Y. N. An, X. Zhang, T. Y. Zhang, M. Y. Zhang, Q. Zhang, X. Y. Deng, F. Zhao, L. J. Zhu, G. Wang, J. Zhang, Y. X. Zhang, B. Liu and X. S. Yao, Sci. Rep., 2016, 6, 27396–27410 CrossRef CAS PubMed.
- E. M. K. Wijeratne, B. P. Bashyal, M. X. Liu, D. D. Rocha, G. M. K. B. Gunaherath, J. M. U'Ren, M. K. Gunatilaka, A. E. Arnold, L. Whitesell and A. A. L. Gunatilaka, J. Nat. Prod., 2012, 75, 361–369 CrossRef CAS PubMed.
- F. W. Wang, R. H. Jiao, A. B. Cheng, S. H. Tan and Y. C. Song, World J. Microbiol. Biotechnol., 2007, 23, 79–83 CrossRef CAS.
- M. Shibazaki, H. Yamaguchi, T. Sugawara, K. Suzuki and T. Yamamoto, J. Biosci. Bioeng., 2003, 96, 344–348 CrossRef CAS PubMed.
- J. Sun, T. Awakawa, H. Noguchi and I. Abe, Bioorg. Med. Chem. Lett., 2012, 22, 6397–6400 CrossRef CAS PubMed.
- B. Yang, J. Dong, X. Lin, X. Zhou, Y. Zhang and Y. Liu, Tetrahedron, 2014, 70, 3859–3863 CrossRef CAS.
- M. A. Fernandez-Herrera, S. Mohan, H. Lopez-Munoz, J. M. V. Hernandez-Vazquez, E. Perez-Cervantes, M. L. Escobar-Sanchez, L. Sanchez-Sanchez, I. Regla, B. M. Pinto and J. Sandoval-Ramirez, Eur. J. Med. Chem., 2010, 45, 4827–4837 CrossRef CAS PubMed.
- M. Liu, G. Luo, Y. Wang, R. Xu, Y. Wang, W. He, J. Tan, M. Xing and J. Wu, Sci. Rep., 2017, 7, 436–450 CrossRef PubMed.
- P. M. Dewick, Medicinal Natural Products: A Biosynthetic Approach, John Wiley Sons Ltd., England, 2nd edn, 2002, pp. 35–96 Search PubMed.
- K. L. Grove, X. Guo, S. H. Liu, Z. Gao and C. K. Chu, Cancer Res., 1995, 55, 3008–3011 CAS.
- S. Anguille, E. Lion, J. Tel, I. J. Vries, K. Couderé, P. D. Fromm, V. F. V. Tendeloo, E. L. Smits and Z. N. Berneman, PLoS One, 2012, 7, 51138–51151 Search PubMed.
- Y. Cheng, L. K. Tsou, J. Cai, T. Aya, G. E. Dutschman, E. A. Gullen, S. P. Grill, A. P. C. Chen, B. D. Lindenbach, A. D. Hamilton and Y. C. Cheng, Antimicrob. Agents Chemother., 2010, 54, 197–206 CrossRef CAS PubMed.
- J. M. Vrolijk, A. Kaul, B. E. Hansen, V. Lohmann, B. L. Haagmans, S. W. Schalma and R. Bartenschlager, J. Virol. Methods, 2003, 110, 201–209 CrossRef CAS PubMed.
- S. L. Doong, C. H. Tsai, R. F. Schinazi, D. C. Liotta and Y. C. Cheng, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 8495–8499 CrossRef CAS.
- Y. Zhong, J. Lv, J. Li, X. Xing, H. Zhu, H. Su, L. Chen and X. Zhou, Antiviral Res., 2012, 93, 185–190 CrossRef CAS PubMed.
- C. J. Wang, W. Lam, S. Bussom, H. M. Chang and Y. C. Cheng, DNA Repair, 2009, 8, 1179–1189 CrossRef CAS PubMed.
- C. Zutz, D. Bandian, B. Neumayer, F. Speringer, M. Gorfer, M. Wagner, J. Strauss and K. Rychli, BioMed Res. Int., 2014, 10, 540292–540305 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07060h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |