
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of an aptamer – based fluorescence displacement biosensor for selective and sensitive detection of kanamycin in aqueous samples†
Long Ma *ab,
Nana Sunab,
Chunhao Tuab,
Qian Zhangc and
Aipo Diao*ab
*ab,
Nana Sunab,
Chunhao Tuab,
Qian Zhangc and
Aipo Diao*ab
aKey Laboratory of Industrial Fermentation Microbiology, Ministry of Education, School of Biotechnology, Tianjin University of Science & Technology, Tianjin 300457, China. E-mail: malong@tust.edu.cn; woshimalong1983@163.com; diaoaipo@tust.edu.cn; Fax: +86 22 60602298; Tel: +86 22 60602948
bTianjin Key Laboratory of Industry Microbiology, School of Biotechnology, Tianjin University of Science & Technology, Tianjin 300457, China
cCollege of Chemical Engineering and Materials Science, Tianjin University of Science & Technology, Tianjin 300457, China
First published on 7th August 2017
Abstract
A label-free detection method for kanamycin A using an aptamer-based biosensor has been developed. To start with, some commonly used DNA G-quadruplex structure sensitive dyes were evaluated and it was verified that the thioflavin T–G-quadruplex DNA complex was useful for constructing a fluorescent displacement biosensor. The limit of detection and the dynamic detection range of this sensor for kanamycin A is 300 pM and 1 nM to 300 μM, respectively, which are comparable to or better than the results for most reported methods, particularly for almost all the optical ones. The kanamycin recovery is in the range from 94% to 105%. The success of this new method is the creation of a new type of displacement biosensor which can be made widely applicable for probing quadruplex DNA–ligand interactions and thus there is the potential that a handful of analytes including small molecules, oligonucleotides and proteins could be detected using it.
1. Introduction
Kanamycin is one type of classic aminoglycoside bacteriocidal antibiotics, which is isolated from Streptomyces species.1 It is widely used as a broad-spectrum antibiotic in veterinary medicine and can be accumulated through the food chain and eventually transferred to humans.2 Overdose and long exposure to kanamycin can cause several problems such as ototoxicity and nephrotoxicity accompanied with symptoms.3 In addition, abuse of antibiotics leads to worldwide aggravation in antibiotic resistance, which creates ‘superbugs’. This is a severe trend and greatly endangers public health.4 It is critical to test whether the residual antibiotics in food products exceed maximum residue limits (MRLs) before they are able to be sold. The European Union has established the following MRLs for kanamycin in animal derived food: 100 μg kg−1 for meat, 600 μg kg−1 for liver, 2500 μg kg−1 for kidney, and 150 μg kg−1 (around 300 nM) for milk.5 Therefore, the development of easy-to-use and accurate techniques to quantify kanamycin A in biological samples is imperative. Aptamers are usually short pieces of RNA or DNA obtained from in vitro selection experiments, which give rise to nucleic acids which will bind targets selectively with high affinity.6 Classically, nucleic aptamers can be generated using systematic evolution of ligands by exponential enrichment (SELEX), in which candidate molecules are isolated from highly diverse (typically 1013 to 1015) nucleic acid pools via several rounds of affinity-based capture and amplification.7 Compared with antibodies, aptamers are demonstrated to have a range of advantages such as: rapid and efficient recognition, easy to handle, cost-effectiveness, less label intense in preparation, and flexible functionalisation. A biosensor based on aptamers as a recognition element can be termed as an aptasensor. Aptasensors can be fabricated using different principles, and produce electrochemical, optical and mass-sensitive biosensors. They have been widely developed and have caused great research interest, because they are able to detect a variety of targets from ions, small molecules, proteins and even prokaryotic and eukaryotic cells.8–12 This technology creates a novel model for expanding nucleic acid small molecule and nucleic acid protein interactions, which have been widely applied for detection, imaging, diagnosis and therapy.13 It is known that a range of DNA aptamers are demonstrated to have G4 structures.14 Meanwhile, it was realised that some organic dyes were useful for lighting up G4 DNA structures and further research is needed to determine their applications. Therefore, a working principle to employ G4 sensitive dye for detecting kanamycin A was hypothesised, as shown in Scheme 1. It is essentially based on a fluorescent displacement biosensor. A previously reported single stranded (ss) kanamycin A DNA apatamer (KanaApt) discovered through in vitro selection was adopted.15 KanaApt has recently been demonstrated to form an intermolecular G-quadruplex by the association of four separate aptamer strands.16 The dye of interest binds KanaApt selectively, and the fluorescence (FL) of this dye can be markedly ‘turned-on’. While in the presence of kanamycin A, the aptamer is prone to bind its ligand kanamycin A and to repel the dye. The free dye unbound to the G4 DNA only displays marginal FL, thus it is anticipated that this target-induced displacement leads to a measurable decrease in FL intensity. This decrease in FL is inversely related to the concentration of kanamycin A, and thus, a standard curve can be plotted for quantification. For the following part of this work, this hypothesis was tested.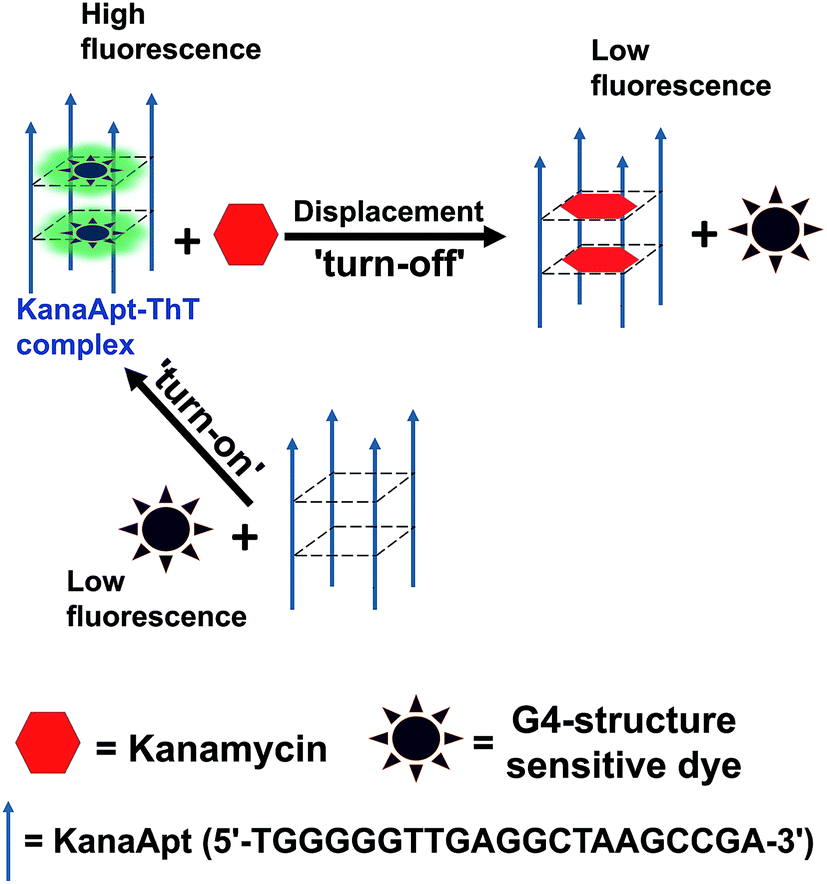 | ||
| Scheme 1 Schematic diagram of the kanamycin A induced fluorescent displacement biosensor. | ||
2. Experimental
2.1. Materials and sample preparations
All DNAs were chemically synthesised and purified by Generay Biotech Co., Ltd (China). One kanamycin A aptamer (KanaApt) and four other sequences were used including three mutated kanamycin A aptamers (named Mut 1, Mut 2 and Mut 3) and one poly A sequence of the same length. Their sequences are listed as follows (5′ to 3′): KanaApt: TGGGGGTTGAGGCTAAGCCGA, poly A: AAAAAAAAAAAAAAAAAAAAA, Mut 1: TGG![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) GGTTGAGCTAAGCCGA, Mut 2: TGGGGTTGAGGCTAAGCCGA, Mut 3: TGGGGTTGAGCTAACCGA, Mut 4: TATTACTAACCA. The nucleobases underlined were mutated. Tris(hydroxymethyl)aminomethane (Tris), sodium chloride (NaCl) and other common chemicals were purchased from Shanghai Sangon Biotech Co., Ltd (China) unless otherwise stated. Thioflavin T [ThT; 4-(3,6-dimethyl-1,3-benzothiazol-3-ium-2-yl)-N,N-dimethylaniline] and antibiotics including ampicillin, chloramphenicol, kanamycin A, sulfadimethoxine, terramycin and tetracycline were purchased from Sigma-Aldrich (USA). Chlortetracycline was purchased from Shanghai Sangon Biotech Co., Ltd (China). All the chemicals were used without further purification. Deionized water was prepared using a Milli-Q® Ultrapure water system. DNA concentrations were quantified at 260 nm absorbance using a UV-1800 ultraviolet-visible (UV-Vis) spectrophotometer (Shimadzu, Japan) and the extinction coefficients were calculated using the online IDT OligoAnalyzer 3.1 (http://www.idtdna.com/calc/analyzer). The ThT was quantified using the molar extinction coefficient at 412 nm in water which was 36
GGTTGAGCTAAGCCGA, Mut 2: TGGGGTTGAGGCTAAGCCGA, Mut 3: TGGGGTTGAGCTAACCGA, Mut 4: TATTACTAACCA. The nucleobases underlined were mutated. Tris(hydroxymethyl)aminomethane (Tris), sodium chloride (NaCl) and other common chemicals were purchased from Shanghai Sangon Biotech Co., Ltd (China) unless otherwise stated. Thioflavin T [ThT; 4-(3,6-dimethyl-1,3-benzothiazol-3-ium-2-yl)-N,N-dimethylaniline] and antibiotics including ampicillin, chloramphenicol, kanamycin A, sulfadimethoxine, terramycin and tetracycline were purchased from Sigma-Aldrich (USA). Chlortetracycline was purchased from Shanghai Sangon Biotech Co., Ltd (China). All the chemicals were used without further purification. Deionized water was prepared using a Milli-Q® Ultrapure water system. DNA concentrations were quantified at 260 nm absorbance using a UV-1800 ultraviolet-visible (UV-Vis) spectrophotometer (Shimadzu, Japan) and the extinction coefficients were calculated using the online IDT OligoAnalyzer 3.1 (http://www.idtdna.com/calc/analyzer). The ThT was quantified using the molar extinction coefficient at 412 nm in water which was 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1.
000 M−1 cm−1.
2.2. Fluorescent measurements
The steady-state fluorescence spectra of ThT were measured using an F-7000 fluorescence spectrophotometer (Hitachi, Japan) with a slit of 5 nm for excitation (425 nm) and a slit of 5 nm for emission (from 450 nm to 600 nm). The scan speed and response time were 60 nm min−1 and 0.1 s, respectively. The solutions used for the FL assay were buffered with 10 mM Tris-hydrochloride (pH 7.0) and 10 mM NaCl. All samples were incubated at 30 °C in a water bath for 20 min and then cooled down to ambient temperature before measurement. The 492 nm emission of ThT was used for curve plotting. For the real-time FL spectra, the excitation and emission were 425 nm (5 nm slit) and 492 nm (5 nm slit), respectively.2.3. Circular dichroism
The circular dichroism (CD) measurements were performed on a MOS-450 CD spectrometer (Bio-Logic, France) at ambient temperature. The scan step was set to 0.125 nm. The concentration of DNA samples used for measurement was 15 μM, kanamycin A and ThT were added where necessary.2.4. Optimisation of conditions
The value of FL intensity (I0/I) was used as an optimisation criterion. I0 and I represent the FL intensities of KanaApt–ThT complex in the absence and presence of kanamycin A. All samples were buffered with 10 mM Tris–HCl (pH 7.0) and 10 mM NaCl. KanaApt DNA was used at a concentration of 1 μM.:1 and an incubation temperature of 30 °C) in the presence of 20 μM kanamycin A were measured at different time points from 0 to 25 min.:1) in the presence of 50 μM of kanamycin A were incubated at 20 °C, 30 °C, 35 °C, 45 °C or 60 °C for 20 min before measurement.2.5. The use of the proposed biosensor for kanamycin A detection
In order to determine kanamycin A, 1 μM KanaApt was complexed with 1 μM ThT and different concentrations of kanamycin A (from 1 nM to 300 μM) were added. Each sample was incubated at 30 °C for 20 min and then cooled down to ambient temperature for FL spectra recording. The FL intensity at 492 nm was used for plotting the calibration curve.2.6. Selectivity of the proposed biosensor
The selectivity of the biosensor for kanamycin A was evaluated using a variety of commonly used antibiotics, including ampicillin, chloramphenicol, chlortetracycline, sulfadimethoxine, terramycin, and tetracycline. The concentration for kanamycin A and the other antibiotics was 20 μM. The concentrations of KanaApt and ThT used were both 1 μM. The decreased FL/original FL after adding kanamycin A into the KanaApt–ThT complex was assigned as a 100% relative FL response.2.7. Detection of kanamycin A in milk sample
Next, the fabricated biosensor was tested to see if it was appropriate for use with water derived samples such as commercial milk. Commercial liquid milk was purchased from the supermarket in Tianjin, China. The milk samples were spiked with different kanamycin A solutions to give final concentrations at three levels (0.10, 2.0, and 20.0 μM). The proteins in milk were precipitated using 300 μL of cold acetonitrile which was added to 100 μL of milk, with gentle mixing. The mixtures were incubated at 4 °C for 20 min. The samples were then centrifuged at 13000 rpm for 10 min at 4 °C. The supernatant was filtered with a 0.22 mm membrane, and adjusted to neutral pH. The filtrate was collected and the fluorescent assay was carried out.
3. Results and discussion
Initially, an appropriate dye was needed for this displacement-based biosensor. As is known, several organic dyes have been reported to selectively bind DNA especially the G4 structure. To start with, several representative dyes were deliberately chosen including N-methyl mesoporphyrin IX (NMM),17 crystal violet (CV),18 malachite green (MG),19 thiazole orange (TO)20 and acridine orange (AO)21 to test their ‘light-up’ property and selectivity toward KanaApt. As shown in Fig. 1A, it can be seen that apart from ThT and TO, none was able to fulfill this purpose. Thus, in order to compare the selectivity of ThT and TO further, a 21-mer poly A ssDNA sequence, and its corresponding double stranded (ds)DNA together with KanaApt were used. As shown in Fig. 1B, it can be seen that TO was highly fluorescent for both dsDNA and G4 DNA structures, thus demonstrating no selectivity. However, the FL for KanaApt/dsDNA and KanaApt/ssDNA in the presence of ThT was more than 10 and 100, respectively, which proved that ThT had a far better selectivity for KanaApt against dsDNA and ssDNA. As ThT was so obviously superior to and it was decided to carry on the investigation using ThT. ThT is a benzothiazole salt and has previously been found to be a specific probe for amyloid fibrils.22 Following this, Mohanty et al. have reported that ThT is also an efficient inducer and selective fluorogenic sensor for the human telomeric G-quadruplex (G4) DNA.23 In 2014, the G4 DNA “light-up” observation was extended and generalised, and researchers indicated that ThT may be used as a convenient and specific nucleic acid G4 probe.24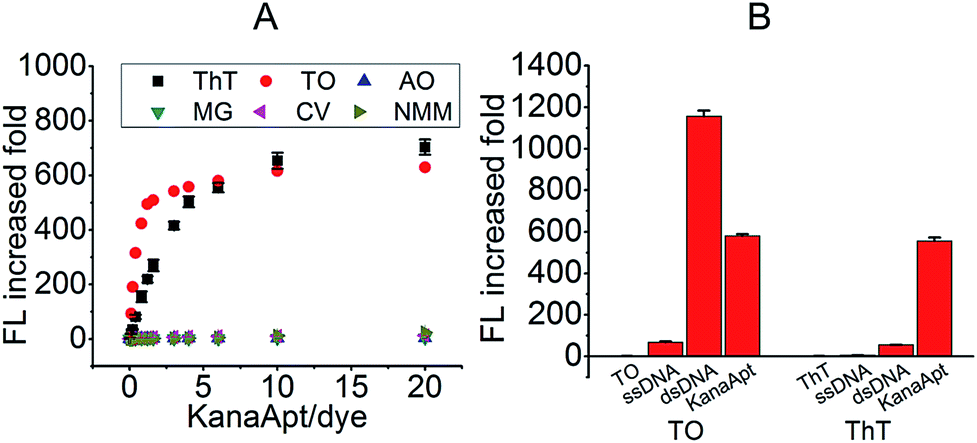 | ||
| Fig. 1 The comparison of several organic dyes to find one which could be selectively lit up by KanaApt. (A) FL response (expressed as increased fold) of different dyes in the presence of KanaApt. (B) Selectivity of TO and ThT for KanaApt against ssDNA and dsDNA. The concentrations of TO and ThT used were both 5 μM and the concentration of all the DNA used was 30 μM. | ||
Next, a preliminary experiment was performed to test if the proposed principle was workable. As shown in Fig. 2A, the FL intensity of free ThT, KanaApt–ThT complex, and KanaApt–ThT complex was measured in the presence of 20 μM of kanamycin A. Clearly, the presence of KanaApt greatly turned on the FL of ThT, whereas the free ThT merely emitted FL. Strikingly, the addition of kanamycin A seemed to displace ThT, which can be shown by the significant decrease of FL. As shown in Fig. 2B, the FL intensity change after addition of kanamycin A can be reflected in real-time. In the absence of kanamycin A, the FL of the KanaApt–ThT complex was rather steady and aligned well with the baseline. However, with the addition of increasing amounts of kanamycin A, it can be noticed that the initial velocity of the ‘displacement’ process increased accordingly, because the ‘displacement’ process was well synchronised with the “turn-off” of the FL of KanaApt–ThT complex. Fig. 2B shows that the “displacement” process had a fast response and was triggered by kanamycin A in a concentration dependent manner, which agreed with the speculation about the proposed biosensing strategy.
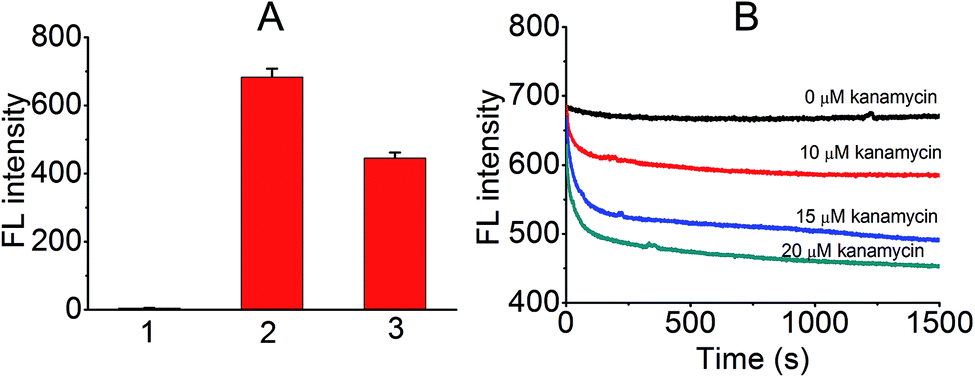 | ||
| Fig. 2 ThT FL analysis for the kanamycin A induced FL displacement biosensor. (A) Depicts the ThT FL in different states. Column 1 = 1 μM ThT, 2 = 1 μM ThT + 1 μM KanaApt, and 3 = 2 + 20 μM kanamycin A. (B) Shows the real-time fluorescence when different concentrations of kanamycin A (0 to 20 μM) were added to the 1 μM KanaApt–ThT complex. | ||
Furthermore, CD experiments were carried out to probe the conformation of KanaApt and KanaApt–ligand interactions, as CD is the most common technique to study the morphology of DNA in aqueous solution. As shown in Fig. 3A, KanaApt DNA displayed a typical G4 structure with parallel conformation, because it is well known that a ‘parallel’ G4 DNA has a positive peak around 260 nm and a negative peak around 240 nm.25,26 The G4 structure is mainly caused by the non-covalent bonding among guanine bases, therefore, the loss of them would result in the breakdown of the G4 secondary structure.27 When systematically mutating guanosines to adenosines in the KanaApt (Fig. 3A), it can be clearly seen that the G4 conformation started to collapse while more guanosines were mutated and the typical CD signals at ∼240 and ∼260 nm became ambiguous for the three mutants. Concomitant with the increase of molar ratio of ThT/KanaApt and kanamycin A/KanaApt, the corresponding CD spectra did not show much change (Fig. 3B and C), indicating that neither ThT nor kanamycin A exerted much disturbance on the dynamic conformation of KanaApt.
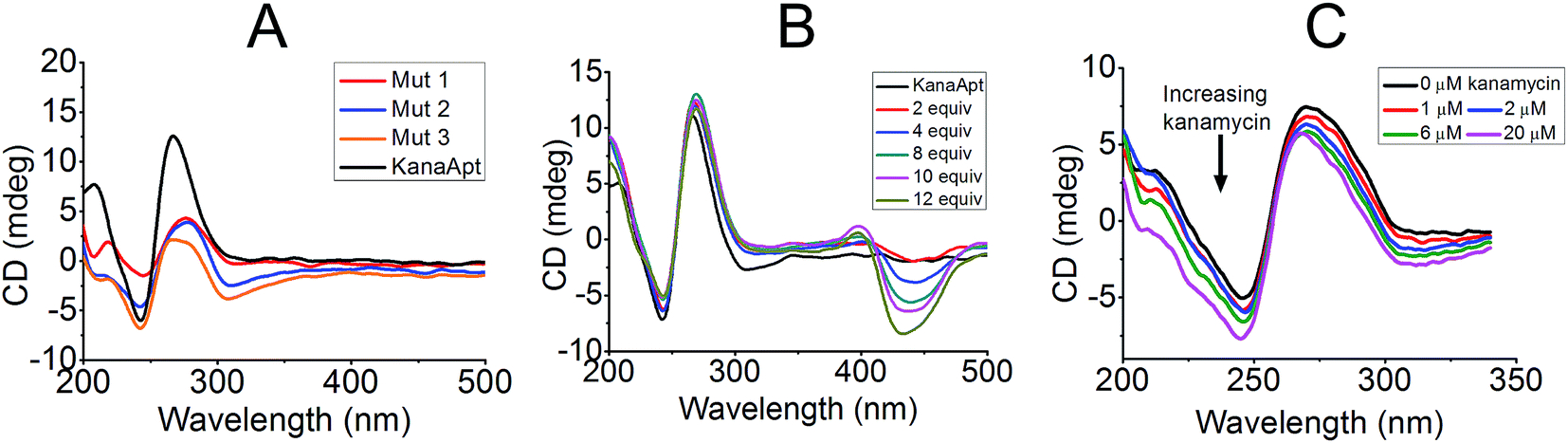 | ||
| Fig. 3 CD spectra recorded to investigate the conformation of KanaApt and its mutations under different circumstances. (A) CD spectra for KanaApt and three other mutated sequences. (B) KanaApt in the presence of different concentrations of ThT (2 to 12 equiv.). (C) KanaApt in the presence of different concentrations of kanamycin A. The black curves are the CD spectra for free KanaApt and all the concentrations of KanaApt used are 15 μM. | ||
Next, the ‘light-up’ effect of KanaApt toward ThT was studied, and as shown in Fig. 4A, free ThT were rather less fluorescent, whereas with the addition of a KanaApt, its FL increased by about 81 fold. However, the same concentrations of a mutated KanaApt (Mut 4, in which all guanosines were replaced with adenosines) and ss poly A enhanced the FL of ThT by only 3.1 and 1.9 fold, respectively. Fig. 4B shows that the FL spectra for KanaApt–ThT complexes with different molar ratios (KanaApt/ThT from 0.02 to 20). These results showed that for KanaApt–ThT complexes with molar ratios of 0.1, 0.4, 3, 6, 10 or 20, each FL was enhanced by 18.9, 81.2, 415.3, 653.2 or 703.4 fold, respectively (Fig. 4C). It is believed that ThT is a specific probe for G4-forming sequences rather than ss- and ds-DNA and this observation agrees with the previous conclusion. Also it proved that KanaApt had a G4 structure and was able to ‘light up’ ThT. Fluorescence titrations (ThT by KanaApt) were carried out and the curve of FL intensities against the concentration of kanamycin A was plotted in Fig. S1 (ESI†).
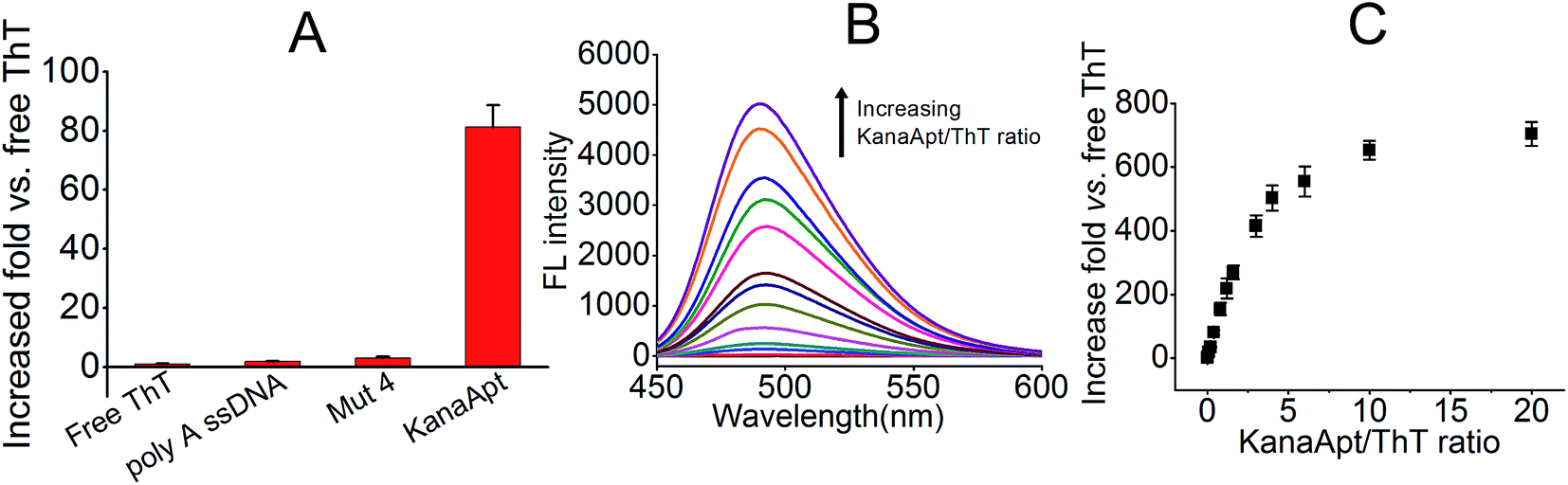 | ||
| Fig. 4 ThT can be specifically lighted up by KanaApt. (A) FL response (expressed as increased fold) of 5 μM ThT in the presence of 2 μM poly A ssDNA, mutated KanaApt (Mut 4) and KanaApt. (B) FL spectra for KanaApt–ThT complexes with different molar ratios. (C) FL when adding increased KanaApt into 5 μM ThT (data derived from B). | ||
It was then decided to fabricate a displacement biosensor for kanamycin A detection. In order to do so, some factors, which correlated well with the performance of the FL measurements, were investigated in detail. The ratio of ThT to KanaApt, incubation time, temperature, and pH were studied. Fig. S2 (ESI†) shows the influence of the ThT/KanaApt molar ratio on the I0/I value in the absence (I0) and presence (I) of kanamycin A, because it was thought that this index could be rather sensitive and be used to obtain a satisfactory signal-to-noise ratio. Interestingly, it was observed that in fact the 1:1 ratio seemed to reach a peak value of I0/I and when using a higher ligand ratio, the I0/I decreased accordingly. Despite counterintuition, the observations made in this study agreed well with the results of Gabelica et al., who found that the FL response of ThT was the highest with the 1:1 complex, and quenched with higher ligand binding ratios.28 In this case, it was decided to use 1:1 ratio for the following study. A discontinuous assay was used, as shown in Fig. S3 (ESI†), and the initial addition of kanamycin A into the KanaApt–ThT complex caused a rise in FL emission intensity as expected. With increase in incubation time, the intensity decreased sharply in the first few minutes. This decrease remained for about 20 min and then reached a plateau. Further incubation of the mixture did not cause an evident change in FL intensity. Whereas in the absence of kanamycin A, when the interaction time (incubation time) was extended up to 25 min, the background signal almost stayed unchanged. Thus, for the following experiments, the incubation time used was 20 min. The dependence of I0/I on the incubation temperature was also investigated, as shown in Fig. S4 (ESI†). The incubation temperature had a considerable affect on the FL response. The strongest response appeared when the temperature was set at 30 °C. Thus, 30 °C was used as the incubation temperature. However, it was found that the linearity between FL intensity and concentration of kanamycin A was less sensitive to the pH value and it was found that pH 7 worked well (data not shown).
Upon the completion of the experimental conditions' optimisation, the relationship between FL intensity and added kanamycin A was tested, as shown in Fig. 5. The addition of kanamycin A decreased the FL of the KanaApt–ThT complex, and this was indicative of a displacement effect for ThT. A range of kanamycin A samples with different concentrations were tested and a curve of FL intensities at 492 nm against different concentrations of kanamycin A was plotted (Fig. 5B). There was a linear relationship (R = 0.990) between FL intensity and kanamycin A concentration (expressed in a logarithmic format) over the range from 1 nM to 300 μM (Fig. 5C), and a limit of detection (LOD) of 300 pM for kanamycin A analysis was achieved, based on the 3σ/slope equation.
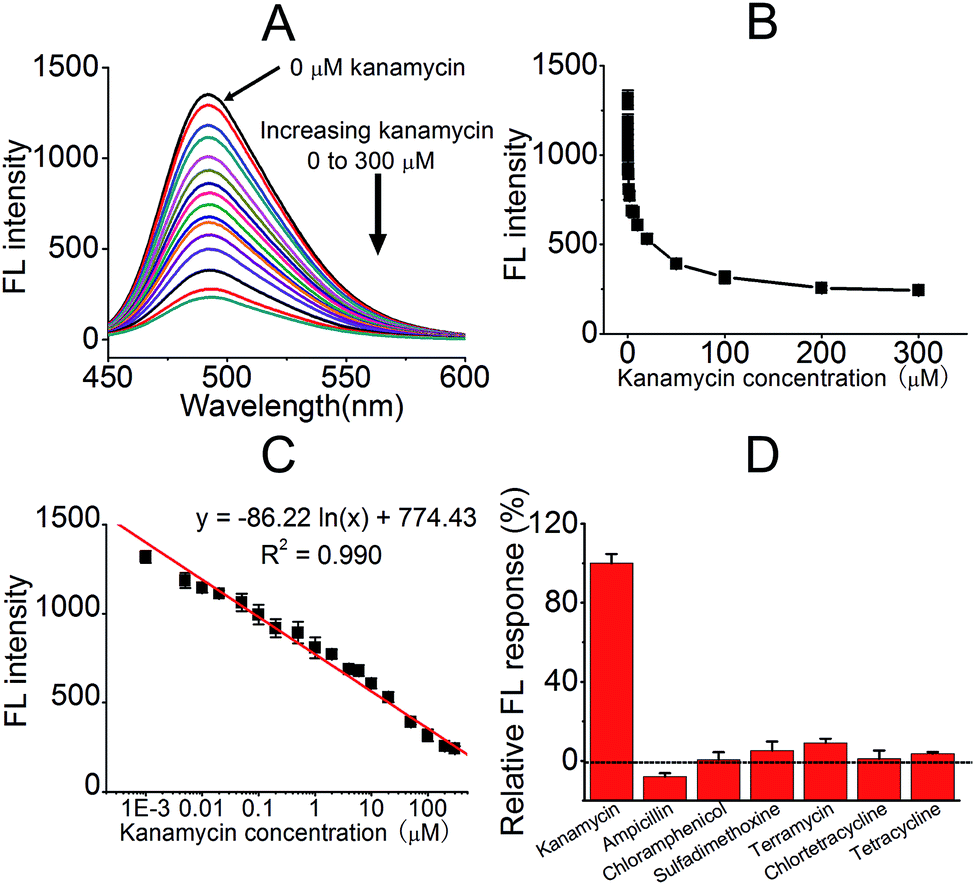 | ||
| Fig. 5 The detection utility of the proposed method for detecting kanamycin A. (A) FL spectra for the KanaApt–ThT complex when adding different concentrations of kanamycin A. The black curve is the spectrum for the free KanaApt–ThT complex without kanamycin A. (B) The dependence of FL on kanamycin A concentrations. (C) The linear relationship between the FL intensity and the concentration of kanamycin A (the calibration curve and fitted equation are both given). (D) The selectivity of the proposed method for kanamycin A compared to six other common antibiotics. Error bars represent the standard deviation in three individual experiments. | ||
The selectivity of kanamycin A compared with six other commonly used antibiotics (ampicillin, chloramphenicol, chlortetracycline, sulfadimethoxine, terramycin, and tetracycline) was evaluated. Fig. 5D presents the histograms of the relative FL responses of kanamycin A and six other antibiotics. It can be observed that compared with kanamycin A, the other antibiotics did not interfere with the system as the proposed biosensor was almost unresponsive (less than 10%). These competing antibiotics had a negligible effect on the detection system. This approach exhibited high selectivity for kanamycin A, which meant it was a possible candidate for future applications.
Lastly, the applicability and reproducibility of the proposed biosensor was further explored by testing some real milk samples. The milk samples were spiked with a series of concentrations of kanamycin A (0.10, 2.0, or 20.0 μM as the final concentration) and then the recovery values were compared with the added values. The analytical results are given in Table S1 (ESI†). The recovery values ranged from 94% to 105% and the relative standard deviation (RSD) for all samples after four independent measurements were less than 7.6%. These results indicate that this proposed sensor is reliable and is capable of detecting kanamycin in water derived samples. It is also worth noting that the required volume of milk for the test was as little as 0.1 mL and the total assay time was less than 2 h for one analyst using a standard protocol.
For kanamycin A detection, a number of methods have been reported previously, including colorimetry,29 high-performance liquid chromatography (HPLC),30 capillary electrophoresis (CE),31 enzyme immunoassays (ELISA),32–34 electrochemical aptasensors35 and immuno-sensors,36 fluorescent aptasensors,16 surface plasmon resonance (SPR),37,38 nanozyme,39 luminescence,40 graphene biosensor41 and a microbiological multi-residue system.42 The methods employed are often complex (e.g., require uncommon and costly instruments), time consuming (e.g., require intricate fabrication of electrodes or preparation of nano-composites), requiring tedious sample pre-treatment and hard to standardise (e.g., home-made materials and antibodies). This proposed method has at least the following advantages: it is label-free and all the chemicals required are commercially available with low prices and thus it is cost-effective and adaptable for practical uses, it only needs miniaturised and inexpensive analytical devices which are affordable for common analysts. The LOD of this sensor for kanamycin A is 300 pM, making it a good platform for kanamycin A quantification. Also the LOD and detection range (1 nM to 300 μM) of the current method is comparable to or better than most reported ones, for almost all of the optical methods. A brief comparison of this approach with others is listed in Table 1 and a through comparison is listed in Table S2 (ESI†). Some methods do have a lower LOD than this one, but they usually have a lot narrower linear range for detection, which discourages their use in actual applications. A low LOD value does not necessarily mean that it will be a desirable biosensor, because a good one has to integrate sensitivity, detection range, cost-effectiveness, selectivity, simplicity and reproducibility for real uses. This method balances all the previously mentioned criteria.
| Detection method/instrument | Linear range | (LOD) limit of detection | Ref. |
|---|---|---|---|
| UV-Vis spectroscopy | 1–500 nM | 1 nM | 17 |
| HPLC | — | ∼78 μM (38 μg mL−1) | 18 |
| FL spectroscopy | 1 nM to 300 μM | 300 pM | This work |
| CE | ∼43 to ∼103 nM | ∼14 nM | 19 |
| ELISA | ∼0.4 to ∼100 nM | ∼0.4 nM | 20 |
| Electrochemistry | 10–150 nM | 5.8 nM | 23 |
| SPR | 20–800 nM | 2 nM | 26 |
| Luminescence | 0.2–150 μM | 143 nM | 28 |
4. Conclusions
The success of the current study not only allows the fabrication of a novel biosensor for kanamycin A detection, but, more importantly, it creates a new type of approach of employing ThT for probing quadruplex DNA–ligand interactions. The greatest part of this work is to highlight the usefulness of ThT in constructing an analyte induced displacement biosensor, and as far as is known, this is the first time such a biosensor has been used. Nucleic acids with specific structures43–47 and enzyme interfaced oligonucleotides have been widely used in bio-sensing and this work also expands the repertoire of biosensors.48–50 To conclude, a displacement strategy has been successfully developed, which is based on an intrinsic fluorogenic dye ThT in a label-free manner. Its use has been demonstrated by the detection of kanamycin A, both sensitively and selectively. This sensor is not only restricted to kanamycin A detection, a number of analytes including small organic molecules, oligonucleotides and proteins could potentially be detected by using this suggested method, when in combination with their respective G4 aptamers.Acknowledgements
This work is supported by National Natural Science Foundation of China (No. 81503086 and 21672161), a starting funding (No. 20140520) from Tianjin University of Science and Technology (TUST), research funding from the ‘1000 Talents Plan’ of Tianjin (to LM) and funding from the Foundation of Key Laboratory of Industrial Fermentation Microbiology of Ministry of Education and Tianjin Key Lab of Industrial Microbiology (No. 2015IM106).References
- C. M. Spahn and C. D. Prescott, J. Mol. Med., 1996, 74, 423–439 CrossRef CAS PubMed.
- K. Hirose, S.-Z. Li, K. K. Ohlemiller and R. M. Ransohoff, J. Assoc. Res. Otolaryngol., 2014, 15, 555–570 CrossRef PubMed.
- G. J. Kaloyanides and E. Pastoriza-Munoz, Kidney Int., 1980, 18, 571–582 CrossRef CAS PubMed.
- C. A. Arias and B. E. Murray, N. Engl. J. Med., 2009, 360, 439–443 CrossRef CAS PubMed.
- European Agency for the Evaluation of Medical Products (EMEA) London, 2003, Regulation No. EMEA/MRL/886/03-FINAL.
- T. Hermann and D. J. Patel, Science, 2000, 287, 820–825 CrossRef CAS PubMed.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Biomol. Eng., 2007, 24, 381–403 CrossRef CAS PubMed.
- H. Hai, F. Yang and J. Li, RSC Adv., 2013, 3, 13144–13148 RSC.
- R. Zou, X. Lou, H. Ou, Y. Zhang, W. Wang, M. Yuan, M. Guan, Z. Luo and Y. Liu, RSC Adv., 2012, 2, 4636–4638 RSC.
- Y. Huang, X. Chen, N. Duan, S. Wu, Z. Wang, X. Wei and Y. Wang, Food Chem., 2015, 166, 623–629 CrossRef CAS PubMed.
- Y.-C. Chang, C.-Y. Yang, R.-L. Sun, Y.-F. Cheng, W.-C. Kao and P.-C. Yang, Sci. Rep., 2013, 3, 1863 CrossRef PubMed.
- D. Shangguan, Y. Li, Z. Tang, Z. C. Cao, H. W. Chen, P. Mallikaratchy, K. Sefah, C. J. Yang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11838–11843 CrossRef CAS PubMed.
- S. Song, L. Wang, J. Li, C. Fan and J. Zhao, TrAC, Trends Anal. Chem., 2008, 27, 108–117 CrossRef CAS.
- W. O. Tucker, K. T. Shum and A. J. Tanner, Curr. Pharm. Des., 2012, 18, 2014–2026 CrossRef CAS PubMed.
- K.-M. Song, M. Cho, H. Jo, K. Min, S. H. Jeon, T. Kim, M. S. Han, J. K. Ku and C. Ban, Anal. Biochem., 2011, 415, 175–181 CrossRef CAS PubMed.
- Y. P. Xing, C. Liu, X. H. Zhou and H. C. Shi, Sci. Rep., 2015, 5, 8125 CrossRef CAS PubMed.
- H. Arthanari, S. Basu, T. L. Kawano and P. H. Bolton, Nucleic Acids Res., 1998, 26, 3724–3728 CrossRef CAS PubMed.
- J. H. Guo, L. N. Zhu, D. M. Kong and H. X. Shen, Talanta, 2009, 80, 607–613 CrossRef CAS PubMed.
- A. C. Bhasikuttan, J. Mohanty and H. Pal, Angew. Chem., Int. Ed., 2007, 46, 9305–9307 CrossRef CAS PubMed.
- I. Lubitz, D. Zikich and A. Kotlyar, Biochemistry, 2010, 49, 3567–3574 CrossRef CAS PubMed.
- J. Nygren, N. Svanvik and M. Kubista, Biopolymers, 1998, 46, 39–51 CrossRef CAS PubMed.
- T. Ban, D. Hamada, K. Hasegawa, H. Naiki and Y. Goto, J. Biol. Chem., 2003, 278, 16462–16465 CrossRef CAS PubMed.
- J. Mohanty, N. Barooah, V. Dhamodharan, S. Harikrishna, P. Pradeepkumar and A. C. Bhasikuttan, J. Am. Chem. Soc., 2012, 135, 367–376 CrossRef PubMed.
- A. R. de la Faverie, A. Guedin, A. Bedrat, L. A. Yatsunyk and J.-L. Mergny, Nucleic Acid Res., 2014, e65 CrossRef PubMed.
- J. Kypr, I. Kejnovska, D. Renciuk and M. Vorlickova, Nucleic Acids Res., 2009, 37, 1713–1725 CrossRef CAS PubMed.
- D. Monchaud, C. Allain and M.-P. Teulade-Fichou, Bioorg. Med. Chem. Lett., 2006, 16, 4842–4845 CrossRef CAS PubMed.
- L. Ma, H. Liu, G. Wu, N. Sun, L. Meng, Y. Li, Z. Liu and A. Diao, Analyst, 2016, 141, 3997–4000 RSC.
- V. Gabelica, R. Maeda, T. Fujimoto, H. Yaku, T. Murashima, N. Sugimoto and D. Miyoshi, Biochemistry, 2013, 52, 5620–5628 CrossRef CAS PubMed.
- N. Zhou, J. Zhang and Y. Tian, Anal. Methods, 2014, 6, 1569–1574 RSC.
- B. Blanchaert, E. Poderós Jorge, P. Jankovics, E. Adams and A. Van Schepdael, Chromatographia, 2013, 76, 1505–1512 CAS.
- C.-Z. Yu, Y.-Z. He, G.-N. Fu, H.-Y. Xie and W.-E. Gan, J. Chromatogr. B, 2009, 877, 333–338 CrossRef CAS PubMed.
- H. Watanabe, A. Satake, Y. Kido and A. Tsuji, Analyst, 1999, 124, 1611–1615 RSC.
- E. E. M. G. Loomans, J. van Wiltenburg, M. Koets and A. van Amerongen, J. Agric. Food Chem., 2003, 51, 587–593 CrossRef CAS PubMed.
- Y. Chen, Z. Wang, Z. Wang, S. Tang, Y. Zhu and X. Xiao, J. Agric. Food Chem., 2008, 56, 2944–2952 CrossRef CAS PubMed.
- X. Sun, F. Li, G. Shen, J. Huang and X. Wang, Analyst, 2014, 139, 299–308 RSC.
- S. Yu, Q. Wei, B. Du, D. Wu, H. Li, L. Yan, H. Ma and Y. Zhang, Biosens. Bioelectron., 2013, 48, 224–229 CrossRef CAS PubMed.
- M. Frasconi, R. Tel-Vered, M. Riskin and I. Willner, Anal. Chem., 2010, 82, 2512–2519 CrossRef CAS PubMed.
- X. Wang, M. Zou, X. Xu, R. Lei, K. Li and N. Li, Anal. Bioanal. Chem., 2009, 395, 2397–2403 CrossRef CAS PubMed.
- T. K. Sharma, R. Ramanathan, P. Weerathunge, M. Mohammadtaheri, H. K. Daima, R. Shukla and V. Bansal, Chem. Commun., 2014, 50, 15856–15859 RSC.
- K.-H. Leung, H.-Z. He, D. S.-H. Chan, W.-C. Fu, C.-H. Leung and D.-L. Ma, Sens. Actuators, B, 2013, 177, 487–492 CrossRef CAS.
- C. Wang, Y. Li, Y. Zhu, X. Zhou, Q. Lin and M. He, Adv. Funct. Mater., 2016, 26, 7668–7678 CrossRef CAS.
- L. Ma and S. L. Cockroft, Biological nanopores for single-molecule biophysics, ChemBioChem, 2010, 11, 25–34 CrossRef CAS PubMed.
- R. Althaus, M. I. Berruga, A. Montero, M. Roca and M. P. Molina, Anal. Chim. Acta, 2009, 632, 156–162 CrossRef CAS PubMed.
- T. Sabir, A. Toulmin, L. Ma, A. C. Jones, P. McGlynn, G. F. Schroder and S. W. Magennis, J. Am. Chem. Soc., 2012, 134, 6280–6285 CrossRef CAS PubMed.
- L. Ma, K. Chen, D. J. Clarke, C. P. Nortcliffe, G. G. Wilson, J. M. Edwardson, A. J. Morton, A. C. Jones and D. T. F. Dryden, Nucleic Acids Res., 2013, 41, 4999–5009 CrossRef CAS PubMed.
- L. Ma, G. Wu, Y. Li, P. Qin, L. Meng, H. Liu, Y. Li and A. Diao, Nanoscale, 2015, 7, 18044–18048 RSC.
- L. Ma, X. Wu, G. G. Wilson, A. C. Jones and D. T. Dryden, Biochem. Biophys. Res. Commun., 2014, 449, 120–125 CrossRef CAS PubMed.
- L. D. Finger, N. Patel, A. Beddows, L. Ma, J. C. Exell, E. Jardine, A. C. Jones and J. A. Grasby, Nucleic Acids Res., 2013, 41, 9839–9847 CrossRef PubMed.
- W. C. Tse and D. L. Boger, Acc. Chem. Res., 2004, 37, 61–69 CrossRef CAS PubMed.
- L. Ma and A. Diao, Chem. Commun., 2015, 51, 10233–10235 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07052g |
| This journal is © The Royal Society of Chemistry 2017 |