
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesoscopic probes in asphaltenes nanoaggregate structure: from perpendicular to paralleled orientation at the water-in-oil emulsions interface†
Jinhui Chena,
Jinliang Chenb,
Chuheng Zhongc,
Shouyu Chena,
Bowen Chend,
Shenwen Fang‡
 *e and
Wenjun Xiangf
*e and
Wenjun Xiangf
aChina University of Geosciences (Wuhan), Wuhan 430074, China
bManagement Center of Sustainable Development Research Institute of Yellow River Delta, Dongying, Shandong 257091, China
cEcole Centrale de Nantes, Pays de la Loire, 44000 Nantes, France
dUniversité Laval, 3 Avenue Des Sciences-Humaines, Sainte-Foy, Québec, Canada
eCollege of Chemistry and Chemical Engineering, Southwest University of Petroleum, 8 Xindu Avenue, Xindu District, Chengdu, Sichuan 610500, P. R. China. E-mail: fangwen1228@yahoo.com.cn; Fax: +86 028 83037346; Tel: +86 028 83037346
fSchool of Chemistry and Chemical Engineering, Sichuan University of Arts and Science, Dazhou, Sichuan 635000, China
First published on 3rd August 2017
Abstract
It is well known that asphaltene molecules play a significant role in stabilizing emulsions of water-in-crude oil or diluted bitumen solutions. Here, the dissipative particle dynamics simulation method was employed to investigate the aggregation and orientation behaviors of asphaltene molecules at the interface of water-in-crude oil emulsions at mesoscale length. Three kinds of asphaltene model molecules with different architectural structures were employed in this work. It was found that the initially disordered asphaltenes quickly self-assembled into ordered nanoaggregates consisting of several molecules, in which the aromatic rings in asphaltenes were reoriented to form nanoaggregate structures. More importantly, the nanoaggregate structure indicates that most of the stacked polycyclic aromatic planes of island architecture asphaltenes preferred to be perpendicular to the O/W interface. However, most of the stacked polycyclic aromatic planes of archipelago architecture asphaltene tend to be parallelled to the O/W interface. Both the perpendicular and parallel nanoaggregate structures of asphaltenes form a steady protective film wrapping the water droplets which hinder the droplet–droplet coalescence. The increasing number of aliphatic chains of asphaltenes results in hindering of the π–π stacking interactions of polycyclic aromatic hydrocarbons, thus asphaltenes prefer to form the parallel orientation at the O/W interface. Our results provide insights into the fundamental understanding of self-assembly mechanisms of asphaltenes at O/W interface and the stabilization behaviors of water-in-oil emulsions with asphaltenes.
1. Introduction
Asphaltenes are practically defined as a solubility class soluble in some solvents, such as aromatics (benzene, toluene, etc.), but insoluble in some others, such as normal alkanes (paraffins, heptane, etc.).1 Asphaltenes are usually presented in petroleum industry and coal liquid as well mine of coalbed gas (e.g. green mine construction). The structure of asphaltenes is complex and normally assumed to be constituted by polycyclic aromatic hydrocarbons (PAHs) substituted with alkyl side chains and some heteroatoms, including nitrogen, sulfur, oxygen.2 Correspondingly, asphaltenes are kinds of interfacially active substances because of their polarizability and some degree of charge separation associated with heteroatoms in the alkyl chains and aromatic ring structures.3 The presence of asphaltenes is critical to many aspects of petroleum utilization, from oil recovery and transportation to refining. The formation of stable water-in-crude oil emulsions in petroleum industry can cause flow assurance problems due to high viscosity of the emulsions and cost efficient methods are required to remove the emulsified water from the emulsions.4 The water emulsified by asphaltenes, in the form of micrometer size water droplets, also enhance corrosion because of the inclusion of salt and brine in the petroleum emulsions, which reduce throughput, and also can lead to equipment failure.5 Therefore, understanding the mechanism of crude oil emulsion stabilized by asphaltenes is crucial for stabilizing desired emulsions and destabilizing undesired emulsions in petroleum industry.Actually, typical asphaltene molecules cannot be considered to be amphiphilic with well-defined hydrophilic and hydrophobic molecular architecture. Moreover, the affinity of asphaltenes toward the O/W interface and their ability of reducing the interfacial tension are lower than the other indigenous crude/heavy oil fractions (such as resins).6 A large number of studies indicate the mechanism of emulsion stabilized by asphaltenes is related to the formation of the nanoaggregate structure at O/W interface. Several researchers7–9 have proposed that a kind of cross-linked gel phase is formed at the O/W interface which results into hindering droplet–droplet coalescence. Building up of 3D network at the interface, the rheological properties of the asphaltenes film were modified to be non-Newtonian with yield stress (gel-like), which is responsible for the stability of crude oil emulsions.8,9 The order asphaltene nanoaggregate structure, typically 7–20 nm, was observed by small-angle X-ray scattering (SAXS) and small-angle neutron scattering (SANS).6,10 Furthermore, the colloidal asphaltene aggregates as compared to individual asphaltene molecules at the O/W interface were also thought to increase the stability of emulsions.11 Liu Juan and co-author12 employed molecular dynamics simulations studied the nanoaggregate structure of asphaltenes. They found most of the stacked polycyclic aromatic planes of asphaltene nanoaggregates tend to be perpendicular to the water surface. These asphaltenes adsorbed at the O/W interface seems as a “fence” and all the fence-like nanoaggregates were twined and knitted together, which pinned them perpendicularly on the water surface to form a steady protective film wrapping the water droplets. The asphaltenes film with nanoscale aggregates at the interface is much stable and rigid.9,12 In our previous studies, it was found that the rigid mechanical film of asphaltenes originates from (i) rigid structure of polycyclic aromatic hydrocarbons (PAHs) and (ii) the π–π bonding interactions between the PAHs of asphaltenes.13 Moreover, a layer-by-layer assembled architecture film was formed at the W/O interface when the system in presence of polyacrylamide (PAM). This layer-by-layer assembled architecture film exhibits great viscoelasticity and rheology properties which enhances the stability of water-in-oil emulsions.
Moreover, the structure of asphaltenes also has a great influence on the nanoaggregate structure at the O/W interface.14 To this end many studies have been carried out on the nanoaggregate structure at the O/W interface with different kinds of asphaltene structure. Chang Chia-Lu and co-authors15 investigated the effect of the chemical structure of amphiphiles on asphaltenes stabilization with tuning the polarity of the asphaltenes's head group and the length of the asphaltenes's alkyl tail. The effects of side-chain length on the aggregation of asphaltenes in water were studied by Jian Cuiying and co-authors.16 Hugo Santos-Silva and co-author17 investigated the effects of the N, O and S heteroatoms on the nano-aggregation of asphaltene mixtures.
Ruiz-Morales Yosadara and co-author5 used the coarse-grained molecular simulations to investigate the effect of peripheral oxygen moieties in coarse-graining asphaltene on the orientation of asphaltene at the O/W interface, they found the asphaltene orients perpendicularly at the oil water interface when the asphaltene contains oxygen atoms. Furthermore, it was observed that the nanoaggregation of asphaltene is dependent on the heteroatom on the conjugated core and is more dependent on the chain-end and conjugated core size. Hossein Rezaei and co-author18 studied the uncharged perylene bisimide-based polyaromatic surfactant molecules, with the same polyaromatic core but with different terminal functional types at the O/W interface. They found that the polyaromatic surfactant with the terminal functional group of aliphatic carboxylic acid absorbed more effectively on the water droplet interface in the crude oil system and can reduce the interfacial tension to facilitate the oil/water separation. Yohei Mikami investigated the nanoaggregate structure of three kinds of asphaltenes with low molecular weights19 at the nanoscale. Though a large number of the aforementioned studies have been reported, a further understanding of the nanoaggregate structure and emulsion stability of asphaltenes at the O/W interface is still needed, especially at mesoscopic scale.13
In this work, we employed dissipative particle dynamics (DPD) simulations for studying the nanoaggregate structures of asphaltenes at water/toluene interface with the mesoscopic scale.13,22 DPD simulations method are an excellent technology for the simulation of coarse-grained systems over considerable length and time scales up to the mesoscopic scale.13,20 DPD simulations have been performed to study static and dynamic interfacial phenomena and the self-assembly behavior of amphiphilic molecule at the interface,20,21 including asphaltenes at liquid/liquid interface22 and the solid/liquid interface.23 In this study, we have used three types of asphaltene molecules with different molecular weights, which was constructed from a statistical representation of the oil sample.24 The stability mechanism of water-in-oil emulsions in presence of asphaltenes was investigates by analyzing the morphologies, film thickness, order parameter and radial distribution function of the crude oil emulsion.
2. DPD simulation
2.1 DPD method
Dissipative particle dynamics (DPD) is a mesoscale particle method that bridges the gap between microscopic and macroscopic simulations. It can be used to simulate the dynamic and rheological properties of simple and complex fluids. In DPD simulation system, several molecules were usually coarse-grained as one or more coarse-grained beads, respectively. The dynamics of the elementary units which are so-called DPD beads, is governed by Newton's equation of motion fi = mdvi/dt. The force between each pair of beads is a sum of a conservative force (FCij), a dissipative force (FDij), and a random force (FRij).25 The conservation force for non-bonded particles is defined by soft repulsion. The dissipative force corresponding to a frictional force depends on both the position and relative velocities of the beads. The random force is a random interaction between bead i and its neighbor bead j. All forces vanish beyond a certain cutoff radius rc, whose value is usually set to one unit of length in simulations. In fact, only the conservation force is related to the intrinsic properties of the bead and it is given by the following formulas: FCij = αijω(rij)eij, where αij = αji > 0, indicating that this force is always repulsive, rij is the distance between beads i and j, and eij is the unit vector (ri − rj)/rij. The function ω(r) determines the radial dependence of the force; the function is continuous, positive for r < rc and zero for r ≥ rc. An extensive description on DPD simulation can be found elsewhere,26,27 more details about DPD technique also can be found in our previous studies.13,22,28To coarse-grain the model of asphaltenes, the bonded potentials including bond potential, angle potential, and inversion angle potential were introduced, as depicted in our previous studies.13,22,28 The bond potential experienced by bead i is 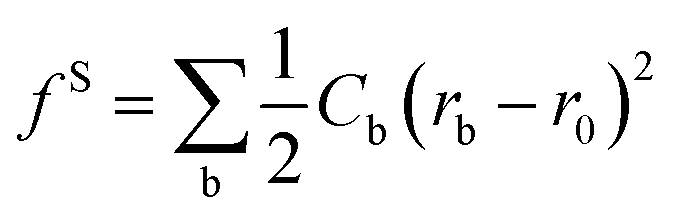 , where r0 is the equilibrium bond length and Cb is the bond spring constant. The equilibrium bond length r0 = 3 Å and a spring constant of 300 kcal (mol Å2)−1 are assigned for all the beads of aromatic rings, while r0 = 4.25 Å and Cb = 3 kcal (mol Å2)−1 are taken for all the alicyclic groups and alkyl chains. The angle potential experienced by bead i due to angular interactions is
, where r0 is the equilibrium bond length and Cb is the bond spring constant. The equilibrium bond length r0 = 3 Å and a spring constant of 300 kcal (mol Å2)−1 are assigned for all the beads of aromatic rings, while r0 = 4.25 Å and Cb = 3 kcal (mol Å2)−1 are taken for all the alicyclic groups and alkyl chains. The angle potential experienced by bead i due to angular interactions is 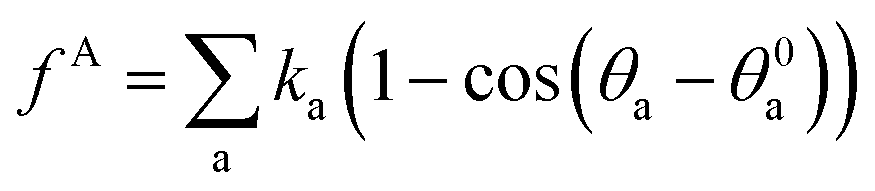 , where θ0 = 120° is the equilibrium angle, and ka = 300 kcal mol−1. Furthermore the inversion angle potentials describe the interaction arising from a particular geometry of three beads (J, K, L) around a central bead I:
, where θ0 = 120° is the equilibrium angle, and ka = 300 kcal mol−1. Furthermore the inversion angle potentials describe the interaction arising from a particular geometry of three beads (J, K, L) around a central bead I: 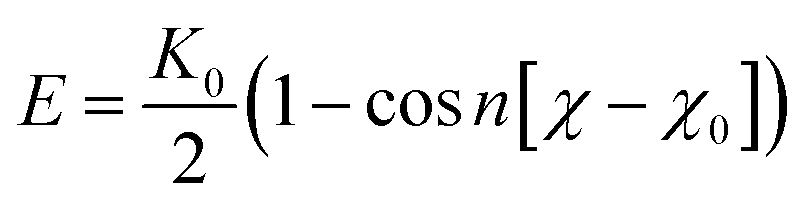 , where K0 = 25 kcal mol−1 is force constant, χ0 = 180° is the improper torsion angle between the KIJ and JIL plane, and n = 2 is the periodicity.
, where K0 = 25 kcal mol−1 is force constant, χ0 = 180° is the improper torsion angle between the KIJ and JIL plane, and n = 2 is the periodicity.
2.2 Coarse-graining model
The coarse-graining process of molecular in DPD simulation system is an essential process. As reported in our previous studies, three water molecules were taken as single bead (W bead). As presented in Fig. 1, the typical beads, B represents the moiety of aromatic rings, which is denoted by benzene molecule; H corresponds to the alkyl chain, which is defined as butane molecule. In DPD the mapping of groups of atoms into beads, all the beads should have same volume, the volume of a water bead is 90 Å3 in this study.5 The volume of one butane molecule is 91.5 Å3 and one benzene molecule is 89.4 Å3 which are close to the volume of a water bead.29 The oil phase is toluene which is clustered into two different coarse-grained particles with B bead and H bead. The fused aromatic rings is constructed by creating the rigid sheet of the hexa-particle ring.13,28 As shown in Fig. 1, T bead is the functional group containing heteroatoms, we take thiourea as T bead which represent the polar groups in asphaltene structure.13,28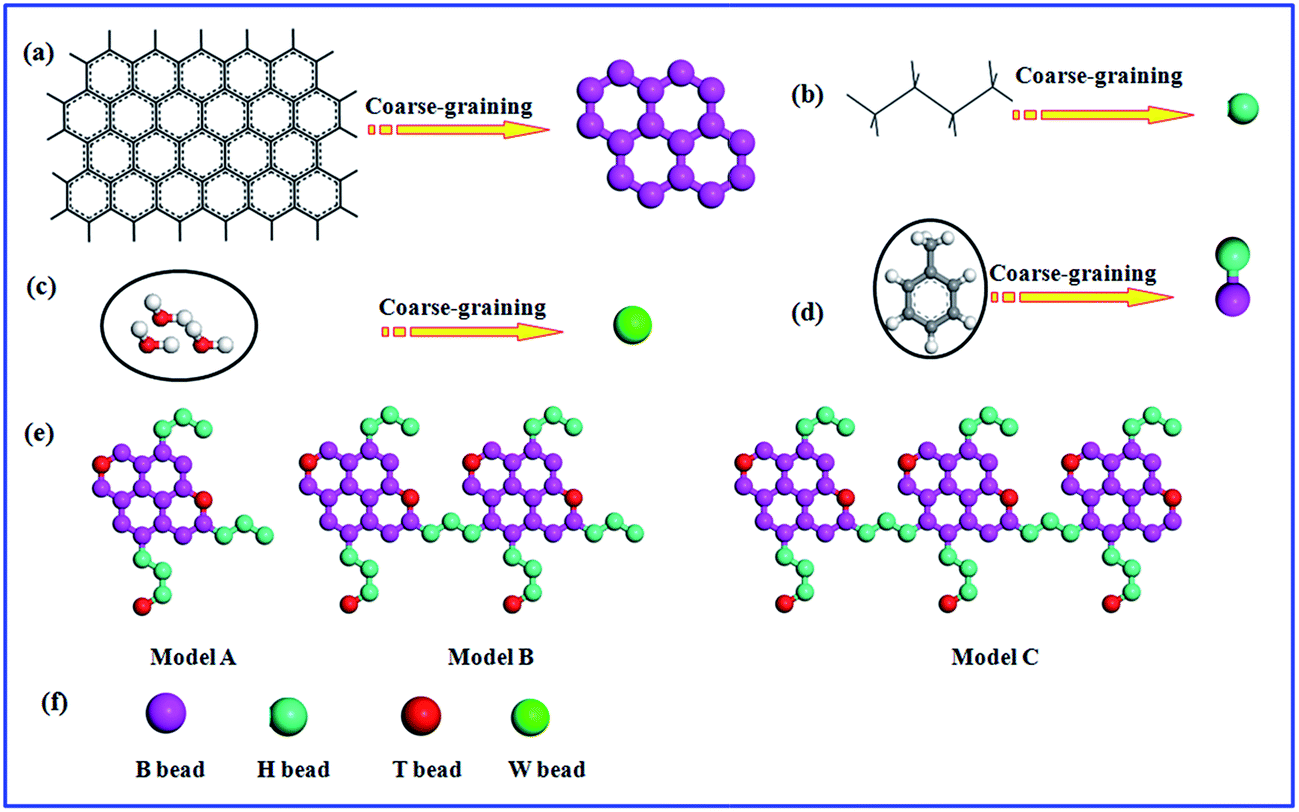 | ||
| Fig. 1 Coarse-grained model molecules of (a) fused aromatic rings (or polycyclic aromatic hydrocarbons), (b) alkyl chain, (c) water molecules, (d) toluene, (e) asphaltenes model architecture: Model A, Model B and Model C, (f) corresponds to the schematic representation of coarse-grained beads in simulations. Model A is island architecture and Model B and Model C are archipelago architecture. | ||
All these coarse-graining models in simulation systems were used and verified.13,28 Three different types of asphaltene models were performed to study the nanoaggregate structure of asphaltenes and their stabilization behaviors of water-in-oil emulsions, as presented in Fig. 1e. These kinds of asphaltene models were reported to study the aggregation of asphaltenes.24,28 Because of the complexity and diversity of asphaltenes in petroleum industry, different model compounds have been employed in various studies. However, the island architecture and archipelago architecture are the widely accepted model. The island architecture, as shown with Model A in Fig. 1e, is usually shaped “like your hand” with a single polycyclic aromatic hydrocarbon core (like palm), which has some aliphatic chains (like fingers). Another widely accepted model for asphaltenes is an archipelago architecture, which was thought to be connected with several polycyclic aromatic hydrocarbon core, as shown with Model B and Model C in Fig. 1e. Nowadays, it remains dubious regarding the archipelago models. Mullins2,24 insisted that their large relative molar mass and colorless character disagree with observations from their experiments. The archipelago model molecules of asphaltene were constructed deliberately for comparison with the island models in this paper. Generally, the archipelago architecture model is more applicable to heavy or ultra-heavy oil rather than to light crude oil. The molecular weight of asphaltenes: Model A (about 1052.85 g mol−1) > Model B (about 2021.93 g mol−1) > Model C (about 3248.55 g mol−1).
2.3 DPD parameters and simulation details
The bead–bead interaction parameters used in this paper are calculated from the Hansen solubility parameters.5,28 Hansen solubility parameters were developed by Charles M. Hansen to predict if one material will dissolve in another and form a solution which has been used to understand issues of solubility dispersion diffusion.29 Moreover, Hansen solubility parameters can be developed to calculate the bead–bead interaction parameters in immiscible liquid/liquid system which has been validated to produce ideal interfacial tensions in experiment method.30 Hansen solubility parameters were also used to calculate the bead–bead interaction parameters in crude oil system. More detailed bead–bead interaction parameters in this study can be found in ESI.† The diffusion coefficient and interlayer distance of asphaltenes in toluene solution were calculated to validate the DPD calculations, as shown in ESI.† From the Fig. S1,† the diffusion coefficient of diluted asphaltene in toluene is 3.41–5.73 × 10−10 m2 s−1 which is consistent with the experimental results 2.2–6.3 × 10−10 m2 s−1.31 Moreover, the interlayer distance value from DPD simulations is about 5.05 Å in our calculation, which is slightly larger than the experimental results (about 3.55 Å).32 On the basis of these results, it is believed that the bead–bead interaction parameters used in the simulations could well reflect the authentic physical properties of petroleum systems.In this study, DPD simulations were performed using the Mesocite module embedded in the Materials Studio 6.1 package from Accelrys, Inc.33 All the simulations were performed in a cubic box with a size of 100 × 100 × 100Rc3 with periodic boundary conditions at three direction. The simulated temperature is set as 298 K. To simulate the water-in-crude oil emulsions, the cubic box was divided into oil phase and water phase, respectively. And water phase is a sphere with radius of 30Rc in the center of box, as reported in ref. 13. The left part of the box is oil phase. The water molecules and toluene molecules as well as asphaltene molecules were randomly filled into oil phase and water phase, respectively. The concentration of asphaltenes is defined as the number ratio of asphaltenes beads to total beads in the oil phase. It has been shown experimentally that emulsions stabilized by individual asphaltene molecules are of little significance, when compared to stabilization by colloidally dispersed asphaltenes.19 In this study, different numbers of asphaltene molecules ranging from 56 to 94 were used to simulate the water-in-crude oil emulsions which is much higher than the number of asphaltenes used in MD simulations. The total number of beads is 1.2428 × 104 in the box when the density of all systems is set to 3.0 in reduced units. A total of 30.16 ns DPD simulation time were carried out with a time step Δt = 0.005τ, where τ is time scale, 3.0158 ps.34
3. Results and discussion
3.1 W/O emulsions morphology
 | ||
| Fig. 2 Morphologies of water-in-oil emulsions at different simulation time with the concentrations of 20% asphaltenes of Model A. Different beads in simulations are represented by different colors as can be illustrated from Fig. 1f (the same as below). The toluene molecules are suppressed for clarity. | ||
The aggregation and orientation of the archipelago architecture asphaltenes at the O/W interface were also investigated. As shown in Fig. 3, some similar results of adsorption process for the asphaltenes of Model B were observed. However, most of the stacked aromatic ring sheets preferred to be paralleled to the water surface. Only a small part of asphaltenes of Model B tend to be perpendicular or slope to the O/W interface. Actually, this orientation of asphaltenes at the O/W interface was also observed in the asphaltenes of Model C, as shown in Fig. S2.† Compared with the asphaltenes of Model A, the transition of orientation at the O/W interface happened in asphaltenes of Model B and Model C was induced by architecture structure of asphaltenes. In the island architecture asphaltenes case (Model A), the asphaltenes reoriented themselves to be perpendicular with the water surface were reported in the previous molecular dynamics simulations.19 The interfacial behavior of island architecture asphaltenes at the O/W interface in our study is in good agreement with the previous simulation results at nano-scale.35,36 However, the paralleled orientation of archipelago architecture asphaltenes at the interface was firstly observed at mesoscopic scale owing to the advantages provided by DPD simulation, such as the simulation of coarse-grained systems over long length and time scales than MD simulation. Different kinds of island architecture of asphaltenes (molecular weights are lower than 800 g mol−1) were investigated using MD simulations with the limited simulation time (lower than 6 ns). It is also observed that the water droplet in oil is wrapped tightly by asphaltenes when the system reaches equilibrium, which produces asphaltene protective films hindering the droplet–droplet coalescence.
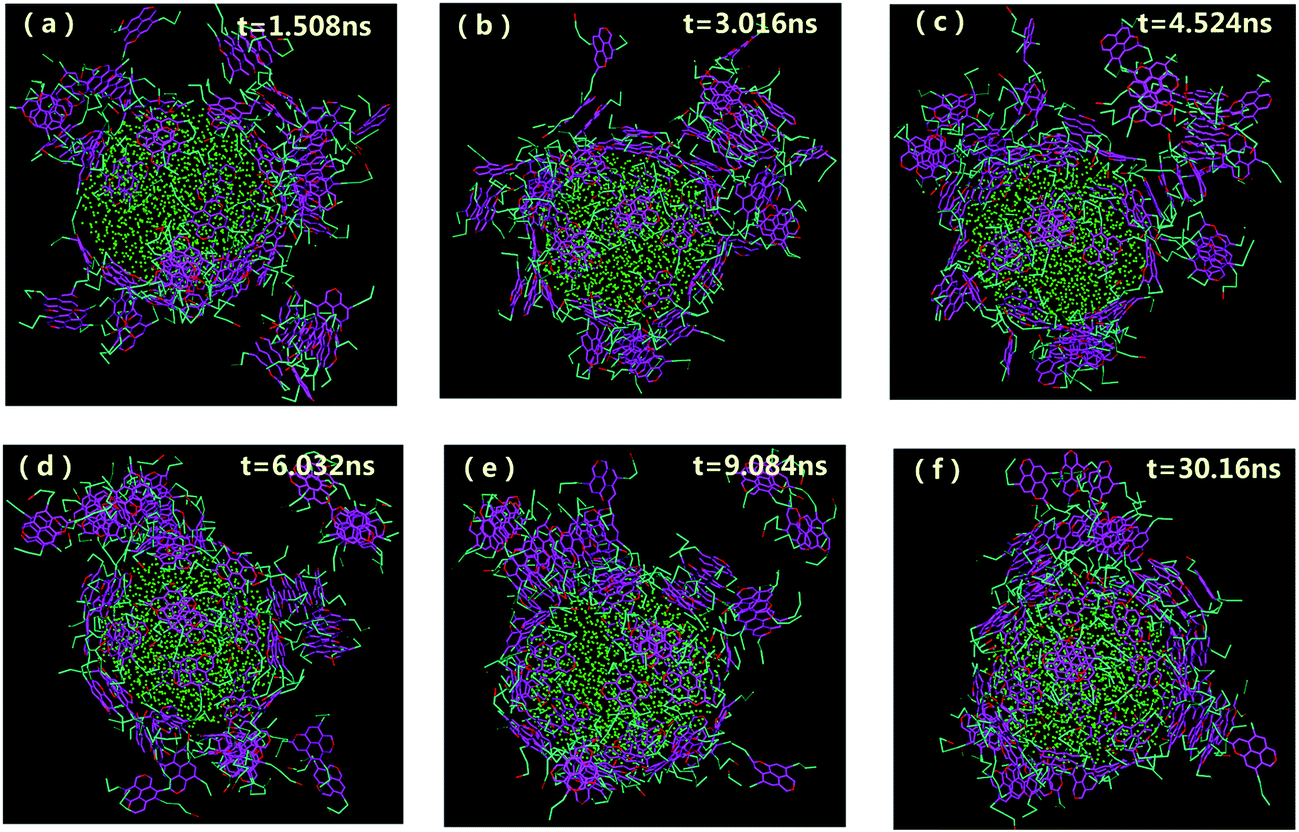 | ||
| Fig. 3 Morphologies of water-in-oil emulsions at different simulation time with the concentrations of 20% asphaltenes of Model B. Different beads in simulations are represented by different colors as can be illustrated from Fig. 1f (the same as below). The toluene molecules are suppressed for clarity. | ||
 | ||
| Fig. 4 Morphologies of water-in-oil emulsions at different concentrations with different asphaltene models. Asphaltenes of Model A were used: (a1) 15%, (a2) 20%, (a3) 25%; asphaltenes of Model B were used: (b1) 15%, (b2) 20%, (b3) 25%; asphaltenes of Model C were used: (c1) 15%, (c2) 20%, (c3) 25%. The toluene molecules are suppressed for clarity. | ||
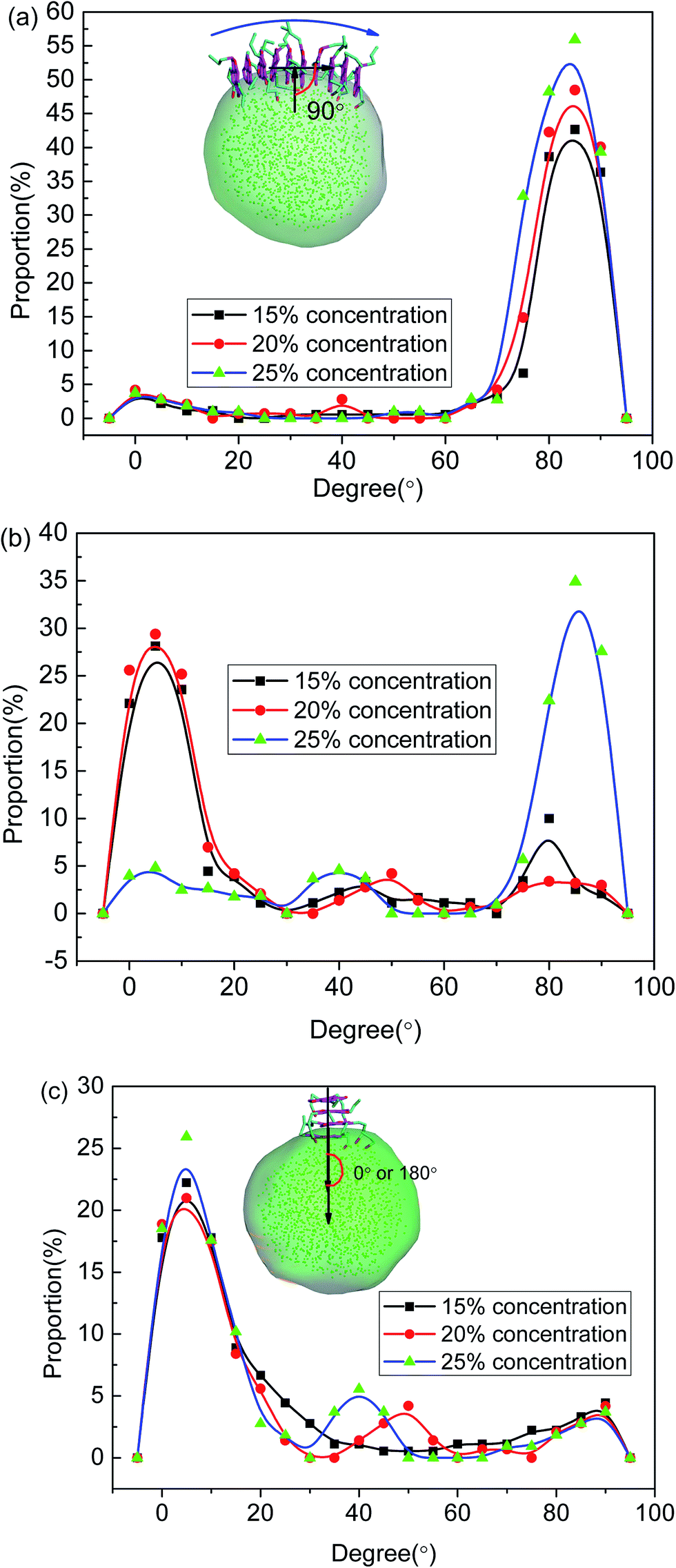 | ||
| Fig. 5 Temporal evolution of angles between the asphaltene aromatic plane and the O/W interface for the systems including (a) asphaltene molecule of Model A, (b) asphaltene molecules of Model B and (c) asphaltene molecules of Model C. The angle is defined on the basis of the normal of the interface and the normal of the aromatic plane of the island-type asphaltene molecule. | ||
3.2 Film properties
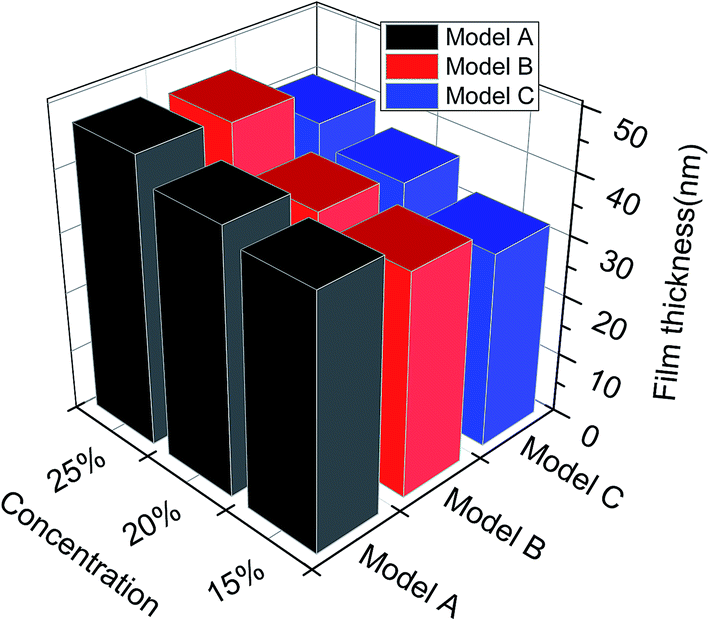 | ||
| Fig. 6 The film thickness for different asphaltene models at different concentrations. | ||
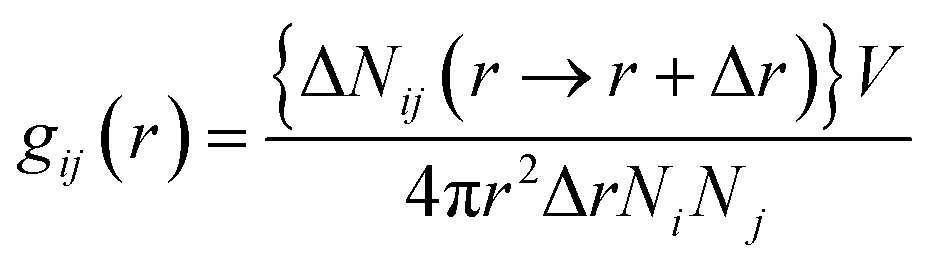 | (1) |
The radial distribution function in this study was calculated for a cut-off radius of 50 Å and an interval distance of 0.05 Å, is shown in Fig. 7. In the asphaltenes of Model A case, it is found that the multi-peaks locates at 5.4 Å, 10.2 Å, 15.1 Å, 21.5 Å, 27.4 Å, respectively. These results depicted that the asphaltene molecules aggregated spontaneously at the interface and formed nanoaggregates with the aromatic rings sheet face-to-face or π–π stacking together. These small peaks reflect that the asphaltenes films adsorbed at the O/W interface are multi-layers due to the intensively π–π bonding between PAHs of asphaltenes which indicates the asphaltene films are much more stable. The nanoaggregates of asphaltenes at the O/W interface were also observed from experiment results42,43 and MD simulations.44 As reported in the previous studies, the nano-aggregates range from 7 to 20 nm in characteristic dimension while the aggregate number ranges from 6 to 14.45 In our simulation results, the aggregates size of 6–22 nm and aggregate number of 7–13 were observed which are qualitatively in good agreement with the previous simulation results. The multi-peaks phenomenon was also observed in the asphaltenes of Model B and C, as shown in Fig. 7b and c. However, the phenomenon of periodic peaks in the radial distribution function for the asphaltenes of Model B and C is not obvious than the asphaltenes of Model A. Moreover, the values of peaks for the asphaltenes of Model B and C are smaller than the asphaltenes of Model A. These results presented the π–π interactions between PAHs of asphaltenes gradually become weaken. Thus, the smaller aggregates size 5–12 nm and aggregate number 3–7 were also observed, as shown in the Fig. 3.
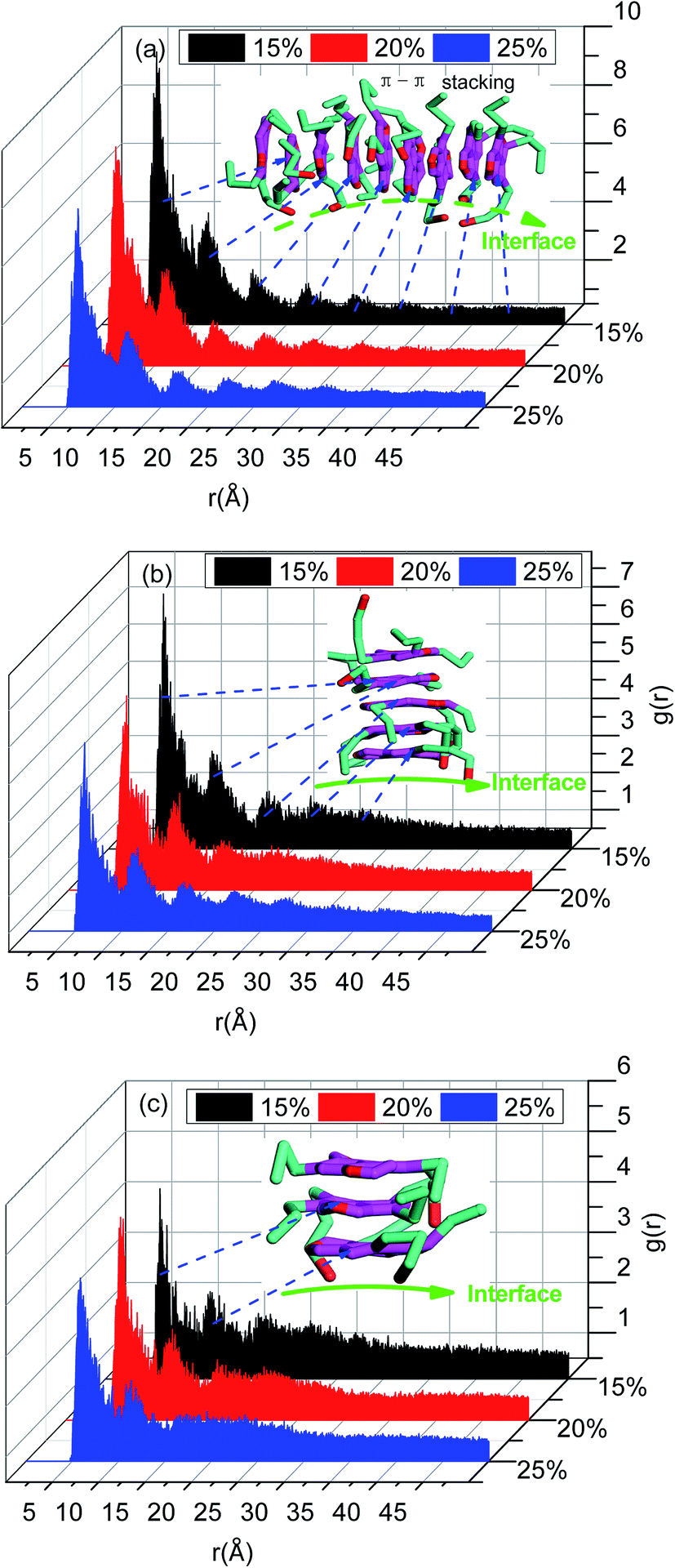 | ||
| Fig. 7 The radial distribution function g(r) for asphaltenes in different simulation systems including (a) asphaltene molecule of Model A, (b) asphaltene molecules of Model B and (c) asphaltene molecules of Model C. | ||
To further investigate the preferred distribution and orientation of asphaltenes at O/W interface, a quantitative analysis on the orientation of the asphaltenes was conducted by calculating the radial distribution function g(r) for different beads in asphaltene structure. As depicted in Fig. 8. The peaks of radial distribution function between W beads and T beads are much higher than the radial distribution function between W beads and B beads as well as H beads. These results are due to the fact that the W beads containing some heteroatoms have stronger affinity for water molecules. What's more, some smaller peaks of radial distribution function between W beads and T beads (marked by green color) were found in Fig. 8. Tough these smaller peaks are not obvious, these smaller peaks can be only found in the radial distribution function between W beads and T beads which illustrates the asphaltene molecules orients at 3.2 Å from the water surface. In the asphaltenes of Model A case, there are two peaks in the radial distribution function between W beads and T beads which is resulted from the perpendicular orientation of asphaltenes at the interface (see the illustration of Fig. 8a). The phenomenon of multi-peaks was observed in the asphaltenes of Model B and C. However, these results are due to the paralleled orientation of asphaltenes at the interface, as shown in illustration of Fig. 8b and c. More importantly, the aliphatic chains without heteroatoms tend to stretch into toluene region with the aliphatic chains with heteroatoms are perpendicular to water region. Jian Cuiying et al.16 proposed that the aliphatic chains hinder the formation of parallel π–π stacking structures of the PHAs while also favoring aggregation through hydrophobic association with enough long side chains. As presented in Fig. 8, the values of peaks between W beads and H beads are smallest in all the beads of asphaltenes, which indicates the side chains of asphaltenes hinder the formation of parallel stacking structures of the polyaromatic cores. In this study, the aliphatic chains of asphaltenes equivalent to dodecane molecule which hinder the π–π stacking interactions of PHAs. Moreover, the number of aliphatic chains in archipelago architecture asphaltenes is bigger than the island architecture asphaltenes. Actually, this result also has an impact on the nanoaggregate structure of asphaltenes at the O/W interface. Because of the increasing aliphatic chains, the PHAs of archipelago architecture asphaltenes preferred parallelling to the O/W interface which increasing the areas of contact with water droplet while decreasing the probability of π–π stacks of PHAs in the oil phases. Consequently, the architecture structure of asphaltenes influenced the nanoaggregate structure configuration of at the O/W interface.
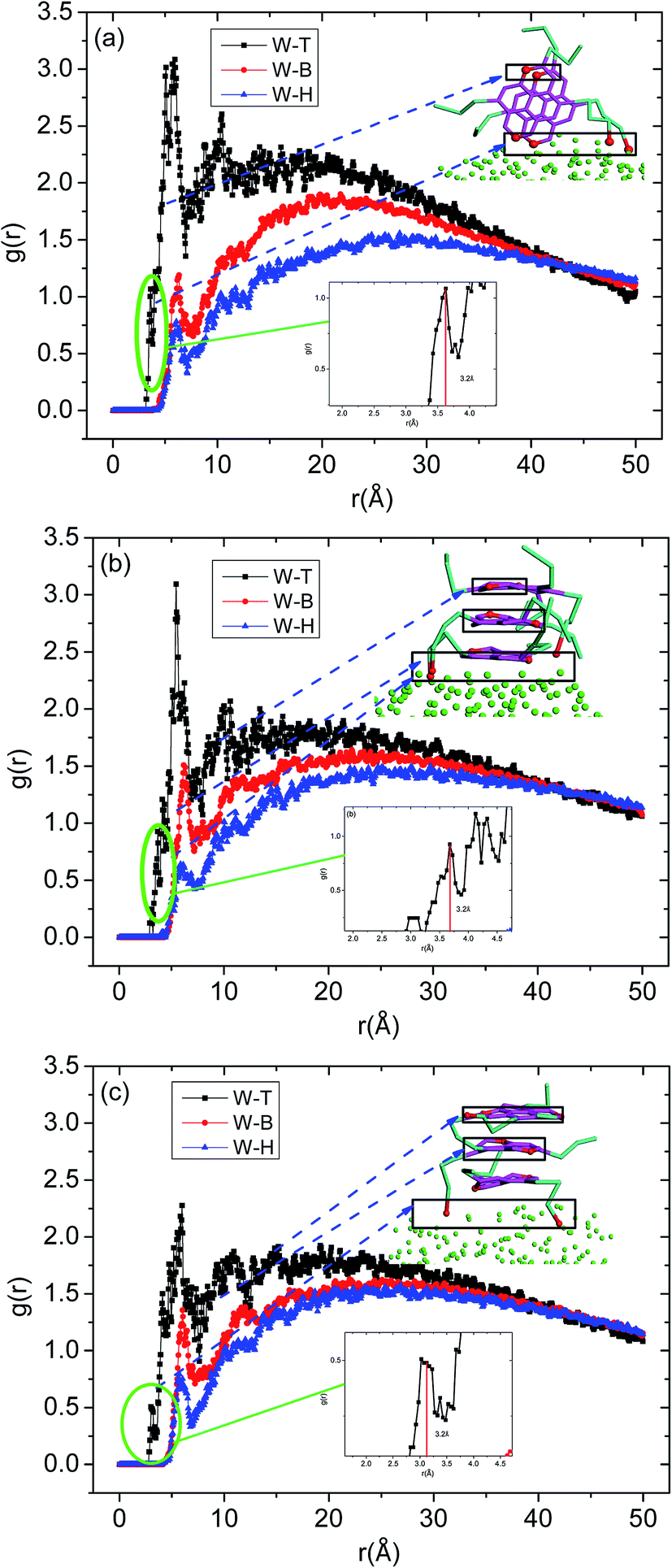 | ||
| Fig. 8 The radial distribution function g(r) for different beads in asphaltenes structure including (a) asphaltene molecules of Model A, (b) asphaltene molecules of Model B and (c) asphaltene molecules of Model C. The concentrations of asphaltenes are 20%. The definition of beads can be found in Fig. 1. | ||
To have a better understanding on the aggregation of asphaltenes at the O/W interface, the nanoaggregate structures were analyzed using order parameter (P). In dynamic simulation processes, an order parameter could be monitored to indicate the changes occurring in the molecular structures, and could thus yielded characteristics of the phase separation and compressibility. The order parameter (P), which was the mean squared deviation from homogeneity for a particular species (A) in volume V, was defined as:46,47
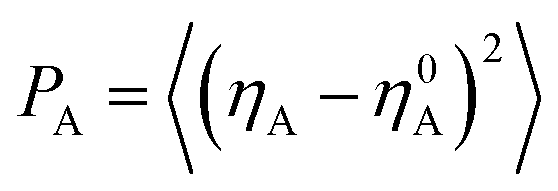 | (2) |
The order parameter versus simulation the time for different asphaltene models at different concentrations were presented in Fig. 9. As time progress, the order parameters of asphaltenes at the O/W interface increase, and finally reach platform. These results showed the asphaltenes films adsorbed at the O/W interface become more ordered and stable which can be reflected from the Fig. 2. The concentrations of asphaltenes in simulation systems also have an influence on the values of order parameters. From the Fig. 9, we can found the order parameters of asphaltenes continuously increase with the increase of concentrations. Thus, higher concentration of asphaltenes could result in the formation of more ordered and stable interfacial films which acts a barrier to droplet coalescence. Moreover, it is also observed that the order parameter for B beads is bigger than H and T beads owing to the multi-layered structure of PAHs. These results also depicted that it is the multi-layered structure of PAHs or π–π staking of PAHs attributes to the stable asphaltene films rather than the alkyl chains with heteroatoms.
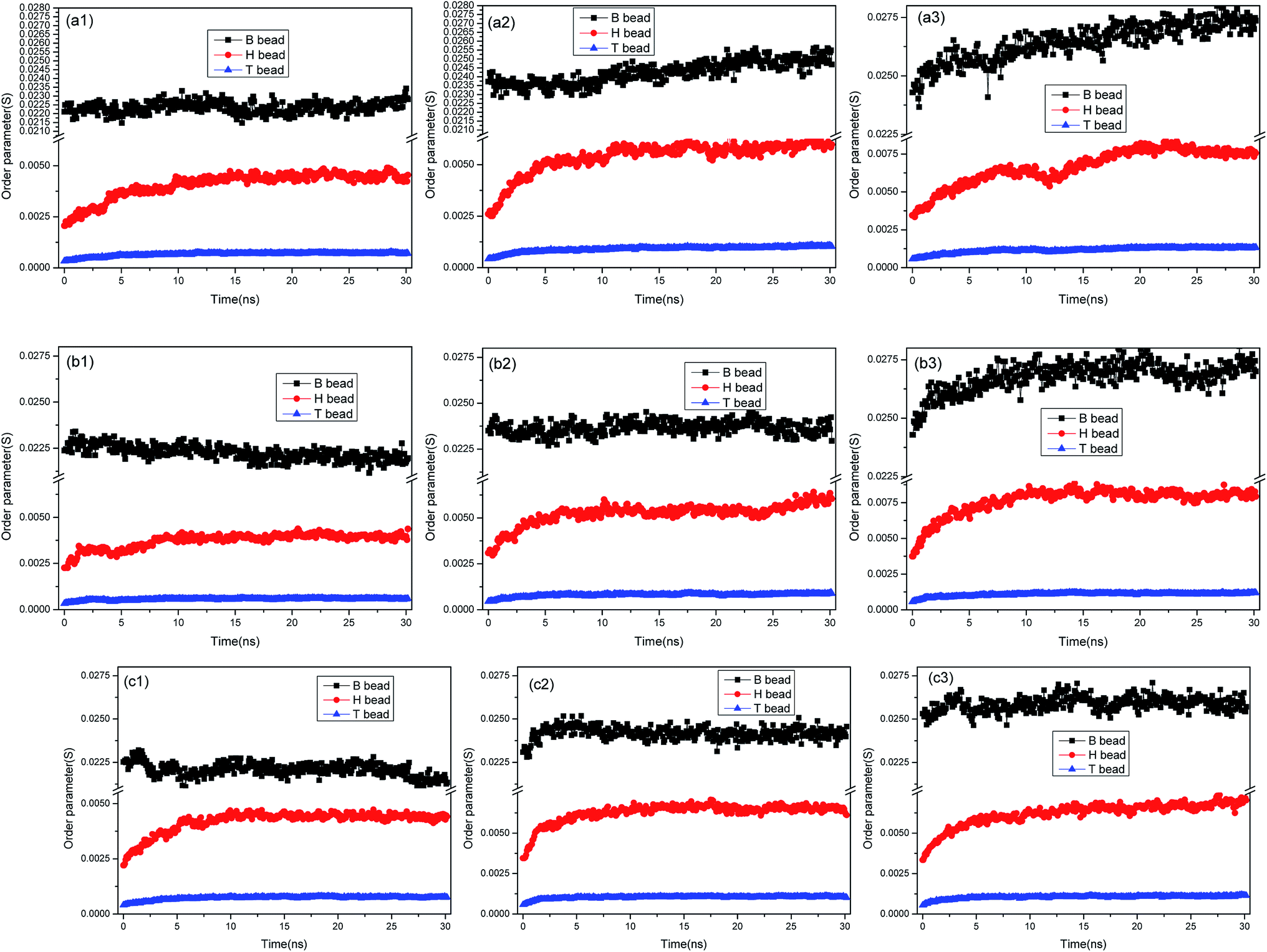 | ||
| Fig. 9 Order parameter versus simulation the time for different asphaltene models at different concentrations. The asphaltenes of Model A were used: (a1) 15%, (a2) 20%, (a3) 25%; the asphaltenes of Model B were used: (b1) 15%, (b2) 20%, (b3) 25%; the asphaltenes of Model C were used: (c1) 15%, (c2) 20%, (c3) 25%. | ||
4. Conclusions
The nanoaggregate structure of asphaltenes at the O/W interface has been studied for decades by different experimental technologies, however, very few studies have been performed on investigating the aggregation and orientation behaviors of asphaltene molecules with different architecture structure at the O/W interface with mesoscale length. Simulation results confirm that the asphaltenes nanoaggregate structures at water-in-oil emulsions interface were majorly mediated by architecture structure as changed from perpendicular to paralleled orientation at the interface with changing from the island architecture to archipelago architecture of asphaltenes. From the simulation results, it is found that the interfacial activity or the driven force of adsorption at interface is attributed from the heteroatoms in asphaltene structures while the formation of nanoaggregate structures at the interface is resulted from the π–π bonding interactions. This nanoaggregate structure transformation of asphaltenes also results into thin-film formation with paralleled orientation of asphaltenes at the interface. From our study, it seems that the integrity of the thin film is formed with an increasing amount of asphaltenes, which might indicate an enhanced stability of water-in-oil emulsions. The resultant effects on the emulsion, including elastic and viscous contributions, will be discussed in the near future.Acknowledgements
This study was supported by Faculty of Earth Resources, China University of Geosciences (Wuhan) and Ecole Central Nantes and Université Laval and the project of integrated exploration project from Chinese Development Research Center of Geological Survey, key fundamental geological study on the integrated exploration area of Baiyinchang polymetallic district, Gansu province (12120114050101). This study was also supported by Special fund for public welfare of the Ministry of Land and Resources: application demonstration and scientific research base construction of three dimensional integrated exploration technology of metal mineral resources (201011002-6). Besides, we acknowledge the supports from the Henan coal-bed methane development and utilization company Ltd. Zhengzhou and Mine companies in Jiaozuo and Yongcheng and Hebi.References
- O. C. Mullins, H. Sabbah, J. Eyssautier, A. E. Pomerantz, L. Barré, A. B. Andrews, Y. Ruiz-Morales, F. Mostowfi, R. McFarlane, L. Goual, R. Lepkowicz, T. Cooper, J. Orbulescu, R. M. Leblanc, J. Edwards and R. N. Zare, Advances in Asphaltene Science and the Yen–Mullins Model, Energy Fuels, 2012, 26, 3986–4003 CrossRef CAS.
- H. Groenzin and O. C. Mullins, Molecular size and structure of asphaltenes from various sources, Energy Fuels, 2000, 14(3), 677–684 CrossRef CAS.
- Z. Hosseini-Dastgerdi, S. A. R. Tabatabaei-Nejad, E. Khodapanah and E. Sahraei, A comprehensive study on mechanism of formation and techniques to diagnose asphaltene structure; molecular and aggregates: a review, Asia-Pac. J. Chem. Eng., 2015, 10(1), 1–14 CrossRef CAS.
- J. G. Speight, Petroleum Asphaltenes-Part 1: Asphaltenes, Resins and the Structure of Petroleum, Oil Gas Sci. Technol., 2004, 59(5), 467–477 CAS.
- Y. Ruiz-Morales and O. C. Mullins, Coarse-grained molecular simulations to investigate asphaltenes at the oil–water interface, Energy Fuels, 2015, 29(3), 1597–1609 CrossRef CAS.
- P. Tchoukov, F. Yang, Z. Xu, T. Dabros, J. Czarnecki and J. Sjöblom, Role of asphaltenes in stabilizing thin liquid emulsion films, Langmuir, 2014, 30(11), 3024–3033 CrossRef CAS PubMed.
- G. Andreatta, C. C. Goncalves, G. Buffin, N. Bostrom, C. M. Quintella, F. Arteaga-Larios, E. Pérez and O. C. Mullins, Nanoaggregates and structure-function relations in asphaltenes, Energy Fuels, 2005, 19(4), 1282–1289 CrossRef CAS.
- H. W. Yarranton, H. Hussein and J. H. Masliyah, Water-in-Hydrocarbon Emulsions Stabilized by Asphaltenes at Low Concentrations, J. Colloid Interface Sci., 2000, 228(1), 52–63 CrossRef CAS PubMed; P. K. Kilpatrick, Water-in-crude oil emulsion stabilization: Review and unanswered questions, Energy Fuels, 2012, 26(7), 4017–4026 CrossRef.
- P. Tchoukov, F. Yang, Z. Xu, T. Dabros, J. Czarnecki and J. Sjöblom, Role of asphaltenes in stabilizing thin liquid emulsion films, Langmuir, 2014, 30(11), 3024–3033 CrossRef CAS PubMed.
- L. Barre, J. Jestin, A. Morisset, T. Palermo and S. Simon, Relation between nanoscale structure of asphaltene aggregates and their macroscopic solution properties, Oil Gas Sci. Technol., 2009, 64, 617–628 CrossRef CAS.
- D. Fenistein, L. Barre, D. Broseta, D. Espinat, A. Livet, J.-N. Roux and M. Scarsella, Viscometric and neutron scattering study of asphaltene aggregates in mixed toluene/heptane solvents, Langmuir, 1998, 14, 1013–1020 CrossRef CAS.
- J. Liu, Y. Zhao and S. Ren, Molecular dynamics simulation of self-aggregation of asphaltenes at an oil/water interface: formation and destruction of the asphaltene protective film, Energy Fuels, 2015, 29(2), 1233–1242 CrossRef CAS.
- M. Duan, X. Song, S. Zhao, S. Fang, F. Wang, C. Zhong and Z. Luo, Layer-by-Layer Assembled Film of Asphaltenes/Polyacrylamide and its Stability of Water-in-Oil Emulsions: A Combined Experimental and Simulation Study, J. Phys. Chem. C, 2017, 121(8), 4332–4342 CAS.
- A. A. Petrov and I. K. Shtof, Investigation of structure of crude oil emulsion stabilizers by means of infrared spectroscopy, Chem. Technol. Fuels Oils, 1974, 10(8), 654–657 CrossRef.
- C. L. Chang and H. S. Fogler, Stabilization of asphaltenes in aliphatic solvents using alkylbenzene-derived amphiphiles. 1. Effect of the chemical structure of amphiphiles on asphaltene stabilization, Langmuir, 1994, 10(6), 1749–1757 CrossRef CAS.
- C. Jian, T. Tang and S. Bhattacharjee, Probing the effect of side-chain length on the aggregation of a model asphaltene using molecular dynamics simulations, Energy Fuels, 2013, 27(4), 2057–2067 CrossRef CAS.
- H. S. Silva, A. C. Sodero, B. Bouyssiere, H. Carrier, J. P. Korb, A. Alfarra, G. Vallverdu, D. Bégué and I. Baraille, Molecular Dynamics Study of Nanoaggregation in Asphaltene Mixtures: Effects of the N, O, and S Heteroatoms, Energy Fuels, 2016, 30(7), 5656–5664 CrossRef CAS.
- H. Rezaei, S. Amjad-Iranagh and H. Modarress, Self-Accumulation of Uncharged Polyaromatic Surfactants at Crude Oil–Water Interface: A Mesoscopic DPD Study, Energy Fuels, 2016, 30(8), 6626–6639 CrossRef CAS.
- Y. Mikami, Y. Liang, T. Matsuoka and E. S. Boek, Molecular dynamics simulations of asphaltenes at the oil–water interface: from nanoaggregation to thin-film formation, Energy Fuels, 2013, 27(4), 1838–1845 CrossRef CAS.
- F. Alvarez, E. A. Flores, L. V. Castro, J. G. Hernández, A. López and F. Vazquez, Dissipative particle dynamics (DPD) study of crude oil–water emulsions in the presence of a functionalized co-polymer, Energy Fuels, 2010, 25(2), 562–567 CrossRef.
- R. Skartlien, S. Simon and J. Sjøblom, A DPD study of asphaltene aggregation: The role of inhibitor and asphaltene structure in diffusion-limited aggregation, J. Dispersion Sci. Technol., 2017, 38(3), 440–450 CrossRef CAS.
- X. Song, P. Shi, M. Duan, S. Fang and Y. Ma, Investigation of demulsification efficiency in water-in-crude oil emulsions using dissipative particle dynamics, RSC Adv., 2015, 5(77), 62971–62981 RSC.
- R. Skartlien, S. Simon and J. Sjöblom, DPD Molecular Simulations of Asphaltene Adsorption on Hydrophilic Substrates: Effects of Polar Groups and Solubility, J. Dispersion Sci. Technol., 2016, 37(6), 866–883 CrossRef CAS.
- S.-F. Zhang, L. L. Sun, J.-B. Xu, H. Wu and H. Wen, Aggregate structure in heavy crude oil: using a dissipative particle dynamics based mesoscale platform, Energy Fuels, 2010, 24, 4312–4326 CrossRef CAS.
- R. D. Groot and P. B. Warren, Dissipative particle dynamics: bridging the gap between atomistic and mesoscopic simulation, J. Chem. Phys., 1997, 107, 4423–4435 CrossRef CAS.
- Y. T. Liu, Y. R. Li and X. Wang, Spontaneous onion shape vesicle formation and fusion of comb-like block copolymers studied by dissipative particle dynamics, RSC Adv., 2017, 7(9), 5130–5135 RSC.
- G. Dorenbos, Pore design within amphiphilic polymer membranes: linear versus Y-shaped side chain architectures, RSC Adv., 2014, 4(92), 51038–51046 RSC.
- X. Song, P. Shi, S. Zhao, M. Duan, C. Wang and Y. Ma, Dissipative Particle Dynamics Study on the Aggregation Behavior of Asphaltenes under Shear Fields, Ind. Eng. Chem. Res., 2016, 55(33), 9077–9086 CrossRef CAS.
- C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- K. Shi, C. Lian, Z. Bai, S. Zhao and H. Liu, Dissipative particle dynamics study of the water/benzene/caprolactam system in the absence or presence of non-ionic surfactants, Chem. Eng. Sci., 2015, 122, 185–196 CrossRef CAS.
- S. F. Zhang, J. B. Xu, H. Wen and S. Bhattacharjee, Integration of Rotational Algorithms into Dissipative Particle Dynamics: Modeling Polyaromatic Hydrocarbons on the Meso-Scale, Mol. Phys., 2011, 109, 1873–1888 CrossRef CAS.
- F. Trejo, J. Ancheyta and M. S. Rana, Structural Characterization of Asphaltenes Obtained from Hydro Processed Crude Oils by SEM and TEM, Energy Fuels, 2009, 23, 429–439 CrossRef CAS.
- Accelrys MS Modeling 6.0, Accelrys, Inc., San Diego, CA, 2011 Search PubMed.
- Y. Li, Y. Guo, G. Xu, Z. Wang, M. Bao and N. Sun, Dissipative particle dynamics simulation on the properties of the oil/water/surfactant system in the absence and presence of polymer, Mol. Simul., 2013, 39, 299–308 CrossRef.
- J. Liu, Y. Zhao and S. Ren, Molecular dynamics simulation of self-aggregation of asphaltenes at an oil/water interface: formation and destruction of the asphaltene protective film, Energy Fuels, 2015, 29(2), 1233–1242 CrossRef CAS.
- T. F. Headen, E. S. Boek and N. T. Skipper, Evidence for asphaltene nanoaggregation in toluene and heptane from molecular dynamics simulations, Energy Fuels, 2009, 23(3), 1220–1229 CrossRef CAS.
- D. M. Sztukowski and H. W. Yarranton, Rheology of asphaltene-toluene/water interfaces, Langmuir, 2005, 21, 11651–11658 CrossRef CAS PubMed.
- J. P. Rane, V. Pauchard, A. Couzis and S. Banerjee, Interfacial rheology of asphaltenes at oil–water interfaces and interpretation of the equation of state, Langmuir, 2013, 29(15), 4750–4759 CrossRef CAS PubMed.
- Y. Li, Y. Guo, M. Bao and X. Gao, Investigation of Interfacial and Structural Properties Of CTAB at the Oil/Water Interface Using Dissipative Particle Dynamics Simulations, J. Colloid Interface Sci., 2011, 361, 573–580 CrossRef CAS PubMed.
- P. Tchoukov, F. Yang, Z. Xu, T. Dabros, J. Czarnecki and J. Sjöblom, Role of asphaltenes in stabilizing thin liquid emulsion films, Langmuir, 2014, 30(11), 3024–3033 CrossRef CAS PubMed.
- Z. Luo and J. Jiang, pH-sensitive drug loading/releasing in amphiphilic copolymer PAE-PEG: integrating molecular dynamics and dissipative particle dynamics simulations, J. Controlled Release, 2012, 162, 185–193 CrossRef CAS PubMed.
- J. Jestin, S. Simon, L. Zupancic and L. Barre, A Small Angle Neutron Scattering Study of the Adsorbed Asphaltene Layer in Water-in-Hydrocarbon Emulsions: Structural Description Related to Stability, Langmuir, 2007, 23, 10471–10478 CrossRef CAS PubMed.
- V. Verruto and P. K. Kilpatrick, Water-in-Model Oil Emulsions Studied by Small-Angle Neutron Scattering: Interfacial Thickness and Composition, Langmuir, 2008, 24, 12807–12822 CrossRef CAS PubMed.
- Y. Mikami, Y. Liang, T. Matsuoka and E. S. Boek, Molecular Dynamics Simulations of Asphaltenes at the Oil–Water Interface: From Nanoaggregation to Thin-Film Formation, Energy Fuels, 2013, 27, 1838–1845 CrossRef CAS.
- V. J. Verruto and P. K. Kilpatrick, Water-in-model oil emulsions studied by small-angle neutron scattering: interfacial film thickness and composition, Langmuir, 2008, 24(22), 12807–12822 CrossRef CAS PubMed.
- H. Liu, Y. Li, W. E. Krause, M. A. Pasquinelli and O. J. Rojas, Mesoscopic Simulations of the Phase Behavior of Aqueous EO19PO29EO19 Solutions Confined and Sheared by Hydrophobic and Hydrophilic Surfaces, ACS Appl. Mater. Interfaces, 2012, 4(1), 87–95 CAS.
- X. Song, S. Zhao, S. Fang, Y. Ma and M. Duan, Mesoscopic Simulations of Adsorption and Association of PEO-PPO-PEO Triblock Copolymers on a Hydrophobic Surface: From Mushroom Hemisphere to Rectangle Brush, Langmuir, 2016, 32(44), 11375–11385 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06717h |
| ‡ Present addresses: 8 Xindu Avenue, Xindu District, Chengdu, Sichuan 610500, P. R. China. |
| This journal is © The Royal Society of Chemistry 2017 |