
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Migration, reactivity, and sulfur tolerance of copper species in SAPO-34 zeolite toward NOx reduction with ammonia†
Xuesong Liu *abd,
Xiaodong Wu*b,
Duan Wengb,
Zhichun Sic and
Rui Ranb
*abd,
Xiaodong Wu*b,
Duan Wengb,
Zhichun Sic and
Rui Ranb
aCollege of Chemistry and Chemical Engineering, Shaoxing University, Zhejiang 312000, P. R. China. E-mail: xuesongliu@usx.edu.cn
bKey Laboratory of Advanced Materials of Ministry of Education, School of Materials Science and Engineering, Tsinghua University, Beijing 100084, P. R. China. E-mail: wuxiaodong@tsinghua.edu.cn
cAdvanced Materials Institute, Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, China
dState Key Laboratory of Green Building Materials, China Building Materials Academy, Beijing, 100024, P. R. China
First published on 1st August 2017
Abstract
Cu/SAPO-34 catalysts were prepared by wet impregnation and ion-exchange methods, and both the catalysts were hydrothermally treated at 750 °C in 10 vol% H2O/air for 24 h. Subsequently, the as-received and hydrothermally treated catalysts were exposed to a sulfur poisoning treatment at 350 °C in 100 ppm SO2/10 vol% H2O/air for 24 h and examined for NOx conversion. Sulfur poisoning considerably decreased the NOx conversion efficiency of the catalysts at low temperatures. In contrast, it improved the high-temperature selective catalytic reduction (SCR) activities of the as-received catalysts. The ion-exchange-prepared catalysts displayed higher sulfur tolerance than the impregnation-prepared catalysts at 150–350 °C. The electron paramagnetic resonance, X-ray photoelectron spectroscopy, energy-dispersive X-ray spectroscopy and H2-temperature-programmed reduction results showed that sulfur poisoning significantly influenced the migration of the copper species and thereby the amount of active isolated Cu2+. More dispersed CuSO4 was produced on the ion-exchange-prepared catalysts than on the impregnation-prepared catalysts after sulfur poisoning. The dispersed CuSO4 showed considerably higher SCR activity than the crystalline CuSO4. Both the dispersed CuSO4 and remaining isolated Cu2+ determined the low-temperature SCR behavior of the sulfur-poisoned catalysts.
1. Introduction
Sulfur is always present in vehicle exhausts arising from residues in the fuel and lubricant oils. It is therefore essential to examine the effect of sulfur on different catalysts involved in NH3-selective catalytic reduction (SCR) after-treatment systems. Despite the considerable reduction in the content of sulfur in diesel fuel in recent years, especially with the implementation of China IV standard diesel in China, sulfur poisoning remains one of the most significant deactivation factors for SCR catalysts employed in heavy-duty diesel motor exhaust after-treatment systems. Although China IV standard diesel contains less than 50 ppm sulfur, the cumulative lifetime exposure of catalysts may amount to kilograms of sulfur.Several studies1–9 have addressed the effects of sulfur poisoning on different NH3-SCR catalysts. Vanadium-based catalysts were found to have a greater tolerance toward SO2 poisoning than metal zeolite catalysts.1,4,5 Among the zeolite-based catalysts, Cu zeolites are more sensitive to sulfur dioxide than Fe zeolites,1–3 and the deactivation effect is more pronounced at low temperatures.2,4
In recent years, Cu-exchanged zeolite catalysts of the chabazite family with unique microporous structures have emerged as a leading candidate among zeolite formulations when compared to Cu/ZSM-5 and Cu/beta owing to their superior NH3-SCR activity and prominent hydrothermal stability at 750 °C.10,11 Previous research studies12–16 demonstrated that isolated Cu2+ species are the active sites for SCR reaction, as determined by extended X-ray absorption fine structure and kinetic tests. Zhang et al. concluded that SO2 inhibited the SCR activity at low temperatures owing to the formation of ammonium sulfates as well as the onset of competitive adsorption between SOx and NOx on the Cu sites.17 Kumar et al.18 compared the effects of SO2 and SO3 on Cu/SAPO-34 catalyst and found that poisoning with SO3 at 400 °C resulted in a more severe deactivation when compared with that with SO2. Because of its strong chemical binding strength and the oxidative conditions in diesel exhaust streams, sulfur can react readily with Cu in a Cu/zeolite SCR catalyst to form stable CuSO4-like compounds.3,6,7 Such a sulfur poisoning phenomenon affects the redox properties of the Cu sites, severely inhibits NO oxidation and subsequent formation of the adsorbed NOx complex, and ultimately decreases the SCR activity of the catalyst.19,50
Although some sulfur poisoning mechanisms of NH3-SCR catalysts have been proposed, little information on the effects of sulfur poisoning on different copper species and the location of sulfur has been reported to date.
In the present study, different preparation methods and a hydrothermal treatment were applied to generate different distributions of Cu species on SAPO-34 support. Various techniques (i.e., X-ray photoelectron spectroscopy, electron paramagnetic resonance, H2 temperature-programmed reduction, and NH3 temperature-programmed desorption) were used to probe the location and state of Cu species before and after sulfur poisoning. The evolution of different copper species was examined, and their sulfur tolerance was determined, which is rarely reported.
2. Experimental
2.1. Catalyst preparation
Cu/SAPO-34 catalysts were synthesized by wet impregnation20 and ion-exchange15 methods, respectively.Cu(CH3COO)2·H2O (Ling Feng Chemical Reagent, China) was dissolved in deionized water and mixed with commercial H-SAPO-34 molecular sieve (Al2O3/SiO2/P2O5 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.3:0.8) powders using a wet impregnation method. The liquid was then dried at 80 °C using a rotary evaporator employing a vacuum pump. The powders were dried at 100 °C for 12 h and calcined at 550 °C for 4 h, and the as-received catalyst was denoted as CuiF.
:0.3:0.8) powders using a wet impregnation method. The liquid was then dried at 80 °C using a rotary evaporator employing a vacuum pump. The powders were dried at 100 °C for 12 h and calcined at 550 °C for 4 h, and the as-received catalyst was denoted as CuiF.
The H-SAPO-34 was ion-exchanged using a NH4NO3 (99% NH4NO3, Aladdin, China) solution at 80 °C for 2 h to obtain the NH4–SAPO-34. Then, the solid was filtered and rinsed with deionized water, and was dried at 100 °C for 12 h. Cu ion-exchange was performed by mixing the NH4–SAPO-34 with a Cu(CH3COO)2 (Ling Feng, China) solution at 60 °C for 4 h. After filtered and washed with deionized water, the powders were dried at 100 °C for 12 h and calcined at 550 °C for 4 h, and the as-received catalyst was denoted as CueF.
The nominal content of Cu in the catalysts was 2 wt%. Additionally, the as-received catalysts were hydrothermally treated at 750 °C in 10 vol% H2O/air for 24 h, and the resulting catalysts were denoted as CuiA and CueA, respectively.
In SCR reactions on many catalysts, the SO2 effects involve competitive adsorption of SO2 and the reactants, physical coverage of active sites by ammonium sulfates and formation of active metal sulfates. The last one was irreversible and paid attention to in the present study. To simplify the study, the accumulation of ammonium sulfates and the introduction of SO2 in SCR reaction were not involved. The as-received and hydrothermally treated catalysts were subjected to a sulfur poisoning treatment in 100 ppm SO2/10 vol% H2O/air for 24 h at 350 °C which was located within the normal diesel exhaust temperature and commonly applied in the literature,6 and the obtained samples were denoted with a “-s” suffix.
For comparison, CuSO4/SAPO-34 catalysts were synthesized by mechanical mixing and wet impregnation methods using CuSO4 solution as the precursor, and the obtained samples were denoted as Cum-s and Cui-s, respectively. The nominal mass content of Cu in the catalysts was also 2 wt%. The comparison of sample and abbreviation were listed in Table 1.
| Sample | Abbreviation | |
|---|---|---|
| Unpoisoned | Sulfur poisoned | |
| SAPO-34 support | SAPO-34 | SAPO-34-s |
| Impregnated Cu/SAPO-34, as-received | CuiF | CuiF-s |
| Ion-exchanged Cu/SAPO-34, as-received | CueF | CueF-s |
| Impregnated Cu/SAPO-34, hydrothermally aged | CuiA | CuiA-s |
| Ion-exchanged Cu/SAPO-34, hydrothermally aged | CueA | CueA-s |
| Impregnated CuSO4/SAPO-34 | — | Cui-s |
| Mechanically mixed CuSO4/SAPO-34 | — | Cum-s |
2.2. Activity measurement
The NH3-SCR activity was evaluated in a fixed bed quartz reactor, and the effluent gases were monitored by infrared spectroscopy (Thermo Nicolet 380). The reaction gas consisted of 500 ppm NO, 500 ppm NH3, 5% O2, 10% H2O, and N2 in balance. The flow rate of the reaction gas was 1000 mL min−1, the amount of catalyst (sieved to 50–80 mesh) used was 300 mg, and the gas hourly space velocity was kept at 100000 h−1. The concentration of NOx (NO, N2O, and NO2) was detected at the ppm level. The NOx conversion of the catalysts was calculated using eqn (1):
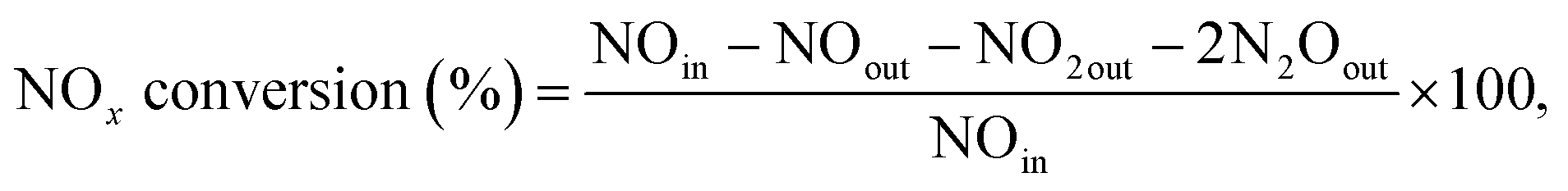 | (1) |
2.3. Characterization
X-ray powder diffraction (XRD) patterns were recorded on a diffractometer (D8 ADVANCE, Bruker, Germany) employing Cu Kα radiation (λ = 0.15418 nm). Data were collected in the range of 5° ≤ 2θ ≤ 50° with a scanning velocity of 6° min−1.Textural properties were determined by nitrogen physisorption at −196 °C using a JW-BK122F (Beijing JWGB, China) instrument. Prior to the measurements, the samples were degassed at 0.01 Pa at 220 °C. The specific surface area and desorption pore volume were assessed using the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda methods, respectively.
The actual contents of Cu, Si, Al, P and S were determined on an Agilent 5100 ICP-OES instrument.
Electron paramagnetic resonance (EPR) spectra in the X-band were recorded on a CW spectrometer JES-FA200 (JEOL) at microwave power of 1 mW, modulation frequency of 100 kHz, and central field of 300 mT. Before the measurements, the samples were heated in vacuum from room temperature to 100 °C for 30 min. The samples were then cooled to −150 °C. Mn2+ was used as an external reference sample, and the g value and EPR spectral intensity of Cu2+ were determined.
Transmission electron microscopy (TEM) image and composition of the catalysts were taken on a JEM-2100F (JEOL, Japan) high resolution transmission electron microscope equipped with energy dispersive X-ray spectroscopy (EDX, Oxford EDX System). The catalysts were suspended in ethanol and dispersed over a carbon-coated holey Ni grid with a film prior to measurements.
Hydrogen temperature-programmed reduction (H2-TPR) was performed on a Micromeritics Auto Chem II. Prior to the H2-TPR experiment, the test sample (50 mg) was treated with He at a total flow rate of 50 mL min−1 at 300 °C for 30 min and subsequently cooled to room temperature and flushed with He for 10 min. The atmosphere was then changed to 10% H2/Ar (50 mL min−1), and the reactor temperature was raised to 600 °C at a heating rate of 10 °C min−1. H2 consumption during the experiment was monitored with a thermal conductivity detector.
Sulfate decomposition tests were performed using a mass spectrometer (Omni-star TM). In a typical test, the test sample (50 mg) was treated with He at a total flow rate of 50 mL min−1 at 300 °C for 30 min and subsequently cooled to room temperature and flushed with He for 10 min. Finally, the atmosphere was changed to 10% H2/Ar (50 mL min−1), and the reactor temperature was raised to 1000 °C using a heating rate of 10 °C min−1. The intensities of the m/z ratios 64 (SO2) and 34 (H2S) in the outlet gas were monitored as a function of temperature.
Ammonia temperature-programmed desorption (NH3-TPD) analysis was performed using a TP 5000-II multiple adsorption apparatus (Xianquan, China). The samples (∼50 mg) were pretreated in He at 500 °C for 30 min. When the temperature decreased to 50 °C, NH3 was introduced until adsorption on the samples was saturated. The samples were then purged with He to remove residual NH3 from the surface of the samples. The samples were then heated in He from 100 to 500 °C at a rate of 10 °C min−1 for subsequent NH3 desorption.
The thermal behavior of the catalysts was evaluated on a Mettler Toledo TG/DSC instrument from room temperature to 1000 °C in N2 (50 mL min−1) at a heating rate of 10 °C min−1. The test powders (30 mg) were loaded in individual alumina sample cells.
X-ray photoelectron spectroscopy (XPS) measurements were performed on a Thermofisher ESCALAB 250Xi high performance electron spectrometer using non-monochromated Al Kα excitation source (hν = 1486.6 eV). Binding energies were calibrated using the contaminant carbon (C 1s = 284.6 eV).
3. Results
3.1. Structural properties
The effects of hydrothermal treatment and sulfur poisoning on the structures of the Cu species and SAPO-34 were investigated by XRD, and the results are shown in Fig. 1. For comparison, the diffraction pattern of SAPO-34 zeolite was included. The diffraction peaks of SAPO-34 were consistent with that of single-phase chabazite.20 The chabazite structure of SAPO-34 remained intact after copper loading, and no copper-related phases (e.g., Cu, Cu2O and CuO) were detected for all the catalysts. These results indicated that the copper species were highly dispersed or existed in the form of Cu2+ ions.15 After sulfur poisoning, the intensity of the diffraction peaks of the catalysts decreased, which was mainly attributed to the slight skeleton damage of SAPO-34 upon the treatment.21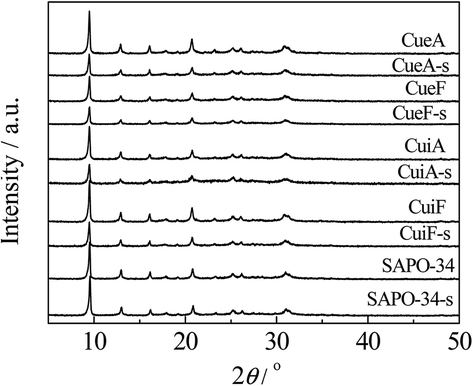 | ||
| Fig. 1 XRD patterns of the SAPO-34 zeolites and Cu/SAPO-34 catalysts. | ||
The specific surface area (SBET), micropore surface area (Smicro), external surface area (Sext), and micropore volume of the catalysts were determined and summarized in Table 2. The Sext of the Cu/SAPO-34 catalysts were lower than that of the SAPO-34 support owing to coverage of CuO on the external surface of the zeolite. Interestingly, the SBET and pore volume of the as-received catalyst CuiF, obtained after copper impregnation of the SAPO-4 support, was higher than those of the support. This increase could be related to the generation of small defects in the SAPO-34 crystals (following Cu impregnation).22 After hydrothermal treatment and sulfur poisoning, the copper species and metal sulfates blocked some of the zeolite channels, thus inducing changes in the textural properties of the zeolite.20,23 Moreover, as indicated by the XRD patterns in Fig. 1, skeleton damage of SAPO-34 led to decreases in the BET surface areas and pore volumes of the poisoned catalysts. Nevertheless, these changes of the textural properties for the catalysts should not be important factors responsible for their different catalytic behaviors.
| Sample | SBET (m2 g−1) | Smicro (m2 g−1) | Sext (m2 g−1) | Pore volume (cm3 g−1) | Concentrationa (wt%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Al | P | Si | Cu | S | |||||
| a Obtained by ICP-OES. | |||||||||
| SAPO-34 | 651 | 607 | 44 | 0.267 | 22 | 21 | 3.9 | — | — |
| SAPO-34-s | 638 | 599 | 39 | 0.262 | 22 | 21 | 3.9 | — | 0.23 |
| CuiF | 676 | 636 | 40 | 0.273 | 22 | 21 | 3.9 | 1.99 | — |
| CuiF-s | 620 | 575 | 45 | 0.253 | 21 | 20 | 3.7 | 1.95 | 0.77 |
| CuiA | 620 | 586 | 34 | 0.241 | 22 | 21 | 3.8 | 1.99 | — |
| CuiA-s | 575 | 525 | 50 | 0.184 | 21 | 20 | 3.7 | 1.96 | 0.81 |
| CueF | 618 | 582 | 36 | 0.238 | 22 | 21 | 3.8 | 2.04 | — |
| CueF-s | 569 | 534 | 35 | 0.219 | 22 | 21 | 3.8 | 2.00 | 0.85 |
| CueA | 621 | 583 | 38 | 0.240 | 22 | 21 | 3.8 | 2.04 | — |
| CueA-s | 560 | 524 | 36 | 0.216 | 22 | 21 | 3.8 | 1.99 | 0.87 |
As indicated by the inductively coupled plasma optical emission spectrometry (ICP-OES) results in Table 2, the obtained Cu contents in the catalysts were very close to the nominal value, and no loss of copper occurred after hydrothermal treatment. The sulfur content in the poisoned Cu/SAPO-34 catalysts ranged from 0.77 to 0.87 wt%.
3.2. Catalytic activity
The temperature dependency of NOx conversion during the NH3-SCR reaction over the catalysts is shown in Fig. 2. The as-received Cu/SAPO-34 catalysts showed superior SCR activity at low temperatures (150–350 °C) as shown in enlarged figure, and the differences were small. The reduced activities at higher temperatures were mainly caused by the competitive reactions between NH3-SCR and NH3 oxidation.24,25 After the hydrothermal treatment, the catalysts showed high SCR activity at 200–500 °C, which are in consistence with the previous report.26 Sulfur poisoning reduced the low-temperature (150–350 °C) activities of the catalysts to different degrees; however, it improved the high-temperature (350–550 °C) activities of the as-received catalysts. The concentration of N2O was always lower than 10 ppm in the whole temperature range (not shown). Such results are discussed further in the following sections.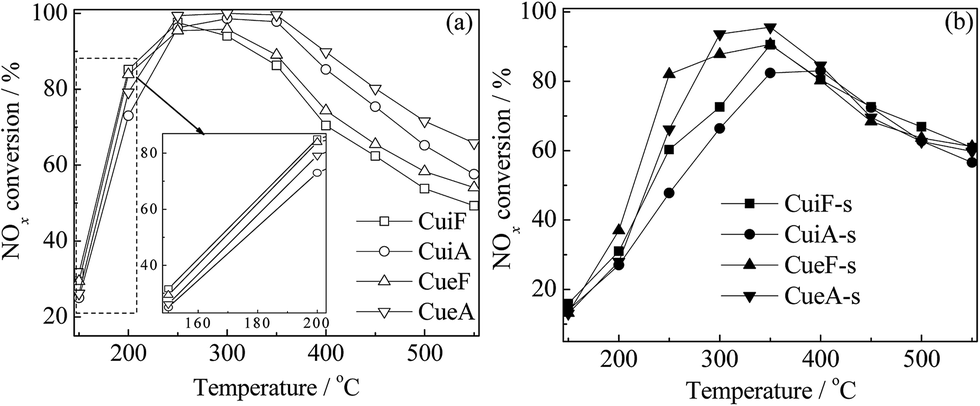 | ||
| Fig. 2 NH3-SCR curves of the (a) as-received and hydrothermally treated and (b) poisoned Cu/SAPO-34 catalysts. Reaction conditions: [NO] = [NH3] = 500 ppm, O2 = 5%, [H2O] = 10%, N2 in balance, and GHSV = 100000 h−1. | ||
3.3. EPR studies
The hyperfine structure of isolated Cu2+ species was characterized by EPR analysis. In contrast, other copper species do not produce any EPR signals.15 Therefore, EPR is an effective method to quantitatively determine the amount of isolated Cu2+ on Cu–zeolite catalysts. Fig. 3 shows the EPR spectra of the non-poisoned (as-received and hydrothermally treated) and poisoned Cu/SAPO-34 catalysts. Four adsorption peaks were detected in the parallel region, and a sharp peak was observed in the vertical region. The EPR signals at g∥ = 2.392, A∥ = 12.958, and g⊥ = 2.074 mT could be ascribed to Cu2+ species on the site (I) displaced from the six-ring structure into the ellipsoidal cavity of the SAPO-34 zeolite.26,27 Furthermore, the intensity of the hyperfine splitting peaks of the sulfur-poisoned samples was lower, indicating a reduction in the number of isolated Cu2+ active species. | ||
| Fig. 3 EPR spectra of the Cu/SAPO-34 catalysts measured at −150 °C. | ||
3.4. EDX studies
In order to examine the effects of the hydrothermal aging and sulfur poisoning on the distributions of copper species on the external surface of SAPO-34, EDX measurements were carried out based on TEM images and spectra in Fig. S1,† and the results were listed in Table 3. It can be seen that the hydrothermal treatment promoted the migration of Cu from the zeolite external surface to the ion-exchanged sites, which was similar to our previous report.16 After the sulfur poisoning, the surface Cu concentration increased again for all the catalysts as well as the accumulation of sulfur, suggesting that the sulfur poisoning can provide the driving force for copper migration.| Catalysts | Concentration (wt%) | |||||
|---|---|---|---|---|---|---|
| O | Al | P | Si | Cu | S | |
| CuiF | 44.09 | 24.98 | 23.82 | 4.25 | 2.86 | — |
| CuiF-s | 46.03 | 23.11 | 22.54 | 4.09 | 3.28 | 0.94 |
| CuiA | 44.23 | 25.06 | 25.30 | 3.84 | 1.57 | — |
| CuiA-s | 45.77 | 22.74 | 23.45 | 3.51 | 4.01 | 0.53 |
| CueF | 46.09 | 23.82 | 23.41 | 3.56 | 3.13 | — |
| CueF-s | 42.87 | 24.30 | 24.04 | 3.26 | 3.65 | 0.93 |
| CueA | 47.60 | 23.32 | 23.42 | 3.70 | 1.96 | — |
| CueA-s | 42.18 | 24.67 | 25.21 | 4.11 | 2.50 | 1.32 |
3.5. H2-TPR studies
H2-TPR is commonly used for determining types of copper species and the redox properties of catalysts. Fig. S2† shows the TPR curves of the unpoisoned Cu/SAPO-34 catalysts. Four reduction peaks were observed in the temperature range of 50–600 °C. The first peak observed at 100–150 °C was attributed to the reduction of surface CuO clusters on the external surface of the zeolite framework, proceeding via a one-step reduction with the stoichiometry of one mole of H2 for one mole of CuO clusters.27 The second peak was attributed to the reduction of isolated Cu2+ in the zeolite framework to Cu+. The third peak was attributed to the reduction of CuO crystallites from CuO to Cu0. The fourth peak was attributed to the reduction of Cu+ to Cu0,11 including that of Cu+ ions obtained from the reduction of isolated Cu2+ and that of original Cu+ ions in the framework of SAPO-34. According to the literature,28 the isolated Cu2+ ions was reducible in the temperature range 200–300 °C, but the reduction of Cu+ ions at the ion-exchange sites to Cu0 occurred at distinctly higher temperatures, or they were too stabile to be reduced in the H2-TPR temperature range. Table 4 lists the amounts of different reducible Cux+ species on the catalysts based on the TPR curves. The H2 consumption was much less than the theoretical value, even if we assumed that all the copper ions existed as Cu+. This indicated that some copper species (Cu ions at the ion-exchange sites) were too stable to be reduced to Cu0 at temperatures below 800 °C. As observed, the isolated Cu2+ ions were more abundant on the ion-exchanged catalysts, and reached the maximum on CueA. Contrarily, more surface CuO clusters were found on the impregnated catalysts, and reached the maximum on CuiF. Furthermore, the strong interaction of copper ions with the zeolite framework, upon the hydrothermal treatment, produced more reducible copper species and especially isolated Cu2+ ions.29| Sample | Amount of coppera (μmol Cu per g cat.) | Amount of reducible copper speciesb (μmol Cu per g cat.) | ||||
|---|---|---|---|---|---|---|
| Surface CuO clusters | Isolated Cu2+ | CuO crystallites | Cu+ | Totalc | ||
| a Calculated based on the ICP results in Table 2.b Obtained by H2-TPR curves.c The data outside and inside the brackets were estimated based on the sum of surface CuO clusters, isolated Cu2+ and CuO crystallites (that of surface CuO clusters, CuO crystallites and Cu+). | ||||||
| CuiF | 310.9 | 47.7 | 30.5 | 61.9 | 56.9 | 140.1 (166.5) |
| CuiA | 310.9 | 10.9 | 65.3 | 96.8 | 148.3 | 173.0 (256.0) |
| CueF | 318.8 | 32.9 | 208.1 | 56.0 | 120.6 | 297.0 (209.5) |
| CueA | 318.8 | 9.9 | 255.1 | 67.2 | 177.4 | 332.2 (254.5) |
Surface CuO clusters are more active toward the oxidation of NO to NO2 than isolated Cu2+ ions,30 which favor low-temperature reactions via the so-called fast SCR reaction.20,30 In contrast, surface CuO clusters inhibit the high-temperature SCR activity owing to their high catalytic activity toward NH3 oxidation.20,30 The isolated Cu ions are always active within a wide temperature range (150–550 °C). Hence, the surface CuO clusters and isolated Cu2+ ions synergistically determine the SCR performance of Cu/SAPO-34 catalysts, resulting in trends in catalytic activity in two temperature regions. The trend in low-temperature activity is in good agreement with the amount of surface CuO clusters as shown in the enlarged figure in Fig. 2a, although the difference is not obvious. For the impregnated catalysts, abundant surface CuO clusters play a more crucial role in the low temperature range than isolated Cu2+.30,31 Isolated Cu2+ ions are abundant in the ion-exchanged catalysts and dominate the activity at low temperatures. Both types of copper species can act as active sites for the low-temperature SCR reaction. The active surface CuO clusters is absent in some studies,32,33 due to the specific SAPO-34 supports and preparation methods adopted, and the hydrothermal treatment will increase the amount of Cu2+ and improve the activity at low temperatures.
H2-TPR analysis was also conducted on the sulfur-poisoned catalysts. Three peaks of H2 consumption were observed (Fig. 4). The peak observed at the lowest temperature (peak A) was attributed to the reduction of isolated Cu2+ to Cu+.34,35 The peaks observed at 230–350 °C (peak B) and 350–450 °C (peak C) were attributed to the reduction of dispersed CuSO4, and overlapping of the reduction of crystalline CuSO4 (ref. 36 and 50) and Cu+ to Cu0, respectively.31,50 Assignments of these reduction peaks were confirmed by the TPR curves of reference samples Cui-s and Cum-s. Specifically, peak A was absent for these mixed catalysts using CuSO4 as the precursor. Cui-s displayed a dominant peak B, which was assigned to the reduction of dispersed CuSO4. Cum-s displayed a bimodal peak distribution that accounts for the reduction of dispersed CuSO4 and crystalline CuSO4 at 320 and 380 °C,31,36 respectively. The reducibility of CuSO4 species followed the order of dispersed CuSO4 > crystalline CuSO4. The results agreed with the XRD results, which indicated that the dispersion of CuSO4 in Cui-s and Cum-s is different. Compared with the unpoisoned catalysts (Fig. S2†), the reduction peak associated with surface CuO clusters (at 100–150 °C) disappeared completely after sulfur poisoning owing to accessibility to SO2 and transformation to copper sulfates. In contrast, some isolated copper ions remained as the small pores (0.43 nm) of SAPO-34 protected the isolated Cu2+ from attack by SO2, which has a similar kinetic diameter of 0.411 nm.37 As determined from the H2-TPR curves, CuSO4 was highly dispersed on the poisoned ion-exchange-prepared catalysts. In contrast, the metal sulfates was much larger on the poisoned impregnation-prepared catalysts, and hence, CuSO4 was not as highly dispersed in the poisoned impregnation-prepared catalysts.
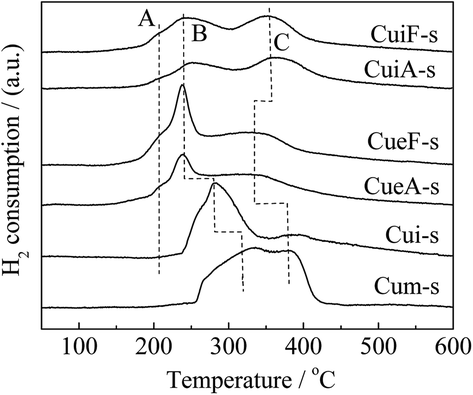 | ||
| Fig. 4 H2-TPR curves of the poisoned catalysts. | ||
Surface CuO clusters and crystallites are more abundant on the impregnation-prepared catalysts. After sulfur poisoning, surface CuO clusters and crystallites tended to transform to dispersed CuSO4 and crystalline CuSO4, respectively, and the generated dispersed CuSO4 would sinter to form crystalline CuSO4, too. Thus, it appeared to be easier for surface copper species to form crystalline sulfates upon the sulfur poisoning treatment since they were exposed on the catalyst surface. As shown in Table 4 and Fig. S2,† the isolated Cu2+ ions were much more abundant on the ion-exchanged catalysts (CueF and CueA). On the other hand, the reduction peak B of the corresponding sulfated catalysts (CueF-s and CueA-s) in Fig. 4, which was attributed to the reduction of dispersed CuSO4, was much intenser than those of the sulfated impregnated catalysts (CuiF-s and CuiA-s). Based on kinetic and thermodynamic theories, it is easier for isolated Cu2+ to migrate onto the catalyst surface to form dispersed CuSO4 than CuSO4 crystallites under the same poisoning treatment condition. Previous studies also have elucidated the impact of sulfur poisoning on Cu dispersion. Cheng et al.7 used XANES/EXAFS analyses to investigate properties of the copper species in Cu/SAPO-34 before and after sulfur poisoning. The Cu K-edge XANES spectra for the Cu/SAPO-34 before and after sulfur poisoning, as well as the spectra for CuSO4 as a reference. The XANES spectra between 8975 and 8985 eV were similar for both the unsulfated and sulfur-poisoned catalysts, but quite different from the spectrum of crystalline CuSO4. This suggests that the sulfate formation resulting from sulfur poisoning does not induce any macroscopic changes in the Cu species; in other words, Cu remains in a highly dispersed state as its initial ion-exchange locations. A similar phenomenon was observed by Hamada et al.38 over a Cu–ZSM-5 catalyst after SO2/O2 treatment.
As observed in Fig. 5, a bimodal SO2 release distribution was detected within 250–500 °C by mass spectrometry analysis. The low-temperature peak was ascribed to the decomposition/reduction of dispersed CuSO4 was centered at 310 and 330 °C for the poisoned ion-exchange- and impregnation-prepared catalysts, respectively. The high-temperature peak was located at 380 °C and ascribed to the decomposition/reduction of crystalline CuSO4. Moreover, the poisoned ion-exchange-prepared catalysts displayed a stronger dispersed CuSO4-related peak. As noted, the temperatures at which the dispersed CuSO4-related SO2 peak was observed were somewhat higher than those of the corresponding H2 peak B. This difference was attributed to the re-adsorption of the SO2 released from the decomposition/reduction of CuSO4 on the catalyst.39,40 No H2S was detected at 50–1000 °C for all the poisoned catalysts. The formation of elemental sulfur on the catalyst surface cannot be completely excluded or some of the reduction products were not detected by MS, which may be responsible for the differences between the sulfate amounts determined by ICP-OES, TGA and MS. In this sense, the ICP-OES results appear to be more reliable.
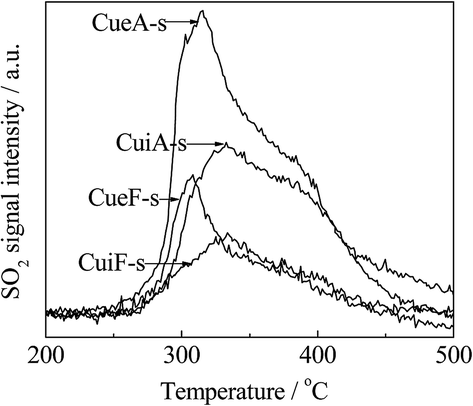 | ||
| Fig. 5 The outlet SO2 signals detected from the H2-TPR tests of the poisoned catalysts. | ||
3.6. NH3-TPD studies
Ammonia is widely used as a probe molecule to identify types of surface acidic sites.41–43 In this study, the acid properties of the non-poisoned and poisoned catalysts were studied by NH3-TPD. Fig. 6 shows the NH3-TPD profiles of the different catalysts. Two broad bands at 120–260 and 320–460 °C could be observed, which could be deconvoluted into three desorption peaks (A, B, and C).44 Peaks B and C observed at the higher temperatures could possibly be assigned to structural Brønsted acid sites, which can be correlated to moderate and strong acidity.37,45–48 After copper loading, considerable changes in peaks A, B, and C were observed. Peak A could be attributed to the adsorption of NH3 molecules on weak Brønsted acid sites on the zeolite and weak Lewis acid sites related to Cu species. Peak B was ascribed to structural Brønsted acid sites on the zeolite and moderate Lewis acid sites related to Cu species. Peak C was related to the adsorption of NH3 molecules on both strong Brønsted acid sites on the SAPO-34 support and new Lewis acid sites created by the Cu species.41 The assignments of different peaks are supported by the DRIFTS results as shown in Fig. S3.† Table 4 lists the amount of the different acid sites calculated from the NH3-TPD results. As observed, the loading of Cu reduced the surface acidity of HSAPO-34 mainly because of the substitution of the support proton (–Si–OH–Al) by Cu2+ ions. Table 5 also shows that more acid sites were created after sulfur poisoning. For the poisoned catalysts, the deposited sulfates could serve as moderate acid sites. Combined with the SCR performance in Fig. 2, the high NH3 adsorption capacity of the poisoned catalysts may facilitate the SCR reaction at high temperatures. When compared with the as-received catalysts, the hydrothermally treated catalysts featured more Lewis acid sites. Sulfur poisoning also led to a decrease in the amount of weak acid sites, which could be related to the reduced amount of Lewis acid sites created by Cu species owing to the formation of copper sulfates.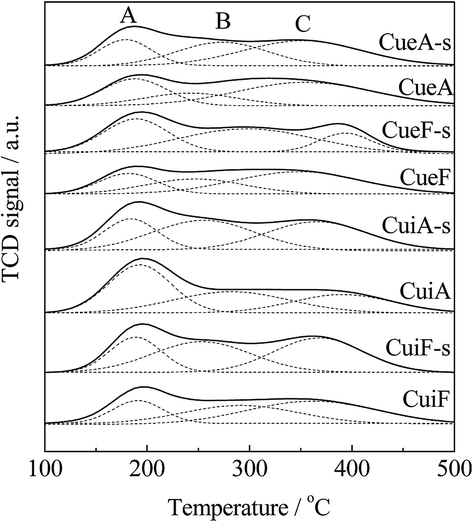 | ||
| Fig. 6 NH3-TPD profiles of the Cu/SAPO-34 catalysts. | ||
| Sample | Acidity (mmol NH3 per g cat.) | |||
|---|---|---|---|---|
| Weak | Moderate | Strong | Total | |
| CuiF | 0.0163 | 0.0302 | 0.0425 | 0.0890 |
| CuiF-s | 0.0255 | 0.0424 | 0.0433 | 0.1112 |
| CuiA | 0.0425 | 0.0350 | 0.0260 | 0.1035 |
| CuiA-s | 0.0226 | 0.0443 | 0.0399 | 0.1068 |
| CueF | 0.0166 | 0.0211 | 0.0399 | 0.0776 |
| CueF-s | 0.0298 | 0.0491 | 0.0134 | 0.0923 |
| CueA | 0.0243 | 0.0168 | 0.0453 | 0.0864 |
| CueA-s | 0.0189 | 0.0308 | 0.0417 | 0.0914 |
| SAPO-34 | 0.0356 | 0.0421 | 0.0391 | 0.1168 |
Thermogravimetric analysis was conducted on the sulfated catalysts. The percentages of sulfur on the poisoned catalysts were calculated from the thermogravimetric analysis obtained in the temperature range of 450–1000 °C shown in Fig. 7 (estimated by SO3), which decreased in the order of CueA-s > CueF-s > CuiA-s > CuiF-s. The TG results are in good agreement with the ICP-OES results, and minor differences determined by two methods may be caused by the incomplete decomposition or dissolution of some sulfate species.19
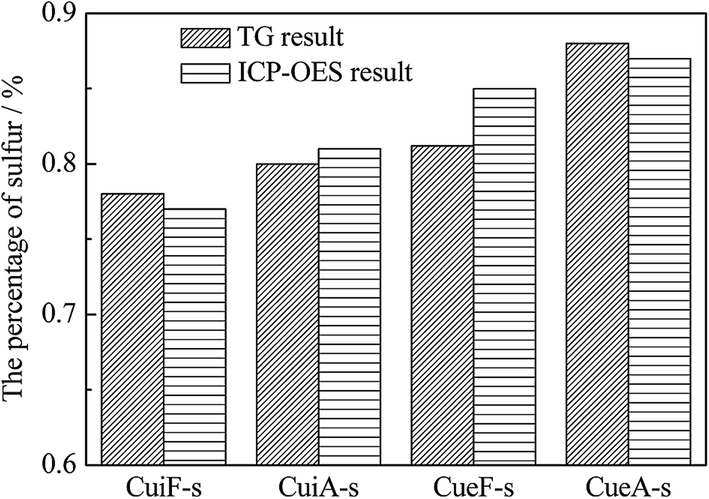 | ||
| Fig. 7 The percentages of sulfur on the poisoned catalysts determined by TG and ICP-OES analyses. | ||
4. Discussion
4.1. Effects of sulfur poisoning on Cu species
The amount of isolated Cu2+ ions was calculated by integrating the EPR curves in the magnetic field range of 0–500 mT and setting the value of CuiF as the standard. As shown in Fig. 8, the amount of isolated Cu2+ decreased after sulfur poisoning to different degrees, and the reduction in the amount of isolated Cu2+ (relative to that prior to sulfur poisoning) decreased in the order of CueA-s > CueF-s > CuiA-s > CuiF-s. This result was consistent with the EPR and H2-TPR results. As the attack of SO2 on the isolated Cu2+ ions is limited owing to the similarity in the pore size of SAPO-34 and kinetic diameter of SO2, the decreased amount of isolated Cu2+ may be due to the migration of isolated Cu2+ to the surface and the formation of the dispersed sulfates.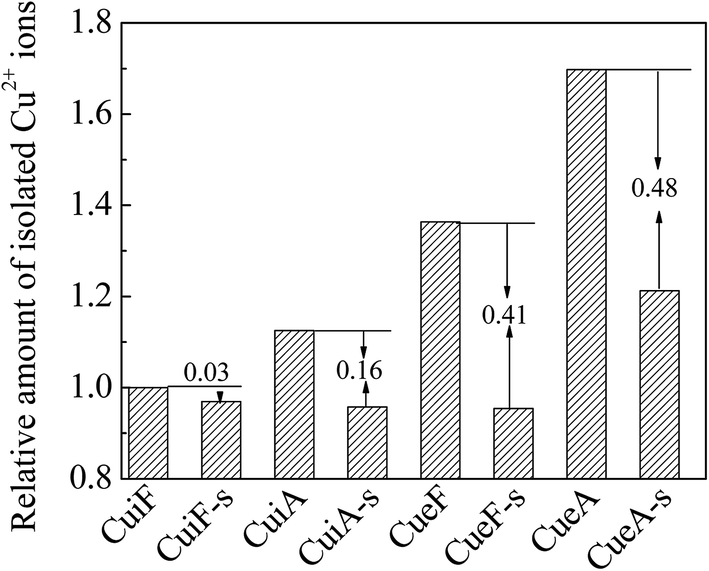 | ||
| Fig. 8 Relative concentration of isolated Cu2+ ions determined from the EPR results. | ||
To confirm the migration of the copper species during the hydrothermal and sulfur poisoning treatments, the Cu/Si atomic ratios on the surface and in the bulk were calculated. As listed in Table 6, the as-received catalysts featured a higher Cu/Si ratio on the surface than that in the bulk, indicating the enrichment of copper on the external surface of SAPO-34. As noted further, the ion-exchange catalyst CueF featured a higher surface Cu concentration than the impregnated sample, CuiF. This result was attributed to the relatively low calcination temperature (550 °C) employed, which did not allow sufficient diffusion of surface CuO into the ion-exchanged sites. After hydrothermal treatment, the surface Cu/Si ratios of the catalysts decreased and were lower than the bulk values. This result confirmed the migration of the surface copper ions to the internal region of SAPO-34 zeolite during hydrothermal treatment.16,49 The sulfur-poisoned catalysts featured considerably higher surface Cu/Si ratios than the non-poisoned catalysts, thus confirming that sulfur poisoning promotes the migration of isolated Cu2+ back to the surface to form copper sulfates. Based on the combined EPR, H2-TPR, EDX and TG results, it is reasonable to suggest that the dispersed CuSO4 species are mainly formed upon migration of isolated Cu2+, whereas the crystalline CuSO4 species are mainly produced from surface CuO clusters upon sulfation.
| Sample | Cu/Si in the bulka | Cu/Si on the surfaceb | S/Si in the bulka | S/Si on the surfaceb |
|---|---|---|---|---|
| a Obtained by ICP-OES.b Obtained by XPS. | ||||
| CuiF | 0.23 | 0.29 | — | — |
| CuiF-s | 0.22 | 0.44 | 0.18 | 0.21 |
| CuiA | 0.23 | 0.21 | — | — |
| CuiA-s | 0.22 | 0.30 | 0.19 | 0.19 |
| CueF | 0.23 | 0.33 | — | — |
| CueF-s | 0.22 | 0.48 | 0.20 | 0.16 |
| CueA | 0.23 | 0.22 | — | — |
| CueA-s | 0.22 | 0.34 | 0.20 | 0.11 |
4.2. Effects of sulfur poisoning on NH3-SCR activity
It has been reported that the types of copper species are critical to the NH3-SCR activity of the zeolite catalysts and the formation of metal sulfates would deactivate the catalysts. Luo et al.29 found that both surface Cu species and isolated Cu2+ were active for SCR reactions, while surface Cu species appeared to be more active for NO oxidation and NH3 oxidation. After sulfur poisoning, both types of Cu species decreased significantly in the population due to the combination with sulfur. In a recent study by Shen et al.,50 both copper sulfate and ammonia sulfate species decrease the SCR reaction rate of the Cu/SAPO-34 catalysts by reducing the number of isolated Cu2+ species, and the copper sulfate is harder to decompose than ammonia sulfate. However, the states of the copper sulfate and their contribution to activity were not involved in these studies.To clarify the roles of the dispersed CuSO4 and crystalline CuSO4 in NH3-SCR reaction, the CuSO4/SAPO-34 catalysts were synthesized by mechanical mixing and wet impregnation methods using CuSO4 solution as the precursor. As observed in the XRD patterns in Fig. 9, Cum-s displayed Cu(SO4)·(H2O) diffraction peaks. In contrast, those peaks were absent in Cui-s. These results agreed with the H2-TPR results (Fig. 4), indicating that CuSO4 is finely dispersed on SAPO-34 for Cui-s. As shown in Fig. 10, Cui-s displays much higher SCR activity at low temperatures (150–450 °C) than Cum-s. This demonstrates the dependency of catalytic activity on the dispersion of copper sulfates. It should be noted that the reference catalyst Cui-s even shows higher SCR activity than CuiF-s and CuiA-s in the whole temperature range measured, which may be due to the different status of the impregnated and in situ formed CuSO4, and the detailed mechanism remains to be investigated.
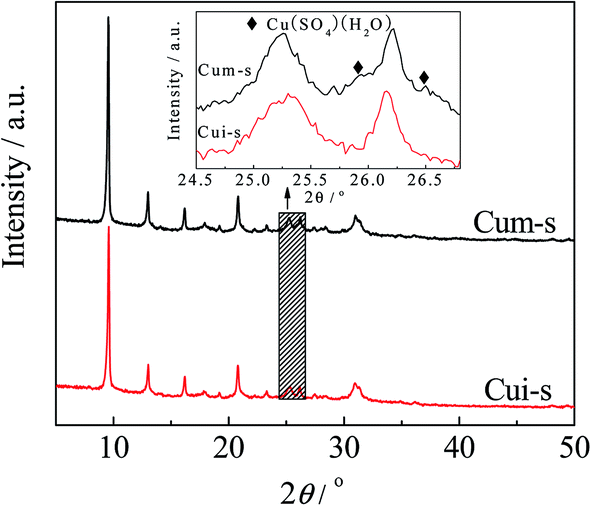 | ||
| Fig. 9 XRD spectra of CuSO4/SAPO-34 catalysts. | ||
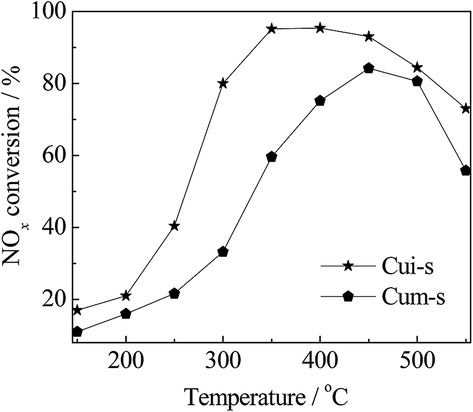 | ||
| Fig. 10 NH3-SCR curves of CuSO4/SAPO-34 catalysts. Reaction conditions: [NO] = [NH3] = 500 ppm, O2 = 5%, [H2O] = 10%, N2 in balance, and GHSV = 100000 h−1. | ||
Thus, the SCR reactions at low and high temperatures over the non-poisoned and poisoned Cu/SAPO-34 catalysts are dominated by different active species as presented in Scheme 1. Surface CuO clusters and isolated Cu2+ act synergistically at low temperatures via fast SCR reaction, which occurs mainly on isolated Cu2+ active sites with NO2 generated from the oxidation of NO on surface CuO clusters. Upon hydrothermal treatment, a decrease in the amount of surface CuO clusters is observed, which leads to reduced low-temperature activities. At high temperatures, the competitive oxidation of NH3 is weakened via sintering of the surface CuO clusters to CuO crystallites, resulting in improved high-temperature SCR activities. In this temperature range, the isolated Cu2+ ions serve as main active sites via the standard SCR reaction. For the sulfur-poisoned catalysts, the catalytic activity depends not only on the sulfur loading but also on the dispersion of the generated CuSO4. As shown in Fig. 10, the dispersed CuSO4 species on Cui-s show much higher activity than the crystalline CuSO4 on Cum-s with the same sulfur loading. Based on the H2-TPR result, the migrated isolated Cu2+ tends to form dispersed CuSO4 species, which are more active for SCR reaction, on the ion-exchanged catalysts after sulfur poisoning. Meanwhile, the surface CuO clusters and crystallites tend to produce crystalline CuSO4 species, which are less active for NH3-SCR. The intensity of reduction peak of dispersed CuSO4 follows the sequence of CueF-s > CueA-s > CuiF-s > CuiA-s, which is in good consistence with the low-temperature activity of the sulfated catalysts. Additionally, the sulfur poisoning improves the high-temperature activities of the as-received catalysts by inhibiting NH3 oxidation.
 | ||
| Scheme 1 Roles of different copper species on the non-poisoned and poisoned Cu/SAPO-34 catalysts in NH3-SCR reactions at different temperatures. | ||
5. Conclusions
The application of different preparation methods and the hydrothermal treatment in this work allowed us to obtain different distributions of Cu species on a zeolite support, and the catalytic activities and sulfur tolerance of those copper species were examined. Sulfur poisoning was found to promote the migration of isolated Cu2+ back to the surface as highly dispersed CuSO4, and the small pores of SAPO-34 appeared to protect the isolated Cu2+ from attack by SO2 to some extent. Crystalline CuSO4 formed mainly from sulfation of surface CuO clusters and crystallites, and contributed little to the low-temperature SCR activity. As a result, the operation window of the Cu/SAPO-34 catalysts shifted toward high temperatures after exposure in 100 ppm SO2 at 350 °C for 24 h. The migration of copper was observed by XPS and EDX analysis, and a possible mechanism of Cu migration in sulfur poisoning was proposed on the basis of this observation.The present study allowed us to learn the relationship between the formation of copper sulfates and the active copper species upon sulfur poisoning, and the contribution of CuSO4 in different dispersion states to the NH3-SCR activity. It provided a strategy to develop anti-sulfur deNOx catalysts for industrial applications.
Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (No. 51372137), Ministry of Science and Technology, PR China (No. 2016YFE0126600), Shaoxing University (No. 2016LG1001), Key Laboratory of Advanced Materials of Ministry of Education of Tsinghua University (2016AML01), the National Key R&D Program of China (No. 2017YFC0210701) and JCYJ20140417115840287.References
- S. J. Schmieg and J. Lee, SAE Tech. Pap. Ser., 2005 DOI:10.4271/2005-01-3881.
- G. Cavataio, J. Girard, J. E. Patterson, C. Montreuil, Y. Cheng and C. K. Lambert, SAE Int., [Spec. Publ.] SP, 2007 DOI:10.4271/2007-01-1575.
- Y. Cheng, C. Montreuil, G. Cavataio and C. Lambert, SAE Int., [Spec. Publ.] SP, 2008, 1, 471–476 Search PubMed.
- J. W. Girard, C. Montreuil, J. Kim, G. Cavataio and C. Lambert, SAE Int., [Spec. Publ.] SP, 2008, 1, 488–494 Search PubMed.
- J. A. Ura, J. Girard, G. Cavataio, C. Montreuil and C. Lambert, SAE Int., [Spec. Publ.] SP, 2009 DOI:10.4271/2009-01-0625.
- Y. Cheng, C. Montreuil, G. Cavataio and C. Lambert, SAE Int., [Spec. Publ.] SP, 2009 DOI:10.4271/2009-01-0898.
- Y. Cheng, C. Lambert, D. H. Kim, J. H. Kwak, S. J. Cho and C. H. F. Peden, Catal. Today, 2010, 151, 266–270 CrossRef CAS.
- M. Castagnola, J. Caserta, S. Chatterjee, H. Chen, R. Conway, J. Fedeyko, W. Klink, P. Markatou, S. Shah and A. Walker, SAE Int., [Spec. Publ.] SP, 2011 DOI:10.4271/2011-01-1329.
- A. Kumar, M. A. Smith, K. Kamasamudram, N. W. Currier, H. An and A. Yezerets, Catal. Today, 2014, 231, 75–82 CrossRef CAS.
- J. H. Kwak, R. G. Tonkyn, D. H. Kim, J. Szanyi and C. H. F. Peden, J. Catal., 2010, 275, 187–190 CrossRef CAS.
- P. G. Blakeman, E. M. Burkholder, H. Y. Chen, J. E. Collier, J. M. Fedeyko, H. Jobson and R. R. Rajaram, Catal. Today, 2014, 231, 56–63 CrossRef CAS.
- D. W. Fickel, E. D'Addio, J. A. Lauterbach and R. F. Lobo, Appl. Catal., B, 2011, 102, 441–448 CrossRef CAS.
- S. T. Korhonen, D. W. Fickel, R. F. Lobo, B. M. Weckhuysen and A. M. Beale, Chem. Commun., 2011, 47, 800–802 RSC.
- F. Gao, E. D. Walter, N. M. Washton, J. Szanyi and C. H. F. Peden, ACS Catal., 2013, 3, 2083–2093 CrossRef CAS.
- J. J. Xue, X. Q. Wang, G. S. Qi, J. Wang, M. Q. Shen and W. Li, J. Catal., 2013, 297, 56–64 CrossRef CAS.
- L. Wang, J. R. Gaudet, W. Li and D. Weng, J. Catal., 2013, 306, 68–77 CrossRef CAS.
- L. Zhang, D. Wang, Y. Liu, K. Kamasamudram, J. H. Li and W. L. Epling, Appl. Catal., B, 2014, 156–157, 371–377 CrossRef CAS.
- A. Kumar, M. A. Smith, K. Kamasamudram, N. W. Currier, H. An and A. Yezerets, Catal. Today, 2014, 231, 75–82 CrossRef CAS.
- X. S. Liu, X. D. Wu, D. Weng and Z. C. Si, Catal. Commun., 2015, 59, 35–39 CrossRef CAS.
- J. Tan, Z. M. Liu, X. H. Bao, X. C. Liu, X. W. Han, C. Q. He and R. S. Zhai, Microporous Mesoporous Mater., 2002, 53, 97–108 CrossRef CAS.
- C. D. Yan, H. Cheng, Z. S. Yuan and S. D. Wang, Environ. Technol., 2015, 36, 169–177 CrossRef CAS PubMed.
- J. Ma, Z. C. Si, D. Weng, X. D. Wu and Y. Ma, Chem. Eng. J., 2015, 267, 191–200 CrossRef CAS.
- F. Bin, C. L. Song, G. Lv, J. N. Song, S. H. Wu and X. D. Li, Appl. Catal., B, 2014, 150–151, 532–543 CrossRef CAS.
- L. Wang, W. Li, G. S. Qi and D. Weng, J. Catal., 2012, 289, 21–29 CrossRef CAS.
- J. Wang, T. Yu, X. Q. Wang, G. S. Qi, J. J. Xue, M. Q. Shen and W. Li, Appl. Catal., B, 2012, 127, 137–147 CrossRef CAS.
- L. Ma, Y. S. Cheng, G. V. Cavataio, R. W. McCabe, L. X. Fu and J. H. Li, Chem. Eng. J., 2013, 225, 323–330 CrossRef CAS.
- M. Calligaris and G. Nardin, Zeolites, 1982, 2, 200–204 CrossRef CAS.
- M. Richter, M. J. G. Fait, R. Eckelt, M. Schneider, J. Radnik, D. Heidemann and R. Fricke, J. Catal., 2007, 245, 11–24 CrossRef CAS.
- J. Y. Luo, D. Wang, A. Kumar, J. H. Li, K. Kamasamudram, N. Currier and A. Yezerets, Catal. Today, 2016, 267, 3–9 CrossRef CAS.
- F. Liu and H. He, J. Phys. Chem. C, 2010, 114, 16929–16936 CAS.
- B. Pereda-Ayo, U. D. L. Torre, M. J. Illán-Gómez, A. Bueno-López and J. R. González-Velasco, Appl. Catal., B, 2014, 147, 420–428 CrossRef CAS.
- D. Wang, Y. Jangjou, Y. Liu, M. K. Sharma, J. Y. Luo, J. H. Li, K. Kamasamudram and W. S. Epling, Appl. Catal., B, 2015, 165, 438–445 CrossRef CAS.
- W. K. Su, Z. G. Li, Y. Peng and J. H. Li, Phys. Chem. Chem. Phys., 2015, 17, 29142–29149 RSC.
- C. Torre-Abreu, M. F. Ribeiro, C. Henriques and G. Delahay, Appl. Catal., B, 1997, 14, 261–272 CrossRef CAS.
- A. Sultana, T. Nanba, M. Haneda, M. Sasaki and H. Hamad, Appl. Catal., B, 2010, 101, 61–67 CrossRef CAS.
- B. Kartheuser, B. K. Hodnett, A. Riva, G. Centi, H. Matralis, M. Ruwet, P. Grange and N. Passarini, Ind. Eng. Chem. Res., 1991, 30, 2105–2113 CrossRef CAS.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- H. Hamada, N. Matsubayashi, H. Shimada, Y. Kintaichi, T. Ito and A. Nishijima, Catal. Lett., 1990, 5, 189–196 CrossRef CAS.
- Y. J. Shi, H. Shu, Y. H. Zhang, H. M. Fan, Y. P. Zhang and L. J. Yang, Fuel Process. Technol., 2016, 150, 141–147 CrossRef CAS.
- S. Kikuyama, A. Miura, R. Kikuchi, T. Takeguchi and K. Eguchi, Appl. Catal., A, 2004, 259, 191–197 CrossRef CAS.
- S. Brandenberger, O. Kröcher, A. Wokaun, A. Tissler and R. Althoff, J. Catal., 2009, 268, 297–306 CrossRef CAS.
- M. Schwidder, M. S. Kumar, U. Bentrup, J. Pérez-Ramírez, A. Brückner and W. Grünert, Microporous Mesoporous Mater., 2008, 111, 124–133 CrossRef CAS.
- R. Q. Long and R. T. Yang, J. Catal., 2001, 198, 20–28 CrossRef CAS.
- D. Z. Zhang, Y. X. Wei, L. Xu, F. X. Chang, Z. Y. Liu, S. H. Meng, B. L. Su and Z. M. Liu, Microporous Mesoporous Mater., 2008, 116, 684–692 CrossRef CAS.
- A. Izadbakhsh, F. Farhadi, F. Khorasheh, S. Sahebdelfar, M. Asadi and Y. Z. Feng, Appl. Catal., A, 2009, 364, 48–56 CrossRef CAS.
- G. A. V. Martins, G. Berlier, S. Coluccia, H. O. Pastore, G. B. Superti, G. Gatti and L. Marchese, J. Phys. Chem. C, 2007, 111, 330–339 CAS.
- G. A. V. Martins, G. Berlier, C. Bisio, S. Coluccia, H. O. Pastore and L. Marchese, J. Phys. Chem. C, 2008, 112, 7193–7200 CAS.
- S. Kieger, G. Delahay, B. Coq and B. Neveu, J. Catal., 1999, 183, 267–280 CrossRef CAS.
- P. N. R. Vennestrøm, A. Katerinopoulou, R. R. Tiruvalam, A. Kustov, P. G. Moses, P. Concepcion and A. Corma, ACS Catal., 2013, 3, 2158–2161 CrossRef.
- C. Wang, J. Wang, J. Q. Wang, T. Yu, M. Q. Shen, W. L. Wang and W. Li, Appl. Catal., B, 2017, 204, 239–249 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06947b |
| This journal is © The Royal Society of Chemistry 2017 |