
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-walled carbon nanotubes as stabilizing agents in red phosphorus Li-ion battery anodes†
Jasmin Smajic ,
Amira Alazmi,
Shashikant P. Patole and
Pedro M. F. J. Costa*
,
Amira Alazmi,
Shashikant P. Patole and
Pedro M. F. J. Costa*
Physical Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia. E-mail: pedro.dacosta@kaust.edu.sa
First published on 16th August 2017
Abstract
Phosphorus boasts extremely high gravimetric and volumetric capacities but suffers from poor electrochemical stability with significant capacity loss immediately after the first cycle. We propose to circumvent this issue by mixing amorphous red phosphorus with single-walled carbon nanotubes. Employing a non-destructive sublimation–deposition method, we have synthesized composites where the synergetic effect between red phosphorus and single-walled carbon nanotubes allows for a considerable improvement in the electrochemical stability of battery anodes. In contrast to the average 40% loss of capacity after 50 cycles for other phosphorus–carbon composites in the literature, our material shows losses of just 22% under analogous cycling conditions.
1. Introduction
By permeating through every aspect of our lives, lithium-ion batteries (LIBs) have proved to be one of the most important energy storage systems available.1–3 They have enabled, for instance, the wireless revolution of mobile phones, laptops and tablets and have the potential to catalyze the transition towards a renewable energy future.4,5 Still, the LIBs currently on the market are not adequate for the challenges ahead and, being the most viable option for the immediate future, there is a pressing need to increase their energy density, decrease production/operational costs and make them more environmentally sustainable.6–8With a theoretical gravimetric capacity of 372 mA h g−1, graphite (either natural or synthetic) is presently being used as the host material for Li+ intercalants in commercial LIBs anodes.9 Alternative materials with higher theoretical capacities and different lithium storage mechanisms continue to be investigated.10 These include Si (3579 mA h g−1), Ge (1384 mA h g−1), Sn (960 mA h g−1) and their various compounds.11 Phosphorus, with theoretical gravimetric and volumetric capacities of 2596 mA h g−1 and 2266 mA h cm−3, respectively, is another promising anode material (by forming the Li3P phase) but its viability has not been properly studied.12 Phosphorus is the 11th most abundant element in the earth's crust and is extracted from geological deposits of phosphates.10,13,14 Additionally, phosphorus is one of the main biomass constituents, so it is naturally recycled into the biosphere and does not present availability or environmental concerns.10 Among the various phosphorus allotropes, there are three main ones: white, black and red. The white phase is toxic and extremely reactive whilst black phosphorus was, until recently, notoriously hard to produce as it requires high temperature and pressure. Conversely, the red phosphorus allotrope is widely available, relatively safe and has the highest commercial value.
From the above, red P could become a strong contender to replace graphite as the preferred anode material for LIBs. However, considerable volume changes take place in phosphorus-based anodes during lithiation/delithiation resulting in their pulverization. Inevitably, this leads to loss of contact between the active material and the current collector, explaining the poor cycling stability and significant capacity loss (immediately after the first cycle) of phosphorus anodes. In addition, the volume changes induce an unstable solid–electrolyte interface (SEI) which, in turn, causes unwanted chemical reactions that consume Li+. One common strategy used to circumvent these issues is to mix phosphorus powders with carbon materials via ball milling or sublimation techniques.15–21 The graphitic particles act as electrically-conductive additives that bridge the alloying phosphorus and the current collector. An example of the ball milling approach is the process described by Yuan et al. that originated a red P – multi-walled carbon nanotube composite with a remarkable initial charge specific gravimetric capacity (2133 mA h g−1).16 Unfortunately, it suffered from low stability resulting in the loss of about 1000 mA h g−1 after 50 cycles. Sublimation is a more popular approach to produce the red P–C composites for LIBs, with different carbon allotropes having been investigated, from activated carbon to reduced graphene oxide.19,20,22,23 Overall, the starting gravimetric capacities reported are high, mostly over 1000 mA h g−1, but the cycling stability continues to be a preponderant challenge. On average, approximately 40% of the initial capacity is lost after 50 cycles (Table S1†).
Interestingly, single-walled carbon nanotubes (SWCNT) have not yet been studied for this purpose. The morphology and exceptional mechanical and electrical properties of SWCNT24–29 could help preserve the structural integrity of red P anodes and thereby prevent the loss of electrical contact with the current collector (Fig. S1†). Following a recent report of sodium-ion batteries mechanically stabilized with SWCNT,30 we synthesized an amorphous red P–SWCNT composite using a non-destructive sublimation–deposition method. Upon its structural and chemical analysis, the composite's electrochemical performance was evaluated.
2. Experimental section
2.1 Material synthesis
SWCNT, with diameters of 0.7–1.1 nm (as per vendor), were purchased from Sigma-Aldrich and used as-received. Red P powder, with purity ≥ 99.99%, was also obtained from Sigma-Aldrich. To prepare the composite, first, the red P was washed in deionized water for 20 min. That was followed by filtering and overnight vacuum drying of the resulting powder at 100 °C. Note that a preliminary washing step is crucial since the red P powder, when exposed to air (oxygen) and humidity for too long, gets covered with hygroscopic oxides. Besides the wet and sticky appearance, the oxide coating may suppress the sublimation and/or introduce impurities into the final material.21 The next step consisted in hand-grinding, for 60 min and using a pestle and mortar, the washed red P and SWCNT in an optimized weight ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. The black-coloured mixture was then sealed in a quartz ampoule under vacuum (<10−2 mbar) and heated in a muffle furnace at 600 °C for 2 hours. This temperature was deliberately chosen to promote the sublimation of red P (Tsubl. = 416 °C) and its subsequent vapor diffusion in-between the bundles and interstices of SWCNT. Ideally, this results in a better dispersion of the re-deposited P hence minimizing strain “hot-spots“ caused by the presence of aggregates and their large volume change during lithiation/delithiation. After the sublimation–deposition process, the temperature was lowered to 300 °C and held there for 48 hours. This likely assisted in the structural conversion to red P of any non-intentional traces of white P.31,32 Finally, the ampoule was allowed to naturally cool down to room temperature, then opened inside a fume hood and the powdered composite material collected.
:3. The black-coloured mixture was then sealed in a quartz ampoule under vacuum (<10−2 mbar) and heated in a muffle furnace at 600 °C for 2 hours. This temperature was deliberately chosen to promote the sublimation of red P (Tsubl. = 416 °C) and its subsequent vapor diffusion in-between the bundles and interstices of SWCNT. Ideally, this results in a better dispersion of the re-deposited P hence minimizing strain “hot-spots“ caused by the presence of aggregates and their large volume change during lithiation/delithiation. After the sublimation–deposition process, the temperature was lowered to 300 °C and held there for 48 hours. This likely assisted in the structural conversion to red P of any non-intentional traces of white P.31,32 Finally, the ampoule was allowed to naturally cool down to room temperature, then opened inside a fume hood and the powdered composite material collected.
2.2 Electrochemical characterization
All electrochemical measurements were done at room temperature. The anodes were fabricated by manually mixing the active material, polyvinylidene fluoride (PVDF, from Sigma-Aldrich) and acetylene black (AB, from Alfa Aesar) in a weight ratio of 85:10:5, respectively. The active material was either P–SWCNT composite, red P or SWCNT. With the addition of 1-methyl-2-pyrrolidone (NMP), a slurry was formed and this was cast on a Cu foil. These electrodes were then dried overnight, under vacuum at 100 °C, and compacted with a hand-operated hydraulic press. That was followed by another drying step, again in vacuum at 100 °C, for one hour and a final pressing. All electrodes were 5 mm × 10 mm and had an active material loading of ∼2 mg cm−2. The electrodes' thickness was ∼50 μm.
The electrochemical assays were carried out on a Biologic Science Instruments VMP3 potentiostat using 2032-type coin cells in a half-cell configuration. The cells were assembled inside an MBraun LABstar Ar-filled glove box with the composite material acting as the working electrode and Li metal foils as the counter and reference electrodes. A Celgard 2325 microporous polymer film was used as the separator and 1.0 M LiPF6 in EC:DEC = 1:1 (v/v) was used as the electrolyte. The galvanostatic charge–discharge profiles were tested at different current densities ranging from 50 mA g−1 to 1000 mA g−1, between 0.001 V and 2 V (vs. Li+/Li). After the cells had reached the cut-off voltage, they were relaxed for 1 second before continuing with the measurements. The cyclic voltammetry (CV) was carried out between 0.001 V and 2.5 V (vs. Li+/Li), at 0.1 mV s−1. Electrochemical impedance spectroscopy (EIS) was done in the frequency range between 100 mHz and 0.1 MHz. All specific charge capacities are reported by the weight of the total P–SWCNT composite.
2.3 Material characterization
The thermal stability and approximate composition of the source and composite materials were studied by thermogravimetric analysis (TGA) on a NETZSCH TG 209 F1 Libra. The sample was heated from room temperature to 1000 °C, in N2 atmosphere, at a rate of 10 °C min−1. The crystal structures were characterized by powder X-ray diffraction (XRD) on a Bruker D8 ADVANCE using a Cu Kα radiation source and operated in the 2θ range of 10° to 90° (scan rate of 5° min−1). The Raman spectra were taken on a Witec alpha 300 RA with a 633 nm solid-state laser. Scanning electron microscopy (SEM) was performed in an FEI Nova NanoSEM, operated at 5 kV and with the sample positioned 5 mm from the polepiece. Transmission electron microscopy (TEM) micrographs were taken on an FEI Titan operated at 300 kV and equipped with EDX. For the electron microscopy analyses, samples were prepared by making a 0.2 mg ml−1 suspension of the material in ethanol. This was then dispersed in a bath sonicator and drop cast on a SEM stub or a TEM grid. X-ray photoelectron spectroscopy (XPS) was performed on Kratos Analytical Amicus equipped with an achromatic Al Kα source (1468.6 eV). The source was operated at a voltage of 10 kV and current of 10 mA. The survey spectra were acquired using a step of 1 eV, whereas 0.1 eV was employed for acquiring narrow scan regions. The pressure in the analysis chamber was less than 5 × 10−6 Pa. Fourier transform infrared spectroscopy (FTIR) was done on a Thermo Scientific Nicolet iS10 spectrometer from 400 cm−1 to 4000 cm−1.3. Results and discussion
To understand the structural characteristics of the P–SWCNT composite and its source materials, Raman and XRD were used. Raman is one of the most powerful characterization methods to study carbon nanotubes. In Fig. 1a, the spectra of the source and composite materials are shown. The radial breathing modes (RBM), as well as the D and G bands, are visible in the SWCNT source material. The RBM region has a dominant peak at 251 cm−1 which is correlated to a nanotube diameter of around 1 nm.33–35 This value is consistent with the product technical specifications provided by the SWCNT supplier. A small diameter is advantageous as the nanotubes become more resistant to radial stress and have smaller Poisson's ratio, which helps to preserve their structural integrity under pressure.36–38 In addition, it favors narrower bundles which in turn are more resistant to shear and axial stress.39 The D band in the Raman spectrum is present at 1303 cm−1.40 It originates from the cumulative effect of the breathing motion of the sp2 bonded C atoms at the edges of graphene layers and the breaking of the symmetry because of defects and stacking disorder in the crystal structure of graphite.19,41,42 As regards the G band, it consists of two separated components. The lower frequency component (1547 cm−1) is commonly labeled as G−, while the higher frequency component (1586 cm−1) is the G+. The G− is due to C atom vibrations in the circumferential direction and the G+ originates from the C atom vibrations in the axial direction of the nanotubes.43 Their presence and shape confirm the single-walled and semiconducting nature of the nanotubes.44 Despite being semiconducting, these nanotubes were selected due to their purity and conformational (n, m) uniformity (as per vendor). Since the intensity ratio of the D and G bands (ID/IG) ≈ 0.08, it can be concluded that the source SWCNT are of high structural quality. Amorphous red P is characterized by three overlapping bands, between 300 cm−1 to 500 cm−1, which are used to distinguish it from other P allotropes.45–50 The Raman spectrum of the source red P (Fig. 1a) shows the three expected peak positions at 349 cm−1, 393 cm−1 and 458 cm−1. Comparing the Raman peaks of the source and composite materials, the ID/IG ratio does not change significantly which confirms the non-destructive nature of the synthesis process. By contrast, it is seen that the G band undergoes a redshift of 5 cm−1 (from 1587 cm−1 to 1582 cm−1). This can be explained by the intercalation of red P particles in SWCNT bundles.51 Also, the reduction of signal intensity for the RBM and red P bands in the composite is noticeable. The first, in particular, could be justified by an extensive P coating on the outer surface of the nanotubes (which would damp the magnitude of the breathing signal).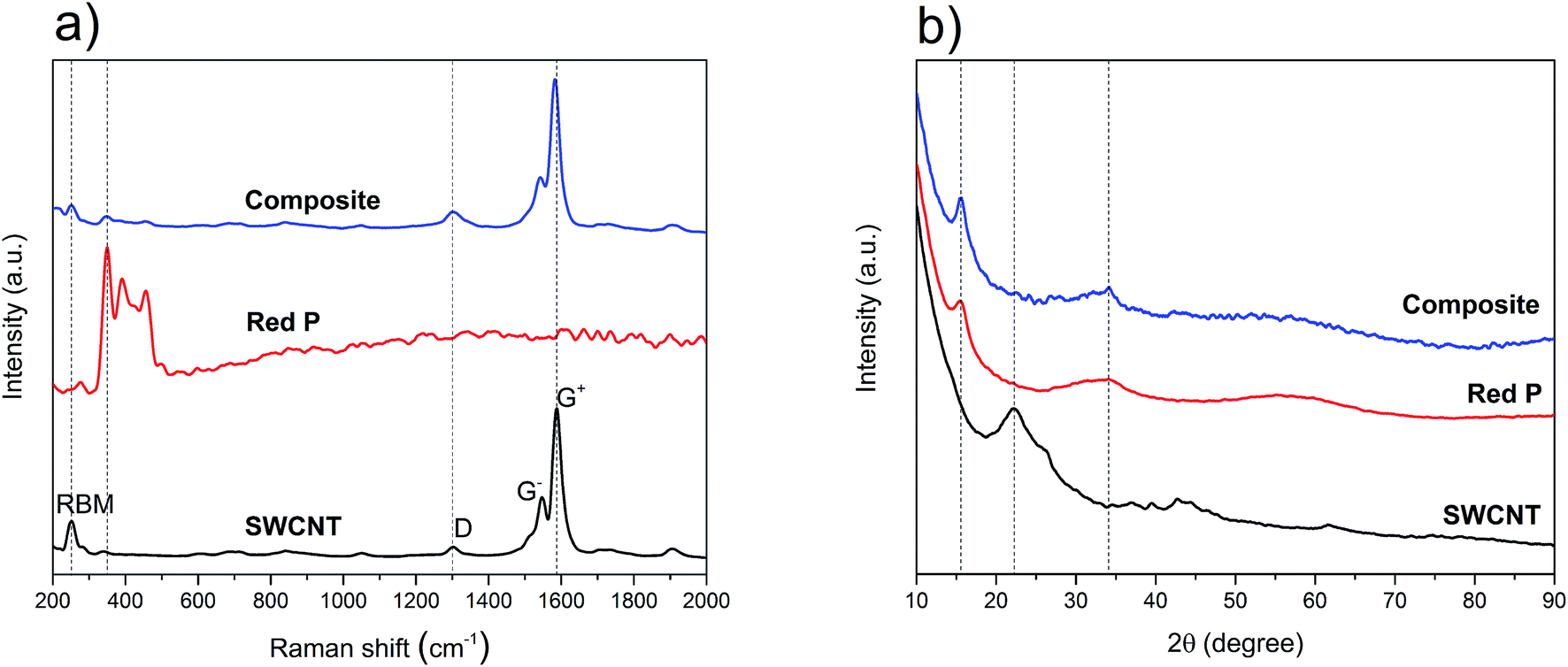 | ||
| Fig. 1 (a) Raman spectra of SWCNT, red P and the composite; (b) powder XRD spectra of SWCNT, red P and the composite; the dashed lines in (a) and (b) are guides for the eye. | ||
From the above, while the presence and structural quality of the SWCNT were confirmed in the composite, asserting the integrity of the red P phase required further analysis. The XRD spectra of the source and composite materials are shown in Fig. 1b. For the SWCNT, the broad peaks at 22° and 43° are attributed to the SWCNT52 while the faint peak at 26° is possibly due to nanotubes as well as small amounts of graphitic particles.53–55 The spectrum of phosphorus, with two broad peaks at 16° and 34°, indicates the presence of a mostly amorphous structure that can only be attributed to the red P allotrope.17,20 With regards to the composite material, and in contrast to the Raman data, the spectrum was dominated by the amorphous red P peaks, with no other phases identified. Given the excess wt% of P used and its expected impregnation throughout the nanotubes' network, the absence of SWCNT peaks in the composite's XRD is not surprising. Overall, the Raman and XRD observations imply that the final composite is a physical mixture of P and SWCNT, corroborating previous reports that also used the sublimation–deposition method.16,30,56
Following the structural characterisation, the final P–SWCNT ratio and thermal stability of these materials were evaluated with TGA (under inert atmosphere). For the pristine SWCNT (Fig. 2a), there is a gradual weight loss, more accentuated from 500 °C onwards. Here, the presence of Co and Mo particles as well as assorted carbonaceous species – all growth by-products that were identified in the as-received SWCNT (not shown), could have contributed in promoting carbonization processes and vaporization of functional groups. Under equal conditions, the red P starts sublimating at around 400 °C and is almost entirely depleted at 500 °C (Fig. 2a). The weight gain at 350–400 °C is expected due to the sudden solid–gas phase transition that takes place.57 The thermal profile of the composite material (Fig. 2a) is clearly the sum of the parts, thereby confirming this is a purely physical mixture. It can also be seen that the 7:3 P–SWCNT mixture used results in a composition that is closer to 6:4. The loss of P is justified by its non-intentional re-deposition on the walls of the quartz ampoule.
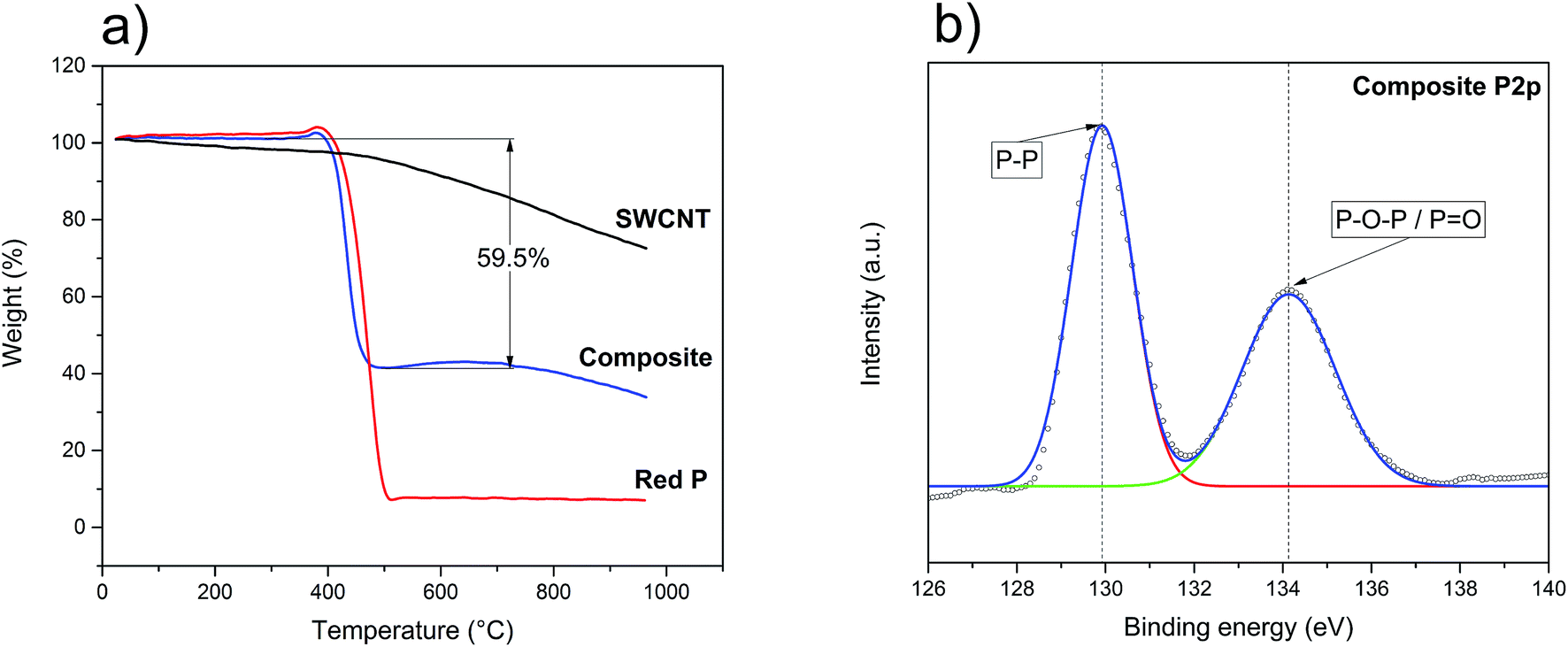 | ||
| Fig. 2 (a) TGA analysis of the source materials and composite; (b) high-resolution XPS P2p spectrum of the P–SWCNT composite. | ||
With both structural integrity and matrix/filler ratio studied, it was then necessary to understand the surface chemistry of the composite. The survey XPS spectrum is shown in Fig. S2a† where it is possible to identify the peaks of P, C and O. The more in-depth analysis of the P2p and O1s signals (Fig. 2b and S2b†) revealed the presence of phosphorus oxide. The P2p spectrum (Fig. 2b) consists of two peaks. One of them corresponds to the P–P bond and is located at 129.9 eV, while the other one, at 134.1 eV, corresponds to phosphorus oxides (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/P–O–P).58 The important conclusion is that there are no P–C peaks further confirming that no chemical reaction occurs and that the composite is held together only by van der Waals forces.59,60 The formation of this oxide, not identified in the above Raman, XRD and TGA analyses, is likely the result of air/moisture exposure previous to the XPS study (e.g. during transport and sample preparation). Knowing phosphorus oxides can be highly hygroscopic,61–63 the stability of an assembled working electrode was tested by exposing it to air. In less than 3 minutes, it was possible to identify the formation of water droplets (Fig. S3a and b†). Accordingly, the complete fabrication of the battery, from the initial washing of red P until the assembly of the half-cells, should ideally be done under an inert atmosphere. Awareness of this issue when working with red P-based anodes is all the more important as moisture may have adverse effects on the LiPF6-based electrolyte.64,65 In the present work, all possible measures were undertaken to minimize the red P exposure to oxygen/moisture prior to the electrochemistry assays. In these circumstances, the oxide concentration should be minimal and not influence the electrochemical behavior of the composite.
O/P–O–P).58 The important conclusion is that there are no P–C peaks further confirming that no chemical reaction occurs and that the composite is held together only by van der Waals forces.59,60 The formation of this oxide, not identified in the above Raman, XRD and TGA analyses, is likely the result of air/moisture exposure previous to the XPS study (e.g. during transport and sample preparation). Knowing phosphorus oxides can be highly hygroscopic,61–63 the stability of an assembled working electrode was tested by exposing it to air. In less than 3 minutes, it was possible to identify the formation of water droplets (Fig. S3a and b†). Accordingly, the complete fabrication of the battery, from the initial washing of red P until the assembly of the half-cells, should ideally be done under an inert atmosphere. Awareness of this issue when working with red P-based anodes is all the more important as moisture may have adverse effects on the LiPF6-based electrolyte.64,65 In the present work, all possible measures were undertaken to minimize the red P exposure to oxygen/moisture prior to the electrochemistry assays. In these circumstances, the oxide concentration should be minimal and not influence the electrochemical behavior of the composite.
SEM and TEM images of the composite and source materials samples are shown in Fig. 3 and S4.† From Fig. 3a and c, it is observed that the composite shows a dense morphology, with the P matrix covering the bundles of nanotubes. From Fig. 3b and d, it is possible to confirm the presence of C, O, P and Co, the latter originating from nanoparticles that were remainders of the SWCNT growth. Further characterisation showed that these catalyst nanoparticles were covered with a thin graphitic shell and were composed of Co and Mo (Fig. S5†). Given their encapsulation and relatively small number,66 these growth by-products are not expected to have influenced the electrochemical behavior of the composite.
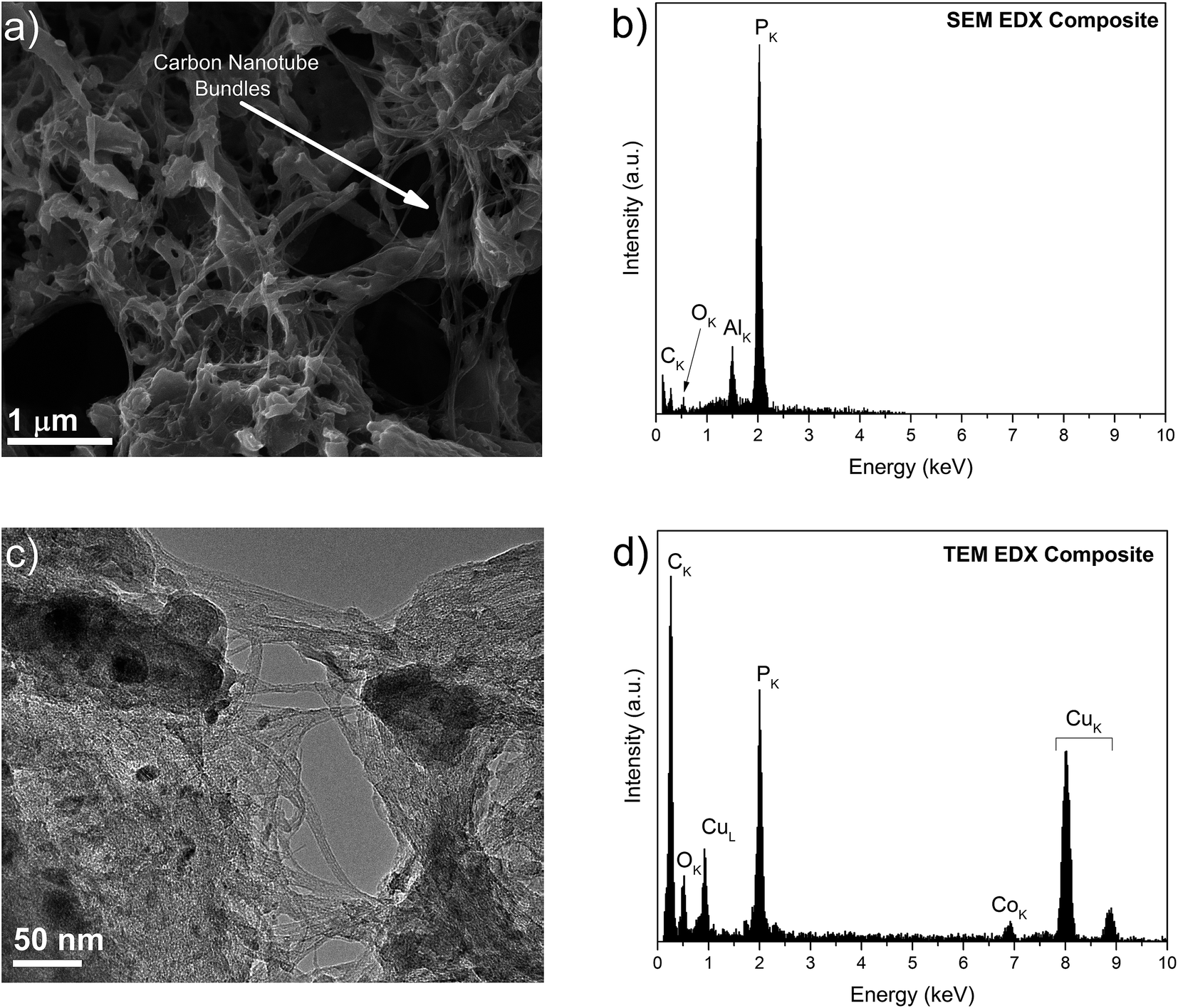 | ||
| Fig. 3 (a) SEM image of the P–SWCNT composite, where the arrow points to a bundle of nanotubes; (b) EDX spectrum of the composite in (a); (c) TEM micrograph of the composite in (a), where the intermixing of P and nanotubes, along with the presence of growth catalyst nanoparticles, is clear; (d) the EDX spectrum of the area shown in (c), confirming the presence of Co. | ||
It is important to note that for the electrochemical experiments (CV and galvanostatic charge/discharge) freshly prepared electrodes should be used. This is to avoid the build-up of surface oxides, as previously explained. Fig. 4a shows the CV of a composite anode. The peaks at ∼2.95 V, ∼2.4 V and ∼1.3 V, present only in the first cathodic scan, are attributed to the decomposition of the electrolyte and the SEI formation. The overlapped peaks between ∼1 V and ∼0.2 V, in the cathodic scan, are due to the sequential lithiation of the red P to form LixP (x = 1–3). The main reduction peak at ∼0.6 V is accompanied, from the second cycle onwards, by two shoulder peaks, at ∼0.8 V and ∼0.9 V, which increase in intensity with the number of cycles (Fig. 4a, also refer to Fig. S6†). This higher visibility of the shoulder peaks (and the widening of the main peaks because of that) is attributed to the activation of red P. The incomplete peak at ∼0.01 V may be attributed to the irreversible intercalation of lithium ions into SWCNT.67 Similarly, from the second cycle onwards, the main oxidation peak at ∼1.2 V, is accompanied by two shoulder peaks at ∼1.3 V and ∼1.4 V. Three oxidation and three reduction peaks are present, indicating a reversible stepwise lithiation/alloying process of red P (Fig. S6†). Due to the absence of significant peaks for Li+ intercalation into SWCNT, it would seem that the vast majority of obtained gravimetric capacities can be attributed to P only.
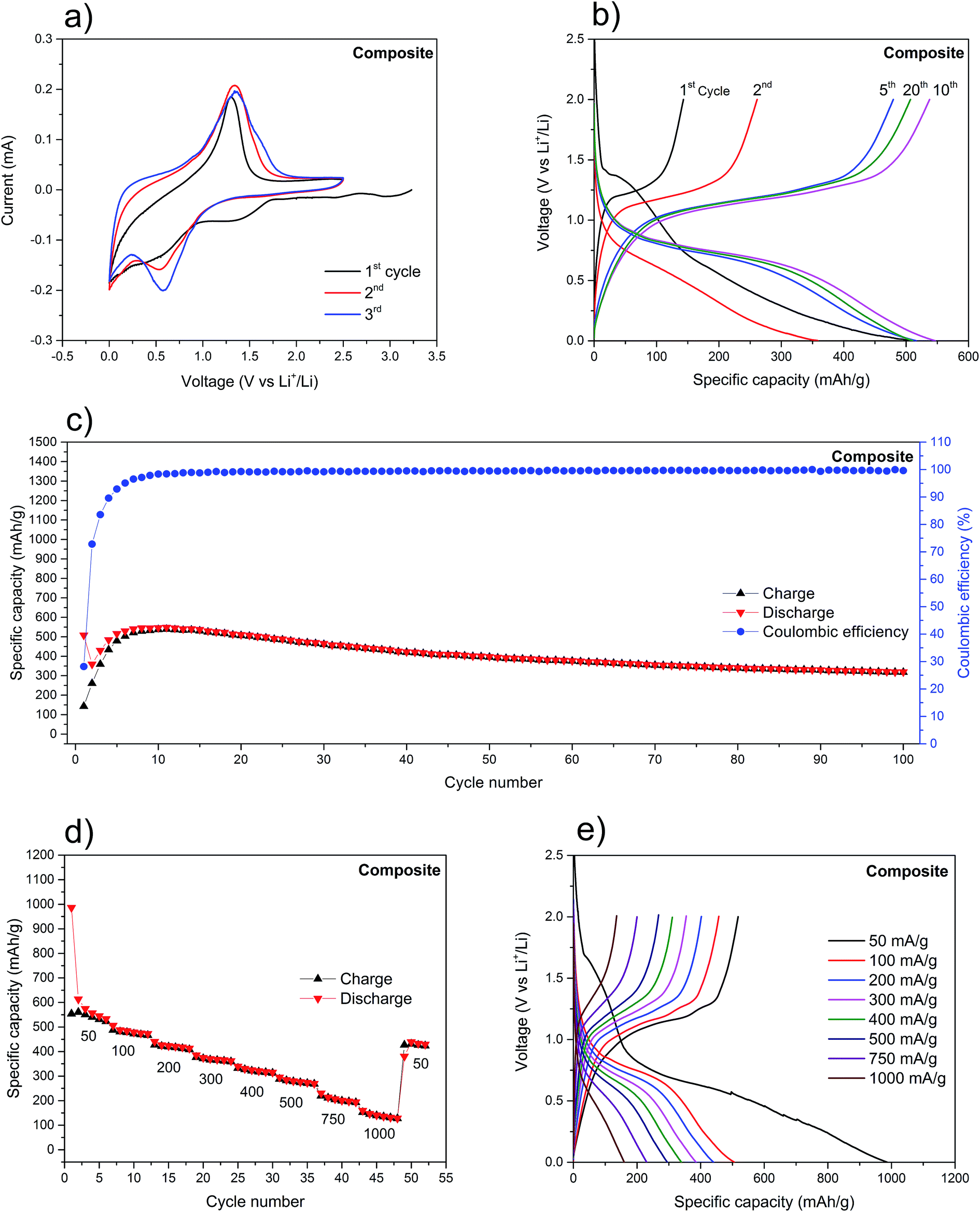 | ||
| Fig. 4 (a) The first three CV curves of the composite at 0.1 mV s−1; (b) voltage profiles of the composite at 150 mA g−1; (c) cycling stability of the composite at 150 mA g−1; (d) cycling stability of the composite cycles at different current densities in mA h g−1; (e) voltage profiles of the composite at different current densities. | ||
The charge/discharge voltage profiles of the composite (Fig. 4b) show the expected voltage plateaus for this type of material: from 0.6 V to 0.8 V (vs. Li+/Li) for the red P lithiation and from 1.0 V to 1.2 V (vs. Li+/Li) for the delithiation.11,16,20 Additionally, there is little variation in plateaus voltages between cycles, indicating a minimal increase in polarization (∼10 mV per cycle), which is mainly attributed to the poor electronic conductivity of red P.30,68 The initial fourteen charge/discharge cycles (Fig. 4c) were characterized by a rapidly rising coulombic efficiency, from 28% to its steady-state at 99%. Because of the tendency of red P to drastically change its volume during lithiation/delithiation, it is expected that the SEI will need several cycles to reach a stable form and subsequently maximize its coulombic efficiency. At the same time, there is a net increase in the discharge specific capacity which is explained by the initial pulverization of the red P and exposure of new surfaces to the electrolyte. Ultimately, this leads to the increase of redox active sites available. Hence, after the first 14 cycles, the anode stabilizes with a maximum discharge specific capacity of 541 mA h g−1 and an average coulombic efficiency of 99.5%. Upon further cycling at 150 mA g−1, the anode retains a discharge capacity of 398 mA h g−1 after 50 cycles, corresponding to a capacity loss of 22%. This value represents a considerable improvement when compared to the source materials (Fig. S8†) and to other reports with similar red P percentage in the composite and cycled at analogous current density (Table S1†). The composite was also cycled at different current densities (Fig. 4d) and, not surprisingly, the capacities were seen to steadily fall as the current increased. Still, the 160 mA h g−1 at 1000 mA g−1 after more than 40 cycles is a remarkable value when considering the poor electron conductivity of red P. Upon restoring the current density to 50 mA g−1, the composite delivers 434 mA h g−1. This clearly demonstrates the stability effect provided by the SWCNT network. Further confirmation was provided by the SEM analysis of the surface morphology of the composite's electrode before and after the electrochemical probing (Fig. S7a and b†).
Contrastingly, the analysis of the voltage plateaus at different current densities (Fig. 4e), reveals a separation of the lithiation and delithiation plateaus which is proportional to the current density. Attributed to polarization phenomena, it further implies a slow kinetic process for the red P lithiation. This is corroborated by the substantial shortening of the lithiation/delithiation plateaus.
While the above results do not represent the best-in-class capacities (Table S1†), there is a vast improvement in electrochemical performance of the composite when compared to pure red P or SWCNT anodes. In fact, for a red P anode, cycled at 150 mA g−1 (Fig. S8a and c†), a remarkably poor specific capacity retention is displayed which is at odds with theoretical predictions (2596 mA h g−1).11 Lowering the current density to 50 mA g−1 seems to improve the performance of the initial cycles considerably (Fig. S9†) with the first discharge reaching 1202 mA h g−1 (compared to 59 mA h g−1 for 150 mA g−1). This further confirms the slow kinetics of the electrochemical alloying reaction between bulk red P and Li+. In both cases, the specific capacity of pure red P rapidly drops below 10 mA h g−1. Similarly, the as-received SWCNT also showed poor electrochemical performance (Fig. S8b and d†). The initial discharge capacity was 1802 mA h g−1, but it rapidly fell to around 130 mA h g−1 (after 50 cycles). This large initial irreversible capacity loss can be explained by the SEI formation on the SWCNT.67
To further explain the improved electrochemical performance of the composite, EIS of the red phosphorus and the composite electrode was carried out after several charge/discharge cycles. The Nyquist plot of the red phosphorus electrode (Fig. S10†) consists of a depressed semicircle at high frequencies, which is assigned to the charge transfer resistance (Rct) and a depressed semicircle at medium frequencies which is attributed to the resistance of the SEI layer (Rs).69 The low frequency sloping line, represented by a Warburg impedance in the equivalent circuit, is due to the diffusion of Li+ ions into the bulk of the active material (W). On the other hand, the Nyquist plot of the composite (Fig. S10†) shows a much smaller high frequency semicircle, indicating lower charge transfer resistance and a sloping line at low frequencies which represents the ionic diffusion into the bulk of the active material. The apparent lack of the second semicircle in the Nyquist plot of the composite points towards the presence of a thinner and more stable SEI layer. In fact, due to the considerable volume changes of the red phosphorus electrode, the SEI layer is repeatedly broken and regenerated, which ends up forming a thick SEI layer for RP only electrode.70 The variations between the two plots indicate that SWCNT result in improved charge transfer kinetics (smaller Rct) and a more stable SEI (smaller Rs).
Taken together, the improved electrochemical performance of the composite is attributed to the synergetic effect between red P and SWCNT. In such a setup, the nanotubes provide fast pathways for electrons to travel from the redox active sites on red P towards the current collector (and vice versa).71 Moreover, the SWCNT can deform elastically to accommodate the matrix volume changes during lithiation/delithiation and, through this, preserve the mechanical integrity of the anode. This is further confirmed in Fig. S11.† While the red P-only electrode (Fig. S11a†) shows significant mass loss due to the P volumetric expansion and subsequent pulverization, the composite electrode (Fig. S11b†) is almost completely preserved, with the majority of the electrode's active material still in contact with the current collector.
All of the above confirms the viability of using SWCNT, with an intact structure, in improving the performance of red P-based anodes for LIB. Although the electrochemical response is improved from the source materials, it is still far below the theoretical prediction of 2596 mA h g−1 for a Li3P phase. This is believed to be due to slow Li+ ion diffusion in red P, most evident when the anode is cycled at higher current densities.72
4. Conclusion
In a bid to make use of the notorious gravimetric and volumetric specific charge capacities of phosphorus, a composite of red P matrix and SWCNT filler was synthesized and for the first time employed as an anode in LIB. Since the electrochemical performance of red phosphorus is hindered by its large volumetric changes upon lithiation and delithiation, the network of SWCNT effectively acted as a structural buffer and provided additional conductive pathways for charge carriers. Accordingly, the cycling stability of the composite – in particular, its capacity retention after the first dozen of cycles – is amongst the best reported to present.Our work indicates that SWCNT are an outstanding additive to improve the electrochemical stability of red P-based anodes, especially at lower current densities. We believe that it is possible to improve the electrochemical performance even further if nanostructured red P is used together with metallic SWCNT.
Conflicts of interest
There are no conflict of interests to declare.Acknowledgements
We acknowledge the financial support from KAUST (BAS/1/1346-01-01) and technical assistance from the Core Laboratories at KAUST.References
- D. Deng, Energy Sci. Eng., 2015, 3, 385–418 CrossRef
.
- M. Broussely, P. Biensan and B. Simon, Electrochim. Acta, 1999, 45, 3–22 CrossRef CAS
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS
- A. Poullikkas, Renewable Sustainable Energy Rev., 2013, 27, 778–788 CrossRef
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed
- J.-M. Tarascon, Philos. Trans. R. Soc., A, 2010, 368, 3227–3241 CrossRef PubMed
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS
- C. M. Park and H. J. Sohn, Adv. Mater., 2007, 19, 2465–2468 CrossRef CAS
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed
- N. Nitta and G. Yushin, Part. Part. Syst. Charact., 2014, 31, 317–336 CrossRef CAS
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS
- M. Heckenmuller, N. Daiju and G. Klepper, Kiel Working Papers, 2014, 26 Search PubMed
- D. Cordell and S. White, Sustainability, 2011, 3, 2027 CrossRef
- J. Qian, D. Qiao, X. Ai, Y. Cao and H. Yang, Chem. Commun., 2012, 48, 8931–8933 RSC
- D. Yuan, J. Cheng, G. Qu, X. Li, W. Ni, B. Wang and H. Liu, J. Power Sources, 2016, 301, 131–137 CrossRef CAS
- Y. Wang, L. Tian, Z. Yao, F. Li, S. Li and S. Ye, Electrochim. Acta, 2015, 163, 71–76 CrossRef CAS
- C. Marino, L. Boulet, P. Gaveau, B. Fraisse and L. Monconduit, J. Mater. Chem., 2012, 22, 22713–22720 RSC
- C. Marino, A. Debenedetti, B. Fraisse, F. Favier and L. Monconduit, Electrochem. Commun., 2011, 13, 346–349 CrossRef CAS
- W. Li, Z. Yang, Y. Jiang, Z. Yu, L. Gu and Y. Yu, Carbon, 2014, 78, 455–462 CrossRef CAS
- L. Wang, X. He, J. Li, W. Sun, J. Gao, J. Guo and C. Jiang, Angew. Chem., Int. Ed., 2012, 51, 9034–9037 CrossRef CAS PubMed
- A. Bai, L. Wang, J. Li, X. He, J. Wang and J. Wang, J. Power Sources, 2015, 289, 100–104 CrossRef CAS
- L. Wang, H. Guo, W. Wang, K. Teng, Z. Xu, C. Chen, C. Li, C. Yang and C. Hu, Electrochim. Acta, 2016, 211, 499–506 CrossRef CAS
- T. W. Odom, J.-L. Huang, P. Kim and C. M. Lieber, Nature, 1998, 391, 62–64 CrossRef CAS
- R. S. Ruoff, D. Qian and W. K. Liu, C. R. Phys., 2003, 4, 993–1008 CrossRef CAS
- J. Z. Liu, Q. S. Zheng, L. F. Wang and Q. Jiang, J. Mech. Phys. Solids, 2005, 53, 123–142 CrossRef CAS
- J.-P. Salvetat, J.-M. Bonard, N. H. Thomson, A. J. Kulik, L. Forró, W. Benoit and L. Zuppiroli, Appl. Phys. A: Mater. Sci. Process., 1999, 69, 255–260 CrossRef CAS
- P. L. McEuen, M. S. Fuhrer and P. Hongkun, IEEE Trans. Nanotechnol., 2002, 1, 78–85 CrossRef
- E. Pop, D. A. Mann, K. E. Goodson and H. Dai, J. Appl. Phys., 2007, 101, 093710 CrossRef
- Y. Zhu, Y. Wen, X. Fan, T. Gao, F. Han, C. Luo, S.-C. Liou and C. Wang, ACS Nano, 2015, 9, 3254–3264 CrossRef CAS PubMed
- M.-T. Averbuch-Pouchot and A. Durif, Topics in Phosphate Chemistry, World Scientific, 2012, DOI:10.1142/9789812812131_0001, pp. 1–9.
- E. N. Rissi, E. Soignard, K. A. McKiernan, C. J. Benmore and J. L. Yarger, Solid State Commun., 2012, 152, 390–394 CrossRef CAS
- C. Thomsen and S. Reich, in Light Scattering in Solid IX, ed. M. Cardona and R. Merlin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, pp. 115–234, DOI:10.1007/978-3-540-34436-0_3
- J. Maultzsch, H. Telg, S. Reich and C. Thomsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 205438 CrossRef
- Y. Miyata, K. Mizuno and H. Kataura, J. Nanomater., 2011, 2011, 7 Search PubMed
- C. Li and T.-W. Chou, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 073401 CrossRef
- T. Chang and H. Gao, J. Mech. Phys. Solids, 2003, 51, 1059–1074 CrossRef CAS
- A. N. Imtani and V. K. Jindal, Comput. Mater. Sci., 2009, 46, 297–302 CrossRef CAS
- P. M. Ajayan, L. S. Schadler, C. Giannaris and A. Rubio, Adv. Mater., 2000, 12, 750–753 CrossRef CAS
- S. D. M. Brown, A. Jorio, M. S. Dresselhaus and G. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 073403 CrossRef
- R. Baddour-Hadjean and J.-P. Pereira-Ramos, Chem. Rev., 2010, 110, 1278–1319 CrossRef CAS PubMed
- L. J. Hardwick, P. W. Ruch, M. Hahn, W. Scheifele, R. Kötz and P. Novák, J. Phys. Chem. Solids, 2008, 69, 1232–1237 CrossRef CAS
- B. Gao, Y. Zhang, J. Zhang, J. Kong and Z. Liu, J. Phys. Chem. C, 2008, 112, 8319–8323 CAS
- R. Graupner, J. Raman Spectrosc., 2007, 38, 673–683 CrossRef CAS
- D. J. Olego, J. A. Baumann, M. A. Kuck, R. Schachter, C. G. Michel and P. M. Raccah, Solid State Commun., 1984, 52, 311–314 CrossRef CAS
- J. Wu, N. Mao, L. Xie, H. Xu and J. Zhang, Angew. Chem., 2015, 127, 2396–2399 CrossRef
- Z. Guo, H. Zhang, S. Lu, Z. Wang, S. Tang, J. Shao, Z. Sun, H. Xie, H. Wang, X.-F. Yu and P.
K. Chu, Adv. Funct. Mater., 2015, 25, 6996–7002 CrossRef CAS
- J. R. Durig and J. M. Casper, J. Mol. Struct., 1970, 5, 351–358 CrossRef CAS
- J. S. Lannin and B. V. Shanabrook, Solid State Commun., 1978, 28, 497–500 CrossRef CAS
- R. A. L. Winchester, M. Whitby and M. S. P. Shaffer, Angew. Chem., Int. Ed., 2009, 48, 3616–3621 CrossRef CAS PubMed
- M. Nath, P. V. Teredesai, D. V. S. Muthu, A. K. Sood and C. N. R. Rao, Curr. Sci., 2003, 85, 956–960 CAS
- G. Liu, Y. Zhao, K. Deng, Z. Liu, W. Chu, J. Chen, Y. Yang, K. Zheng, H. Huang, W. Ma, L. Song, H. Yang, C. Gu, G. Rao, C. Wang, S. Xie and L. Sun, Nano Lett., 2008, 8, 1071–1075 CrossRef CAS PubMed
- B. D. Che, B. Q. Nguyen, L.-T. T. Nguyen, H. T. Nguyen, V. Q. Nguyen, T. Van Le and N. H. Nguyen, Chem. Cent. J., 2015, 9, 10 CrossRef PubMed
- L.-Y. Meng and S.-J. Park, Bull. Korean Chem. Soc., 2012, 33, 209–214 CrossRef CAS
- G. Sun, X. Li, Y. Qu, X. Wang, H. Yan and Y. Zhang, Mater. Lett., 2008, 62, 703–706 CrossRef CAS
- W.-J. Li, S.-L. Chou, J.-Z. Wang, H.-K. Liu and S.-X. Dou, Nano Lett., 2013, 13, 5480–5484 CrossRef CAS PubMed
- R. Bottom, in Principles and Applications of Thermal Analysis, Blackwell Publishing Ltd, 2008, pp. 87–118, DOI:10.1002/9780470697702.ch3
- A. G. Kannan, N. R. Choudhury and N. K. Dutta, Polymer, 2007, 48, 7078–7086 CrossRef CAS
- J. Sun, H.-W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980–985 CrossRef CAS PubMed
- J. Sun, G. Zheng, H.-W. Lee, N. Liu, H. Wang, H. Yao, W. Yang and Y. Cui, Nano Lett., 2014, 14, 4573–4580 CrossRef CAS PubMed
- D. Stachel and A. Barz, J. Phys. Chem. Solids, 2007, 68, 1021–1023 CrossRef CAS
- W. Sung, J. Won, J. Lee and H. Kim, Mol. Cryst. Liq. Cryst., 2009, 499, 234 Search PubMed
- M. Kawashita, F. Miyaji, T. Kokubo, G. H. Takaoka, I. Yamada, Y. Suzuki and M. Inoue, J. Non-Cryst. Solids, 1999, 255, 140–148 CrossRef CAS
- S. F. Lux, I. T. Lucas, E. Pollak, S. Passerini, M. Winter and R. Kostecki, Electrochem. Commun., 2012, 14, 47–50 CrossRef CAS
- C. Lin, A. Tang, H. Mu, W. Wang and C. Wang, J. Chem., 2015, 2015, 11 Search PubMed
- F. R. F. Simoes, N. M. Batra, B. H. Warsama, C. G. Canlas, S. Patole, T. F. Yapici and P. M. F. J. Costa, Anal. Chem., 2016, 88, 11783–11790 CrossRef CAS PubMed
- B. J. Landi, M. J. Ganter, C. D. Cress, R. A. DiLeo and R. P. Raffaelle, Energy Environ. Sci., 2009, 2, 638–654 CAS
- J. Song, Z. Yu, M. L. Gordin, S. Hu, R. Yi, D. Tang, T. Walter, M. Regula, D. Choi, X. Li, A. Manivannan and D. Wang, Nano Lett., 2014, 14, 6329–6335 CrossRef CAS PubMed
- J. Xu, I.-Y. Jeon, J. Ma, Y. Dou, S.-J. Kim, J.-M. Seo, H. Liu, S. Dou, J.-B. Baek and L. Dai, Nano Res., 2017, 1–14, DOI:10.1007/s12274-016-1383-4
- H. Wu, G. Chan, J. W. Choi, I. Ryu, Y. Yao, M. T. McDowell, S. W. Lee, A. Jackson, Y. Yang, L. Hu and Y. Cui, Nat. Nanotechnol., 2012, 7, 310–315 CrossRef CAS PubMed
- L. David, D. Asok and G. Singh, ACS Appl. Mater. Interfaces, 2014, 6, 16056–16064 CAS
- G. Ning, B. Haran and B. N. Popov, J. Power Sources, 2003, 117, 160–169 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06601e |
| This journal is © The Royal Society of Chemistry 2017 |