
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evolution analysis of V2O5·nH2O gels for preparation of xerogels having a high specific surface area and their replicas†
Kanji Ishii a,
Yuki Kimurab,
Tomoya Yamazakib,
Yuya Oakia and
Hiroaki Imai*a
a,
Yuki Kimurab,
Tomoya Yamazakib,
Yuya Oakia and
Hiroaki Imai*a
aDepartment of Applied Chemistry, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan. E-mail: hiroaki@applc.keio.ac.jp
bInstitute of Low Temperature Science, Hokkaido University, Kita-19, Nishi-8, Kita-ku, Sapporo, Hokkaido 060-0819, Japan
First published on 19th July 2017
Abstract
The evolution of a V2O5·nH2O gel skeleton through the hydration of vanadium alkoxide was monitored using ex situ and in situ transmission electron microscopy and X-ray diffractometry. We successfully observed the morphological evolution of the gel skeleton by vacuum drying after replacing the pore liquid of acetone with cyclohexane. The gel skeleton comprised of nanofibers 10–20 nm wide and over 300 nm long was formed from ultrathin films 1–3 nm thick through thin fibrils 3–7 nm wide and about 100 nm long. Fixation of the thin fibrils by a simple vacuum-drying technique provided xerogels having a specific surface area as high as ∼320 m2 g−1 at ambient temperature. Highly porous polypyrrole frameworks were obtained as replicas of the V2O5·nH2O xerogels.
1. Introduction
V2O5·nH2O and orthorhombic V2O5 crystals have been applied as semiconductors,1 redox catalysts,2 intercalation hosts for various compounds (including ions, organics, and polymers),3,4 and electrochromism5 by utilizing their layered crystal structure and redox properties of vanadium ions. The vanadium compounds have also been studied as cathode materials for lithium ion batteries (LIBs) with a high reversible capacity of 300 mA h g−1 (ref. 6–8) and promising cathode materials for sodium, potassium, and polyvalent batteries.9–12 Because of their higher surface areas and flexibility originating from layered structures, V2O5·nH2O xerogels have better electrochemical properties than orthorhombic V2O5 crystals.8,9 Therefore, V2O5·nH2O xerogels having high specific surface areas have been important in recent years.V2O5·nH2O forms a gel consisting of nanometer-wide fibers. The gel structures of V2O5·nH2O have been well investigated by many researchers, as reviewed by Livage.13 Petkov et al. reported the detailed crystal structure of V2O5·nH2O, which consists of V2O5 bilayers separated by water layers. The V2O5 layers, which are made up of orderly assembled VO6 octahedral units, spread in the a–b plane and stack in the c direction (Fig. S1†).14
The synthesis of V2O5·nH2O gel has been explored thoroughly: pouring melted orthorhombic V2O5 into water;13 adding hydrogen peroxide to vanadium metal;15 the ion-exchange processing of sodium meta-vanadate;16,17 and hydrolysis and subsequent condensation of alkoxide.7,8,18–20 Xerogels are produced from the wet gels through several drying methods. In the synthesis by ion-exchanging sodium meta-vanadate, the growth process of a V2O5·nH2O skeleton had been observed in the 1990s using cryo-transmission electron microscopy (TEM) to study the mechanism of gel formation. In previous works, two growing mechanisms were proposed: one is growth by the addition of VO2+ cations,16 and the other is growth by the self-assembly of small threads.13,21–23 In the process using alkoxide, the morphological evolution of V2O5·nH2O xerogels has not been traced due to a high reactivity of the vanadium sources.
In general, pores in xerogels are collapsed by the surface tension during the drying process, and their surface areas are very low (<10 m2 g−1).20 Supercritical drying,19,20 freeze-drying, and ambient pressure drying after exchanging the pore liquid for one with small surface tension7,8,18,20 are used to obtain V2O5·nH2O xerogels having higher pore volumes and specific surface areas, which are called aerogel, cryogel, and ambigel, respectively. A high surface area of 450 m2 g−1 was reported for aerogel prepared by supercritical drying.17 The specific surface areas of cryogels and ambigels are limited to within 280 m2 g−1.18
In the present work, V2O5·nH2O gels were synthesized using alkoxide as a vanadium source, and xerogels were obtained through a simple process of ambient temperature vacuum drying. The ambient temperature vacuum drying after exchanging the pore liquid for one with low surface tension successfully maintained the skeleton of the wet gel without collapse. The growing process of V2O5·nH2O gels was clarified using ex situ and in situ fluid reaction TEM observation. Examining the effect of the period of aging and the ratio of the solvent and alkoxide, we proposed a growing mechanism of the gel skeleton. Afterward, on the basis of the evolutional process of the gel framework, xerogels having the highest specific surface area ever reported were successfully synthesized without supercritical drying.
Here, we produced replicas of V2O5·nH2O xerogels with a functional polymer. Kuwabara et al. used several crystals of oxidizing agents with a nanostructure as templates for the polymerization of conductive polymers having enhanced electrochemical properties.24 V2O5·nH2O xerogels were applied for an oxidizing agent and template to utilize their oxidizability and high surface areas at the same time. We tried to synthesize replicas of V2O5·nH2O xerogels with polypyrrole. This is a novel application of V2O5·nH2O xerogels.
2. Experimental
2.1. Synthesis of V2O5·nH2O xerogels
All reagents were used as purchased without purification. V2O5·nH2O gels were prepared by the hydrolysis of vanadyl triisopropoxide (VO(OiPr)3, Tokyo Chemical Industry) in a water/acetone solution. The volume of VO(OiPr)3 was 1.2 cm3, and the molar ratio of [VO(OiPr)3]/[H2O] varied from 1/5 to 1/40, while the molar ratio of [VO(OiPr)3]/[acetone] was fixed at 1/20. Prior to mixing, both alkoxide and the water/acetone mixture were cooled separately in an ice bath for 20 min. After cooling, the water/acetone mixture was poured into VO(OiPr)3, and the color of the solution became deep red. The resultant deep red solution was shaken vigorously from 0.5 to several minutes until gelation occurred. The gelation time increased with the decreasing ratio of water to alkoxide. The wet gels were aged at 25 °C for a period from 0.5 to 192 h. After aging, water, acetone, isopropanol, and unreacted VO(OiPr)3 as pore liquids were replaced with acetone and cyclohexane in order. The exchange for acetone was performed 4 times for samples of the ratio [VO(OiPr)3]/[H2O] = 1/5 and 1/10 and 6 times for samples of [VO(OiPr)3]/[H2O] = 1/40 in 4 days, and then the replacement with cyclohexane was made 4 times in 4 days. Xerogels were obtained by vacuum drying of the wet gels at ambient temperature or freeze-drying using liquid nitrogen.2.2. Synthesis of V2O5·nH2O xerogel replicas
Replicas of V2O5·nH2O xerogels were synthesized with polypyrrole (PPy). The synthesis was conducted based on the synthesis method reported by Kuwabara et al.24 A vial with 2.0 cm3 of pyrrole (Py, Tokyo Chemical Industry) and a Petri dish with 100 mg of V2O5·nH2O xerogels were placed in a 600 cm3 sealed container. The container was kept standing at 60 °C for a predetermined time (4–72 h). The color of the V2O5·nH2O xerogels changed from green to black during standing. This implied the polymerization of PPy on the surface of the V2O5·nH2O xerogels. The V2O5·nH2O xerogels were removed from the complex by immersion into 1 mol dm−3 HClaq. After filtration and washing with water and ethanol, the sample was dispersed in acetone and cyclohexane in this order. The PPy replicas of the V2O5·nH2O xerogels were then obtained by drying at 60 °C at ambient pressure.2.3. Characterization
Identification and analysis of the crystal structure were conducted using X-ray powder diffraction (XRD, Rigaku MiniFlex II). The scan range was 2θ = 3–60 degrees, and the scan rate was 2 degrees per min. Specific surface areas were evaluated using the Brunauer–Emmett–Teller (BET) method from the results of nitrogen physisorption (Shimadzu 3Flex-3MP) at 77 K. The morphologies of xerogels were observed with scanning electron microscopy (SEM, Hitachi S-4700, operated at 5 kV) and TEM (FEI Tecnai F20, operated at 200 kV). The dispersion was dropped on a copper grid with a cellulose acetobutyrate microgrid for TEM observation. Composition analysis was performed using the energy-dispersive X-ray (EDX) spectroscopy attached to the Tecnai F20.We directly observed the morphological evolution of gel skeletons in the wet gel using in situ fluid reaction TEM (in situ FR-TEM, JEOL JEM-2100F, operated at 200 kV). For the observation, we used a “Poseidon” fluid reaction cell holder (Protochips). The fluid reaction cell consists of a pair of semiconductor-based plates with an amorphous silicon nitride window and 150 nm-thick spacer. For the in situ FR-TEM observation, gel was synthesized with 0.6 cm3 of VO(OiPr)3, 7.2 cm3 of water, and 7.5 cm3 of acetone ([VO(OiPr)3]/[H2O]/[acetone] = 1/160/40 (molar ratio)). In order to minimize the effect of the electron beam on the growth of gel skeletons, the electron beam was irradiated only at shooting at the interval of 10 min.
3. Results and discussion
3.1. Morphological evolution of V2O5·nH2O gel skeletons
We explored the morphological evolution of the gel skeleton of V2O5·nH2O by increasing the aging time. Fig. 1 shows XRD patterns, SEM and TEM images, and the schematic illustration of V2O5·nH2O xerogels synthesized at [VO(OiPr)3]/[H2O] = 1/40. Two-dimensional ultrathin films were initially formed as shown in SEM and TEM images (first stage). One-dimensional fibrils (second stage) and, finally, smoother nanofibers were then produced (third stage) with the disappearance of the ultrathin films (Fig. 1b–d).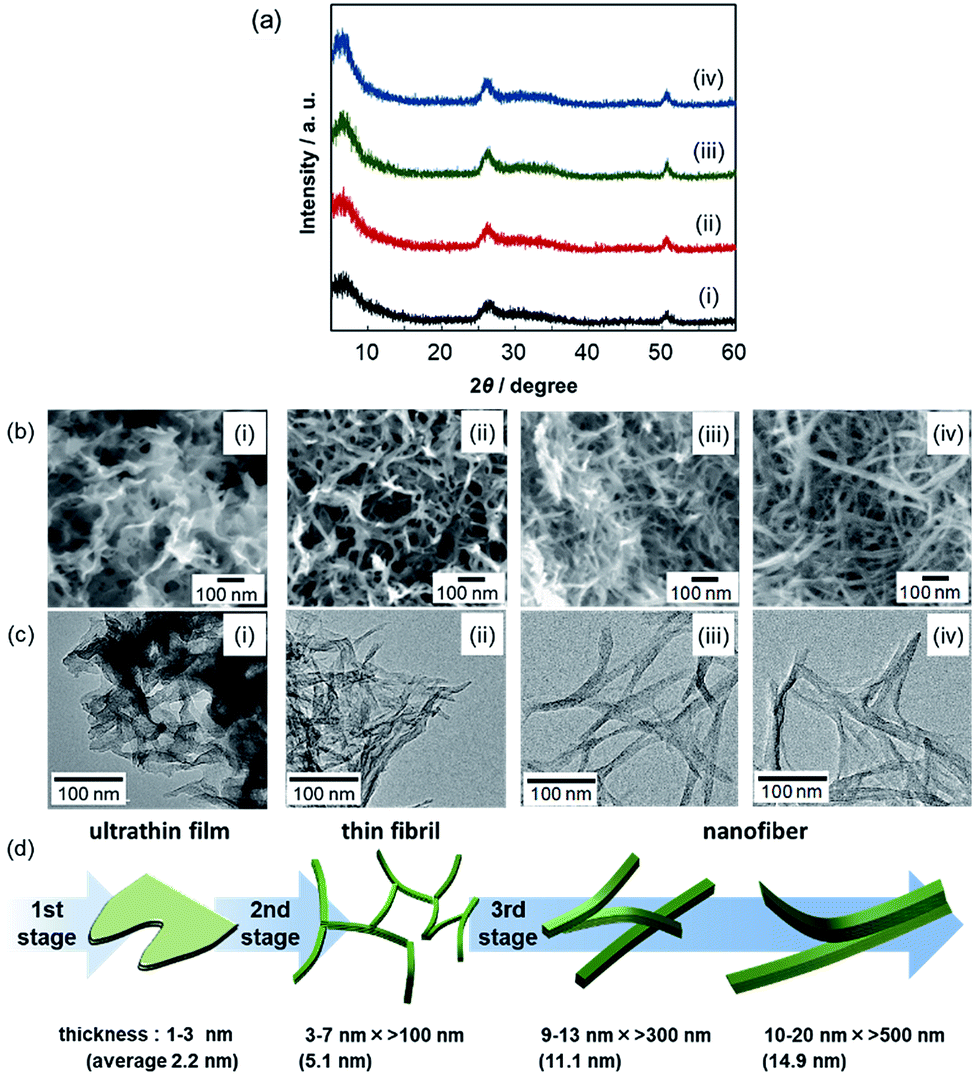 | ||
| Fig. 1 XRD patterns (a) and SEM (b) and TEM (c) images of V2O5·nH2O xerogels ([VO(OiPr)3]/[H2O]/[acetone] = 1/40/20) after aging for 0.5 (i), 2.0 (ii), 24 (iii), and 48 h (iv). Schematic illustration of the evolution of a V2O5·nH2O gel skeleton (d). | ||
In the XRD patterns of V2O5·nH2O xerogels (Fig. 1a), all the samples exhibited broad signals at around 2θ = 7, 26, and 51°. According to the literature,17,18 the first band corresponds to the 001 direction giving an interlayer spacing, while the other peaks originate from the internal layer structure spreading in the a–b plane. A single sheet of V2O5·nH2O consists of a bilayer structure similar to that of orthorhombic V2O5, with interlayer water (Fig. S1†). As the aging time increased, the structures observed in the electron micrographs (Fig. 1b–d) thickened upon increasing the intensity of the 001 signal. Ultrathin films 1–3 nm thick (0.5 h) evolved to nanofibers 10–20 nm wide (48 h). This indicates that the stacking of the bilayers in the c direction proceeds through the transition from ultrathin films to thick fibers by the aging of the gel.
The ultrathin films 1–3 nm thick (Fig. 1(i)) produced in the first stage (<0.5 h at [VO(OiPr)3]/[H2O]/[acetone] = 1/40/20) are inferred to be constructed by the stacking of a few V2O5 layers along the c-axis. The direct observation using in situ FR-TEM was conducted in order to research the dynamic morphological evolution in the early stages of the sol–gel reaction (Fig. 2). For the in situ FR-TEM observation, gel was synthesized at [VO(OiPr)3]/[H2O]/[acetone] = 1/160/40 (molar ratio). The formation of ultrathin films was observed within 2.0 h (Fig. 2a).
 | ||
| Fig. 2 In situ FR-TEM images of a V2O5·nH2O wet gel ([VO(OiPr)3]/[H2O]/[acetone] = 1/160/40) after aging for 2.0 (a), 4.0 (b), and 6.5 h (c). | ||
In the second stage (0.5–2.0 h at [VO(OiPr)3]/[H2O]/[acetone] = 1/40/20), thin fibrils, 3–7 nm wide and ∼100 nm long, were produced (Fig. 1(ii)). In in situ FR-TEM observation, the networks of thin fibrils with films were observed over 4.0 h at [VO(OiPr)3]/[H2O]/[acetone] = 1/160/40 (Fig. 2b and c). As the reaction time increased, growth of the fibrils proceeded in the surrounding films.
In the third stage (>2.0 h at [VO(OiPr)3]/[H2O]/[acetone] = 1/40/20), nanofibers 9–13 nm wide and >300 nm long (24 h) and 10–20 nm wide and >500 nm long (48 h) were produced (Fig. 1(iii and iv)). Y-Shaped nanofibers seen in the TEM images imply the stacking or side-by-side attachment of thin fibrils. The growth in the direction of stacking (c-axis) was supported by an increase in the 001 signal intensity. This indicates that the stacking is an “oriented attachment”. The oriented attachment of multiple fibrils also contributes to the growth in the direction of length (b-axis).
We explored the effect of water on the growth of the gel skeleton. The XRD patterns and SEM and TEM images of V2O5·nH2O xerogels synthesized with various ratios of water after the same aging period of 48 h are shown in Fig. 3. The 001 peak intensity increased as the ratio of water increased. Thus, growth in the direction of the c-axis is accelerated by water. As discussed above, nanofibers 10–20 nm wide and >500 nm long were observed at [VO(OiPr)3]/[H2O] = 1/40 (Fig. 3(iii)). In contrast, thin fibrils 4–9 nm wide and ∼100 nm long and ultrathin films 2–3 nm thick were observed at [VO(OiPr)3]/[H2O] = 1/10 (Fig. 3(ii)) and 1/5 (Fig. 3(i)), respectively. The structural evolution from ultrathin films to thin fibrils and nanofibers occurs in the aging process. The evolution was accelerated by increasing the ratio of water ([VO(OiPr)3]/[H2O] = 1/5, 1/10, and 1/40) at the same aging period.
 | ||
| Fig. 3 XRD patterns (a) and SEM (b) and TEM (c) images of V2O5·nH2O xerogels after aging for 48 h. [VO(OiPr)3]/[H2O] = 1/5 (i), 10 (ii), and 40 (iii). | ||
The effects of water for the evolution from the thin films to nanofibers were investigated by changing [VO(OiPr)3]/[H2O]. At [VO(OiPr)3]/[H2O] = 1/5, the film morphology did not change to fibrils or fibers even at aging period of 192 h (Fig. S2†). At [VO(OiPr)3]/[H2O] = 1/10, the evolution needed a longer aging period (96 h) (Fig. S3†). These results support the acceleration of morphological evolution by water.
3.2. Growth mechanism of V2O5·nH2O gel skeletons
Based on the in situ FR-TEM images (Fig. 2), the V2O5·nH2O wet gel showed the same evolution process as shown in the ex situ TEM observation of V2O5·nH2O xerogel (Fig. 1c); two-dimensional films changed to the networks of one-dimensional fibrils. This indicates that xerogels obtained from ambient temperature vacuum drying after replacing the pore liquid with one with low surface tension maintain the gel skeleton of wet gels. Thus, the morphological evolution of the gel skeleton can be discussed from the ex situ TEM observation of xerogels. Because supercritical drying, which is the common method to obtain high surface area xerogels, has risks of changing the crystal structure and morphology due to high temperature and high pressure,25 the ambient drying method is preferable to study the evolution of the gel skeleton. Two growth mechanisms of the V2O5·nH2O gel skeleton have been already proposed for synthesis by the ion exchange of sodium metavanadate on the basis of direct observation using cryo-TEM: small particles ∼3 nm wide and 30–40 nm long are initially formed and then grow to fibers ∼3 nm × ∼25 nm × >1 μm due to the supply of VO2+ from solution;16 nanometer-scale threads ∼2 nm wide and ∼100 nm long are formed and then evolve into ribbons ∼10 nm wide and >1 μm long via self-assembly of the threads.13 However, on the formation of V2O5·nH2O gels by the hydrolysis of alkoxides, the morphological evolution is much different from that in the ion-exchange method. Thus, we must discuss the growing mechanism for V2O5·nH2O gels from alkoxides as a source of vanadium.In sol–gel synthesis using alkoxides, growth of the gel skeleton during aging generally proceeds with the condensation of clusters or monomers and modification of the gel network structure by subsequent condensation of M–OH or M–OR (M: metal) groups.26 These growth mechanisms are suggested mainly based on the silica gel. The dissolution of precipitated particles is not considered in the growth of the silica gel skeleton. On sol–gel synthesis of ZnO2, water was reported to accelerate the particle growth by increasing the concentration of Zn2+ ions.27 Whereas silica and ZnO2 particles form a random accumulation of isotropic particles, V2O5·nH2O fibrils accumulate anisotropically. The growth mechanism of anisotropic fibrils in sol–gel methods has not been discussed sufficiently. Here, we suggest a growth mechanism of the oriented attachment assisted by water which promotes the partial dissolution and reprecipitation and stacking of layers.
Fig. 4 illustrates the growth mechanism of a V2O5·nH2O gel skeleton based on our observation in the current work. According to direct observation by in situ FR-TEM, ultrathin films are formed in the first stage. The V2O5 layers expand through deposition of VO(OH)3, which is produced by hydrolysis of VO(OiPr)3, at the edges of the ultrathin films (Fig. 4a and S4†).
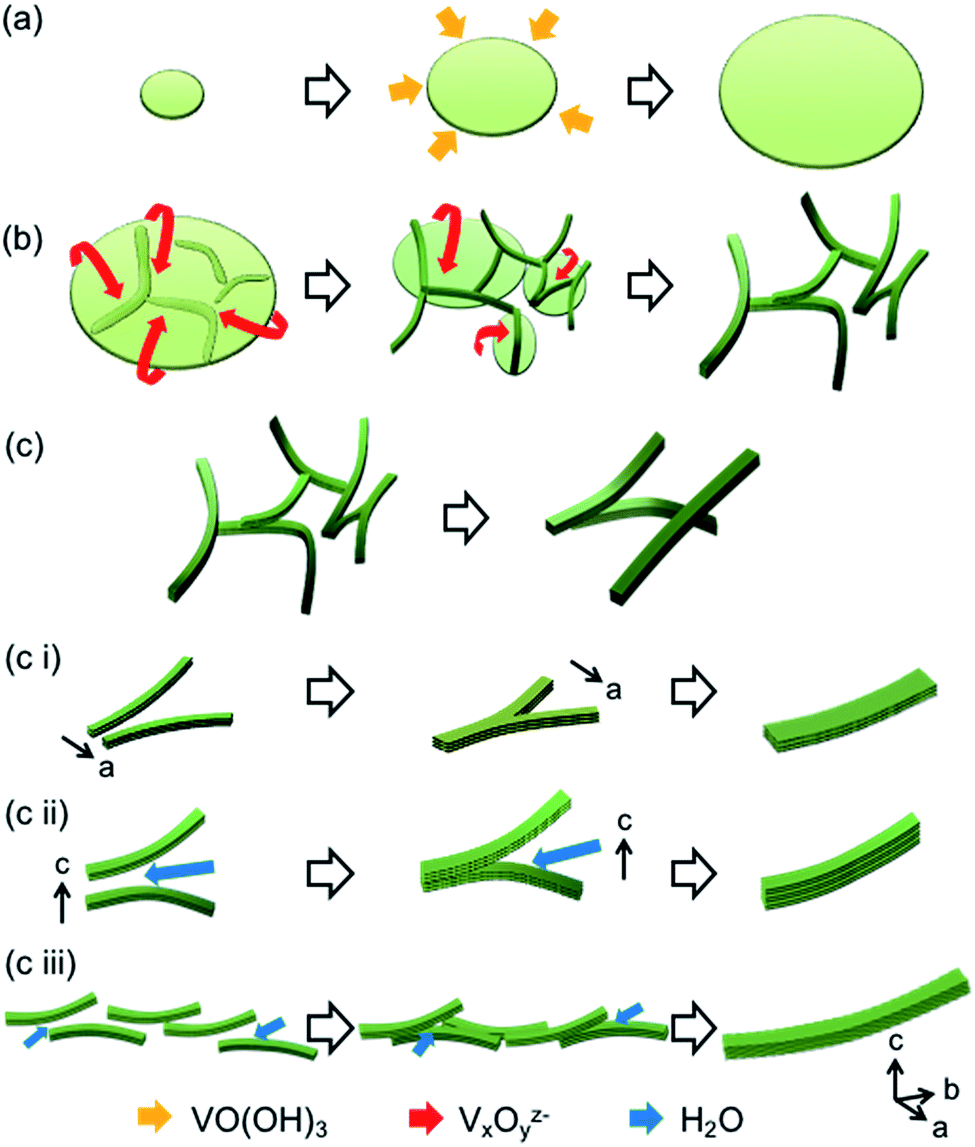 | ||
| Fig. 4 Schematic illustrations of the growth mechanism of the V2O5·nH2O gel skeleton. Formation of ultrathin films in the first stage (a), change from ultrathin films to thin fibrils in the second stage (b) and growth of fibrils to nanofibers in the third stage (c). Growth modes in several directions in the third stage (c(i–iii)): side-by-side attachment of fibrils in the a direction by partial dissolution and reprecipitation (c(i)), stacking of V2O5 layers in the c direction with interlayer water molecules (c(ii)), and elongation in the b direction by side-by-side oriented attachment of multiple fibrils (c(iii)). Orange, red, blue arrows indicate movements of VO(OH)3, VxOyz− polyanion, and H2O, respectively. | ||
In the second stage, ultrathin films change into thin fibrils. Because the growth is accelerated by water, this change from ultrathin films to thin fibrils proceeds through the partial dissolution and reprecipitation of ions enhanced by the presence of water (Fig. 4b and S4†). When the pH of the pore liquid was 3–5, V2O5·nH2O dissolved in water as some kinds of polyvanadate anions (VxOyz−).13 The variation of the growth mode from two-dimensional expansion of the thin film to the stacking of V2O5 layers with interlayer water is ascribed to change of the growth units from VO(OH)3 to polyvanadate anions.
The evolution from thin fibrils to nanofibers in the third stage is ascribed to the oriented attachment according to TEM images of V2O5·nH2O xerogels at [VO(OiPr)3]/[H2O] = 1/40 (Fig. 1c(iii and iv)). The acceleration of the evolution by water implies that water assists the oriented attachment. The mechanism of the attachment assisted by water is considered as follows. Side-by-side attachment of the fibrils in the a direction is promoted by partial dissolution and reprecipitation (Fig. 4c(i)). In contrast, for another side-by-side attachment in the c direction, thin fibrils stack with interlayer water molecules (Fig. 4c(ii)). Thin fibrils grow in the b direction by water-assisted side-by-side oriented attachment of multiple fibrils in the direction of both a- and c-axes (Fig. 4c(iii)). The formation of nanofibers with different widths and lengths can be explained using these mechanisms.
Some previous reports have shown the oriented attachment of nanofibrils on the formation of vanadate nanofibers by adding orthorhombic V2O5 powder to a H2O2 aqueous solution21,22 or pouring melted orthorhombic V2O5 into water.23 The former reports showed that side-by-side attachment of nanofibrils occurs on the growth process of V2O5·nH2O under a hydrothermal condition. On the other hand, the elongation mechanism of the fibrous forms has not been explained sufficiently by the side-by-side mode. In the latter report, ultralong nanobelts of anhydrous V2O5 was found to be formed through the oriented attachment of the nanofibrils under a hydrothermal condition. The elongation process of the nanobelts with a staggered side-by-side attachment mode similar to Fig. 4c(iii) was proposed. However, these processes were promoted only at a high temperature above 100 °C. In the present study, we have shown the oriented attachment of V2O5·nH2O thin fibrils under the ambient condition. The characteristic growth mode on the sol–gel process from alkoxides as a vanadium source proceeds easily at room temperature and is assisted by water.
3.3. Fabrication of V2O5·nH2O xerogels having a high specific surface area
Thin fibrils easily produce finest branching structures in the whole growing process. Ultrathin films are relatively likely to stack with each other due to the capillary force of the pore liquid. Basically, nanofibers have a thicker structure than thin fibrils. The structure of gel skeletons was maintained by ambient temperature vacuum drying after replacing the pore liquid with cyclohexane. The key to obtaining V2O5·nH2O xerogels having a high specific surface area is halting the evolution of thin fibrils in the gel skeleton. The specific surface areas of obtained xerogels are listed in Table S1.† When [VO(OiPr)3]/[H2O] = 1/40, the highest surface area of 320 m2 g−1 was obtained with thin fibrils by aging for 2.0 h (Fig. S5†). By aging for 48 h, the xerogel from [VO(OiPr)3]/[H2O] = 1/10 obtains the finest fibril structure, with 4–9 nm width (Fig. 3(ii)), and the higher surface area. The surface area of xerogels obtained by a normal drying method is generally below 10 m2 g−1.20 Aerogels obtained by a supercritical drying method exhibited high specific surface areas of 420–450 m2 g−1.17,19 Cryogels and ambigels were reported to have specific surface areas of 280 m2 g−1 and 210 m2 g−1, respectively.18 This is the first report to obtain V2O5·nH2O xerogels with a specific surface area over 300 m2 g−1 without supercritical drying, on the basis of the growth mechanism of the gel skeleton.3.4. Application of V2O5·nH2O gel skeletons for fabrication of porous polymer
We applied V2O5·nH2O xerogels having a high specific surface area as an oxidizing agent and template for the fabrication of porous polymer frameworks. PPy was synthesized with V2O5·nH2O xerogels ([VO(OiPr)3]/[H2O] = 1/10 after aging for 48 h) as a template. From EDX analysis, almost all vanadium was confirmed to be removed by dissolution of the gel skeleton (Fig. S6†). SEM images and FT-IR spectra of PPy replicas of V2O5·nH2O xerogels are shown in Fig. 5.28 The synthesized PPy reflected the structure of the V2O5·nH2O xerogel when the reaction time was longer than 24 h (Fig. 5a and b). The specific surface area of the PPy replicas of V2O5·nH2O xerogels was 465 m2 g−1 when the reaction time was 24 h. PPy has been mainly synthesized by electropolymerization. The specific surface area of the PPy specimen (Sigma-Aldrich) and PPy chemically polymerized in water (the preparation procedure is described in the ESI†) is about 5 m2 g−1. PPy with a high specific surface area of 90 m2 g−1 is reported by using porous coordination polymers as a template.29 The PPy replicas of V2O5·nH2O xerogels have the highest surface area ever reported. The width of the fibril-like structures of PPy replicas was about 20 nm. The high specific surface area suggests that the fibrils of PPy were hollow. Strong signals of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and OH bonds in the FT-IR spectrum (Fig. 5c) are attributed to the overoxidation reaction. Thus, unfortunately, the electrical conductivity of the PPy replicas would be relatively low due to their poorly conjugated system.30–32
O and OH bonds in the FT-IR spectrum (Fig. 5c) are attributed to the overoxidation reaction. Thus, unfortunately, the electrical conductivity of the PPy replicas would be relatively low due to their poorly conjugated system.30–32
 | ||
| Fig. 5 SEM images of PPy replicas of V2O5·nH2O xerogels prepared by aging for 24 h (a, b). FT-IR spectra of PPy samples (c); a PPy specimen (i) and a PPy replica (ii). The absorption bands (A–E) correspond to the following characteristic vibrations in PPy:27 CO stretching (A), CC stretching (B), CN stretching (C), CH in-plane bending (D), and CH out-of-plane bending (E). | ||
4. Conclusions
The evolution of the V2O5·nH2O gel skeleton was investigated in the sol–gel method using vanadium alkoxide as a precursor. Ultrathin films a few nanometers thick were initially formed and then evolved into nanofibers 10–20 nm wide and >500 nm long through thin fibrils 3–7 nm wide and ∼100 nm long. The evolution of the V2O5·nH2O gel skeleton is ascribed to side-by-side oriented attachment with partial dissolution and reprecipitation enhanced by the presence of water. By adjusting the reaction and the drying condition, V2O5·nH2O gels with a surface area as high as 320 m2 g−1 were obtained under the ambient condition. Highly porous polypyrrole was fabricated using the gel skeleton as an oxidizing agent and template.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Advanced Low Carbon Technology Research and Development Program – Specially Promoted Research for Innovative Next Generation Batteries (ALCA-SPRING) of the Japan Science and Technology Agency (JST).Notes and references
- J. Bullot, O. Gallais, M. Gauthier and J. Livage, Appl. Phys. Lett., 1980, 36, 986 CrossRef CAS.
- S. B. Kristensen, A. J. Kunov-Kruse, A. Riisager, S. B. Rasmussen and R. Fehrmann, J. Catal., 2011, 284, 60 CrossRef CAS.
- M. G. Kanatzidis, C. Wu, H. O. Marcy and C. R. Kannewurf, J. Am. Chem. Soc., 1989, 111, 4139 CrossRef CAS.
- S. Kittaka, H. Yamamoto, S. Higuma and T. Sasaki, J. Chem. Soc., Faraday Trans., 1992, 88, 715 RSC.
- Y. Fujita, K. Miyazaki and C. Tatsuyama, Jpn. J. Appl. Phys., 1985, 24, 1082 CrossRef CAS.
- X. Huang, X. Rui, H. H. Hng and Q. Yan, Part. Part. Syst. Charact., 2015, 32, 276 CrossRef CAS.
- J. S. Sakamoto and B. Dunn, J. Electrochem. Soc., 2002, 149, A26 CrossRef CAS.
- H. Li, P. He, Y. Wang, E. Hosono and H. Zhou, J. Mater. Chem., 2011, 21, 10999 RSC.
- G. Gershinsky, H. D. Yoo, Y. Gofer and D. Aurbach, Langmuir, 2013, 29, 10964 CrossRef CAS PubMed.
- I. Shterenberg, M. Salama, Y. Gofer, E. Levi and D. Aurbach, MRS Bull., 2014, 39, 453 CrossRef CAS.
- G. G. Amatucci, F. Badway, A. Singhal, B. Beaudoin, G. Skandan, T. Bowmer, I. Plitz, N. Pereira, T. Chapman and R. Jaworski, J. Electrochem. Soc., 2001, 148, A940 CrossRef CAS.
- P. E. Tang, J. S. Sakamoto, E. Baudrin and B. Dunn, J. Non-Cryst. Solids, 2004, 350, 67 CrossRef CAS.
- J. Livage, Chem. Mater., 1991, 3, 578 CrossRef CAS.
- V. Petkov, P. N. Trikalitis, E. S. Bozin, S. J. L. Billinge, T. Vogt and M. G. Kanatzidis, J. Am. Chem. Soc., 2002, 124, 10157 CrossRef CAS PubMed.
- M. Hibino, M. Ugaji, A. Kishimoto and T. Kudo, Solid State Ionics, 1995, 79, 239 CrossRef CAS.
- J. K. Bailey, G. A. Pozarnsky and M. L. Mecartney, J. Mater. Res., 1992, 7, 2530 CrossRef CAS.
- D. B. Le, S. Passerini, J. Guo, J. Ressler, B. B. Owens and W. H. Smyrl, J. Electrochem. Soc., 1996, 143, 2099 CrossRef CAS.
- G. Sudant, E. Baudrin, B. Dunn and J.-M. Tarascon, J. Electrochem. Soc., 2004, 151, A666 CrossRef CAS.
- F. Chaput, B. Dunn, P. Fuqua and K. Salloux, J. Non-Cryst. Solids, 1995, 188, 11 CrossRef CAS.
- V. Augustyn and B. Dunn, C. R. Chim., 2010, 13, 130 CrossRef CAS.
- W. Avansi, C. Ribeiro, E. R. Leite and V. R. Mastelaro, J. Cryst. Growth, 2010, 312, 3555 CrossRef CAS.
- W. Avansi, C. L. P. Oliveira, C. Ribeiro, E. R. Leite and V. R. Mastelaro, CrystEngComm, 2016, 18, 7636 RSC.
- M. Li, F. Kong, H. Wang and G. Li, CrystEngComm, 2011, 13, 5317 RSC.
- K. Kuwabara, Y. Oaki, R. Muramatsu and H. Imai, Chem. Commun., 2015, 51, 9698 RSC.
- S. V. Balakhonov, S. Z. Vatsadze and B. R. Churagulov, Russ. J. Inorg. Chem., 2015, 60, 9 CrossRef CAS.
- N. Hüsing and U. Schubert, Angew. Chem., Int. Ed., 1998, 37, 22 CrossRef.
- E. A. Meulenkamp, J. Phys. Chem. B, 1998, 102, 5566 CrossRef CAS.
- K. Majid, R. Tabassum, A. F. Shah, S. Ahmad and M. L. Singla, J. Mater. Sci.: Mater. Electron., 2009, 20, 958 CrossRef CAS.
- T. Uemura, Y. Kadowaki, N. Yanai and S. Kitagawa, Chem. Mater., 2009, 21, 4096 CrossRef CAS.
- H. Ge, G. Qi, E.-T. Kang and K. G. Neoh, Polymer, 1994, 35, 504 CrossRef CAS.
- S. Ghosh, G. A. Bowmaker, R. P. Cooney and J. M. Seakins, Synth. Met., 1998, 95, 63 CrossRef CAS.
- I. Rodríguez, B. R. Scharifker and J. Mostany, J. Electroanal. Chem., 2000, 491, 117 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06850f |
| This journal is © The Royal Society of Chemistry 2017 |