
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRemarkable enhancement of Fe–V–Ox composite metal oxide to gold catalyst for CO oxidation in the simulated atmosphere of CO2 laser†
Qingquan Lin‡
 a,
Chun Han‡a,
Huijuan Sua,
Libo Suna,
Tamao Ishidab,
Tetsuo Honmac,
Xun Suna,
Yuhua Zhenga and
Caixia Qi*a
a,
Chun Han‡a,
Huijuan Sua,
Libo Suna,
Tamao Ishidab,
Tetsuo Honmac,
Xun Suna,
Yuhua Zhenga and
Caixia Qi*a
aShandong Applied Research Center of Nanogold Technology(Au-SDARC), School of Chemistry & Chemical Engineering, Yantai University, Yantai 264005, China. E-mail: Qicx@ytu.edu.cn; Fax: +86 535 6911732; Tel: +86 535 6911732
bResearch Center for Gold Chemistry, Graduate School of Urban Environmental Sciences, Tokyo Metropolitan University, Tokyo 192-0397, Japan
cJapan Synchrotron Radiation Research Institute (JASRI)/SPring-8, Hyogo, 679-5198, Japan
First published on 8th August 2017
Abstract
A novel Au/Fe–V–Ox/Al2O3 catalyst displayed remarkable activity and good stability for CO oxidation in the simulated atmosphere of a CO2 laser with high CO2 levels up to 60 vol% and stoichiometric or even lower O2/CO ratio. The improved surface labile lattice oxygen on the FeOx and VOx modified Al2O3 plays a crucial role in its outstanding performance for the CO oxidation reaction in an oxygen-poor atmosphere.
The carbon dioxide laser (CO2 laser) which can be called the “invisible man”, emitting a laser with a wavelength of 10.6 μm in the infrared “body”, has become one of the most efficient and powerful lasers and has been widely used in laser cutting, welding, drilling and surface treatment. For CO2 lasers, a persistent problem is the decomposition of CO2 molecules in the discharge,1–3 accompanied by a host of gas products, such as CO, O2, O, O3, NO, and N2O.4 With the increase of these species, especially CO as the main composition, the CO2 laser will lose its output power or is even unable to work.5,6
The catalytic oxidation of CO over the internal solid catalysts is a way to expand the life of sealed CO2 laser via recombining CO and O2. The main challenges for the catalyst are to work at the conditions with high CO2 level and stoichiometric or even less O2/CO ratio since some oxygen formed by CO2 dissociation is probably consumed in other reactions. Some supported noble metal catalysts, such as Pt/SnO2, Pt/MnO, Pd/SnO2, Rh/SnO2, Ru/MnO2, have been investigated as promising catalysts for use in sealed CO2 lasers. Among them, SnO2 supported Pt and Pd catalysts showed the best performance. However, their deactivation with time was continuously observed in the process.7
Compared with supported Pt, Pd, Ru and Rh nanoparticles, supported gold nanoparticles, well-known for their excellent catalytic performance towards many heterogeneous catalytic reactions, have exhibited unexpectedly higher catalytic activity for CO oxidation, especially at lower temperatures.8 This brings a possibility for the Au catalysts to serve in CO2 laser and a number of works have shown some positive results. An increase in output power of a CO2 laser was observed after gold was coated on the inside wall of a discharge tube in the presence of a discharge.9 Better activity and less deactivation of CO oxidation in CO2 laser over Au/MnO2 than Pt/SnO2 were reported by Upchurch et al.10 A two-week constant output power of 10 W was observed in a transversely excited atmospheric pressure CO2 laser (TEA CO2 laser) when the Au/Fe2O3 catalyst was applied in its external body.11 An even long-time life, 5 month's constant output power was obtained in a compact low-power (4 W) sealed CO2 laser by incorporating a supported gold catalyst which was coated on outer jacket.12
Due to a large amount of CO2 in the reaction feed gases, acidic supports are likely to be more durable, which could be helpful to maintain the activity of the catalysts during the reaction, for less carbonate species formatted on the surface of the support.13 Alumina is one of those kinds of suitable supports, owing to its neutral property, thermal stability, mechanical strength, etc.14 Our laboratory has prepared a series of very active and stable Au catalysts which are supported by metal oxides modified Al2O3 for the CO oxidation in oxygen-rich atmosphere.15–17 The output power of 160 W in a radio frequency excited CO2 laser (RF CO2 laser) can be maintained for more than two weeks via a continuous usage18 and for 4 months with a day-time operation mode by inputting one of our alumina-based Au catalysts. The output power (24 W) of a 600 mm long sealed CO2 laser can be maintained for about 4.5 months when our coated Au catalysts working on the inside wall of a discharge tube with day-time operation model too (unpublished data). Au catalysts are so obviously to deserve an in-depth study in the atmosphere of CO2 laser which features in high level of CO2 and low O2/CO ratio. An attempt has been made to clarify the working mechanism of Au catalysts in this specific situation and hope to finally meet the need from industries to provide a qualified catalyst for CO2 laser with more than one year life.
In order to improve and maintain the catalytic activities for CO oxidation under lean or rich conditions, lots of studies have done on oxygen storage materials (OSM), e.g., Prof. M. S. Hegde developed the CeO2 or SnO2 based OSM supported catalysts19,20 using their own creative solution combustion method.21 They found that the high rates of CO oxidation by O2 over Pd ion substituted OSM are shown to be due to highly labile lattice oxygen and the oxide ion vacancies. Similarly, the main point for supported gold nanoparticles to be more efficient relies in the adsorption, activation rate of O2 and its reaction rate with CO during the reaction. For iron and vanadium oxides possess a very rich redox behavior associated with its various oxidation states (Fe3+, Fe2+, and V5+, V4+, V3+, V2+), the employment of these oxides to Al2O3 could highly increase the surface oxygen species of the catalyst. In this communication, we successfully developed for the first time FeOx and VOx modified γ-Al2O3 (denoted as Fe–V–Ox/Al2O3) supported Au catalysts, which were not only much more active in the simulated atmosphere of CO2 laser than Au/Al2O3 but also stable. Moreover, further studies revealed that compared with Au/Al2O3, the higher dispersion of Au nanoparticles and the increased concentration of surface oxygen on the support caused by the modification of FeOx and VOx accounted for their good performance. A comparison with the performance of Au/Al2O3 was made simultaneously in order to shed a light on the catalyst design for the application in CO2 laser.
The Fe–V–Ox/Al2O3 support was prepared by a two step of incipient wetness impregnation of γ-Al2O3 with Fe(NO3)3 and NH4VO3 solution, respectively, and followed by calcination at 600 °C for 4 h. Au targeted at 1 wt% was deposited on both γ-Al2O3 and Fe–V–Ox/Al2O3 supports using the deposition precipitation method as reported previously16 (details see ESI†). The resultant Au/γ-Al2O3 and Au/Fe–V–Ox/Al2O3 catalysts have actual Au loading of 0.77 wt% and 0.23 wt% (Table S1†), respectively, which were both lower than the target value due to gold loss in adsorption process.
Fig. 1a illustrates the profiles of CO conversions as a function of reaction temperature over Au/Al2O3 and Au/Fe–V–Ox/Al2O3 catalysts at a high space velocity (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL gcat−1 h−1). Considering the obvious difference of the two curves, a significant support effect is observed. The Au/Al2O3 catalyst, with 0.77 wt% gold loading, gave a CO conversion of 52% at 60 °C and the CO conversion increased slowly with elevating the reaction temperature. Even though the temperature was elevated at 240 °C and reached 300 °C, the CO conversion over Au/Al2O3 catalyst was still lower than 75%, increased only c.a. 20%. However, for Au/Fe–V–Ox/Al2O3 catalyst, although its gold loading was only 0.23 wt%, the CO conversion was another story, unexpectedly, it was maintained at 100% from 60 °C to 300 °C in the simulated atmosphere of CO2 laser.
000 mL gcat−1 h−1). Considering the obvious difference of the two curves, a significant support effect is observed. The Au/Al2O3 catalyst, with 0.77 wt% gold loading, gave a CO conversion of 52% at 60 °C and the CO conversion increased slowly with elevating the reaction temperature. Even though the temperature was elevated at 240 °C and reached 300 °C, the CO conversion over Au/Al2O3 catalyst was still lower than 75%, increased only c.a. 20%. However, for Au/Fe–V–Ox/Al2O3 catalyst, although its gold loading was only 0.23 wt%, the CO conversion was another story, unexpectedly, it was maintained at 100% from 60 °C to 300 °C in the simulated atmosphere of CO2 laser.
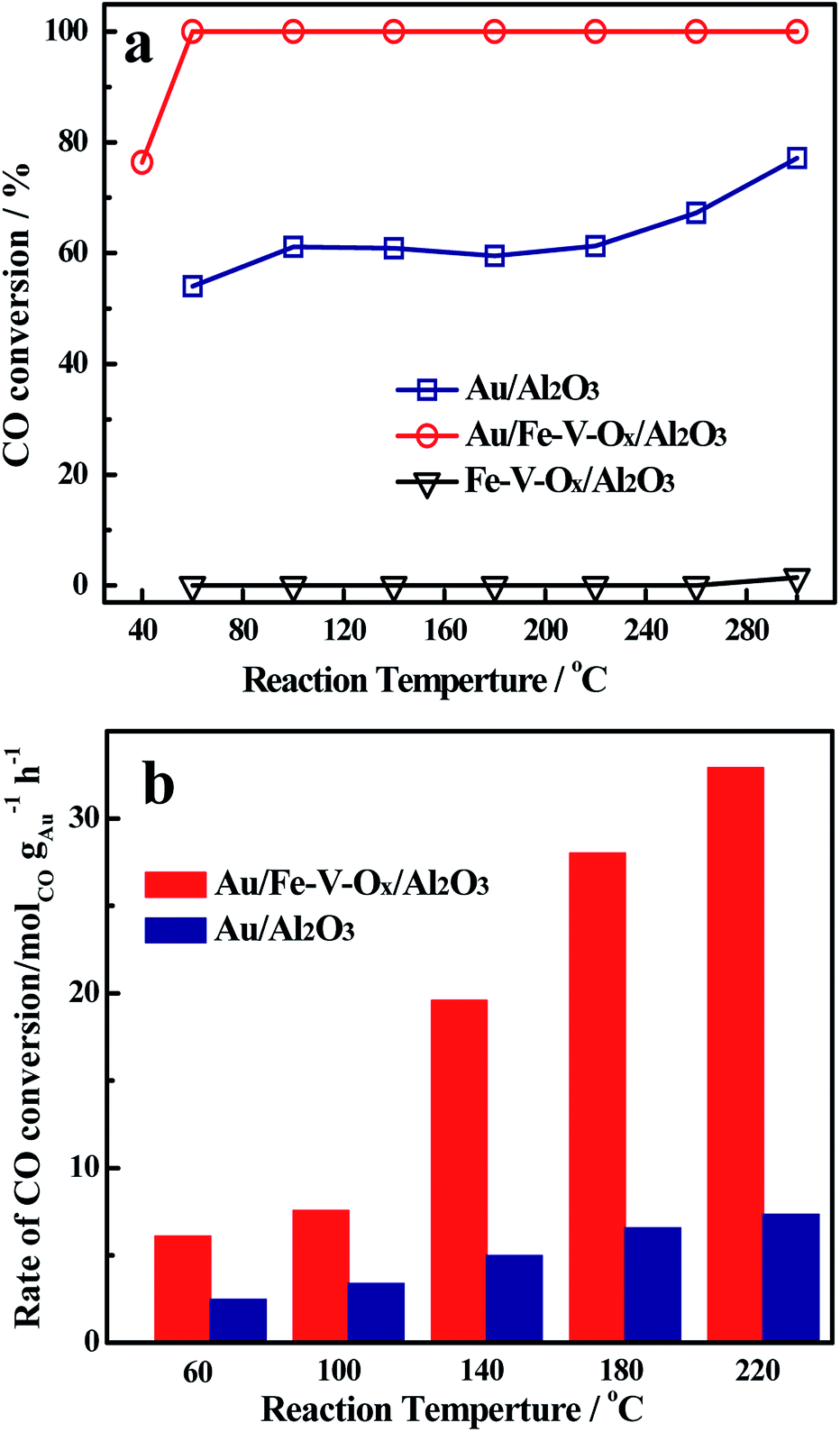 | ||
| Fig. 1 (a) CO conversions as a function of the reaction temperatures over Au/Al2O3 and Au/Fe–V–Ox/Al2O3 catalyst in the simulated atmospheres of CO2 laser and (b) their specific rates at different temperatures. Reaction conditions of (a): 60 vol% CO2 + 1 vol% CO + 0.5 vol% O2 + 0.5 vol% H2O and balance N2. Weight hourly space velocity (WHSV): 60000 mL gcat−1 h−1. | ||
In order to further explore the intrinsic catalytic activity, the specific rates at different temperatures of the investigated catalysts were also evaluated (Fig. 1b). The reaction rates over Au/Fe–V–Ox/Al2O3 catalysts were calculated to be 6.2 and 7.4 molCO gAu−1 h−1 at 60 °C and 100 °C, respectively: c.a. 2.5 times higher than the results of Au/Al2O3 (2.5 and 3.1 molCO gAu−1 h−1). When the temperatures were elevated to 140, 180, and 220 °C, the more striking observation is that the reaction rate of Au/Fe–V–Ox/Al2O3 catalyst reached 19.8, 27.9, and 32.9 molCO gAu−1 h−1, respectively, which were nearly four times as high as those of Au/Al2O3 (5.1, 6.8, and 7.2 molCO gAu−1 h−1).
It is interesting that in the feed of 1% CO in air, 0.1 g of Au/Al2O3 can completely convert 1% CO at −11 °C with a space velocity of 60, 000 mL gcat−1 h−1, at where only the conversion of CO over same amount of Au/Fe–V–Ox/Al2O3 is 27% (Fig. S1†). The reaction rates at this temperature for Au/Al2O3 and Au/Fe–V–Ox/Al2O3 are 3.41 and 3.96 molCO gAu−1 h−1, respectively. This means that the modified catalyst is slightly active than unmodified one in oxygen-rich atmosphere, but much more active in CO2-rich (or oxygen-poor) atmosphere.
In addition, the Au/Fe–V–Ox/Al2O3 catalyst presented similar stability to Au/Al2O3 with time on stream of the simulated atmosphere of CO2 laser at space velocity of 154, 200 mL gcat−1 h−1, only minor activity loss was observed at the beginning of the test and then the CO conversion reached a plateau (Fig. S2†). Furthermore, we chose a well-known active and stable gold catalyst 1 wt% Au/Fe2O3–HGI and 1 wt% Au/MnO2–HGI provided by Haruta Gold Incorporation and prepared by M. Haruta's laboratory to benchmark and compare with our catalysts. As shown in Fig. S2 (ESI†), even at a high space velocity of 154, 200 mL gcat−1 h−1, the CO conversion at 220 °C over our Au/Al2O3 and Au/Fe–V–Ox/Al2O3 was higher than that of 1 wt% Au/Fe2O3–HGI and 1 wt% Au/MnO2–HGI tested at a low space velocity of 120, 000 mL gcat−1 h−1, and the Au/Fe–V–Ox/Al2O3 catalyst performed much more active during the 750 min's long time test.
To reveal the unique properties of Au/Fe–V–Ox/Al2O3 catalyst for CO oxidation in the simulated atmosphere of CO2 laser, further studies were performed to discuss the influence of the modification of FeOx and VOx on the structure of the Au active sites. The Au L3-edge X-ray absorption near-edge structure spectra (Fig. S3†) indicated that the chemical state of gold nanoparticles in Au/Fe–V–Ox/Al2O3 was mainly metallic Au, which is the same as the chemical state of Au/Al2O3 prepared by a same manner as reported previously,14 so we could know that the increased activity was not ascribed to the change of the chemical state of Au.
There was no Au diffraction peaks observed in the XRD patterns of both Au/Al2O3 and Au/Fe–V–Ox/Al2O3 presented in Fig. S4,† implying the presence of Au nanoparticles smaller than 2 nm in a diameter as major species. This point can be verified from the typical images HAADF-STEM images (Fig. 2a and b, S5 and S6†) and their Au nanoparticle size distributions (Fig. 2c and d). For Au/Al2O3 catalysts, most of the Au nanoparticles were dispersed well on the surface of Al2O3 (Fig. 2a), but severely sintered Au nanoparticles larger than 10 nm were observed (Fig. S5a and b†). But we could not find any visible large Au nanoparticles for Au/Fe–V–Ox/Al2O3 catalyst despite our careful observations (Fig. 2b, S6a and b†), indicating that the Au nanoparticles deposited on the surface of Fe–V–Ox/Al2O3 support were fairly uniform than those of the Au/Al2O3 catalysts. Fig. 2c and d also clearly showed that the particle size distributions of Au/Fe–V–Ox/Al2O3 catalysts was much more uniform with a mean particle size of 2.0 ± 0.7 nm. These results demonstrated that the modification of FeOx and VOx on the surface of Al2O3 supports could stabilize small Au nanoparticles during the preparation process. The FeVO4 identities formed on alumina surface due to the modification of FeOx and VOx, which is evidenced by the Raman peak at 804 cm−1 in Fig. S7,† probably play certain role in hindering aggregation of small Au nanoparticles.
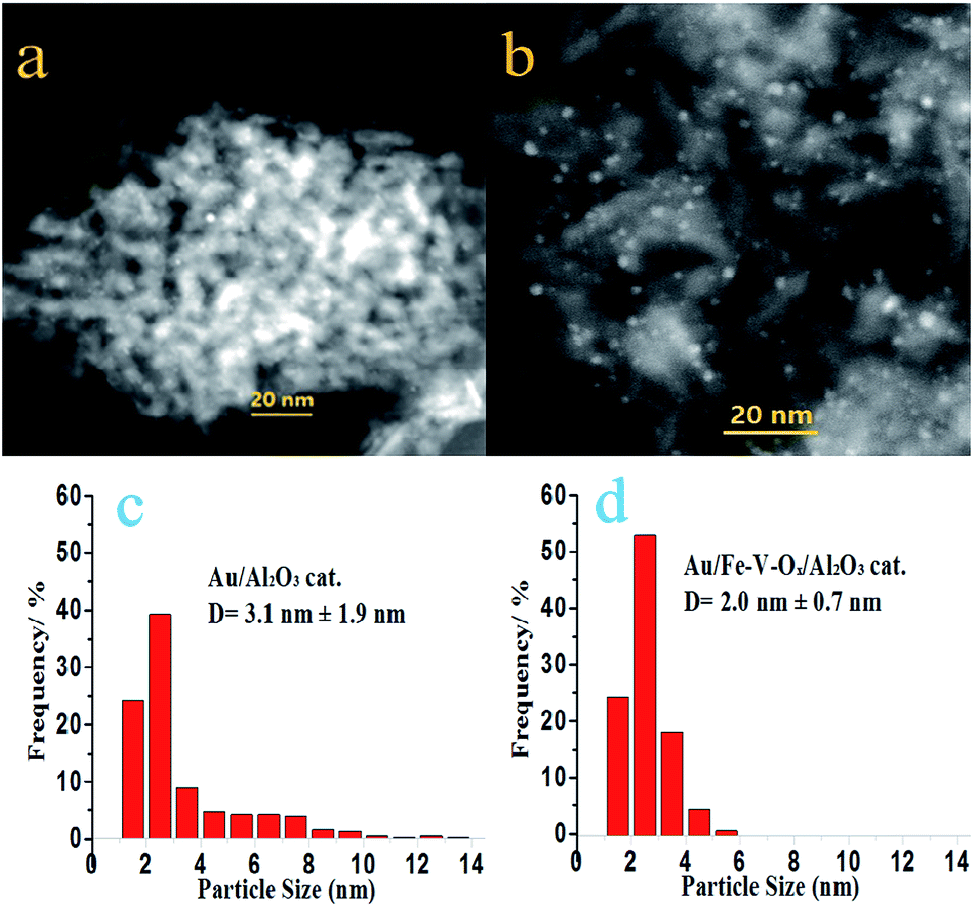 | ||
| Fig. 2 HAADF-STEM images of Au/Al2O3 (a) and Au/Fe–V–Ox/Al2O3 (b) and their Au particle size distributions (c and d). | ||
To ascertain the influence of the chemical properties of the supports on their catalytic activity, we further focused on the concentration of surface oxygen species, which has pronounced effect for CO oxidation, through the temperature-programmed desorption of oxygen (O2-TPD) over Au/Al2O3 and Au/Fe–V–Ox/Al2O3 catalysts. Generally, the desorption of the adsorbed oxygen was activated via the following procedures: O2 (adsorbed) → O2− (adsorbed) → O− (adsorbed) → O2− (lattice).22 During the characterization of O2-TPD, if the more adsorbed oxygen species be desorbed from surface of the catalyst at low temperatures, it indicates the more labile lattice oxygen could be attend the CO oxidation in the range of reaction temperatures we tested. By means of the O2-TPD analysis of the Au/Al2O3 and Au/Fe–V–Ox/Al2O3 catalysts, two different desorption peaks of oxygen species were observed in Fig. 3 and could be distinguished by their desorption temperatures as reported in the literature.23 For both of Au/Al2O3 and Au/Fe–V–Ox/Al2O3 catalysts, the peaks at low temperatures are ascribed to the desorbed oxygen species, which are weakly chemisorbed surface oxygen, and the peaks at the temperatures higher than the 400 °C are attributed to the release of oxygen in the lattice of the supports, which is hard to be reacted and the difference in the peaks at higher temperature for two catalysts is somehow week. In contrast, we can obviously see that the weakly chemisorbed surface oxygen species of Au/Fe–V–Ox/Al2O3 catalyst desorbed at the temperature of 231 °C, which was 22 °C lower than that of Au/Al2O3 catalyst (253 °C). Its peak area which represents the amount of the chemisorbed surface oxygen species is much larger. It suggests that the modification of FeOx and VOx on the surface of Al2O3 support truly increases the concentration of the chemisorbed surface oxygen (labile lattice oxygen), which could highly promote the oxidation of CO to CO2 with the labile lattice oxygen on the support similar as reported in the literature before.24,25 This point was further confirmed by the higher formation rate of CO2 on Au/Fe–V–Ox/Al2O3 than that of Au/Al2O3 at 200 °C in the atmosphere of 97%CO in N2 (Fig. S8†). The oxidation of CO could only happen with the chemisorbed oxygen of the supports because of no or very little O2 in the feed gas. Based on the above analysis, we can conclude that after modified by FeOx and VOx, even though its total content is only 2 wt%, the concentration of the labile lattice oxygen increase and thereby its catalytic activity can be markedly improved for the oxidation of CO in the simulated atmosphere of CO2 laser.
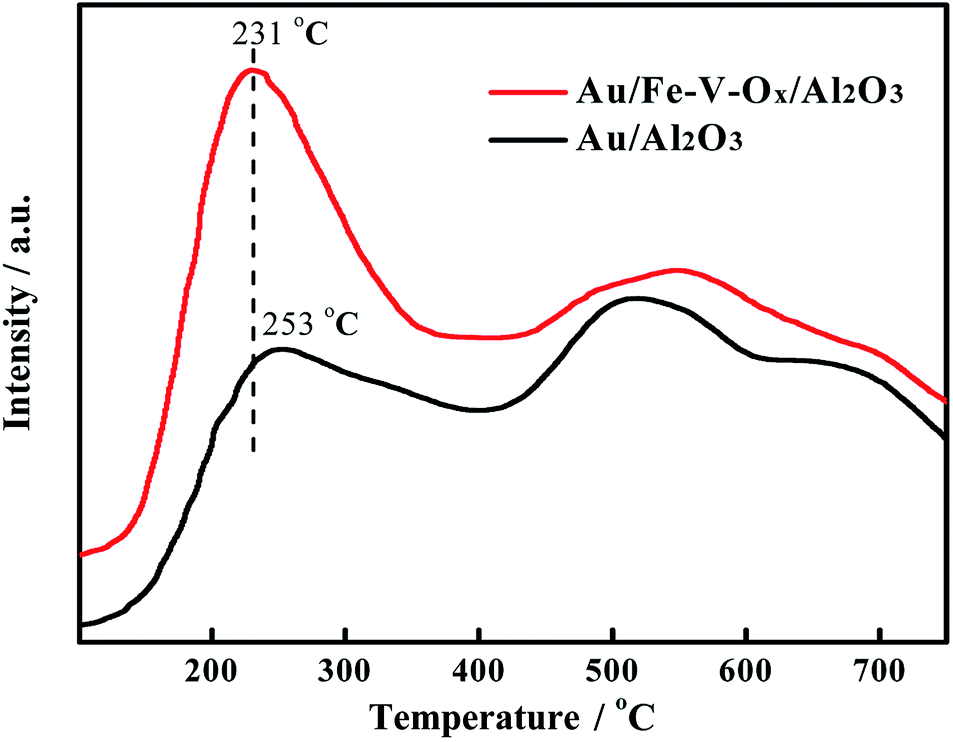 | ||
| Fig. 3 O2-TPD profiles of the Au/Fe–V–Ox/Al2O3 and Au/Al2O3 catalysts. | ||
In conclusion, we reported a highly active and stable Au/Fe–V–Ox/Al2O3 catalyst for CO oxidation in the simulated atmosphere of CO2 laser. The improved labile lattice oxygen caused by the modification of FeOx and VOx on the surface of Al2O3 is of great importance for the CO oxidation at the conditions with low O2/CO ratio.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21606189, 21603181) and Shandong Provincial Natural Science Foundation, China (ZR2015BQ011, ZR2015BM006). The synchrotron radiation experiments were performed at the BL14B2 in the SPring-8 with the approval of the JASRI (2016A1521).Notes and references
- P. K. Cheo and H. G. Cooper, IEEE J. Quantum Electron., 1967, QE3, 79 CrossRef.
- V. S. Aleinikov, V. K. Sysoev and Y. F. Bondarenko, Soviet Journal of Quantum Electronics, 1979, 9(10), 2160 CrossRef.
- D. J. Biswas and U. K. Chatterjee, IEEE J. Quantum Electron., 1981, 17, 1964 CrossRef.
- K. Smith and R. M. Thomson, Computer Modeling of Gas Lasers, Plenum press, New York, 1978, p. 211 Search PubMed.
- A. L. S. Smith, T. H. BETT and P. G. Browne, IEEE J. Quantum Electron., 1975, QE-11, 335 CrossRef.
- P. Bletzinger, D. A. LaBorde, W. F. Bailey, W. H. Long Jr, P. D. Tannen and A. Garscadden, IEEE J. Quantum Electron., 1974, QE11, 314 Search PubMed.
- S. D. Gardn, G. B. Hoflund, D. R. Schryer, J. Schryer and D. R. Brown, NASA Conf. Publ., 1989, 3076, 123 Search PubMed.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301 CrossRef CAS.
- J. A. Macken, S. K. Yagnik and M. A. Samis, IEEE J. Quantum Electron., 1989, 25, 1695 CrossRef CAS.
- B. T. Upchurch, D. R. Schryer, K. G. Brown, E. J. Kielin, G. B. Hoflund and S. D. Gardner, Proc. SPIE, 1991, 1416, 21 CrossRef CAS.
- A. K. Tripathin, Indian J. Technol., 1992, 30(2), 107 Search PubMed.
- A. K. Tripathi, N. M Gupta, U. Chatterjee and D. Bhawalkar, Rev. Sci. Instrum., 1995, 65(12), 3853 CrossRef.
- G. C. Bond and D. T. Thompson, Catal. Rev.: Sci. Eng., 1999, 41, 319 CAS.
- Q. Lin, B. Qiao, Y. Huang, L. Li, J. Lin, X. Liu, A. Wang, W. Li and T. Zhang, Chem. Commun., 2014, 50, 2721 RSC.
- C. Qi, S. Zhu, H. Su, H. Lin and R. Guan, Appl. Catal., B, 2013, 138–139, 104 CrossRef CAS.
- X. Sun, H. Su, Q. Lin, C. Han, Y. Zheng, L. Sun and C. Qi, Appl. Catal., A, 2016, 527(1), 19 CAS.
- M. Haruta, Chin. J. Catal., 2016, 37(9), 1441 CrossRef.
- Z. Weng, C. Qi, S. Shou, H. Su, W. Song, R. Gui, L. Song, X. Sun, Y. Zheng, H. Lin and L. An, CN Patent 103623842 A, 2014-03-12.
- M. S. Hegde, G. Madras and K. C. Patil, Acc. Chem. Res., 2009, 42(6), 704 CrossRef CAS PubMed.
- T. Baidya, A. Gupta, P. A. Deshpandey, G. Madras and M. S. Hegde, J. Phys. Chem. C, 2009, 113, 4059 CAS.
- P. Bera, K. C. Patil, V. Jayaram, M. S. Hegde and G. N. Subbanna, J. Mater. Chem., 1999, 9, 1801 RSC.
- L. Zhang, L. Dong, W. Yu, L. Liu, Y. Deng, B. Liu, H. Wan, F. Gao, K. Sun and L. Dong, J. Colloid Interface Sci., 2011, 355, 464 CrossRef CAS PubMed.
- L. Luo, G. Shao and Z. Duan, Turk. J. Chem., 2005, 29(6), 597 CAS.
- A. Gupta, A. Kumar, U. V. Waghmare and M. S. Hegde, Chem. Mater., 2009, 21(20), 4880 CrossRef CAS.
- P. Singh and M. S. Hegde, Dalton Trans., 2010, 39, 10768 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: The characterization data of XRD, HAADF-STEM, XANES and Raman spectra. See DOI: 10.1039/c7ra06826c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |