
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of polymethylene-b-poly(vinyl acetate) block copolymer via visible light induced radical polymerization and its application†
HeYing Wangab,
Fang Xuab,
Kun Cuib,
Hao Zhangab,
Jin Huangb,
QiaoLing Zhaob,
Tao Jiang*a and
Zhi Ma *b
*b
aCollege of Chemical Engineering and Materials Science, Tianjin University of Science & Technology, Tianjin, 300457, People's Republic of China. E-mail: jiangtao@tust.edu.cn
bKey Laboratory of Synthesis and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, People's Republic China. E-mail: mazhi728@sioc.ac.cn
First published on 1st September 2017
Abstract
New well-defined amphiphilic polymethylene-b-poly(vinyl acetate) (PM-b-PVAc) diblock copolymers were synthesized via a tandem strategy combining polyhomologation of ylides, chain-end functionalization and a visible light induced degenerative iodine transfer polymerization using decacarbonyldimanganese (Mn2(CO)10). Firstly, an iodo terminated polymethylene (PM-I) was prepared by polyhomologation of ylides initiated by a new organic borane based on catecholborane followed by functional group transformation using iodine as the end-capping reagent. Then, a series of amphiphilic PM-b-PVAc diblock copolymers with controlled molecular weight (Mn = 5530–17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 020 g mol−1) and relatively narrow molecular weight distribution (Đ = 1.31–1.45) were obtained efficiently through a visible light induced degenerative iodine transfer polymerization of vinyl acetate (VAc) using PM-I as macroinitiator in the presence of Mn2(CO)10 under weak visible light irradiation at 40 °C. The micelles of such an amphiphilic diblock copolymer formed in tetrahydrofuran were observed by transmission electron microscopy (TEM). Moreover, the fabrication of porous films using PM-b-PVAc diblock copolymers via a static breath-figure process was investigated.
020 g mol−1) and relatively narrow molecular weight distribution (Đ = 1.31–1.45) were obtained efficiently through a visible light induced degenerative iodine transfer polymerization of vinyl acetate (VAc) using PM-I as macroinitiator in the presence of Mn2(CO)10 under weak visible light irradiation at 40 °C. The micelles of such an amphiphilic diblock copolymer formed in tetrahydrofuran were observed by transmission electron microscopy (TEM). Moreover, the fabrication of porous films using PM-b-PVAc diblock copolymers via a static breath-figure process was investigated.
Introduction
The low cost, excellent physical properties, easy processability and recyclability of polyolefins have led to their commercial success.1 However, the poor compatibility of polyolefins with other polar materials limits their applications. To overcome this deficiency, the development of polyolefin-based (co)polymeric materials with novel architectures and components is needed.2 To explore the highly active and efficient strategies targeting the functionalization of non-polar polyolefins at their main-chain ends or branches was of active interest. By virtue of such functional groups or their derivatives formed by chemical transformation, novel functionalized polyolefin copolymers were synthesized via a variety of strategies and had a wide range of applications.1–6Recently, Shea and coworkers7 developed polyhomologation of ylides which can be used to build up a carbon backbone polymers one carbon at a time giving rise to a polyethylene analog named polymethylene. Various main-chain end functionalized polymethylene can be prepared by polyhomologation of ylides using different boranes followed by functional group transformation. Recently, a variety of polymethylene-based copolymers with different architectures and chemical components have been synthesized via a combination of polyhomologation of ylides with nitroxide-mediated radical polymerization (NMP),8 atom transfer radical polymerization (ATRP),9 reversible addition–fragmentation chain transfer polymerization (RAFT),10 ring-opening polymerization (ROP)11 ring-opening metathesis polymerization (ROMP),12 living cationic polymerization,13 Diels–Alder reaction14 and “click” chemistry.15
Light induced polymerization has been widely employed in conventional radical polymerization as a key technique in various applications, such as coatings, adhesives, medicine, optics and microelectronics.16 The use of light in controlled/living radical polymerization also brings several distinct advantages, including temporal and spatial control over chain growth, easy preparation of well-defined polymers at room temperature and minimization of possible higher temperature side reactions.17,18 Although extensive efforts have been made in this area, it is still crucial to explore an effective light induced polymerization in synthetic block polymers.
Among monomers for constructing block copolymer, vinyl acetate (VAc) has gained extensive attention due to the wide applications of poly(vinyl acetate) (PVAc) in different areas, such as paints, adhesives, additives to pharmaceuticals and so on.19 Furthermore, the largest volume water-soluble polymer poly(vinyl alcohol) is also made commercially available by the hydrolysis of PVAc. In contrast to a large variety of the polymerization systems for conjugated vinyl monomers, the controlled/living radical copolymerizations of VAc are difficult because of its low copolymerizability20 and the lack of effective systems for both monomers. Although the reversible addition–fragmentation chain transfer (RAFT) polymerization or macromolecular design via interchange of xanthate systems are effective for various monomers including VAc and other conjugated monomers by designing the dithiocarbonyl compounds, there are no effective agents for controlling the copolymerizations.21
Koumura and coworkers20 reported the controlled/living radical polymerization of VAc and its copolymerization with methyl acrylate in bulk or fluoroalcohols using dimanganese decacarbonyl [Mn2(CO)10] in conjunction with an alkyl iodide as an initiator under weak visible light. The dinuclear complex itself is stable and inactive in the dark; in conjunction with halogen containing compounds, it is capable of generating free radicals and initiating polymerization under visible light. In addition, since alkyl halides are also used as coinitiators to generate initiating radicals, so it provides molecular design flexibility in macromolecular synthesis.
Iodo functionalized polyethylene, prepared by the addition of iodine after catalyzed polyethylene chain growth on magnesium, acted as an efficient macroinitiator for the thermal-induced controlled radical polymerization of styrene and methyl methacrylate using Mn2(CO)10 at 80 °C was reported by Ciftci and coworkers.22 The proposed mechanism involved metal-catalyzed homolysis of the C–I bond of the initiator and dormant polymer chain end, resulting in polymers with controlled molecular weights.
Herein, we'd like to report a combined synthetic strategy targeting the PM-b-PVAc diblock copolymers with relatively narrow molecular weight distribution. Firstly, iodo terminated polymethylene (PM-I) was prepared by polyhomologation of dimethylsulfoxonium methylide using a new organic borane based on catecholborane followed by the functional group transformation using iodine as the end-capping reagent. Then, a visible light induced degenerative iodine transfer polymerization of VAc was performed using PM-I as macroinitiator in the presence of Mn2(CO)10 under weak visible light at 40 °C, obtaining amphiphilic PM-b-PVAc diblock copolymers with relatively low molecular weight distribution. The properties of such diblock copolymer such as the micellization and the formation of ordered porous films were also investigated.
Experimental
Materials
All manipulations involving air- and/or moisture-sensitive compounds were carried out in a N2 filled dry box or using Schlenk techniques. Mn2(CO)10 (99%, Aldrich) was purified by sublimation and stored in a refrigerator in the dark. Toluene (Sinopharm Chemical Reagent Co., Ltd., ≥99.5%) was refluxed over sodium and distilled under nitrogen before use. Sodium hydride (Beidouxing Company, Tianjin, 95%) was stirred in n-hexane for 6 h and standing for 1 h, then n-hexane drew off by a needle, in this way washed three times with n-hexane, dried in a vacuum, and stored under nitrogen. Trimethylsulfoxonium iodide (Acros Organics, 98%), benzyltributylammonium chloride (TCI, >98%), dichloromethane (Sinopharm Chemical Reagent Co., Ltd., ≥99.5%) was refluxed over sodium and distilled under nitrogen before use. Styrene (Lingfeng Company, Shanghai, ≥99%) was degassed with nitrogen prior to use. Vinyl acetate (VAc, 99%, Acros) was distilled under reduced pressure and stored under nitrogen. Catecholborane (Aldrich, 1.0 M in THF), iodine (Sinopharm Chemical Reagent Co., Ltd., >99.8%), tetrahydrofuran (Tianlian Company, Shanghai, ≥99%) was refluxed over sodium and distilled under nitrogen before use, chloroform (Shenbo Company, Shanghai, ≥99%).Synthesis of PM-I
In a typical procedure to obtain a new organic borane, this round-bottom flask was then capped with a rubber septum and deoxygenated by degassing and backfilling with nitrogen for three times, styrene (1.5 equiv., 1.5 mmol) was freeze-dried three times in liquid nitrogen, and then dried toluene (10 mL) were introduced into a nitrogen-purged, 500 mL, round-bottom flask equipped with a stirring bar. Then the solution was dried with nitrogen for 0.5 h. The solution was cooled to 0 °C, adding dropwise of catecholborane solution (1.1 equiv., 1.1 mmol), then was stirred at 80 °C for 3 h. Such reaction mixture was added via syringe under nitrogen protection to the solution of dimethylsulfoxonium methylide (72 mmol, synthesized in a similar procedure described in literature23) to operate the polyhomologation in a similar procedure as described in our previous work24 at 80 °C for 3 h. Cooling to room temperature, an iodine solution (20 mL saturated solution of iodine in dried toluene) was added and the resulting mixture was stirred under the condition of dark for 3 h. The polymer was precipitated in methanol under nitrogen atmosphere and collected by filtration, washed with methanol for several times, and dried under vacuum. PM-I, 1H NMR (400 MHz, C6D4Cl2, 50 °C) δ: 3.09 (2H, –CH2–I), 2.60 (2H, C6H5–CH2–), 1.06–1.53 (94H, –CH2–), 0.67–1.05 (5H,–CH3) ppm. FT-IR (KBr): 3082, 3059, 3025 (ν, C–H on phenyl), 2849–2918 (ν, –CH2–), 560 (ν, C–I) cm−1.Synthesis of block copolymers using VAc monomer with polymethylene macroinitiators
In a typical polymerization, PM-I (0.1 g, 0.1 mmol) and Mn2(CO)10 (0.02 g, 1.282 mmol) was added to a 100 mL round-bottom flask equipped with a stirring bar. This flask was then capped with a rubber septum and deoxygenated by degassing and backfilling with nitrogen for three times. Then, VAc was placed in another flask equipped with a stirring bar. This flask was capped with a rubber septum, purged with nitrogen for about 3 times, and charged with deoxygenated VAc (5 mL, 54.3 mmol) via syringe. The VAc was purged with nitrogen at room temperature for about 30 min, then transfer it into round-bottom flask with a syringe under a nitrogen atmosphere. The formulation was stirred and exposed to light continuously at 40 °C for several minutes under a blue LED, in which the sample was surrounded by silver paper. At the end of the photo irradiation, the reaction was terminated with liquid nitrogen and the resultant polymer was washed with hexane for three times under nitrogen atmosphere and dried under vacuum then analyzed by 1H NMR, GPC and FT-IR. 1H NMR (400 MHz, C6D4Cl2, 50 °C) δ: 5.11 (88H, –CH2–CH–), 2.06 (264H, –OCCH3), 1.33 (94H, –CH2–), 0.93 (5H, –CH3) ppm. FT-IR (KBr): 3082, 3059, 3025 (ν, C–H on phenyl), 2849–2918 (ν, –CH2), 1740 (ν, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1148 (ν, –C(O)–O–C) cm−1.
O), 1148 (ν, –C(O)–O–C) cm−1.
Formation of honeycomb films via the breath figure method
The porous polymer films were fabricated by a static breath-figure process in 50 mL wide-mouth glass bottle with a cap. The glass slides (1.0 × 1.0 cm2) were cleaned sequentially in an ultrasonic bath of acetone, ethanol and deionized water, and dried overnight at 40 °C in vacuo finally. Relative humidity (R. H.) in the glass vessel were achieved by adding saturated aqueous solutions of deionized water (R. H. = 95%). A piece of glass slide was placed into the glass vessel, which was 1 cm higher than the liquid level. The PM-b-PVAc (run 10 in Table 1) were used to prepare the films. Polymer solutions with different concentrations were prepared by dissolving PM-b-PVAc in THF, CHCl3 and CH2Cl2, respectively. A certain amount of solution (10 μL) was dropped and spread on the substrate with a micro-syringe in a static humid condition, which was controlled at 20 °C, respectively. The polymer films were dried at room temperature after complete evaporation of solvents.| Run | Time [min] | Conv. [%] | Mn,theoryb [g mol−1] | Mn,NMRc [g mol−1] | Mn,GPC [g mol−1] | Đ |
|---|---|---|---|---|---|---|
| a Polymerization conditions: PM-I/[VAc]0/[Mn2(CO)10] = 1/470/1, Vtoluene/VVAc = 1/1.5, samples were irradiated by blue LED light at 40 °C.b Mn,theory = [VAc]0/[PM-I]0 × MVAc × conv.c The Mn,NMR was calculated by 1H NMR Mn,NMR = (integral area of –CH2–/integral area of –CH2–I) × M–CH2– + M–C6H5 + M–I. | ||||||
| 1 | 10 | 12.0 | 5180 | 4540 | 5530 | 1.31 |
| 2 | 20 | 17.4 | 6590 | 6600 | 6790 | 1.32 |
| 3 | 30 | 22.4 | 8410 | 8500 | 8520 | 1.45 |
| 4 | 35 | 24.7 | 9400 | 9500 | 9490 | 1.38 |
| 5 | 40 | 26.6 | 10200 |
10100 |
10600 |
1.39 |
| 6 | 50 | 31.6 | 11950 |
12000 |
12060 |
1.41 |
| 7 | 60 | 35.9 | 13350 |
13600 |
13690 |
1.45 |
| 8 | 90 | 41.5 | 15400 |
15700 |
15780 |
1.45 |
| 9 | 150 | 42.2 | 16390 |
16000 |
16780 |
1.43 |
| 10 | 240 | 44.7 | 16900 |
17000 |
17020 |
1.41 |
Instruments and measurements
Blue LED (light-emitting diode, Shenzhen Tengyi, 220 V, 40 W). The 1H NMR analysis was performed on a Bruker Avance III 400 MHz NMR spectrometer using C6D4Cl2 as the solvent. GPC measurements: molecular weights (Mw and Mn) and molecular weight distributions (Đ) were determined using GPC equipped with a Waters 1515 Isocratic HPLC pump, a Waters 2414 refractive index detector, and a set of Waters Styragel columns (HR3, HR4, and HR5, 7.8 × 300 mm2). GPC measurements of polymers were run at 35 °C using THF as eluent with a flow rate of 1.0 mL min−1. The data were calibrated with linear polystyrene standards. FT-IR spectra were recorded on a Nicolet AVATAR-360 FT-IR spectrophotometer with a resolution of 4 cm−1. Transmission Electron Microscope (TEM) observations were performed on a TEM (JEOL JEM-1230) instrument operated at 80 kV. Scanning electron microscopy (SEM) was performed on a SEM (JEOL JSM-6390LV) instrument operated at 10 kV.Results and discussion
The PM-b-PVAc copolymer was synthesized according to a procedure illustrated in Scheme 1. Firstly, PM-I was prepared via the strategy of polyhomologation of ylides combining with chain-end functionalization. Then, a series of well-defined PM-b-PVAc copolymers were synthesized via visible light induced degenerative iodine transfer polymerization of VAc in toluene at 40 °C using Mn2(CO)10 as initiator and PM-I as a macromolecular chain transfer agent. | ||
| Scheme 1 Synthetic routes for the preparation of well-defined PM-b-PVAc diblock copolymers. | ||
Synthesis and characterization of iodo terminated polymethylene (PM-I)
The PM-I were successfully synthesized by polyhomologation of dimethylsulfoxonium methylide following the functional group transformation using iodine as the end-capping reagent. The chain structure of PM-I was confirmed by 1H NMR, 13C NMR and FT-IR spectra.Fig. 1 showed the 1H NMR spectrum of PM-I. The characteristic resonances of aryl unit in the PM-I can be observed at 7.22–7.08 ppm. The resonance of the methylene group adjacent to the iodine group appeared at 3.09 ppm. And the triplet peak at 2.60 ppm was assigned to methylene protons next to the aryl group, indicating that the halogenated compound based on polymethylene was formed. The signal at 0.93 ppm assigns to the side methyl groups for the existence of dimethylsulfoxonium ethylide impurity in the dimethylsulfoxonium methylide. The integral ratio of peak (a) to (e) is 2:2 and the ratios (aromatic protons:peak (a)) is 5:2 indicating the formation of PM-I. Meanwhile, the integral ratio of peak (d) to peak (c) was 2:120, which is agreement with GPC information. All of the information shown in 1H NMR spectrum of PM-I proved that the functionalization proceeded quantitatively. Furthermore, by 13C NMR analysis, the chemical shifts at 128.32 and 6.33 ppm indicated the presence of carbon atoms of aryl and next to the iodine. It was note worthy that the signals at 142.67 and 33.77 ppm prove the aryl connect to the chain of methylene evidently. In the FT-IR spectrum of PM-I, the peak of C–I stretching vibration absorption had formed at 560 cm−1 and the peaks at 3000–3150 cm−1 proved the existence of the benzene ring.
 | ||
| Fig. 1 1H NMR spectrum of PM-I. | ||
The readily precipitated PM-I chains are simply recovered by filtration and dried. Well-defined PM-I (Đ = 1.10) of relatively low molar mass (Mn, GPC = 1000 g mol−1) with high functionality (∼100%) was employed as macroinitiator in the following polymerization of VAc.
Synthesis and characterization of PM-b-PVAc diblock copolymer
A series of amphiphilic PM-b-PVAc diblock copolymers were obtained via a visible light induced degenerative iodine transfer polymerization of VAc using PM-I/Mn2(CO)10 system under weak visible light at 40 °C.For the PM-I can solve in the VAc completely, we first have a test for bulk polymerization, the polymerization showed typical radical polymerization behaviour, uncontrolled molecular weight (Mn = 62000 g mol−1) and a wide molecular weight distribution (Đ = 1.75). The conversion of VAc reached 88.7% after 30 min under visible light at 40 °C.
The polymerization could not take place within 24 h at 40 °C under light irradiation without Mn2(CO)10, which could initiate the quick polymerization of VAc. When the polymerization was conducted in toluene with both PM-I and Mn2(CO)10 under visible light, the obtained polymers showed controlled molecular weight and a narrow molecular weight distribution, which are shown in Table 1.
The GPC traces (Đ = 1.31–1.45) of PM-b-PVAc diblock copolymers moved to higher molecular weight in comparison with PM-I indicating the successful chain extension and the formation of PM-b-PVAc diblock copolymers. There are no shoulder and no weight tails in GPC. According to the comparison of the elution time of PM-I and diblock polymers, no self coupling of PM-I was observed.
High reaction temperatures are undesirable for visible light induced degenerative iodine transfer polymerization with alkyl iodides due to the possibility of decomposition of the chain end of iodo-terminated polymers. Because alkyl-I/Mn2(CO)10 can decompose at low temperature (t = 40 °C)25 under visible light, so the polymerization of vinyl acetate can be performed at 40 °C, which should also result in the reduced decomposition rate of the iodo-terminated poly(vinyl acetate).
The system is sensitive to moisture, because traces of water could result in the hydrolysis of C–I end groups,26 leading to the decomposition of the iodo-terminated polymer with the formation of aldehyde end groups and acetyl iodide.27 Increased polarity of the reaction medium may accelerate this decomposition reaction, so toluene was selected as the medium solvent. Because alkyl iodides are UV and light sensitive, all the polymerization reactions were performed in the dark.
Fig. 2 shows the GPC curves of the PM-b-PVAc diblock copolymers (a) and the dependence of Mn and Đ on conversion (b) in visible light induced degenerative iodine transfer polymerization of VAc with PM-I/Mn2(CO)10 at 40 °C. The Mn increased in direct proportion to the monomer conversion and agreed with the calculated values assuming that one initiator molecule generates one polymer chain. As the polymerization proceeded, the GPC curves shifted to high molecular weights though the Đs became broader. One of the main reasons for the broadening of the Đs in the later stages of the polymerization would be the accumulation of the less reactive primary C–I terminal originating from the head-to-head addition, which is inherent in VAc radical polymerization.25,28,29 These results indicate that Mn2(CO)10 is effective for the fast and controlled radical polymerization of VAc under weak light irradiation, which induces slow and continuous generation of the highly active manganese pentacarbonyl radical species [˙Mn(CO)5]. To the best of our knowledge, this is the first example of visible light induced degenerative iodine transfer polymerization of VAc at 40 °C with an iodo-terminated polymethylene as the initiator in the presence of Mn2(CO)10.
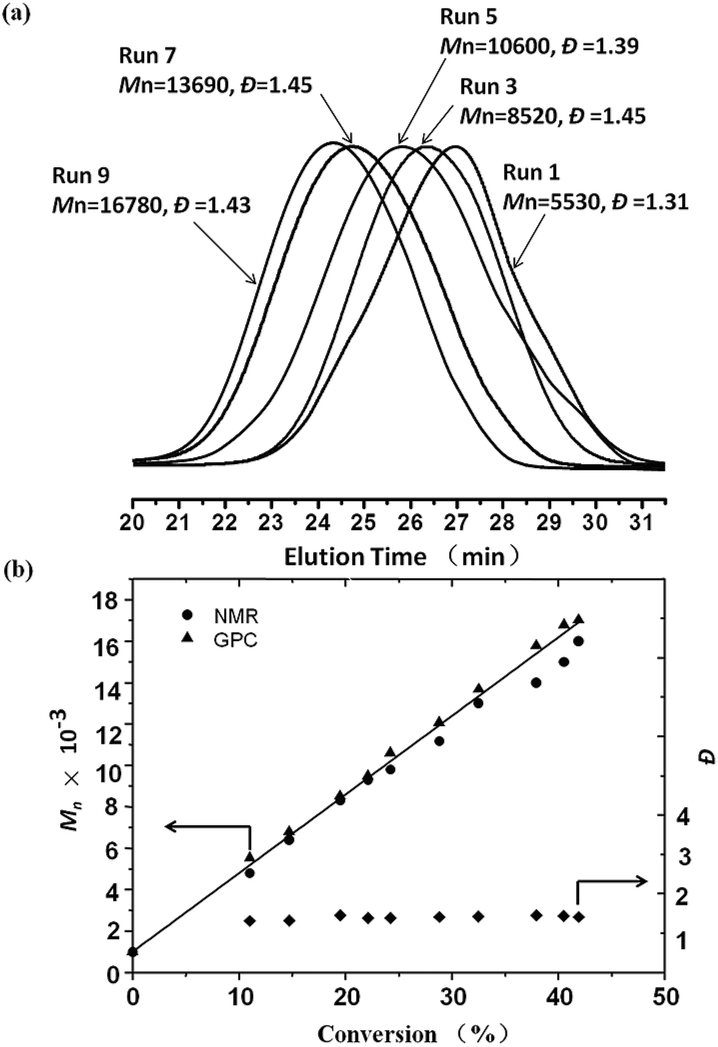 | ||
| Fig. 2 GPC curves of PM-b-PVAc diblock copolymers (a) and dependence of Mn and Đ on conversion (b) in visible light induced degenerative iodine transfer polymerization of VAc. | ||
As shown in Fig. 3, the main absorption signals of poly(vinyl acetate) were assigned as shown. The methine proton (a) in the polymer backbone absorbs at 5.11 ppm, and the methylene (b) group appears at 1.77–1.93 ppm. Methyl groups in polymer (c) absorb at 2.06 ppm. The signals (e and e′) at 3.38 ppm and 6.71 ppm indicates the internal connectivity of the end VAc with head to head and head to tail two different styles. Chain end fidelity is an important issue not only to show the controlled character of the polymerization but also further use of the obtained polymers for chain extension and preparation of block copolymers. The PM-b-PVAc was purified and analyzed by 1H NMR using 1,2-dichlorobenzene-D4 as the deuterium reagent. The iodide chain-end functionality (82.4%) is calculated on the basis of the ratio of (f) (next to benzene ring) to (e) and (e′) (the iodide chain-end functionality = (Ie + Ie′/2)/(If/2) × 100%).
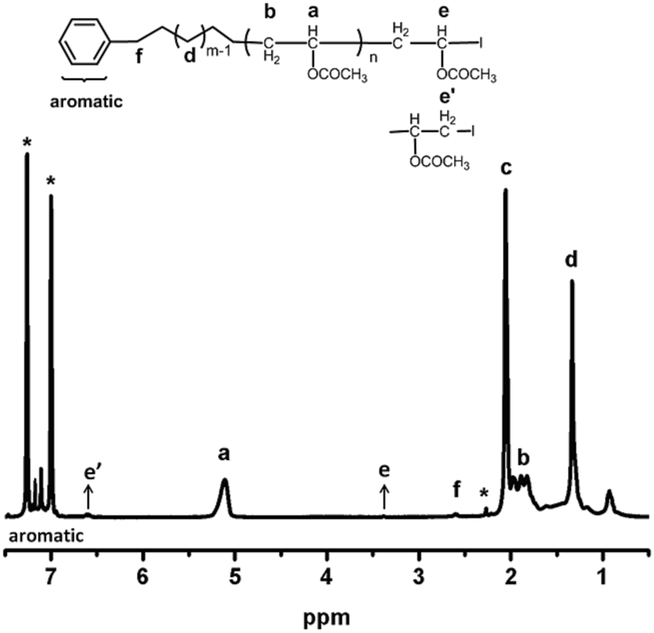 | ||
| Fig. 3 1H NMR spectrum of PM-b-PVAc diblock copolymer (run 3 in Table 1). | ||
Micellization of PM-b-PVAc diblock copolymer
The amphiphilic copolymers in a selective solvent can form aggregates with different morphologies such as spheres, vesicles, rod like, ribbon like, and so forth.30–32 The amphiphilicity of the resultant PM-b-PVAc diblock copolymer was investigated through its micellization in aqueous media. The polymer (run 3) was dissolved in THF to prepare 1 mg mL−1 solutions. A solution of PM-b-PVAc/THF (0.5 mL), then 5 mL of membrane-filtered water was added with stirring at a flow rate of 0.5 mL min−1. THF was then evaporated for 2 days at 25 °C in a water bath. Opaque dispersions of particles were obtained after evaporation.Then, the aggregates of diblock copolymers deposited on a carbon-coated copper grid were observed by TEM after the complete evaporation of water at room temperature and atmosphere.
As shown in Fig. 4, the amphiphilic PM-b-PVAc diblock copolymer aggregated into spheres with diameters of 10–30 nm. Consulted from the study of the model of diblock copolymer micelles,33,34 such tendency may be explained by the increasing PVAc corona layer thickness because of the longer PVAc segment in PM-b-PVAc.
 | ||
| Fig. 4 TEM image of the precipitated aggregates of PM-b-PVAc. | ||
Fabrication of honeycomb porous PM-b-PVAc films
Ordered honeycomb structured films can be prepared by a simple bottom-up process via the breath-figure (BF) technique, which takes advantage of the condensation of water droplets on a cold surface.35The regularity of pores on the surface of film is influenced by various factors in BF process, including polymer structure, polymer molecular weight, polymer concentration, solvent, temperature and humidity. In this work, the effects of solvent and polymer concentration on the surface morphology of PM-b-PVAc (run 10, Table 1) film were investigated in the fabricating porous films via a static BF process developed by Li and co-workers.36 It was found that PM-b-PVAc diblock copolymer can be readily dissolved in chloroform (CHCl3), dichloromethane (CH2Cl2) and tetrahydrofuran (THF). So, the polymer solutions in such three solvents were employed to fabricate porous polymer films in a static BF process at 20 °C under a relative humidity of 95%. All of the PM-b-PVAc solutions in THF, CH2Cl2 and CHCl3 can form regular porous films at the concentration of 5 mg mL−1 using the static BF process.
Polymer concentration is an important influencing factor.35c,37 The porous films of PM-b-PVAc diblock copolymer (run 10 in Table 1) were fabricated at 20 °C under R. H. of 95% with different concentration of 3, 5, 10 and 15 mg mL−1, respectively. SEM images of such films were shown in Fig. 5. The formation of disordered porous film was probably due to the combined effects of lower solution viscosity and lower concentration in 3 mg mL−1 polymer solution, which cannot inhibit the coalescence of water droplets efficiently, resulting in larger pores (D = average pore size, DCHCl3 = 9.66 μm, DCH2Cl2 = 1.99 μm, DTHF = 1.69 μm) and less regularity. With the polymer concentration rising to 5 mg mL−1, the porous films with ordered pore structures were observed (DCHCl3 = 2.79 μm, DCH2Cl2 = 2.07 μm, DTHF = 2.15 μm). The porous films with irregular pore structures and smaller pore sizes were formed with the increasing of polymer concentration to 10 mg mL−1 or 15 mg mL−1, respectively. Such results are probably attributed to the weak Marangoni convection and the increased difficulty of water-droplets sinking into the polymer solution with higher viscosity. At the same time, the average pore sizes of films fabricated from the polymer solution in CHCl3 and CH2Cl2 are becoming smaller as DCHCl3 = 1.39/1.02 μm and DCH2Cl2 = 0.77/0.41 μm at the concentration of 10 mg mL−1/15 mg mL−1, respectively. Being a better candidate, CH2Cl2 will be employed as solvent in the forthcoming systematic investigation on the fabrication of highly ordered porous PM-b-VAc films.
 | ||
| Fig. 5 SEM images of porous films fabricated from PM-b-PVAc (run 10 in Table 1) solutions in various solvents at different concentrations. | ||
Conclusions
New organic borane was prepared by the hydroboration of styrene using catecholborane and initiated in situ the polyhomologation of ylides. Subsequently, the post functionalization was carried out directly in the reaction system to achieve terminal –I group forming the iodo terminated polymethylene. The polymerization of vinyl acetate was successfully carried out in toluene using PM-I as macroinitiator in the presence of Mn2(CO)10 under weak visible light at 40 °C. The amphiphilic PM-b-PVAc diblock copolymers with a series of molecular weights and narrow molecular weight distributions were synthesized successfully. The capabilities of such amphiphilic diblock copolymer in the formation of micelles and highly ordered porous films were presented in this work. It is worthy for PM-b-PVAc diblock copolymers to be investigated further in the application fields of functional micelles and highly ordered porous films.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly appreciate the financial support from the National Natural Science Foundation of China (no. 21374130, 21074146).Notes and references
- T. C. Chung, Prog. Polym. Sci., 2002, 27, 39–85 CrossRef CAS.
- M. J. Yanjarappa and S. Sivaram, Prog. Polym. Sci., 2002, 27, 1347–1398 CrossRef CAS.
- J. Y. Dong and Y. Hu, Coord. Chem. Rev., 2006, 250, 47–65 CrossRef CAS.
- R. Gody-Lopez, F. D'Agosto and C. Boisson, Prog. Polym. Sci., 2007, 32, 419–454 CrossRef.
- J. Mazzolini, E. Espinosa, F. D'Agosto and C. Boisson, Polym. Chem., 2010, 1, 793–800 RSC.
- Y. Zhao, L. Wang, A. Xiao and H. Yu, Prog. Polym. Sci., 2010, 35, 1195–1216 CrossRef CAS.
- (a) K. J. Shea, J. W. Walker, H. Zhu, M. Paz and J. Greaves, J. Am. Chem. Soc., 1997, 119, 9049–9050 CrossRef CAS; (b) K. J. Shea, Chem.–Eur. J., 2000, 6, 1113–1119 CAS; (c) J. Luo and K. J. Shea, Acc. Chem. Res., 2010, 43, 1420–1433 CrossRef CAS PubMed.
- (a) X. Z. Zhou and K. J. Shea, Macromolecules, 2001, 34, 3111–3114 CrossRef CAS; (b) N. Alkayal, H. Durmaz, U. Tunca and N. Hadjichristidis, Polym. Chem., 2016, 7, 2986–2991 RSC.
- (a) J. Z. Chen, K. Cui, S. Y. Zhang, P. Xie, Q. L. Zhao, J. Huang, L. P. Shi, G. Y. Li and Z. Ma, Macromol. Rapid Commun., 2009, 30, 532–538 CrossRef CAS PubMed; (b) H. C. Lu, Y. Xue, Q. L. Zhao, J. Huang, S. G. Xu, S. K. Cao and Z. Ma, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3641–3647 CrossRef CAS; (c) Y. Xue, H. C. Lu, Q. L. Zhao, J. Huang, S. G. Xu, S. K. Cao and Z. Ma, Polym. Chem., 2013, 4, 307–312 RSC; (d) Z. Zhang, H. Zhang, Y. Gnanou and N. Hadjichristidis, Chem. Commun., 2015, 51, 9936–9938 RSC; (e) Z. Zhang, M. Altaher, H. Zhang, D. Wang and N. Hadjichristidis, Macromolecules, 2016, 49, 2630–2638 CrossRef CAS.
- X. Wang, J. P. Gao, Q. L. Zhao, J. Huang, G. L. Mao, W. Wu, Y. N Ning and Z. Ma, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2892–2899 CrossRef CAS.
- (a) C. Yuan, H. C. Lu, Q. Z. Li, S. Yang, Q. L. Zhao, J. Huang, L. H. Wei and Z. Ma, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2398–2405 CrossRef CAS; (b) Q. Z. Li, G. Y. Zhang, J. Z. Chen, Q. L. Zhao, H. C. Lu, J. Huang, L. H. Wei, F. D'Agosto, C. Boisson and Z. Ma, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 511–517 CrossRef CAS.
- (a) H. Zhang, Y. Gnanou and N. Hadjichristidis, Polym. Chem., 2014, 5, 6431–6434 RSC; (b) H. Zhang, Z. Zhang, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2015, 48, 3556–3562 CrossRef CAS; (c) D. Zhang, H. Zhang, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2016, 49, 2484–2492 CrossRef CAS.
- H. Zhang, S. Banerjee, R. Faust and N. Hadjichristidis, Polym. Chem., 2016, 7, 1217–1220 RSC.
- N. Alkayal and N. Hadjichristidis, Polym. Chem., 2015, 6, 4921–4926 RSC.
- N. Alkayal, H. Durmaz, U. Tunca and N. Hadjichristidis, Polym. Chem., 2016, 7, 2986–2991 RSC.
- (a) B. Aydogan, Y. Yagci, L. Toppare, S. Jockusch and N. J. Turro, Macromolecules, 2016, 45, 7829–7834 CrossRef; (b) S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016, 116, 10212–10275 CrossRef CAS PubMed; (c) X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS; (d) M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- S. Yamago and Y. Nakamura, Polymer, 2013, 54, 981–994 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed.
- (a) K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS PubMed; (b) K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed; (c) H. Lee, R. Mensire, R. E. Cohen and M. F. Rubner, Macromolecules, 2012, 45, 347–355 CrossRef CAS.
- (a) K. Koumura, K. Satoh and M. Kamigaito, Macromolecules, 2008, 41, 7359–7367 CrossRef CAS; (b) K. Koumura, K. Satoh and M. Kamigaito, Macromolecules, 2009, 42, 2497–2504 CrossRef CAS; (c) K. Koumura, K. Satoh and M. Kamigaito, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1343–1353 CrossRef CAS.
- (a) G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed; (b) G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- M. Ciftci, S. Norsic, C. Boisson, F. D'Agosto and Y. Yagci, Macromol. Chem. Phys., 2015, 216, 958–963 CrossRef CAS.
- B. B. Busch, M. M. Paz, K. J. Shea, C. L. Staiger, J. M. Stoddard, J. R. Walker, X. Z. Zhou and H. Zhu, J. Am. Chem. Soc., 2002, 124, 3636–3646 CrossRef CAS PubMed.
- F. Xu, P. Dong, K. Cui, S. Z. Bu, J. Huang, G. Y. Li, T. Jiang and Z. Ma, RSC Adv., 2016, 6, 69828–69835 RSC.
- M. C. Iovu and K. Matyjaszewski, Macromolecules, 2003, 36, 9346–9354 CrossRef CAS.
- B. Boutevin, J. P. Hugon, Y. Pietrasanta and A. Sideris, Eur. Polym. J., 1978, 14, 353–356 CrossRef CAS.
- E. K. Euranto, A. Noponen and T. Kujanpaa, Acta Chem. Scand., 1966, 20, 1273–1280 CrossRef CAS.
- M. Wakioka, K. Y. Baek, T. Ando, M. Kamigaito and M. Sawamoto, Macromolecules, 2002, 35, 330–333 CrossRef CAS.
- K. Koumura, K. Satoh, M. Kamigaito and Y. Okamoto, Macromolecules, 2006, 39, 4054–4061 CrossRef CAS.
- I. Astafieva, X. F. Zhong and A. Eisenberg, Macromolecules, 1993, 26, 7339–7352 CrossRef CAS.
- Y. Yu, L. Zhang and A. Eisenberg, Langmuir, 1997, 13, 2578–2581 CrossRef CAS.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- G. S. Kwona and K. Kataoka, Adv. Drug Delivery Rev., 1995, 16, 295–309 CrossRef.
- J. Z. Chen, Q. L. Zhao, H. C. Lu, J. Huang, S. K. Cao and Z. Ma, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1894–1900 CrossRef CAS.
- (a) G. Widawski, M. Rawiso and B. Francois, Nature, 1994, 369, 387–389 CrossRef CAS; (b) U. H. F. Bunz, Adv. Mater., 2006, 18, 973–989 CrossRef CAS; (c) M. H. Stenzel, C. Barner-Kowollik and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2363–2375 CrossRef CAS.
- (a) L. Li, C. K. Chen, A. J. Zhang, X. Y. Liu, K. Cui, J. Huang, Z. Ma and Z. H. Han, J. Colloid Interface Sci., 2009, 331, 446–452 CrossRef CAS PubMed; (b) A. J. Zhang, H. Bai and L. Li, Chem. Rev., 2015, 115, 9801–9868 CrossRef CAS PubMed.
- (a) L. W. Zhu, B. H. Wu, L. S. Wan and Z. K. Xu, Polym. Chem., 2014, 5, 4311–4320 RSC; (b) X. Y. Li, Q. L. Zhao, T. T. Xu, J. Huang, L. H. Wei and Z. Ma, Eur. Polym. J., 2014, 50, 135–141 CrossRef CAS; (c) J. P. Gao, W. Wu, L. Rong, G. L. Mao, Y. N. Ning, Q. L. Zhao, J. Huang and Z. Ma, Eur. Polym. J., 2014, 59, 171–179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06908a |
| This journal is © The Royal Society of Chemistry 2017 |