
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn in situ iodine-doped graphene/silicon composite paper as a highly conductive and self-supporting electrode for lithium-ion batteries†
Cheng Chen ,
Mengqiang Wu*,
Sizhe Wang,
Jian Yang,
Jingang Qin,
Zhi Peng,
Tingting Feng* and
Feng Gong
,
Mengqiang Wu*,
Sizhe Wang,
Jian Yang,
Jingang Qin,
Zhi Peng,
Tingting Feng* and
Feng Gong
Center for Advanced Electric Energy Technologies (CAEET), School of Energy Science and Engineering, University of Electronic Science and Technology of China, Chengdu 611731, China. E-mail: mwu@uestc.edu.cn; fengtt@uestc.edu.cn
First published on 7th August 2017
Abstract
A graphene/silicon composite paper is considered as a promising anode material for flexible batteries. Herein, a highly conductive, flexible, self-supporting, and binder-free graphene/Si composite paper has been prepared via in situ iodine doping and simultaneous reduction of a graphene oxide/silicon composite slice with a solution of hydrohalic (HI) acid as a reducing agent. The in situ iodine doping not only increases the electrical conductivity of the graphene/silicon composite paper, but also improves the strength of the graphene matrix; this results in high capacity and enhanced cycling stability. The in situ iodine-doped composite paper is used as a flexible, self-supporting, and binder-free electrode. The composite paper exhibits a stable capacity retention of 805 mA h g−1 after 100 cycles and an enhanced rate capability, which shows superior performance as compared to the common thermally reduced rGO/Si composites. The high flexibility and high conductivity as well as improved electrochemical performance of this binder-free self-supporting paper anode make it attractive for LIB applications in flexible storage devices.
Introduction
Currently, soft portable electronic devices show a dynamic development trend in a wide range of applications such as in folding screens, artificial electronic skin, flexible sensors, etc. Most importantly, wearable equipment should to be softer and thinner, and it may even become a part of the human body in the future.1 However, many existing energy storage devices are still too heavy or bulky. This means that it is imperative that lithium-ion batteries (LIBs)2 should be lightweight, flexible, have high energy density and long cycle life, and play key roles in lightweight and highly efficient flexible devices.3–5With the emergence of graphene,6,7 known as a one-atom-thick, two-dimensional material, industrialized soft LIBs have become most likely achieved. Graphene, which is the basic building block for all sp2 carbons including graphite, fullerene, and CNTs, serves as an excellent carbon source for the preparation of carbon-based compound anodes due to its high mechanical strength, superior chemical and thermal stability, and remarkable electrical conductivity. Graphene paper-like flexible electrodes for LIBs have attracted significant attention from researchers due to their outstanding performances. However, the pure graphene paper delivers a low capacity of ∼90 mA h g−1, which greatly limits its application in LIBs. Therefore, many graphene-based composite materials such as graphene/polyaniline,8 graphene/metallic oxides,9 graphene/organic salt,10 and graphene/silicon11–13 have been widely studied. Among the anode materials, silicon achieves highest specific capacity for LIBs (as high as 4200 mA h g−1 for Li22Si5 alloy).14–16 However, the application of silicon anodes is seriously hindered due to their volume expansion (∼320%), low conductivity, and unstable solid electrolyte interphase (SEI) problems.17 Thus, a combination of Si and graphene is a smart choice to retain high conductivity and flexibility while enhancing the electrochemical performances.
Moreover, graphene was modified via the introduction of foreign elements (e.g. N,18,19 B,20 P,21 S,22 and Se23), and the modified graphene showed improved properties. Iodine doping, as a halogen-mixed method, has been successfully applied to various materials such as carbon nanotubes (CNTs),24 TiO2,25 polymeride26 etc., which can enhance the conductivity of materials. Iodine-doping into graphene was realized by exposing the graphene sample to iodine vapors, and the iodine-doped graphene showed metallic behavior and high electronic conductivity.27 As an anode material for LIBs, iodine-doped graphene was achieved via the pyrolysis of a mixture of GO and I2 (GO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :I2 = 1:1), and it exhibited excellent electrochemical performance.28 An iodine-doped rGO paper prepared using HI reducing agents was found to have an electronic conductivity as high as 298 S cm−1 and retained a high tensile strength and an outstanding ductility.29 Similarly, mixed acid (HI–AcOH) was reported as a reducing agent system that allowed an efficient reduction of the rGO paper and also exhibited high conductivity.30
:I2 = 1:1), and it exhibited excellent electrochemical performance.28 An iodine-doped rGO paper prepared using HI reducing agents was found to have an electronic conductivity as high as 298 S cm−1 and retained a high tensile strength and an outstanding ductility.29 Similarly, mixed acid (HI–AcOH) was reported as a reducing agent system that allowed an efficient reduction of the rGO paper and also exhibited high conductivity.30
Herein, a sandwich structure of rGO/silicon was fabricated via vacuum filtration of a GO/Si composite suspension through a wet paper and subsequent in situ iodine doping during the reduction process with hydrohalic (HI) acid. The in situ iodine doping not only increases the electrical conductivity of the graphene/silicon composite paper, but also improves the strength of the graphene matrix; this results in a highly conductive, highly flexible, self-supporting, and binder-free rGO/Si composite paper with superior electrochemical performances as electrodes for flexible LIBs.
Experimental
Sample preparation
:Si = 1:0, 2:1, 1:1, and 1:2 composite paper in a solution of HI acid in an autoclave reactor heated in an oven for 1 h at 100 °C. After being cooled down to room temperature, the wet composite paper was gently soaked and rinsed in distilled water to remove the adsorbed excessive HI. After drying for 20–30 min at 80 °C in an oven, the final sample was kept in a vacuum oven overnight.Structural characterization methods
X-ray diffraction (XRD) analyses of the prepared composite paper were carried out using a Bruker-D8 Advance X-ray Diffractometer (Cu Kα radiation: λ = 1.5406 Å) at a scanning speed of 5 deg min−1. X-ray photoelectron spectroscopy (XPS) analysis was performed via a PHI 5000 Versa probe system, operating at 25 W, using monochromatic Al Kα radiation (1486.6 eV). The morphology and microstructure of the as-prepared composite paper were characterized using field emission scanning electron microscopes (JSM-65900LV, JSM-7500F, JEOL) with an energy-dispersive spectrometer (EDS) accessory for elemental measurement. TEM images were taken on a JEOL JEM-2100F transmission electron microscope. TGA was conducted using a STA449C TGA-DSC tester under an air atmosphere from 40 to 1000 °C at the heating rate of 5 °C min−1. The sheet resistance (Rs, X sq−1) of the as-prepared composite paper was obtained by a 4-probe method, and the corresponding volume conductivity (r, S cm−1) was calculated using the formula: r = 1/(RsT), where T (unit: cm) is the paper thickness. The tensile tests were performed using a Hounsfield H5K-S materials tester.Electrochemical measurement
To investigate the electrochemical performance of free-standing electrodes, the prepared I-doped rGO/Si composite paper (I-rGO/Si P) and thermal rGO/Si composite paper (rGO/Si P) were directly used as working electrodes of a coin-type half-cell (CR2032) without any conductive agent and bonding material. The weight of the paper electrodes is about 1.4–1.7 mg cm−2. The pure silicon electrodes were prepared by mixing 80 wt% active materials, 10 wt% super P, and 10 wt% PVDF dissolved in NMP to form a homogeneous slurry. The slurry was then coated on a current collector (Cu foil) and dried for 10 h in vacuum at 80 °C. A lithium metal foil was used as a counter/reference electrode, and 1 M solution of LiPF6 in a 1:1 v/v mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) was used as the electrolyte. The coin-half cells were assembled in an argon-filled glove box (Vigor 1G1200/750TS). Cyclic voltammetry curves were obtained using a CHI660E electrochemical workstation at the scanning rate of 0.1 mV s−1 in the voltage range of 0.01–1.5 V (versus Li+/Li). The galvanostatic charge/discharge curves were obtained using a LAND battery-channel tester (CT2001A) in a voltage range of 0.01–1.5 V (versus Li+/Li) under a current range of 100–1000 mA g−1.
Results and discussion
Fig. S1† shows a schematic of the in situ iodine-doped reduction process of the GO/Si composite paper by hydroiodic acid. We prepared a sandwich structure of the graphene oxide/Si composite paper via vacuum filtration, subsequent immersion of GO/Si P in a HI acid solution, and then homogeneous dispersion of the HI acid solution in the structure. After hydrothermal process, the GO substrate reduced into rGO, and residual HI decomposed into I2 and H2O: 4HI + O2 = 2I2 + 2H2O. Finally, the iodine atoms was homogeneously adsorbed on the surface of the rGO substrate (in Fig. 1b). The reaction mechanism of GO reduction by HI is exhibited in Fig. 1a: first, the ring opening reaction of an epoxy group on the GO substrate occurs. Next, iodine atom substitution reaction happens on the hydroxyl group of the GO substrate.29 Fig. 1d presents the X-ray diffraction patterns obtained for the in situ iodine-doped rGO/Si composite paper (I-rGO/Si P) and GO/Si composite paper (GO/Si P). The diffraction peaks located at 28°, 47°, 56°, and 69° for the in situ I-rGO/Si P and GO/Si P can match well with silicon lattice planes (JCPDS card no. 27-1402) of (111), (220), (311), and (400).32 For GO/Si P, the small diffraction peak (001) at 10.6° is a typical characteristic peak of graphene oxide. Furthermore, an additional broadened peak (002) at 25.5° of in situ I-rGO/Si P confirms the successful reduction of graphene oxide to graphene after the hydroiodic acid treatment, as described in the previous literature.33,34 The variation in the composition of the precursor GO/Si P and final products (in situ I-rGO/Si P) was also proven via Raman spectra analyses.11,35 As shown in Fig. 1d, both the red and black curves display a sharp characteristic peak at around 509 cm−1, which corresponds to silicon particles.32,36,37 On the other hand, the two observed peaks at around 1349 cm−1 and 1588 cm−1 should be identified, respectively, as the D band and G band of graphene-based materials. The intensity ratio of G and D bands (ID/IG), a parameter that cannot be neglected, has been used to evaluate the degree of graphitization and the density of defects in graphene-based materials.31,34 It can be clearly observed in Fig. 1d that there is an increase in the intensity ratio of G and D bands (ID/IG) that is attributed to the presence of localized defects within the hexagonal network upon reduction of the GO layer. Two-dimensional (∼2677 cm−1) and S3 (∼2920 cm−1) peaks30,38 were found in the spectra of in situ I-rGO/Si P, which again prove the better graphitization of this composite paper. The two Raman peaks at 116 cm−1 and 154 cm−1 (Fig. S2†) are similar to those reported by G Kalita et al.39 and signify triiodide (I3−) and pentaiodide (I5−) formation, respectively.40 All results showed that the iodine-doped graphene/Si composite paper was successfully prepared from the graphene oxide/Si composite paper via in situ I doping and reduction.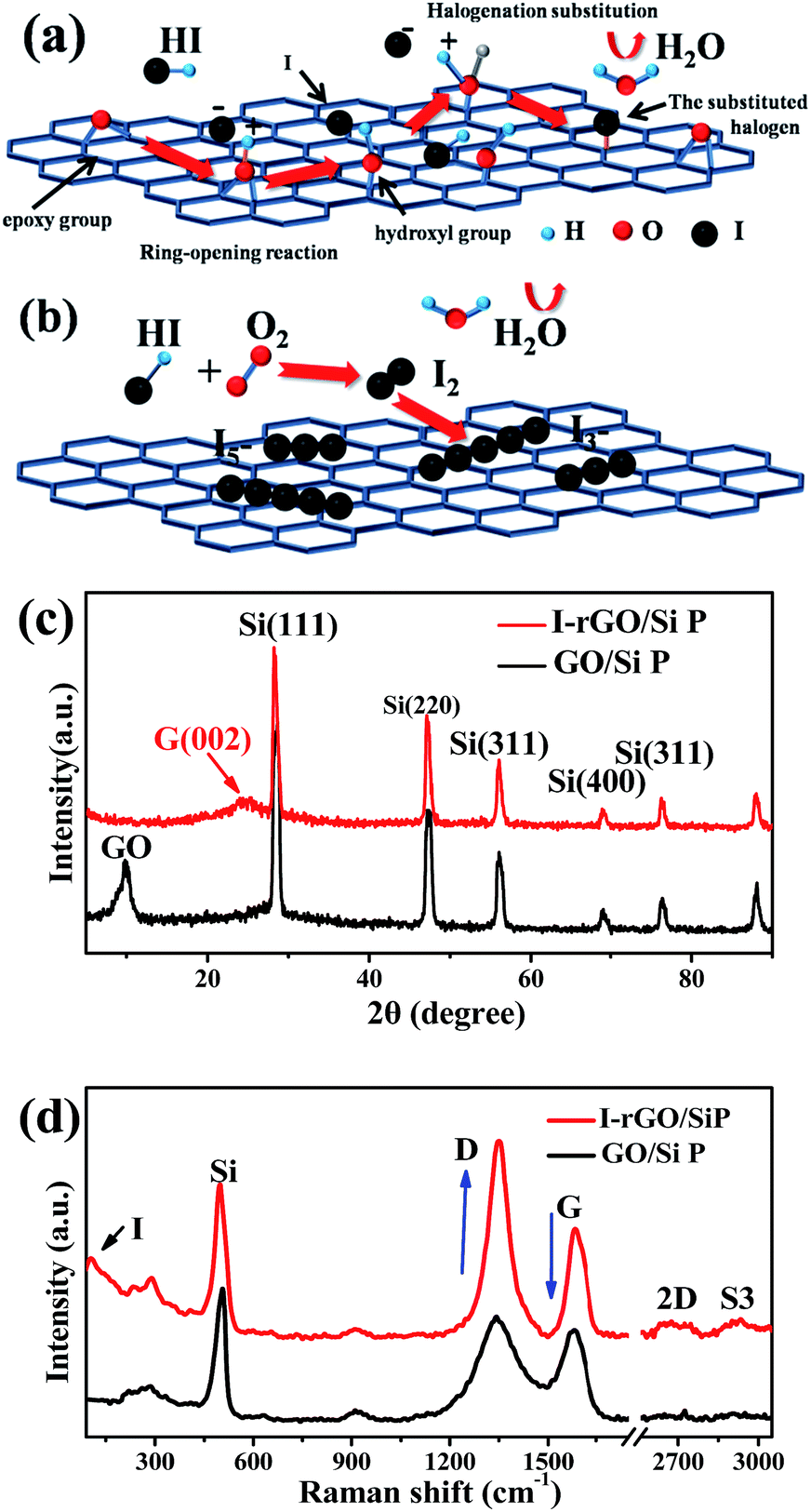 | ||
| Fig. 1 (a) Reaction mechanism of GO reduction by HI. (b) Iodine doping mechanism of the GO/Si composite paper (GO/Si P) by hydrohalic acids. (c) XRD patterns of GO/Si P and I-rGO/Si P. (d) The Raman spectra of GO/Si P and I-rGO/Si P. | ||
Furthermore, to investigate the structure of the I-rGO/Si P specimen, the morphologies of the samples were affirmed using FE-SEM. We can direct the control over the thickness of I-rGO/Si P using different dosages of the precursor solution (Fig. S3†). The cross-sectional SEM images for the 2 μm, 8 μm, and 17 μm thick I-rGO/Si P are presented in Fig. S4A, 2a and S4B,† respectively. Fig. 2b is an enlarged image based on Fig. 2a and shows a typical 3D sandwich structure of silicon, rGO sheet composition, and the existing space between the rGO substrates and silicon particle that forms a better buffer since it provides more space to ease the volume variation of silicon particles during the lithium-ion intercalation and deintercalation processes. The planform SEM images of I-rGO/Si P before and after HI-reduction were obtained to check encapsulation between the Si particles and rGO sheet. For comparison, Fig. S5A† and 2c show the top-view SEM images of GO/Si P and I-rGO/Si P (after spraying gold). Through the comparative study of these two images, we can see that the I-rGO/Si P surface is smooth, dense, and crack-free, but the surface of I-rGO/Si P is rough. The same results appear in a wide range of SEM images (Fig. S5B†). This may be because graphene oxide sheet splices together into a large reduced graphene oxide film on a composite paper surface during the HI reduction process. On the other hand, the SEM images of I-rGO/Si P before gold spraying (Fig. 2d) displayed the encapsulation of the Si particles by rGO. All Si particles are entirely covered by the gauze-like graphene sheets. It is also proved by the TEM images of I-rGO/Si P (Fig. 2e).
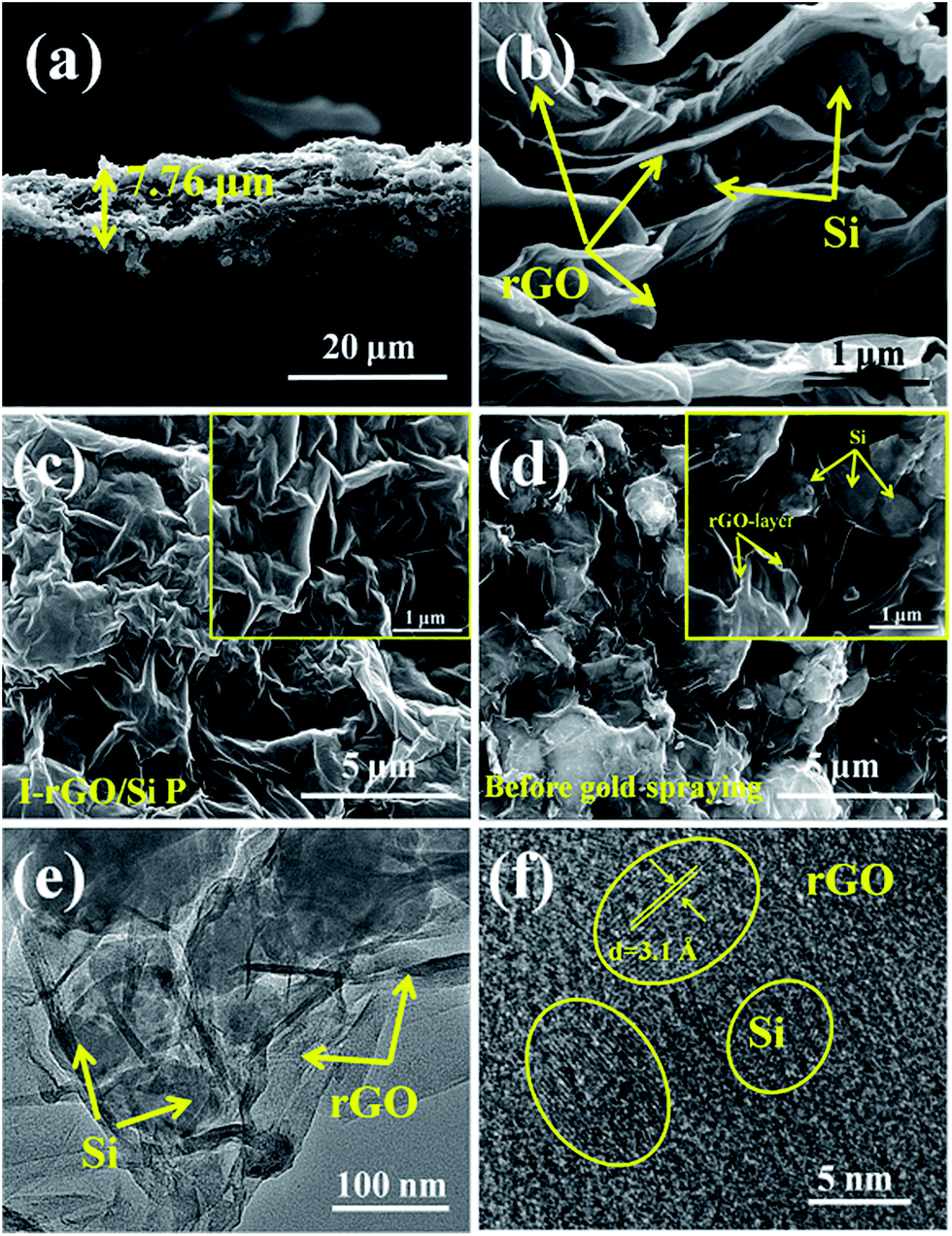 | ||
| Fig. 2 (a, b) The cross-sectional SEM images of I-rGO/Si P show the sandwich, layer-by-layer structure. The planform SEM images of I-rGO/Si P: (c) after spraying gold (d) before spraying gold. (e) The TEM images and (f) the HRTEM images of I-rGO/Si P. | ||
The surface electronic state and composition of I-rGO/Si P were analyzed via XPS, as shown in Fig. 3a–c. For I-rGO/Si P, the C 1s peak (Fig. 3a) contains four group waves. The peaks of C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–OH, C–O, and CO of graphene layers are observed at 284.5, 285.5, 286.5, and 289.5 eV, respectively.31,41 The peak of C–C/CC showed higher strength than other peaks. The C–C bonds become dominant after HI acid reduction because the majority of the hydroxyl and epoxy groups in GO P are removed. However, some oxygen-containing functional groups still remains in the rGO sheets, which are typical for chemically reduced GO. It is difficult to fully recover the carbon network using chemical reduction methods alone due to some of the missing carbon atoms in the sp2 carbon network of GO. The Si 2p3/2 was detected at 99.9 eV, Si 2pl/2 peaks were detected at 99.5 eV, and Si/SiOx (x < 2) was detected at 103.5 eV,42 as shown in the Si 2p XPS spectrum (Fig. 4b). The SiOx layer as an intermediate phase formed during the preparing process, which may improve the adhesion force between the Si particles and rGO.17,43,44 XPS spectra is also used to characterize the doping state of carbon-based materials. As shown in Fig. 4c, the iodine bands are divided into two types: I 3d5/2 (631.1 eV and 630.1 eV) and I 3d3/2 (619.5 eV and 618.5 eV).39,40,45 The matching of the I 3d5/2 peak provides two peaks: the peak near 620 eV was often considered as pentaiodide (I5−) and another at 618 eV was often considered for triiodide (I3−).28,40 The former peak is attributed to the introduction of iodine into the GO substrate reduced by HI acid and the latter peak is attributed to iodine anion from excess of HI.
C, C–OH, C–O, and CO of graphene layers are observed at 284.5, 285.5, 286.5, and 289.5 eV, respectively.31,41 The peak of C–C/CC showed higher strength than other peaks. The C–C bonds become dominant after HI acid reduction because the majority of the hydroxyl and epoxy groups in GO P are removed. However, some oxygen-containing functional groups still remains in the rGO sheets, which are typical for chemically reduced GO. It is difficult to fully recover the carbon network using chemical reduction methods alone due to some of the missing carbon atoms in the sp2 carbon network of GO. The Si 2p3/2 was detected at 99.9 eV, Si 2pl/2 peaks were detected at 99.5 eV, and Si/SiOx (x < 2) was detected at 103.5 eV,42 as shown in the Si 2p XPS spectrum (Fig. 4b). The SiOx layer as an intermediate phase formed during the preparing process, which may improve the adhesion force between the Si particles and rGO.17,43,44 XPS spectra is also used to characterize the doping state of carbon-based materials. As shown in Fig. 4c, the iodine bands are divided into two types: I 3d5/2 (631.1 eV and 630.1 eV) and I 3d3/2 (619.5 eV and 618.5 eV).39,40,45 The matching of the I 3d5/2 peak provides two peaks: the peak near 620 eV was often considered as pentaiodide (I5−) and another at 618 eV was often considered for triiodide (I3−).28,40 The former peak is attributed to the introduction of iodine into the GO substrate reduced by HI acid and the latter peak is attributed to iodine anion from excess of HI.
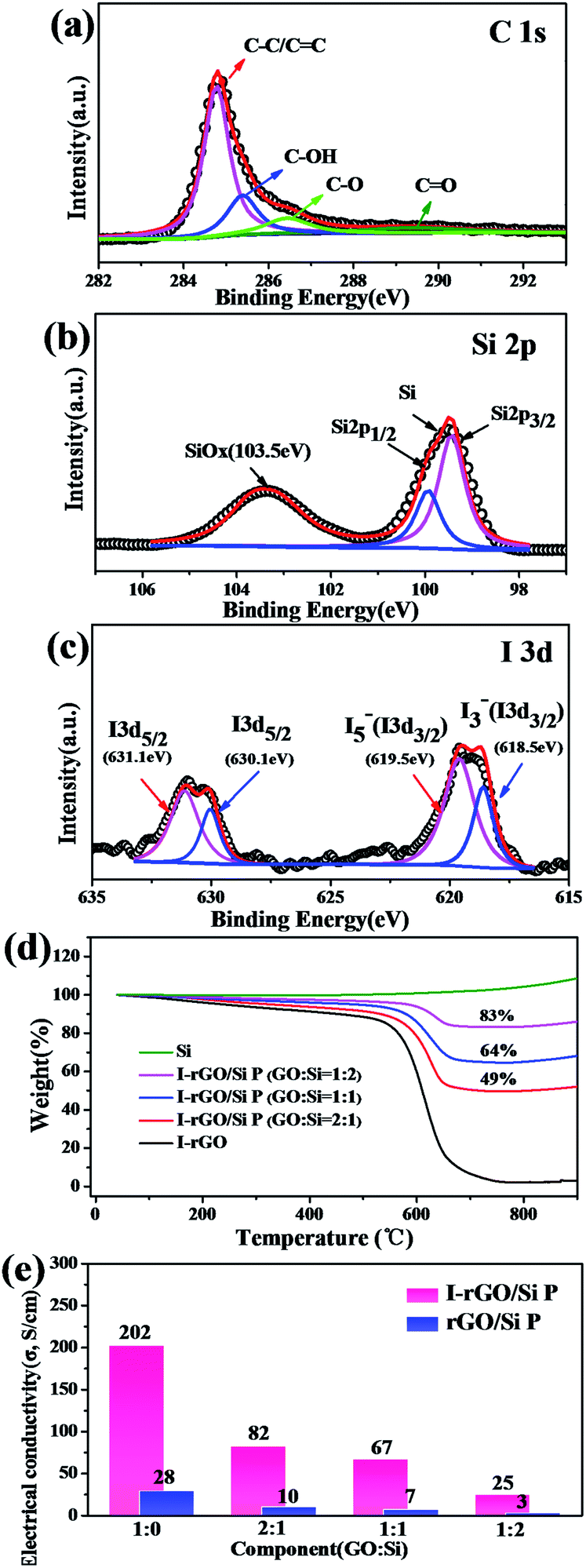 | ||
| Fig. 3 (a) XPS C 1s, (b) XPS Si 2p, and (c) XPS I 3d curve of in situ I-rGO/Si P. (d) TG curves of I-rGO/Si P, I-rGO, and pure Si tested in air. (e) Electrical conductivity of thermal rGO/Si P and I-rGO/Si P. | ||
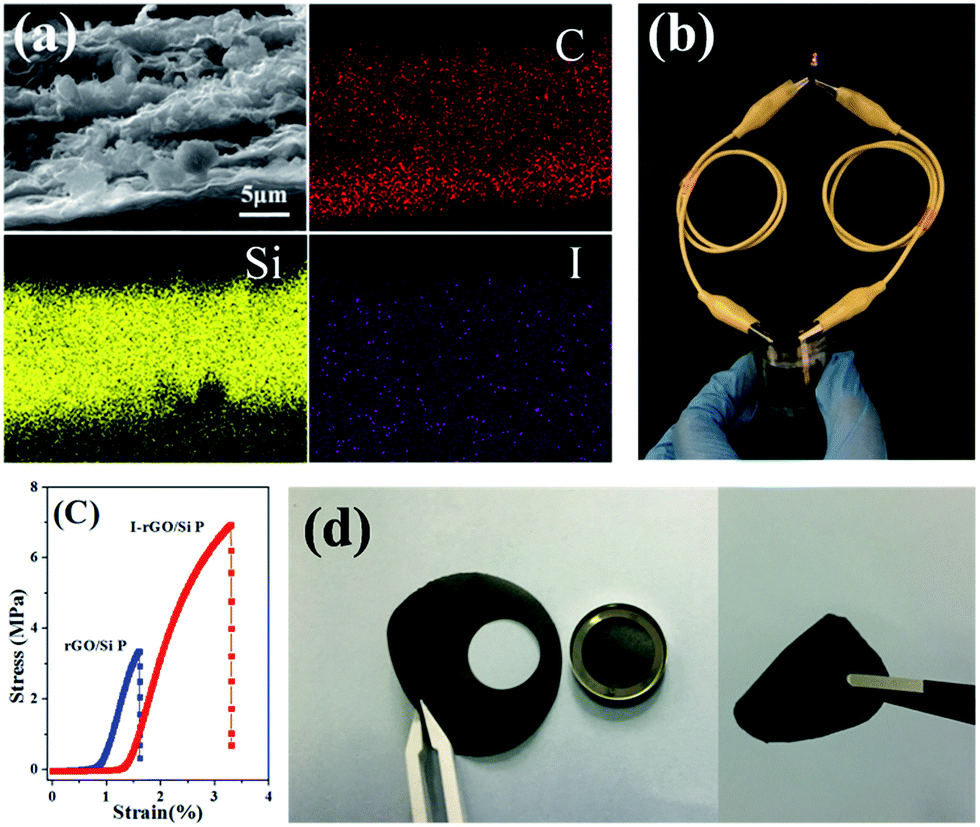 | ||
| Fig. 4 (a) The cross-sectional SEM image of I-rGO/Si P and corresponding carbon, silicon, and iodine elemental mapping. (b) I-rGO/Si P as a flexible battery light a red LED. (c) Engineering stress–strain curve of thermal rGO/Si P and I-rGO/Si P. (d) Images showing in situ I-rGO/Si P directly used as working electrodes of coin-type half cell (CR2032) without any conductive agent and bonding material. | ||
To further investigate the distribution of iodine ions, EDS elemental mappings based on the cross-section of I-rGO/Si P were obtained. In Fig. 4a, the elemental mappings for carbon, silicon, and iodine indicate that the location of carbon, silicon, and iodine overlap. It is worth noting that the density of carbon is less than that of silicon due to graphene that exists in its layer-by-layer form, and the points of iodine are evenly distributed according to the surface shape. Around 1.43 wt% of atomic iodine was found in the EDS elemental analysis based on the cross-section of I-rGO/Si P (Fig. S6†). All this suggests that iodine ions distribute uniformly in the sandwich structure of the composite paper. A high conductivity graphene/Si composite paper obtained using HI to reduce the GO/Si composite paper is expected. We prepared I-rGO/Si P by immersing GO:Si = 1:0, 2:1, 1:1, and 1:2 composite paper in a solution of HI acid. TGA of I-rGO/Si P was performed under an air atmosphere from 40 to 1000 °C (Fig. 3d). The content of silicon in I-rGO/Si P is about 0%, 49%, 64%, and 83% corresponding to GO:Si = 1:0, 2:1, 1:1, and 1:2 and are denoted as I-rGO P, I-rGO/Si21 P, I-rGO/Si11 P, and I-rGO/Si12 P, respectively. Fig. 3e demonstrates electrical conductivity of Si loading in two types of composite paper. I-rGO/Si P showed the electrical conductivities of approximately 202, 82, 67, and 25 S cm−1 corresponding to I-rGO P, I-rGO/Si21 P, I-rGO/Si11 P, and I-rGO/Si12 P, respectively, as compared to those of the thermal rGO/Si P (approximately 28, 10, 7, and 3 S cm−1) composite paper. Thus, this improved trend of electrical conductivity is coincident with the iodine doping effect. It is clear that the in situ iodine-doped rGO was layered with Si; this formed a layered-structure with good electronic conductivity and also a self-support skeleton for the composite paper. For any flexible battery electrode, the tensile strength and strain to failure are important parameters. Stress–strain curve derived from the load-displacement data are shown in Fig. 4c. The in situ I-rGO/Si11 P showed higher fracture strength (a tensile strength of 7 MPa with a strain of 3.4%) than rGO/Si11 P (a tensile strength of 3.8 MPa with a strain of 1.6%). As can be seen in Fig. 4d, I-rGO/Si P can bend 180 degrees and edge of fracture is smooth after cutting of the electrodes. The in situ I-rGO/Si P has high flexibility; thus, it can directly be used as working electrodes of a coin-type half cell (CR2032) without any conductive agent and bonding material. The feasibility of in situ I-rGO/Si P as a flexible battery was demonstrated by a home-made pouch cell (Fig. S7†) and the flexible device light a red LED, as shown in Fig. 4b. The abovementioned results provide good evidence that a sandwich structure of graphene/Si matrix is obtained by iodine-doped rGO sheets encapsulated by the Si particles; this structure can form a highly conductive skeleton and greatly improve the flexibility of the whole composite paper.
As self-supporting anode materials, the in situ iodine-doped rGO/Si composite paper prepared through reducing GO/Si composite paper with HI has been studied using coin-type half cells. Fig. 5a shows the cyclic voltammograms of in situ I-rGO/Si P at a scan rate of 0.1 mV s−1. For the first cycle, the main potential plateau becomes quite large below 0.1 V (vs. Li+/Li) that corresponds well to the lithiation process of crystallized Si. In addition, a broad peak located at about 0.4–0.7 V (vs. Li+/Li) is different from that of subsequent cycles and attributed to the formation of an SEI-layer on the electrode.46,47 In the second cycle, the peak at around 0.2 V in the discharge process, which is accepted as the phase change of Si from crystalline to amorphous during the first cycle. During the subsequent charging process, two anodic branches are located at 0.3 and 0.5 V corresponding to the de-lithiation process of amorphous LixSi.48 This provides shape similar to that of the CV curves of pure silicon and iodine-doped rGO paper (Fig. S8†).
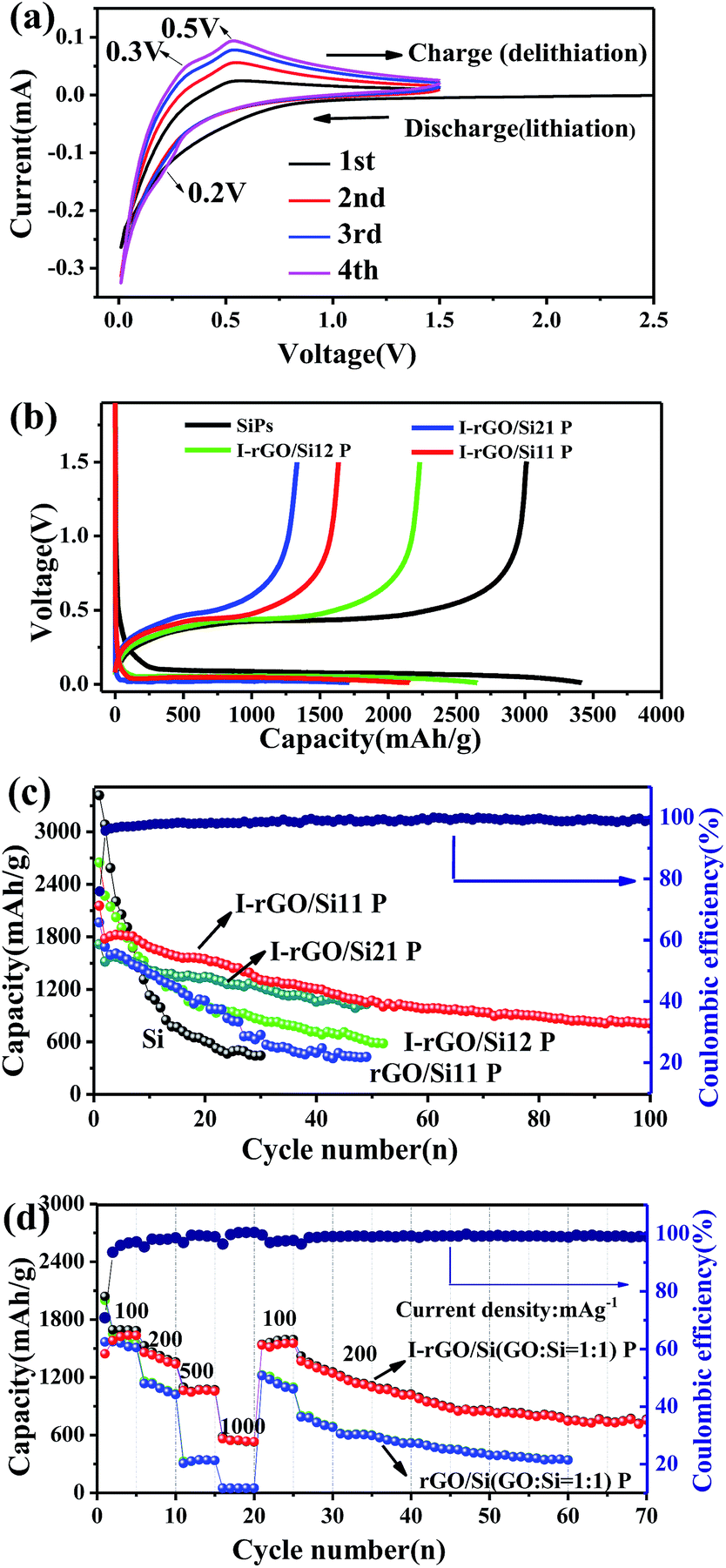 | ||
| Fig. 5 (a) CV curves of the self-supporting I-rGO/Si P in the initial four cycles at a scan rate of 0.1 mV s−1 in the potential range from 0 to 1.5 V (vs. Li+/Li). (b) The galvanostatic charge/discharge profiles of I-rGO/Si21 P, I-rGO/Si11 P, and I-rGO/Si12 P for comparison. (c) The cycling performance of the I-rGO/Si P with different compositions, and rGO/Si11 P for comparison. (d) The charge–discharge capacities versus the cycle number for I-rGO/Si P and rGO/Si P at different current rates of 100–1000 mA g−1 and then 200 mA g−1 for 70 cycles. | ||
Cycling performance of the in situ I-rGO/Si11 P is tested at a current density of 100 mA h g−1, as shown in Fig. 5c. The first discharge/charge capacity of I-rGO/Si11 P is 2154.2/1635.5 mA h g−1, corresponding to the first coulombic efficiency (CE) of 75.9%. The initial irreversible capacity loss can be associated with consumption of a number of Li+ ions during the formation of an SEI-layer on the surface of the composite paper from the decomposition of the electrolyte, as abovementioned in the CV section. After the first cycle, the in situ I-rGO/Si11P exhibits higher coulombic efficiency during the first 10 cycles and more stable coulombic efficiency of 99% during subsequent cycles. After 100 cycles, in situ I-rGO/Si11 P still delivers a reversible capacity of 805 mA h g−1. For comparison, pristine silicon is cast on a Cu current collector and self-supporting rGO/Si P to demonstrate the effect of self-supporting I-rGO/Si P (the proportion of different silicon content) on the electrochemical performance. As is shown in Fig. 5c, the in situ I-rGO/Si P (21, 11, and 12) exhibit a discharge capacity of 594, 1033, and 1005 mA h g−1, respectively, after 50 cycles. In Fig. S9B,† the coulombic efficiency of I-rGO/Si P (21, 11, and 12) and bare Si are shown. It clearly demonstrates that the coulombic efficiency of I-rGO/Si P (21, 11, and 12) is more stable than that of bare Si. Although bare Si and rGO/Si P deliver higher initial capacity of 3415 mA h g−1 and 1963 mA h g−1, respectively, the electrode suffers from rapid capacity fade in the following cycles. The capacity of bare Si drops to 540 mA h g−1 after only 30 cycles, and that of rGO/Si P drops to 428 mA h g−1 after 50 cycles. To offer more intuitionistic information of the comparison, the galvanostatic charge/discharge profiles of rGO/Si P and I-rGO/Si P at different cycles (Fig. S10†) deliver similar results. This result may be due to the large volume expansion of silicon and the falling of active material from Cu collector.
Fig. 5d compares the rate performance of in situ I-rGO/Si P and rGO/Si11 P at various current densities. The coin-type half was first cycled at 100 mA g−1 for 5 cycles, in which a stable reversible capacity of about 1582 mA h g−1 was observed. At the current densities of 200, 500, 1000, 100, and 200 mA g−1, the in situ I-rGO/Si11 P can still deliver the high reversible specific capacities of 1457, 1062, 561, 1535, and 1367 mA h g−1, respectively. The rGO/Si11 P only delivers the capacities of 1145, 318, 60, 1220, and 784 mA h g−1 under the same current densities. A relatively stable capacity of about 754 mA h g−1 was obtained at a current density of 200 mA g−1 after 70 cycles even if it went through large current density charge and discharge. Fig. 5b, as the counterpart, shows the galvanostatic charge/discharge profiles of the in situ I-rGO/Si11 P at the current densities from 100 to 1000 mA h g−1 in the voltage range of 0.01–1.5 V, which is in accordance with the results shown in Fig. S9A.† The in situ iodine doping not only increases the electrical conductivity of the graphene/silicon composite paper, but also improves the strength of the graphene matrix; this results in high capacity and enhanced cycling stability. Thus, we concluded that the homogeneous iodine doped into the sandwich structure of the graphene/silicon matrix as well as the effective encapsulation and formation of the sandwich structure of graphene/Si are the key factors contributing to the outstanding cycle performance of the rGO/Si composite papers.
The Nyquist plots of I-rGO/Si P are tested after 5 cycles and 100 cycles from 10000 Hz to 0.01 Hz in the full delithiation state. The data of Fig. 6a clearly demonstrates two semicircles on the Nyquist plots of I-rGO/Si P. In general, the 1st semicircle at higher frequency corresponding to the formation of an SEI film on the surface of I-rGO/Si P and the 2nd semicircle at lower frequency is related to the Li ion charge transfer at the interface.49,50 For comparison, Nyquist plots of rGO/Si P in the 5th and the 50th cycle are shown in the Fig. S11.† There is only a single broad semicircle in the high frequency region in the 5th full delithiation state, whereas two semicircles are observed in the 50th full delithiation state. The difference can be attributed to the formation of the SEI film. Similar results are also reported in previous literature.49–52 After long cycles, it is clearly demonstrated that the diameter of two semicircles for the in situ I-rGO/Si P electrode is much smaller than that of the rGO/Si P electrode. We attributed this phenomenon to the iodine doping effect. The I-rGO/Si P has better electrical conductivity than rGO/Si P and lower electron-transfer resistance. The rGO sheets exhibit electrical conductivity, high tensile strength, and outstanding ductility via HI reduction. Due to high ductility and layer-by-layer self-assembly, the HI-rGO sheets can form a better buffer for the Si particles than the general thermal-rGO sheets; thus, they have a good elasticity and can deform upon the subsequent volume expansion-contraction of Si during cycling processes (schematic in Fig. 6c). The cells of the I-rGO/Si P were disassembled after 100 cycles, and the free-standing electrode was recovered for further characterization. Fig. 6b and c demonstrate the planform and the cross-sectional SEM images of the iodine-doped rGO/Si composite electrode after 100 cycles. Obviously, a free-standing electrode surface without cracks was observed from the planform SEM image. The thickness of the I-rGO/Si composite paper increases from 7.8 μm (Fig. 2a) to 16.6 μm (Fig. 6c); this is due to the volume expansion of the composite paper and the formation of SEI-layers. The structural integrity of the electrode specimen remains favorable after 100 cycles; this suggests high mechanical structural strength of the in situ iodine-doped rGO/Si composite paper.
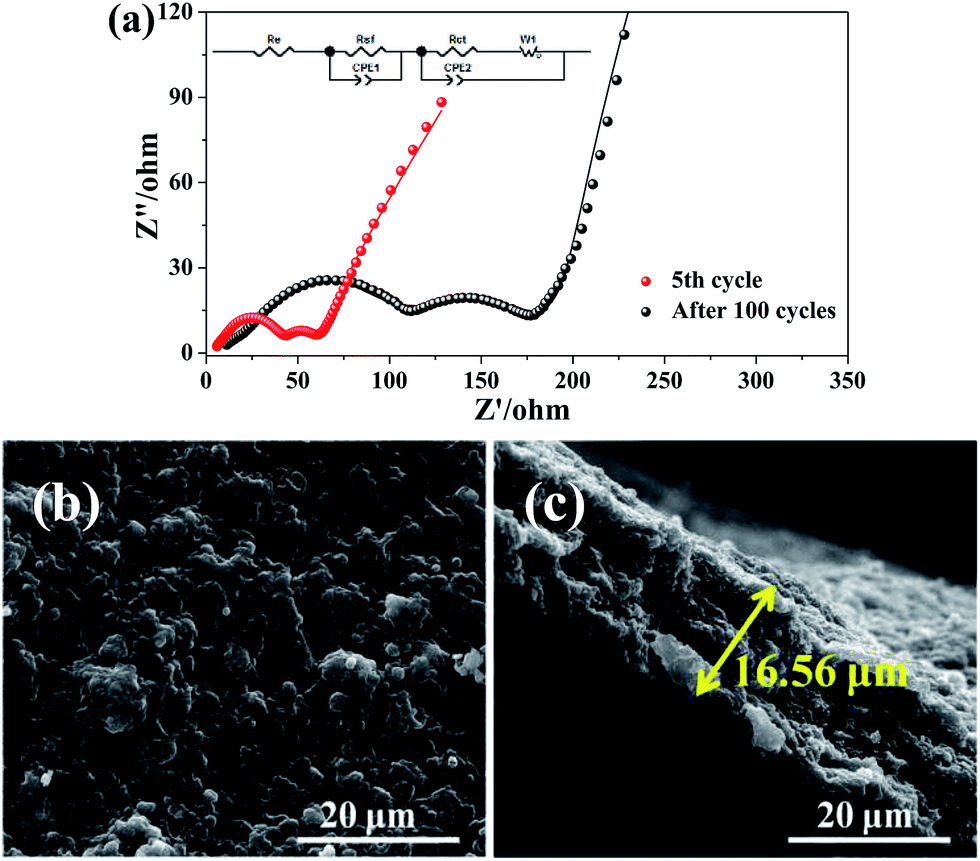 | ||
| Fig. 6 (a) Nyquist plots of the in situ I-rGO/Si P after 5 cycles and 100 cycles from 100 kHz to 0.01 Hz in the fully charged state. (b) The planform and (c) cross-sectional SEM images of I-rGO/Si P after 100 cycles. | ||
In a previous report, iodine-doped graphene was achieved via the pyrolysis of a mixture of GO and I2 (GO:I2 = 1:1) and it exhibited excellent electrochemical performance.28 Iodine-doped rGO paper using HI reducing agents was found to have an electronic conductivity as high as 298 S cm−1 and maintained a high tensile strength and an outstanding ductility.29 In a recent report, iodine-doped red phosphorus has been used as an anode material for LIBs.53 Moreover, the significantly improved conductivity of RPNPs showed more excellent electrochemical performance than that obtained without doping. Thus, we believe that the superior lithium-storage performance of the graphene/silicon composite paper may be attributed to the direct reduction of the GO/Si composite paper to highly conductive and flexible rGO/Si composite paper via in situ iodine doping reduction method.
Conclusions
In summary, we report the direct reduction of GO/Si composite paper to highly conductive and flexible in situ iodine-doped rGO/Si composite paper by HI acid. The preparation of the in situ iodine-doped rGO/Si composite paper showed high electrical conductivity (approximately 67 S cm−1 corresponding to GO:Si = 1:1) and high fracture strength (a tensile strength of 7 MPa with a strain of 3.4%), and this in situ iodine-doped rGO/Si composite paper is more preferable as compared to the usual thermal-rGO/Si composite paper. As a self-supporting and free-binder electrode, the composite paper displays a good cyclic retention of 805 mA h g−1 after 100 cycles and an enhanced rate capability. The superior electrochemical performance of the in situ iodine-doped rGO/Si composite paper is attributed to the synergistic effect of the iodine doping effect, high ductility of the HI-rGO substrate, and sandwiched structure of rGO/Si via in situ iodine doping method. It is further suggested that this composite paper used in real flexible battery applications for electronic devices is feasible.
Acknowledgements
The authors would like to appreciate the financial support received from the National Natural Science Foundation of China (201503036, 6131052) and Sichuan Science and Technology Support Program (2015GZ0130, 2015GZ0132, 2016GZ0025).References
- Z. Song, T. Ma, R. Tang, Q. Cheng, X. Wang, D. Krishnaraju, R. Panat, C. K. Chan, H. Yu and H. Jiang, Nat. Commun., 2014, 5, 3140 Search PubMed.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- G. Zhou, F. Li and H.-M. Cheng, Energy Environ. Sci., 2014, 7, 1307–1338 CAS.
- N. S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y. K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angewandte Chemie, 2012, 51, 9994–10024 CrossRef CAS PubMed.
- Y. Xu, Y. Zhu, F. Han, C. Luo and C. Wang, Adv. Energy Mater., 2015, 5, 1400753 CrossRef.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- D. W. Wang, F. Li, J. Zhao, W. Ren, Z. G. Chen, J. Tan, Z. S. Wu, I. Gentle, G. Q. Lu and H. M. Cheng, ACS Nano, 2009, 3, 1745–1752 CrossRef CAS PubMed.
- H. Gwon, H.-S. Kim, K. U. Lee, D.-H. Seo, Y. C. Park, Y.-S. Lee, B. T. Ahn and K. Kang, Energy Environ. Sci., 2011, 4, 1277 CAS.
- C. Luo, Y. Zhu, Y. Xu, Y. Liu, T. Gao, J. Wang and C. Wang, J. Power Sources, 2014, 250, 372–378 CrossRef CAS.
- H. Tang, Y. J. Zhang, Q. Q. Xiong, J. D. Cheng, Q. Zhang, X. L. Wang, C. D. Gu and J. P. Tu, Electrochim. Acta, 2015, 156, 86–93 CrossRef CAS.
- Y. Q. Zhang, X. H. Xia, X. L. Wang, Y. J. Mai, S. J. Shi, Y. Y. Tang, L. Li and J. P. Tu, Electrochem. Commun., 2012, 23, 17–20 CrossRef CAS.
- H. Tang, X. H. Xia, Y. J. Zhang, Y. Y. Tong, X. L. Wang, C. D. Gu and J. P. Tu, Electrochim. Acta, 2015, 180, 1068–1074 CrossRef CAS.
- X. Li and L. Zhi, Nanoscale, 2013, 5, 8864–8873 RSC.
- B. Liang, Y. Liu and Y. Xu, J. Power Sources, 2014, 267, 469–490 CrossRef CAS.
- X. Chen, X. Li, F. Ding, W. Xu, J. Xiao, Y. Cao, P. Meduri, J. Liu, G. L. Graff and J. G. Zhang, Nano Lett., 2012, 12, 4124–4130 CrossRef CAS PubMed.
- X. Zhou, Y.-X. Yin, L.-J. Wan and Y.-G. Guo, Adv. Energy Mater., 2012, 2, 1086–1090 CrossRef CAS.
- T. Van Khai, H. G. Na, D. S. Kwak, Y. J. Kwon, H. Ham, K. B. Shim and H. W. Kim, J. Mater. Chem., 2012, 22, 17992 RSC.
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS PubMed.
- Z. H. Sheng, H. L. Gao, W. J. Bao, F. B. Wang and X. H. Xia, J. Mater. Chem., 2011, 22, 390–395 RSC.
- C. Zhang, N. Mahmood, H. Yin, F. Liu and Y. Hou, Adv. Mater., 2013, 25, 4932 CrossRef CAS PubMed.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. A. Chen and S. Huang, ACS Nano, 2012, 6, 205 CrossRef CAS PubMed.
- Z. Jin, H. Nie, Z. Yang, J. Zhang, Z. Liu, X. Xu and S. Huang, Nanoscale, 2012, 4, 6455–6460 RSC.
- Y. Zhao, J. Wei, R. Vajtai, P. M. Ajayan and E. V. Barrera, Sci. Rep., 2011, 1, 83 CrossRef PubMed.
- W. Su, Y. Zhang, Z. Li, L. Wu, X. Wang, J. Li and X. Fu, Langmuir, 2008, 24, 3422–3428 CrossRef CAS PubMed.
- X. R. Zeng and T. M. Ko, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 1993–2001 CrossRef CAS.
- Z. Wu, Y. Han, R. Huang, X. Chen, Y. Guo, Y. He, W. Li, Y. Cai and N. Wang, Nanoscale, 2014, 6, 13196–13202 RSC.
- Y. Zhan, B. Zhang, L. Cao, X. Wu, Z. Lin, X. Yu, X. Zhang, D. Zeng, F. Xie, W. Zhang, J. Chen and H. Meng, Carbon, 2015, 94, 1–8 CrossRef CAS.
- S. Pei, J. Zhao, J. Du, W. Ren and H.-M. Cheng, Carbon, 2010, 48, 4466–4474 CrossRef CAS.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 1–6 CrossRef PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- N. Lin, Y. Han, J. Zhou, K. Zhang, T. Xu, Y. Zhu and Y. Qian, Energy Environ. Sci., 2015, 8, 3187–3191 CAS.
- J.-Z. Wang, C. Zhong, S.-L. Chou and H.-K. Liu, Electrochem. Commun., 2010, 12, 1467–1470 CrossRef CAS.
- H.-C. Tao, L.-Z. Fan, Y. Mei and X. Qu, Electrochem. Commun., 2011, 13, 1332–1335 CrossRef CAS.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- S.-J. Kim, M.-C. Kim, S.-B. Han, G.-H. Lee, H.-S. Choe, D.-H. Kwak, S.-Y. Choi, B.-G. Son, M.-S. Shin and K.-W. Park, Nano Energy, 2016, 27, 545–553 CrossRef CAS.
- B. Li, S. Yang, S. Li, B. Wang and J. Liu, Adv. Energy Mater., 2015, 5, 1500289 CrossRef.
- P. Cui, J. Lee, E. Hwang and H. Lee, Chem. Commun., 2011, 47, 12370–12372 RSC.
- G. Kalita, K. Wakita, M. Takahashi and M. Umeno, J. Mater. Chem., 2011, 21, 15209 RSC.
- Z. Yao, H. Nie, Z. Yang, X. Zhou, Z. Liu and S. Huang, Chem. Commun., 2012, 48, 1027–1029 RSC.
- A. Y. Romanchuk, A. S. Slesarev, S. N. Kalmykov, D. V. Kosynkin and J. M. Tour, Phys. Chem. Chem. Phys., 2013, 15, 2321 RSC.
- H. Kobayashi, Y. Yamashita, T. Mori, Y. Nakato, K. H. Park and Y. Nishioka, Surf. Sci., 1995, 326, 124–132 CrossRef CAS.
- H. Fukui, H. Ohsuka, T. Hino and K. Kanamura, ACS Appl. Mater. Interfaces, 2010, 2, 998–1008 CAS.
- J. Yang, Y. Takeda, N. Imanishi, C. Capiglia, J. Y. Xie and O. Yamamoto, Solid State Ionics, 2002, 152, 125–129 CrossRef.
- Y. Zhan, J. Huang, Z. Lin, X. Yu, D. Zeng, X. Zhang, F. Xie, W. Zhang, J. Chen and H. Meng, Carbon, 2015, 95, 930–939 CrossRef CAS.
- L. Xiao, Y. H. Sehlleier, S. Dobrowolny, H. Orthner, F. Mahlendorf, A. Heinzel, C. Schulz and H. Wiggers, ChemElectroChem, 2015, 2, 1983–1990 CrossRef CAS.
- H. Tang, J. Zhang, Y. J. Zhang, Q. Q. Xiong, Y. Y. Tong, Y. Li, X. L. Wang, C. D. Gu and J. P. Tu, J. Power Sources, 2015, 286, 431–437 CrossRef CAS.
- Y. Zhu, W. Liu, X. Zhang, J. He, J. Chen, Y. Wang and T. Cao, Langmuir, 2013, 29, 744–749 CrossRef CAS PubMed.
- Q. Q. Xiong, J. P. Tu, X. Ge, X. L. Wang and C. D. Gu, J. Power Sources, 2015, 274, 1–7 CrossRef CAS.
- X. Wu, Z. Wang, L. Chen and X. Huang, Electrochem. Commun., 2003, 5, 935–939 CrossRef CAS.
- F. Wang, S. Xu, S. Zhu, H. Peng, R. Huang, L. Wang, X. Xie and P. K. Chu, Electrochim. Acta, 2013, 87, 250–255 CrossRef CAS.
- A. Ponrouch and M. R. Palacín, J. Power Sources, 2012, 212, 233–246 CrossRef CAS.
- W. C. Chang, K. W. Tseng and H. Y. Tuan, Nano Lett., 2017, 17, 1240–1247 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06871a |
| This journal is © The Royal Society of Chemistry 2017 |