
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modeling alveolar injury using microfluidic co-cultures for monitoring bleomycin-induced epithelial/fibroblastic cross-talk disorder†
Jiarui Hea,
Weixing Chenb,
Shijie Dengc,
Lan Xieb,
Juan Fengb,
Jing Gengd,
Dingyuan Jiangd,
Huaping Dai*d and
Chen Wang *ad
*ad
aBeijing Key Laboratory of Respiratory and Pulmonary Circulation Disorders, Beijing Chao-Yang Hospital-Beijing Institute of Respiratory Medicine, Capital Medical University, Beijing, P. R. China. E-mail: cyh-birm@263.net
bMedical Systems Biology Research Center, School of Medicine, Tsinghua University, Beijing, P. R. China
cState Key Laboratory of Precision Measurement Technology and Instruments, Tsinghua University, Beijing, P. R. China
dDepartment of Pulmonary and Critical Care Medicine, Center for Respiratory Diseases, China-Japan Friendship Hospital, National Clinical Research Center for Respiratory Diseases, Beijing, China. E-mail: daihuaping@ccmu.edu.cn
First published on 4th September 2017
Abstract
Epithelial/fibroblastic cross-talk is a significant disorder leading to pulmonary fibrosis, but its pathogenesis remains unclear because the current in vitro and in vivo models do not allow for optimal visualization of the complex disease processes at the human lung epithelial–interstitial interface. In this study we aimed to develop a microfluidic co-culture device that mimics a human disease model of pulmonary fibrosis. Our microfluidic device consists of two parallel side channels separated by a middle channel, recapitulating the physiological microenvironment of cell growth and the native microenvironment of the epithelial–interstitial interface in the human lung. We designed this device to represent bleomycin (BLM)-induced pulmonary fibrosis in human lungs. We recorded fibroblast and inflammatory cell migration in real-time and showed that epithelial cells became apoptotic or developed morphological changes in response to BLM. In summary, we established a co-culture device that models the native microenvironment of the lung epithelial–interstitial interface. Our data provide evidence of the role of epithelial cells and fibroblasts in pulmonary fibrosis, suggesting that fibrosis occurs as a result of injury to epithelial cells. Thus, epithelial injury plays an essential role in the pathogenesis of epithelial/fibroblastic cross-talk disorder.
Introduction
Pulmonary fibrosis, particularly idiopathic pulmonary fibrosis (IPF) is a significant lung disease that is associated with a high mortality rate and is increasingly resistant to treatment. It belongs to a heterogeneous group of lung disorders characterized by progressive and irreversible damage to epithelial cells, cell apoptosis, and disruption of basement membrane and gas exchange, leading to destruction of lung architecture and, consequently, death from respiratory failure.1 Epithelial/fibroblastic cross-talk disorder has been hypothesized to be one of the key mechanisms underlying IPF pathogenesis.2,3 The primary events include an initial epithelial cell injury followed by fibroblast activation due to changes in fibroblast phenotype (e.g. transform into a-smooth muscle actin – expressing myofibroblasts that secrete extracellular matrix (ECM) components (e.g. collagen I)), migration and proliferation.4,5 However, limited animal and cellular models exist to support this hypothesis. The current animal models do not provide a direct and real-time evaluation of epithelial/fibroblastic cross-talk, and the current in vitro cellular models do not convincingly represent the microenvironment of epithelial cells and fibroblasts, resulting in limited understanding of this pathological process. In other words, no universal experimental model is available to study epithelial/fibroblastic cross-talk, both temporally and spatially, mainly due to difficulties in generating a well characterized and sustained inducer (e.g. drugs or cytokines) gradient in mimicking the in vivo environment while allowing dynamic cell imaging; moreover, few in vitro lung models report the inclusion of interstitial cells that are essential for sensing environmental changes as well as exerting a crucial role in the pathogenesis of pulmonary.Development of a meaningful disease model that recapitulates the physiological microenvironment of cell grow is necessary to better understand tissue-specific functions and pathological changes arising due to epithelial/fibroblastic cross-talk disorder. Recently, advancement in microscale engineering combined with our ability to culture living cells has resulted in the development of microfluidic devices and biomimetic microsystems.6–16 These devices have brought new opportunities for controlling the cellular microenvironment with high spatiotemporal precision and studying the complex human disease pathogenesis in an organ or tissue-specific context.8,13–16 For example, Huh et al. developed a microdevice containing two closely apposed microchannels separated by a porous and flexible membrane made of PDMS, to reconstitute the alveolar capillary interface of the human lung by culturing human alveolar epithelial cells and human pulmonary microvascular endothelial cells on opposite sides of the membrane, and to further develop this device to be a disease model of pulmonary edema.15 Zhou et al. established a microfluidic device, in which hepatocytes and stellate cells could be 2D cultured in adjacent microfluidic compartments divided by a retractable wall, to recapitulate local concentrations of signaling molecules produced during liver injury.16 However, these cell-based co-culture micro-devices mostly focused on 2D co-cultures, which fail to maintain cell–ECM interactions. For example, epithelial–interstitial interface constitutes of a cavity-like, single layer of epithelium and interstitial cells that are grown within a three-dimensional (3D) ECM, including cell–cell and cell–matrix interactions. Therefore, a in vitro co-culture micro-device that could provide such cavity-like 2D and 3D culture simultaneously (2D–3D co-culture), and maintain cell–cell cross talk and cell–ECM interactions is necessary. Moreover, significant challenges remain with regards to the fabrication and maintenance of biomimetic tissue-interface models in vitro. For example, most of in vitro tissue-interface models are the membrane-embedded co-culture systems,17–22 which is difficult to fabricate and thus have not been tested for scalable applications. As such, a co-culture microdevice with simpler design and fabrication is needed.
The pulmonary fibrosis hypothesis supposes that fibroblasts migration in 3D ECM is an essential process involved with fibrosis formation. Presently, cell migration assay based on 2D substrate has been allowed by various microfluidic devices.23,24 These devices can precisely configure chemoattractant gradients and allow quantitative cell analysis.7,8 However, these approaches are not applicable to the migration analysis of interstitial cells (e.g. fibroblasts and macrophages) migration in 3D substrate. Also some researchers focused on the cell migration assay by fabricating a sandwich chip in vertical direction,25,26 which might bring some challenges caused by gravity during the verification process of the migration paths and the fabrication of the micro-devices. In order to capture the whole migration path, the micro-device must be fabricated thin enough. Whereas for the thin micro-device, the mechanic strength of PDMS might not be strong enough to hold the entire channel, and the top of the channel may collapse to reach the bottom of the channel. Thus, the aim of the present work is to develop a novel microfluidic-based chip that allows real-time observation of interstitial cells horizontally migrating across the 3D ECM without the interference of gravitational force.
In this study, we constructed a simple microfluidic co-culture device that replicates the microstructure and microenvironment of an epithelial–interstitial interface of the human lung and investigated the dynamic changes of cellular phenotypes and behaviors during epithelial/fibroblastic cross-talk disorder. Our in vitro epithelial–interstitial interface is designed in such a way that epithelial cells grow on the surface of the basement membrane to form a 3D epithelial cell-lined cavity-like structure (left channel), more precisely mimic the lining of the alveolar air space, and fibroblasts and macrophage grow within an extracellular matrix (ECM) (right channel), mimic pulmonary mesenchyme. An interface in the middle channel was used to link the left and right channels to monitor cell migration in real time. Therefore, the microfluidic co-culture device consists of three interconnected channels.
Bleomycin (BLM) can cause fatal pulmonary toxicity leading to lung fibrosis, and thus is extensively used to induce fibrogenesis for purposes of studying human pulmonary fibrosis.27–29 We used BLM to induce epithelial injury and then examined the changes of epithelial cells in phenotype expression, viability and properties. We also assess the activation of interstitial cells under pathological conditions by quantifying the number of cell migration and the expression of activation-relative biomarkers (α-SMA and collagen I). And on this basis, we discussed the role of epithelial cells, fibroblasts, and macrophages in BLM-induced epithelial/fibroblastic cross-talk disorder. Our data strongly demonstrate the feasibility of our microfluidic co-culture model to study the pathogenesis of pulmonary fibrosis.
Materials and methods
Design and fabrication of microfluidic co-culture device
The microfluidic co-culture device is illustrated in Fig. 1A. This device was fabricated using polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning, U.S.A.) based on standard soft lithography methods.30 PDMS and a curing agent were mixed at a weight ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Each channel includes an inlet (Φ = 1.5 mm) and an outlet (Φ = 2.0 mm). The two side channels, containing either epithelial cells or interstitial cells, respectively, were designed to be 2000 μm long, 1500 μm wide, and 1000 μm high. The interface (2000 × 1000 × 200 μm) enables cell communication and migration between the two side channels. The device was treated with oxygen plasma to allow the PDMS to bind to the glass surface (inside of a glass-bottom Petri dish; MatTek, Ashland, ME, USA) with or without the plugs, and the thickness of the PDMS device is 5–7 mm (Fig. 1B).
:1. Each channel includes an inlet (Φ = 1.5 mm) and an outlet (Φ = 2.0 mm). The two side channels, containing either epithelial cells or interstitial cells, respectively, were designed to be 2000 μm long, 1500 μm wide, and 1000 μm high. The interface (2000 × 1000 × 200 μm) enables cell communication and migration between the two side channels. The device was treated with oxygen plasma to allow the PDMS to bind to the glass surface (inside of a glass-bottom Petri dish; MatTek, Ashland, ME, USA) with or without the plugs, and the thickness of the PDMS device is 5–7 mm (Fig. 1B).
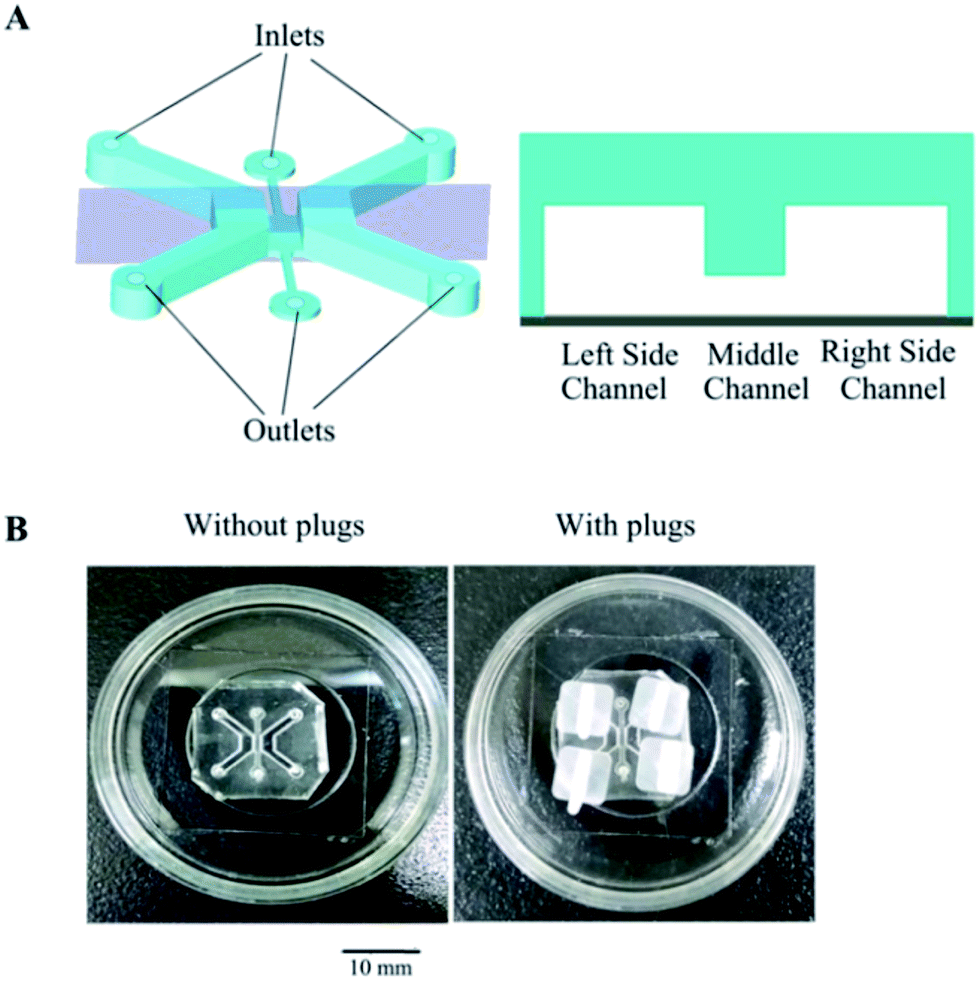 | ||
| Fig. 1 Design and fabrication of microfluidic co-culture device. (A) Illustration of our triple channel design. (B) A typical microfluidic co-culture device after treatment with plasma to allow bonding with a glass-bottom Petri dish (Φ = 35 mm) with or without plugs. Notes: scale bar represents 10 mm (B). | ||
Propagation of epithelial cells and fibroblasts
All materials were purchased from Gibco, USA, unless stated otherwise. The epithelial (A549) and fibroblast (MRC-5) cell lines (ATCC, USA) were routinely cultured at 37 °C and 5% CO2 in RPMI 1640 medium or MEM media, respectively. Both culture media were supplemented with fetal calf serum (FCS) (10% (v/v)), and an antibiotic solution (1% v/v) comprised of penicillin (10000 units per mL) and streptomycin sulfate (100 mg mL−1).
Generation of macrophages
Macrophages were generated from human monocyte line, m cells. Briefly, human monocyte line THP-1 cells (ATCC) were maintained at 2 × 105 cells per mL in RPMI 1640 medium supplemented with 10% FCS and 2 mmol L−1 L-glutamine. THP-1 cells (2 × 105 cells per mL) were differentiated using 200 nM phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich, USA) for 3d. Differentiation of PMA treated cells was enhanced after the initial 3d stimulus by removing the PMA containing media then incubating the cells in fresh RPMI 1640 (10% FCS, 1% L-glutamine) for a further 5d.31Construction of epithelial-interstitial cells co-cultures
The microfluidic device was sterilized under UV light for 30 min before use. To facilitate adhesion of the epithelial cells to the channel walls, the insides of the channels were coated with 100 mg mL−1 fibronection (FN) and subsequently removed after incubating the microfluidic device at room temperature for 60 min. Then a hydrogel containing ECM proteins was prepared at a final concentration of 25% Matrigel (BD Biosciences, Bedford, MA, USA) and 75% RPMI 1640 medium. The hydrogel was then loaded into the middle channel through an inlet, keeping the two side channels closed, and the microfluidic device was incubated at 37 °C for 10 min to allow for complete polymerization of the hydrogel. Subsequently, keeping the left channel closed, a solution consisting of 7.5 μL Matrigel, 7.5 μL RPMI 1640 medium, 7.5 μL MRC-5 cells (3 × 106 cells per mL), and 7.5 μL macrophages cells (2 × 106 cells per mL) was loaded into the right channel and maintained at 37 °C for 15 min until gelation. An epithelial cell solution at 2 × 106 cells per mL was prepared and 30 μL of this solution was added to the left channel coated with FN followed by incubation for 30 min at 37 °C to allow the epithelial cells to attach to the bottom wall of the channel. The cell solution (30 μL) was slowly added to the channel with a pipette followed by rotating the microfluidic device to 90°, incubation for 30 min at 37 °C to allow the epithelial cells to attach to the side wall of the channel, and then perfused with fresh media to wash away unattached cells (Fig. 2). Between cell seeding steps, the cell solution was preserved on ice. Subsequently, the device was righted, incubated for 96 h (37 °C, 5% CO2). The cell culture media composition remained as a 50:50 mixture of A549 and MRC-5 cell media.
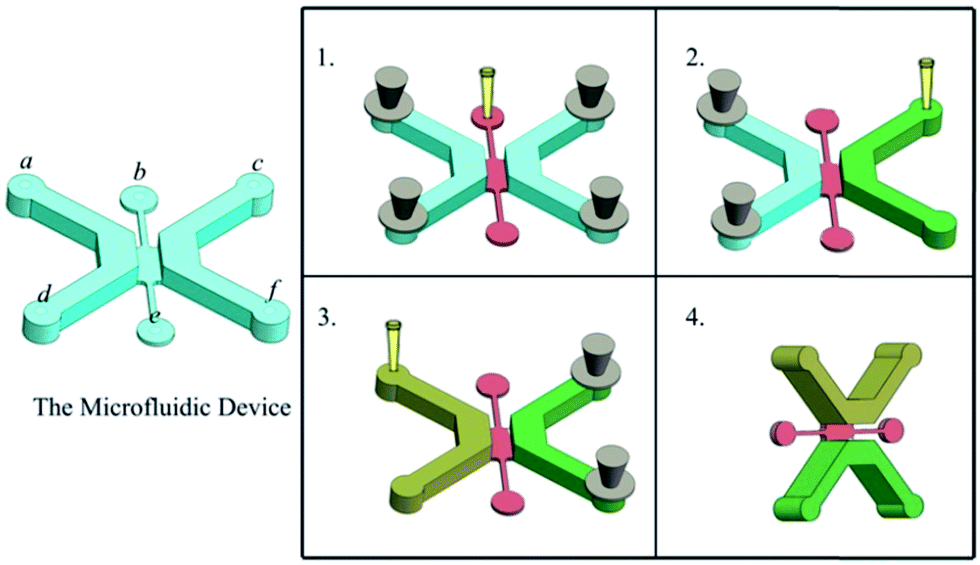 | ||
| Fig. 2 Generation of an alveolar cavity-like structure and culturing of fibroblasts and macrophages within a 3D ECM: (1) plug a, c, d, f, fill the middle channel; (2) open c, f, fill the right channel (the interstitial channel); (3) plug c, f, open a, d, and then fill the left channel (the epithelial channel); (4) rotate the device to 90°, remain the left channel on the up, and line the channel with cells. Notes: a–c and d–f represent the inlets and outlets of the microfluidic device, respectively. | ||
Immunofluorescence staining
All antibodies were purchased from Abcam, USA. E-Cadherin, α-smooth muscle actin (α-SMA), and collagen I expression levels in A549 and MRC-5 cells were measured by immunofluorescence assays. After 96 h of BLM treatment, the cells were fixed in phosphate buffered saline (PBS) supplemented with 4% paraformaldehyde and 0.3% Triton-X100 for 15 min and incubated in 1% bovine serum albumin for 1 h to prevent nonspecific binding. Subsequently, the cells were rinsed with PBS and incubated overnight along with the following primary antibodies: mouse anti-human E-cadherin (no. ab1416), rabbit anti-α-SMA (no. ab5694), and rabbit anti-collagen I (no. ab34710), each at a 1:200 dilution. On the following day, the cells were rinsed with PBS (3 × 5 min) and incubated with an appropriate IgG matched secondary antibody at a dilution of 1:200 for 1 h. Immunofluorescence images were acquired using an A1RSi confocal microscope (Nikon Instruments, Tokyo, Japan).
Cell apoptosis assay
After staining with DAPI (Roche Reinach, Switzerland), nuclei size was measured to determine cell apoptosis.32,33 After treatment with BLM for 48 and 96 h, A549 cells were washed three times with PBS and fixed in 4% paraformaldehyde for 15 min at room temperature. Subsequently, the cells were incubated with DAPI for 15 min at room temperature and photographed using an A1RSi confocal microscope.Migration assay
Before using the epithelial channel in our experiments, we first performed a diffusion study with FITC-dextran (10 kDa) (Sigma, St. Louis, MO, USA). Fluorescence microscopy indicated that FITC-dextran gradually diffused into the hydrogel, spreading toward the right channel after 1 h and forming a stable concentration gradient (ESI Fig. S1†), confirming a stimulus mediated directional cell response towards chemoattractant gradients in the hydrogel.Experiments were performed using cultured cells under normal conditions for 24 h. A gradient was formed by adding 20 μg mL−1 BLM (NIPPON KAYAKU Co, Japan) to the epithelial channel and maintained for a total of 96 h. Under different conditions, MRC-5/macrophages migration were observed by time-lapse microscopy and quantified by cell tracking based on image analysis techniques.
Enzyme linked immunosorbent assay (ELISA)
Tumor growth factor-β1 (TGF-β1) ELISA kits were purchased from Abcam, USA (no. ab100647). All reagents, samples, and standards were prepared by strictly following the manufacturer's instructions. Briefly, the supernatant collected from channels were added to each well. Following incubation, the plate was washed and biotin-conjugated anti-TGF-β1 antibody was added to the appropriate wells. The plate was again incubated and washed followed by the addition of streptavidin HRP conjugated to the appropriate wells. The plate was incubated at room temperature and then washed. The tetramethylbenzidine (TMB) substrate was then added to each well. Finally, a stop solution was added to each well and the plates were immediately read at 450 nm by Microplate Reader (Thermo Scientific, USA).Statistical analysis
All data are expressed as mean ± standard deviation (SD). For immunofluorescence and migration analyses, quantitation was performed using Imaris 8.0 software (Bitplane, Zurich, Switzerland). Statistical data analysis was performed based on Student's t test and ANOVA analysis using SPSS 13.0 software (SPSS, Chicago, IL, USA). A P value < 0.05 was considered statistically significant.Results
Construction of a 3D co-culture microfluidic device
We designed a microfluidic device consisting of three channels. A549 cells, MRC-5 cells, and macrophages were loaded into the left and right channels while the interface was filled with ECM as described earlier (Fig. 2). Interestingly, the A549 cells grew into a 3D cavity and the device facilitated the formation of a cavity-like structure from multiple cells. The right channel, consisting of fibroblasts (MRC-5) and macrophages, recapitulated the in vivo pulmonary interstitium microenvironment. After culturing the three types of cells in the device for 48 hour analysis demonstrated evidence of cell growth within the microfluidic device (Fig. 3). | ||
| Fig. 3 Construction of epithelial–interstitial cell co-cultures. (A) Illustration of epithelial–interstitial cell co-cultures (B) visualization of cells in the co-culture in the microfluidic device after seeding for 48 h, as evidenced after staining with CellTracker Green CMFDA (A549), CellTracker Red CMTPX (MRC-5), and CellTracker Deep Red Dye (Macrophages). Note: scale bar represents 300 μm. The coronal section of the microfluidic device (A and B). Abbreviations 3D, three-dimensional; ECM, extracellular matrix. | ||
Morphology and apoptosis of A549 cells
We observed pathological changes to epithelial cells in the co-culture after treatment with BLM. Treatment with 20 μg mL−1 BLM for 4 days caused time-dependent morphological changes in A549 cells, whereas a control group of A549 cells maintained the classic cobblestone morphology and growth pattern (Fig. 4A). Table 1 reports the changes in area (μm2) and roundness of A549 cells. In agreement with the morphological data, our immunofluorescence results showed an increased expression of α-SMA and a decreased expression of E-cadherin in the BLM-treated group (Fig. 4B–C). Specifically, the apoptosis rate of A549 cells treated with BLM (20 μg mL−1) significantly increased in a time-dependent manner (Fig. 5). | ||
| Fig. 4 BLM-induced morphological changes in epithelial cells. (A) Incubation of co-cultures with 20 μg mL−1 BLM at different time points. (B) Expression of E-cadherin and α-SMA in A549 cells treated with BLM for 96 h. (C) Quantification of fluorescence intensity is shown for the two proteins. Notes: magnification, 100×, scale bar represents 30 μm (A); green: E-cadherin, red: α-SMA, blue: cell nuclei, scale bar represents 200 μm (B); data are plotted as average ± SD from three separate experiments (C). Abbreviations: BLM, bleomycin. | ||
| Sample | Time, h | Area (μm2) | Roundness |
|---|---|---|---|
| a Notes: Imaris software was used to randomly read ∼20 cells per image. Mean values ± SD are presented. (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). | |||
| Control | 24 | 116.90 ± 23.15 | 0.61 ± 0.13 |
| 48 | 109.60 ± 25.92 | 0.61 ± 0.13 | |
| 96 | 94.30 ± 27.07 | 0.64 ± 0.14 | |
| BLM (20 μg mL−1) | 24 | 192.50 ± 25.79* | 0.50 ± 0.11* |
| 48 | 283.00 ± 24.22** | 0.40 ± 0.08** | |
| 96 | 303.00 ± 21.93*** | 0.25 ± 0.12*** | |
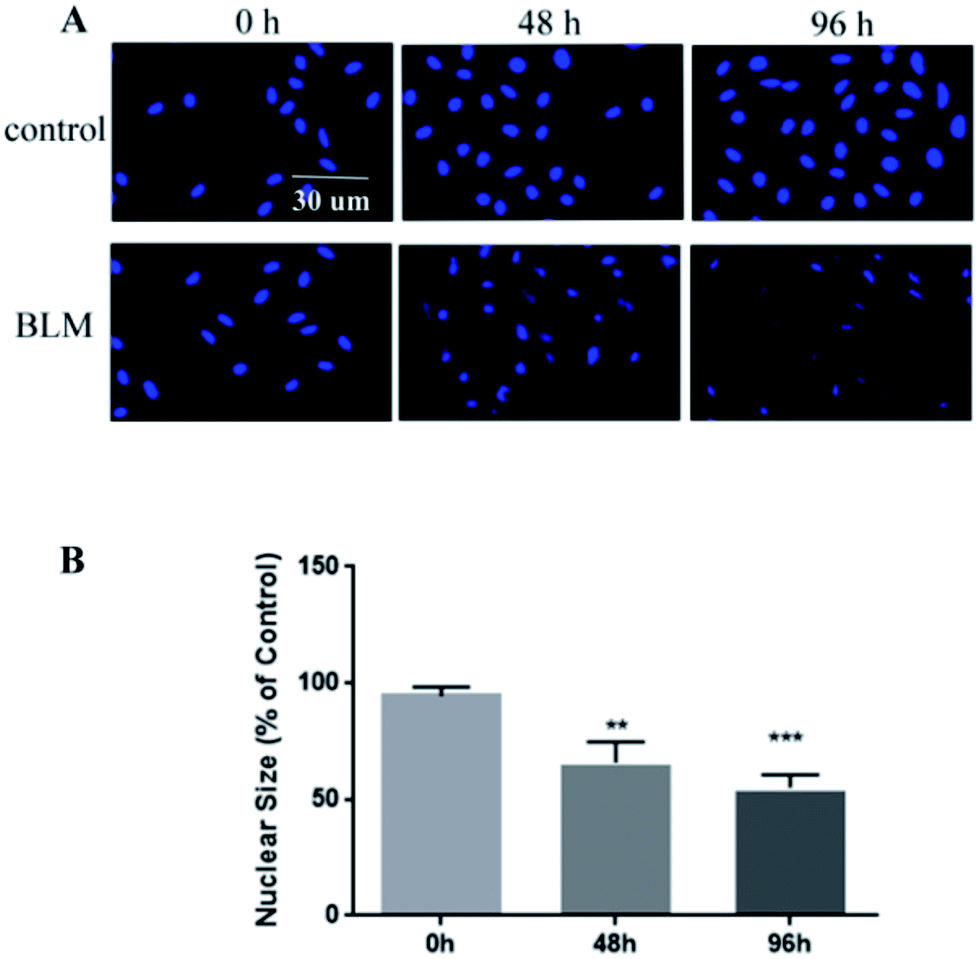 | ||
| Fig. 5 BLM-induced apoptosis in A549 cells. (A) A549 cells were incubated with 20 μg mL−1 BLM at different time points. Representative images of DAPI-stained nuclei are shown. (B) Quantification of apoptosis in BLM-treated A549 cells. Notes: magnification, 200×, and scale bar represents 30 μm (A); data are plotted as average ± SD from three separate experiments, **P < 0.01, ***P < 0.001 vs. 0 h group (B). Abbreviations: BLM, bleomycin. | ||
Activation of fibroblasts
We next investigated if BLM induces fibroblast activation in vitro. We added 20 μg mL−1 BLM into the microfluidic co-culture system to obtain a stable concentration gradient. Fibroblasts (MRC-5) and macrophages responses were recorded using an inverted confocal microscope over a period of 96 h (Fig. 6A). We observed that both MRC-5 and macrophages moved toward the epithelial channel where the concentration of BLM was the highest (Fig. 6B–C; ESI Video S1†). This migratory behavior was significant after 48 h treatment with BLM. Interestingly, BLM treatment of the A549/MRC-5 co-culture (without macrophages), significantly increased MRC-5 migration, but did not significantly affect migratory behavior in the MRC-5/macrophages co-culture (without A549) after treatment with BLM (Fig. 6B–C). Consistent with our migratory data, our immunofluorescence staining of activation-related markers of fibroblasts showed an increased expression of myofibroblast markers (α-SMA) and collagen I in BLM-treated A549/MRC-5/macrophages co-culture compared to the BLM-treated MRC-5/macrophages co-culture. Of note, the collagen I deposition was significantly closer to the left channel where epithelial cells were injured (Fig. 7). | ||
| Fig. 6 Migration assay of MRC-5 and macrophages. (A) A representative image of MRC-5 and macrophages in the microfluidic co-culture device. (B) Imaging of different co-cultures after seeding for a period of 24 h, and BLM-induced migratory behavior of MRC-5 and macrophages at different time points. (C) Quantitative data representing the number of migrating cells. Notes: scale bar represents 1000 μm (A); green: A549; red: MRC-5; deep red: macrophages. Scale bar represents 300 μm (B); data are plotted as average ± SD from three separate experiments, *P < 0.05, **P < 0.01, ***P < 0.001 vs. 96 h control; +P < 0.05, ++P < 0.01 vs. 48 h control (C). | ||
 | ||
| Fig. 7 Expression levels of activation-related markers in cultured fibroblasts after BLM treatment for 96 h. (A) Immunofluorescence analysis of α-SMA and collagen I in MRC-5 cells. (B) Quantification of fluorescence intensity of the two proteins. Notes: scale bar represents 500 μm (left) and 300 μm (right), respectively. Magnification (100×), red: α-SMA (up row)/collagen I (bottom row), blue: cell nuclei (A); data are plotted as average ± SD from three separate experiments (B). | ||
TGF-β1 levels in BLM-treated co-culture
BLM treatment of A549/MRC-5/macrophages, A549/MRC-5 co-culture and A549 supernatants for 96 h significantly increased the TGF-β1 concentration compared to the control (P ≤ 0.001) as determined by ELISA analysis. However, TGF-β1 levels did not increase in the BLM-treated MRC-5/macrophages co-culture compared to normal co-cultures (Fig. 8). | ||
| Fig. 8 TGF-β1 levels in co-culture supernatants. Notes: experiments were performed in triplicate, and mean ± SD are presented (***P ≤ 0.01 vs. control). | ||
Discussion
Epithelial/fibroblastic cross-talk disorder is highly correlated with the development of pulmonary fibrosis, which is characterized by successive events: insults and stimulation provoke epithelial damage. Injured epithelial cells release factors inducing fibroblasts activated, they transform into myofibroblasts that secrete extracellular matrix (ECM) components (e.g. collagen I) and migration/proliferation toward the injured epithelium through the ECM in an attempt to repair the damaged epithelium instead of appropriate re-epithelialization. However, no model exists that can visualize the process. For example (1) the use of commercially available two-dimensional (2D) platforms (e.g. Transwell) upon which epithelial cells can be cultured at the bottom, and fibroblasts can be cultured on the top is widely practiced. Although information regarding epithelial–interstitial cellular interaction can be identified using these methods, the 2D platforms fail to represent the cellular microenvironment seen in vivo (e.g. fibroblasts should be thrive within the 3D environment of ECM), and also in which inflammatory/immune cells could not be included; (2) improved tissue organization can be promoted by growing cells in three dimensional ECM gels to provide more in vivo-like cell morphology, function and intercellular interactions enabling greater resemblance to physiological conditions.24,25 However, they do not to construct efficient tissue interface models that recapitulates the specific structural and cellular architecture of tissue–tissue interface between the epithelium and neighboring interstitial tissues (e.g. encapsulating epithelial cells whose primary function is monolayer barrier formation could be counterintuitive), nor do they permit observation of dynamic pathologic changes (e.g. migration of cells) that are required for the development of meaningful models for effectively mimic complex disease processes and drug responses. Thus we developed a microfluidic cell co-culture device that simulates the epithelial/interstitial environment and mimics the pathogenesis of BLM-induced pulmonary fibrosis.First, we developed a 3D epithelial–interstitial tissue interface in a microfluidic device (Fig. 1–3). This device is small-scale, convenient, and significantly enhances the physiological relevance of in vitro tissue microenvironment, allowing for the incorporation of different cell types in vivo. Epithelial cells, fibroblasts, and macrophages were cultured in parallel channels to mimic the in vivo epithelial–interstitial tissue interface of the lung. The middle channel was filled with ECM and it linked the two side channels to allow easy visualization of cell migration without interference of gravitational force. As shown in Fig. 1D, epithelial cells formed an intact monolayer lining the fibronectin-coated channel, representing the alveolar space and alveolar wall (left channel); fibroblasts and macrophages cultured within the 3D ECM mimicked the microenvironment of the lung interstitium (right channel). Culture medium treated with BLM flowed from the left channel to the right channel, which induced paracrine communication between the cell types. Thus, our disease model demonstrated that BLM induces epithelial injury and phenotype changes, as well as migration of fibroblasts and macrophages with a significantly higher expression of α-SMA and deposition of ECM (collagen I).
In the first set of experiments, we studied the changes in alveolar epithelium after BLM treatment. Our results showed that BLM induced morphological changes in A549 cells in a time-dependent manner, evidenced by the presence of cuboidal cells and reactive large and elongated epithelial cells. Consistent with this morphological data, immunofluorescence studies showed that BLM treatment increased α-SMA expression and decreased E-cadherin expression in A549 cells, suggesting that epithelial properties (e.g. cellular morphology and adhesiveness) were changed (Fig. 4). Moreover, we observed BLM-induced apoptosis in epithelial cells (Fig. 5), which is characteristic of the changes in the alveolar epithelium under IPF.26 Activation of fibroblasts is a central wound-healing response in the lung. In addition, fibroblast activation is associated with an accelerated clinical course, since fibroblast chemoattractant activity in the air space of individuals with IPF has been previously reported.27,28 Although the activation of interstitial cells has been extensively studied, several issues, including the migratory behavior of fibroblasts and the relationship between migration and epithelial injury, remain unclear. The present study has constructed a microfluidic co-culture device that simulates the cavity-like structure of pulmonary alveoli and the interstitial environment and also facilitates a real-time observation of the migratory behavior of fibroblasts (MRC-5) and macrophages, both spatially and temporarily. Quantification of cellular migration after treatment of co-cultures with BLM for 96 h showed that fibroblasts moved through the ECM toward the epithelial channel, caused by epithelial injury (Fig. 6) as evidenced by increased expression α-SMA and continued ECM production (collagen I) compared to healthy co-cultures (Fig. 7). These findings confirm that fibroblasts are activated in response to epithelial injury. A similar migratory behavior was also observed in macrophages (Fig. 6C), suggesting that inflammation and fibroblast activation occurs following the initial lung injury. Interestingly, fibroblast migration and activation also occured in the absence of macrophages (A549/MRC-5 co-culture), suggesting that fibrotic changes could occur both in the presence and the absence of inflammatory cells, indicating that a fibrotic response is possible without inflammation. Therefore, fibrosis may not be driven by inflammation alone. Research has also suggested that an active inflammatory response is not a strict prerequisite for IPF,27 which is supported by the fact that most patients with IPF do not respond to anti-inflammatory drugs.1,28 However, fibroblast and inflammatory cells (MRC-5/macrophages) in our co-culture system treated with BLM did not undergo migration (Fig. 6C), confirming our hypothesis that migration occurs primarily in response to epithelial injury, which partly explains the pathogenesis of most interstitial lung diseases.
To further study this pathological process, we performed ELISA to measure TGF-β1, a key driver of cell differentiation, apoptosis, and ECM production in pulmonary fibrosis.29 In agreement with our data related to fibroblast activation, TGF-β1 levels significantly increased level in the BLM-treated co-cultures with or without inflammatory cells compared to healthy co-cultures. However, the level of TGF-β1 in BLM-treated co-cultures without epithelial cells was similar to that of healthy co-cultures (Fig. 8), suggesting that type II alveolar epithelial cells might be the primary source of TGF-β1 synthesis. Khalil et al. reported similar findings.30,31 Two important interpretations can be derived from our study: first, alveolar epithelial injury appears to be responsible for the release of TGF-β1, and second, fibroblasts might be activated in response to the release of TGF-β1 from injured epithelial cells. Therefore, we conclude that, the release of TGF-β1 promotes alveolar epithelial/fibroblastic cell cross-talk disorder.
A potential limitation of our study is that the pulmonary alveolar epithelial (A549) cells used in our experiments were isolated from a lung tumor, and therefore these cells might not fully recapitulate the critical characteristics of a healthy alveolar epithelium in vivo. Addressing this challenge directly would require reproducing these experiments with primary human alveolar epithelial cells, but long-term culturing of these primary cells while maintaining physiological functions and proliferative capacity in vitro is not yet possible.
In conclusion, our co-culture system recapitulates the critical pathological changes characteristic of pulmonary fibrosis, including injury and morphological changes of epithelial cells, activation of interstitial cells, and up-regulation of TGF-β1 production, underscoring the utility of our microfluidic co-culture device as a useful model for studying BLM-induced fibrosis. This study strongly suggests that the fibrotic process is orchestrated by a complex exchange of paracrine signals among various cell types involved in the wound healing response. However, a more refined understanding of this cross-talk phenomenon is necessary to extend the scope of this platform into the future. For example, the device could include more cell types involved with pulmonary fibrosis. Moreover, our device presents the potentiality for the tests for screening of fibrogenic agents, disease modeling, and academic and pharmaceutical research within the fields of lung biology.
Results and discussions
Epithelial/fibroblastic cross-talk disorder is highly correlated with the development of pulmonary fibrosis, which is characterized by successive events: insults and stimulation provoke epithelial damage. Injured epithelial cells release factors inducing fibroblasts activated, activated fibroblasts transform into myofibroblasts that secrete extracellular matrix (ECM) components (e.g. collagen I) and migration/proliferation toward the injured epithelium through the ECM in an attempt to repair the damaged epithelium instead of appropriate re-epithelialization. However, no model exists that can visualize the process. For example (1) the commercially available two-dimensional (2D) platforms (e.g. Transwell) fail to represent the cellular microenvironment seen in vivo (e.g. fibroblasts should be thrive within the 3D environment of ECM), and also in which inflammatory/immune cells could not be included; (2) traditional 3D culture by growing cells in three dimensional ECM gels34,35 could not to construct efficient tissue interface models that recapitulates the specific structural and cellular architecture of epithelial–interstitial interface (e.g. encapsulating epithelial cells whose primary function is monolayer barrier formation could be counterintuitive), nor do they permit observation of pathologic changes (e.g. migration of cells) that are required for the development of meaningful models for effectively mimic complex disease processes and drug responses. Thus we developed a microfluidic cell co-culture device that simulates the epithelial/interstitial environment and mimics the pathogenesis of BLM-induced pulmonary fibrosis.Mimicking the human epithelial-interstitial interface on a chip
In vivo, the human epithelial-interstitial interface consists of epithelium and interstitial cells separated by the ECM. Thus, we designed a microfluidic device consisting of three channels (Fig. 1A–B). A549 cells, MRC-5 cells and macrophages were loaded into the left and right channels while the middle channel was filled with ECM as described previously, and it linked the two side channels to allow easy visualization of cell migration without interfering of gravitational force (Fig. 2). Interestingly, epithelial cells formed an intact monolayer lining the fibronectin-coated channel in the left channel, representing the alveolar space and alveolar wall. It is noted that the device also facilitated the formation of a cavity-like structure from multiple cells with this simple manipulations. The right channel, consisting of fibroblasts and macrophages cultured within the 3D ECM, mimicked the microenvironment of the lung interstitium (Fig. 3A). Culture medium flowed from the left channel to the right channel, which induced paracrine communication between the cell types. After culturing the three types of cells in the device for 48 hours, the analysis provided evidence of cell growth within the microfluidic device (Fig. 3B). This device is small-scale, convenient, and significantly enhances the physiological relevance of in vitro tissue microenvironment, allowing for the incorporation of different cell types in vivo.Pathological changes to epithelial–interstitial interface under bleomycin treatment
A potential limitation of our study is that the pulmonary alveolar epithelial (A549) cells used in our experiments were isolated from a lung tumor, and therefore these cells might not fully recapitulate the critical characteristics of a healthy alveolar epithelium in vivo. Addressing this challenge directly would require reproducing these experiments with primary human alveolar epithelial cells, but long-term culturing of these primary cells while maintaining physiological functions and proliferative capacity in vitro is not yet possible.
In conclusion, our co-culture system recapitulates the critical pathological changes of pulmonary fibrosis, including injury and morphological changes of epithelial cells, activation of interstitial cells, and up-regulation of TGF-β1 production, underscoring the utility of our microfluidic co-culture device as a useful model for studying BLM-induced fibrosis. This study strongly suggests that the fibrotic process is orchestrated by a complex exchange of paracrine signals among various cell types involved in the wound healing response. However, a more refined understanding of this cross-talk phenomenon is necessary to extend the scope of this platform in the future. For example, the device could include much more cell types involved with pulmonary fibrosis. Moreover, our device presents the potentiality for the tests for screening of fibrogenic agents, disease modeling, academic and pharmaceutical research within the fields of lung biology.
Conclusion
In this study, we developed a microfluidic co-culture device to monitor the fibrotic process via investigating epithelial–interstitial cell communication. We observed cell apoptosis and phenotypic changes in A549 cells accompanied by abnormal levels of protein expression in the injury response. Our data indicate that the epithelium sends paracrine signals to activate interstitial cells, resulting in the migration of fibroblast and inflammatory cells with a subsequent increase in ECM (collagen I) deposition. Our device provides new insights into the complex and dynamic interactions between epithelial and fibroblastic cells. Our study confirms that epithelial injury is a source of epithelial/fibroblastic cross-talk during BLM-induced fibrosis.Author contributions
C. W., H. D., J. H., and W. C. conceived and designed the experiments. J. H. and W. C. performed the experiments. C. W., H. D., J. H., W. C., S. D., L. X., J. F., J. G. and D. J. analyzed the results. C. W., H. D., and J. H. wrote the manuscript.Conflicts of interest
The authors report no conflicts of interest in this work.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China No. 81430001(to C. Wang). All authors would like to acknowledge the Imaging Core Facility of Protein Research Center for Technology Development of Tsinghua University for providing assistance with Imaris software.References
- T. A. Wynn, Integrating mechanisms of pulmonary fibrosis, J. Exp. Med., 2011, 208, 1339–1350 CrossRef CAS PubMed.
- R. Perez-Padilla, J. Salas and R. Chapela, et al., Mortality in Mexican patients with chronic pigeon breeder's lung compared with those with usual interstitial pneumonia, Am. Rev. Respir. Dis., 1993, 148, 49–53 CrossRef CAS PubMed.
- M. Selman and A. Pardo, Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder, Respir. Res., 2002, 3, 3 CrossRef PubMed.
- M. Selman, T. E. King and A. Pardo, et al., Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy, Ann. Intern. Med., 2001, 134, 136–151 CrossRef CAS PubMed.
- A. L. Katzenstein and R. F. Fiorelli, Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance, Am. J. Surg. Pathol., 1994, 18, 136–147 CrossRef CAS PubMed.
- M. Shin, K. Matsuda and O. Ishii, et al., Endothelialized networks with a vascular geometry in microfabricated poly(dimethyl siloxane), Biomed. Microdevices, 2004, 6, 269–278 CrossRef CAS PubMed.
- J. W. Song, W. Gu and N. Futai, et al., Computer-controlled microcirculatory support system for endothelial cell culture and shearing, Anal. Chem., 2005, 77, 3993–3999 CrossRef CAS PubMed.
- D. Huh, H. Fujioka and Y. C. Tung, et al., Acoustically detectable cellular-level lung injury induced by fluid mechanical stresses in microfluidic airway systems, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18886–18891 CrossRef CAS PubMed.
- A. Carraro, W. M. Hsu and K. M. Kulig, et al., In vitro analysis of a hepatic device with intrinsic microvascular-based channels, Biomed. Microdevices, 2008, 10, 795–805 CrossRef PubMed.
- S. R. Khetani and S. N. Bhatia, Microscale culture of human liver cells for drug development, Nat. Biotechnol., 2008, 26, 120–126 CrossRef CAS PubMed.
- R. Baudoin, L. Griscom and M. Monge, et al., Development of a renal microchip for in vitro distal tubule models, Biotechnol. Prog., 2007, 23, 1245–1253 CAS.
- K. J. Jang and K. Y. Suh, A multi-layer microfluidic device for efficient culture and analysis of renal tubular cells, Lab Chip, 2010, 10, 36–42 RSC.
- N. J. Douville, P. Zamankhan and Y. C. Tung, et al., Combination of fluid and solid mechanical stresses contribute to cell death and detachment in a microfluidic alveolar model, Lab Chip, 2011, 11, 609–619 RSC.
- D. D. Nalayanda, C. Puleo and W. B. Fulton, et al., An open-access microfluidic model for lung-specific functional studies at an air-liquid interface, Biomed. Microdevices, 2009, 11, 1081–1089 CrossRef CAS PubMed.
- D. Huh, D. C. Leslie and B. D. Matthews, et al., A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice, Sci. Transl. Med., 2012, 4, 159ra47 Search PubMed.
- Q. Zhou, D. Patel and T. Kwa, et al., Liver injury-on-a-chip: microfluidic co-cultures with integrated biosensors for monitoring liver cell signaling during injury, Lab Chip, 2015, 15, 4467–4478 RSC.
- D. Huh, B. D. Matthews and A. Mammoto, et al., Reconstituting organ-level lung functions on a chip, Science, 2010, 328(5986), 1662–1668 CrossRef CAS PubMed.
- K. J. Jang and K. Y. Suh, A multi-layer microfluidic device for efficient culture and analysis of renal tubular cells, Lab Chip, 2010, 10(1), 36–42 RSC.
- J. A. Brown, S. G. Codreanu and M. Shi, et al., Metabolic consequences of inflammatory disruption of the blood-brain barrier in an organ-on-chip model of the human neurovascular unit, J. Neuroinflammation, 2016, 13, 306 CrossRef PubMed.
- L. J. Chen, S. Ito and H. Kai, et al., Microfluidic co-cultures of retinal pigment epithelial cells and vascular endothelial cells to investigate choroidal angiogenesis, Sci. Rep., 2017, 7, 3538 CrossRef PubMed.
- C. Blume, R. Reale and M. Held, et al., Cellular crosstalk between airway epithelial and endothelial cells regulates barrier functions during exposure to double-stranded RNA, Immun., Inflammation Dis., 2017, 5, 45–56 CrossRef CAS PubMed.
- Y. Du, N. Li and H. Yang, et al., Mimicking liver sinusoidal structures and functions using a 3D-configured microfluidic chip, Lab Chip, 2017, 17, 782–794 RSC.
- J. Sai, M. Rogers and K. Hockemeyer, et al., Study of Chemotaxis and Cell–Cell Interactions in Cancer with Microfluidic Devices, Methods Enzymol., 2016, 570, 19–45 Search PubMed.
- H. J. Chiang, S. L. Yeh and C. C. Peng, et al., Polydimethylsiloxane-polycarbonate Microfluidic Devices for Cell Migration Studies Under Perpendicular Chemical and Oxygen Gradients, J. Visualized Exp., 2017, 120, e55292 Search PubMed.
- K. H. Lee, K. H. Lee and J. Lee, et al., Integration of microfluidic chip with biomimetic hydrogel for 3D controlling and monitoring of cell alignment and migration, J. Biomed. Mater. Res., Part A, 2014, 102, 1164–1172 CrossRef PubMed.
- C. T. Kuo, C. L. Chiang and C. H. Chang, et al., Modeling of cancer metastasis and drug resistance via biomimetic nano-cilia and microfluidics, Biomaterials, 2014, 35, 1562–1571 CrossRef CAS PubMed.
- S. Sleijfer, Bleomycin-induced pneumonitis, Chest, 2001, 120, 617–624 CrossRef CAS PubMed.
- Y. Dong, Y. Geng and L. Li, et al., Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice, J. Exp. Med., 2015, 212, 235–252 CrossRef CAS PubMed.
- H. He, H. Tang and L. Gao, et al., Tanshinone IIA attenuates bleomycin-induced pulmonary fibrosis in rats, Mol. Med. Rep., 2015, 11, 4190–4196 CrossRef CAS PubMed.
- B. H. Jo, L. M. Van Lerberghe, K. M. Motsegood and D. J. Beebe, Three-dimensional micro-channel fabrication in polydimethylsiloxane (PDMS) elastomer, J. Microelectromech. Syst., 2000, 9, 76–81 CrossRef CAS.
- M. Daigneault, J. A. Preston and H. M. Marriott, et al., The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages, PLoS One, 2010, 5, e8668 Search PubMed.
- E. C. Filippi-Chiela, M. M. Oliveira and B. Jurkovski, et al., Nuclear morphometric analysis (NMA): screening of senescence, apoptosis and nuclear irregularities, PLoS One, 2012, 7, e42522 CAS.
- N. Ye, J. Qin and W. Shi, et al., Cell-based high content screening using an integrated microfluidic device, Lab Chip, 2007, 7, 1696–1704 RSC.
- A. J. Carterson, K. Honer zu Bentrup and C. M. Ott, et al., A549 lung epithelial cells grown as three-dimensional aggregates: alternative tissue culture model for Pseudomonas aeruginosa pathogenesis, Infect. Immun., 2005, 73, 1129–1140 CrossRef CAS PubMed.
- S. Ravindran, Y. Song and A. George, Development of three-dimensional biomimetic scaffold to study epithelial-mesenchymal interactions, Tissue Eng., Part A, 2010, 16, 327–342 CrossRef CAS PubMed.
- M. Kasper and G. Haroske, Alterations in the alveolar epithelium after injury leading to pulmonary fibrosis, Histol. Histopathol., 1996, 11, 463–483 CAS.
- M. Demedts, J. Behr and R. Buhl, et al., High-dose acetylcysteine in idiopathic pulmonary fibrosis, N. Engl. J. Med., 2005, 353, 2229–2242 CrossRef CAS PubMed.
- V. J. Thannickal, G. B. Toews and E. S. White, et al., Mechanisms of pulmonary fibrosis, Annu. Rev. Med., 2004, 55, 395–417 CrossRef CAS PubMed.
- B. C. Willis and Z. Borok, TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2007, 293, L525–L534 CrossRef CAS PubMed.
- N. Khalil, R. N. O'Connor and H. W. Unruh, et al., Increased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosis, Am. J. Respir. Cell Mol. Biol., 1991, 5, 155–162 CrossRef CAS PubMed.
- N. Khalil, R. N. O'Connor, K. C. Flanders and H. Unruh, TGF-beta 1, but not TGF-beta 2 or TGF-beta 3, is differentially present in epithelial cells of advanced pulmonary fibrosis: an immunohistochemical study, Am. J. Respir. Cell Mol. Biol., 1996, 14, 131–138 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06752f |
| This journal is © The Royal Society of Chemistry 2017 |