DOI:
10.1039/C7RA06560D
(Paper)
RSC Adv., 2017,
7, 36705-36713
Hierarchical h-, m- and n-BiPO4 microspheres: facile synthesis and application in the photocatalytic decomposition of refractory phenols and benzene†
Received
12th June 2017
, Accepted 20th July 2017
First published on 25th July 2017
Abstract
BiPO4 microspheres with three kinds of crystal phase (h-BiPO4, m-BiPO4 and n-BiPO4) were prepared by a facile hydrothermal-calcination process. The crystalline phase, microstructures and photoelectrochemical properties were studied by various techniques. The results indicated that h-BiPO4 could transform into m-BiPO4 at 600 °C. Interestingly, h-BiPO4 accompanied with a little bit of n-BiPO4 would transform into n-BiPO4. After UV irradiation for 2 h, conversion of 94.6% and a mineralization rate of 76.8% could be achieved on n-BiPO4 for photocatalytic degradation of bisphenol A, which is almost 1.33 times as high as that of m-BiPO4 and 3.14 times that of h-BiPO4. Moreover, the n-BiPO4 also displayed the highest photocatalytic activity for degradation of other phenols (phenol and p-chlorophenol) in water and gaseous benzene. The enhancement of activity could be attributed to the improvement of the photogenerated electron–hole separation efficiency, which would be conducive to the formation of active species (hydroxyl radicals (˙OH) and superoxide radicals (˙O2−)). The results suggest that n-BiPO4 shows potential applications as an efficient and stable photocatalyst for the photocatalytic decomposition of persistent organic pollutants in water and in the gas phase.
1. Introduction
As one of the most promising technologies for environmental remediation (wastewater and exhaust gas), photocatalysis has been intensively studied because of its environmental merits and the capability to be driven by solar light.1–4 Since Fujishima5 and Carey6 found that H2 could be generated and organic pollutants could be degraded over TiO2 by a photocatalytic process, TiO2 has been deemed as a candidate due to its nontoxicity, stability and high activity.1 However, the shortcomings of rapid recombination of photogenerated carriers and low quantum efficiency hinder its extensive application in environmental remediation.7–9 To solve the problem, exploitation of new photocatalysts with excellent activity is one of the developing directions of photocatalysis.10–12 In numerous photocatalysts, BiPO4 has drawn much great attention in recent years.13–24 For example, Zhu's group13 reported that the photocatalytic activity of BiPO4 is superior to P25 (TiO2, Degussa) under UV light irradiation. Moreover, the photocatalytic activity of BiPO4 could be enhanced remarkably by decreasing the grain size,14 building phase junction,15 exposing high-energy facet16 or coupling with other semiconductors.17 It is not only efficient for decoloration of dyes, but also diverse in crystal phase (hexagon (h-BiPO4) and monoclinic phase with space group P21/m (m-BiPO4) and P21/n (n-BiPO4)).18–20 What's more, Zhu's group has demonstrated that n-BiPO4 displayed higher activity than m-BiPO4 and h-BiPO4 due to the most distorted PO4 tetrahedron.20 Therefore, it is a promising photocatalyst and has much potential for environmental remediation in air and water by the direct absorption of light.21 However, the present studies mainly focus on the degradation of dyes (methylene blue,13–17 methyl orange22 and rhodamine B17,23), while dyes are not suitable used to test the photocatalytic activity of photocatalyst because dyes have the dye-sensitized effect and an additional and more substantial problem.4 On the other hand, the colorless, highly toxic and refractory environmental endocrine disruptors and gaseous benzene were less reported.24 And the photocatalytic activity is not high enough, for example, less than 20% of benzene was degraded on BiPO4 photocatalyst under UV light irradiation for 12 h.24a Therefore, fabrication of BiPO4 with excellent photocatalytic activity on degradation of environmental endocrine disruptors and gaseous benzene are significant foci on BiPO4 photocatalyst. It should be noted that the reason why the UV-light driven BiPO4 photocatalyst (the band-gap energies are in the range of 3.8–4.6 eV) was chosen in our work is not only because what we have introduced on the above but also because UV-light driven photocatalysis has potential use in the actual water industry.3 This is primarily due to the exorbitant occupied area that would result in replacing UV lamp reactors with solar irradiation. For instance, how much solar collection area is required to replace one 1000 W mercury lamp? 2.6 m2 of solar exposure would be needed to achieve the treatment power of one lamp. If for industrialization, this area would quickly add up due to the energy input requirements.3
Herein, BiPO4 with three different crystal phases (h-BiPO4, m-BiPO4 and n-BiPO4) and similar morphology (spherically hierarchical structure) were controllably synthesized by a facile hydrothermal-calcination method. Various characterization techniques were carried out on the as-prepared BiPO4 to obtain the information of crystal phase, micro-structure and optical properties. Bisphenol A (BPA) was selected as a model pollutant, because it is a representative material among environmental endocrine disruptors, ubiquitous in natural water, colorless, hard to be decomposed and has attracted much attention in recent years.25–27 For example, A. Omoike reported Heliscus lugdunensis, an aquatic fungus, could degrade over 70% of BPA after 12 days (microorganism method).27c Zhu's group reported that the maximum adsorption capacity of graphene for decontamination of BPA could reach up to 128 mg g−1, whereas the adsorption capacity of graphene is sensitive to pH and temperature of the solution (adsorption method).27d For photocatalytic method, although some photocatalysts such as Pd/mpg-C3N4 (ref. 27a) and Ag3PO4 (ref. 27b) exhibited high activity with 93.9% and 100% of BPA removal from the solution within 180 min and 10 min, respectively. The removal efficiency of noble metal-free photocatalysts is still low. For instance, only 6% and 6.7% of BPA was degraded over g-C3N4 and TiO2 after irradiation for 180 min, respectively.27a Therefore, it calls for the development of more economical, powerful and durable photocatalyst for the removal of BPA. Up to now, there is scarce report about BPA degradation over BiPO4 photocatalyst. The results showed that n-BiPO4 photocatalyst displayed the highest activity and separation efficiency of photogenerated electron–hole pairs among these three BiPO4 photocatalysts. Moreover, the mechanism of influence on the activity of BiPO4 photocatalyst was also discussed. This work attempts to explore the potential application of BiPO4 for photocatalytic degradation of refractory phenols (BPA, including phenol and 4-chlorophenol (4-CP)) and decomposition of benzene in the gas phase.
2. Experimental
2.1. Synthesis
All the chemicals used in this work were analytical grade reagents without any further purification. In a typical synthesis, 1.03 g polyphosphoric acid and 0.97 g bismuth nitrate were successively dissolved in 20 mL deionized water. Then, the resulting solution dealt with two different heating ways to get BiPO4 samples. The first one is hydrothermal way that the beaker was heated to 160 °C and kept at this temperature for 1 h, which was marked with S-OH. The second was heated by microwave (rated output of microwave is 800 W), denoted as S-MH. All the obtained BiPO4 samples (S-OH and S-MH) were washed several times with distilled water and fully dried at 80 °C in oven. For further analysis, the prepared products S-MH and S-OH were calcined in a muffle furnace at 600 °C for 2 h. The obtained samples were called S-MHT and S-OHT, respectively.
2.2. Characterization
X-ray powder diffraction (XRD, Bruker D8 Advance, Cu Kα radiation) was used to characterize the crystalline and phase constitutions of the samples. The morphology was analyzed by scanning electron microscopy (SEM, FEL Noval NanoSEM 230). Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images were taken on a JEOL-2100 electron microscope operating at an accelerating voltage of 200 kV. The UV-vis diffuse reflection spectroscopy (DRS) patterns were measured by a Hitachi UV-365 spectrophotometer equipped with an integrating sphere, and BaSO4 as a reference sample coated standard pattern. The photoluminescence (PL) spectra were recorded on a JASCO FP-6500 with an excitation wavelength of 256 nm. The photoelectrochemical analysis was performed in a conventional three-electrode cell (CHI-660E, Chenhua Instruments Co., Shanghai, China), using a Pt wire and an Ag/AgCl electrode as the counter electrode and reference electrode, respectively. The working electrode was prepared on indium-tin oxide (ITO) glass which was covered by 0.5 cm × 0.5 cm samples. The electrolyte was 0.1 M KCl aqueous solution containing 0.01 M K3[Fe(CN)6]–K4[Fe(CN)6].
2.3. Photocatalytic tests
The photocatalytic activities of the synthesized samples were tested by the degradation of phenol, p-chlorophenol (4-CP) and bisphenol A (BPA) aqueous solution and degradation of gaseous benzene (C6H6). The photocatalytic reaction system has been reported in our previous work.12 The degradation of phenols was operated in a tubular reactor, which was surrounded by a water cooling jacket. A low pressure mercury UV light (H-shaped, 16 W, a monochromatic emission at 254 nm) was assembled at the center of the tubular reactor. 0.1 g sample was put into 150 mL phenol, p-chlorophenol or bisphenol A aqueous solution (20 mg L−1) and then magnetically stirred in dark for 30 min to establish an adsorption–desorption equilibrium. After that, a 4 mL solution was sampled at various illumination time intervals and analyzed through a UV-vis spectrophotometer (Persee TU-1950). Photocatalytic degradation of benzene was carried out in a fixed-bed tubular quartz reactor (250 mm × 4 mm), in which 0.1 g sample mixed evenly with 1 g 50–70 mesh high purity quartz sands was loaded, surrounded by four UV-254 nm lamps (TUV 6W/G6 T5, Philips) and operated in a single-pass mode. Benzene vapor with a constant concentration of 250 ppm was supplied by a gas cylinder (Dalian Da'te gas Co. China) and fed to the sample a total flow rate of 30 mL min−1. The reactor temperature was kept at 30 ± 1 °C by an air-cooling system and circulating water. Simultaneous determination of the concentrations of residual C6H6 and generated CO2 was performed on an online gas chromatograph (Agilent 7820A) with a flame ionization detector (FID) and a thermal conductivity detector (TCD), respectively.
3. Results and discussion
3.1. Characterization
Fig. 1 shows the XRD patterns of S-MH, S-MHT, S-OH, S-OHT and the standard diffraction peaks of h-BiPO4 (JCPDS 45-1370), m-BiPO4 (JCPDS 43-0637) and n-BiPO4 (JCPDS 15-0767). It can be seen that the crystalline phase of S-MH is hexagonal phase with the main diffraction peaks at about 14.6°, 20.1°, 25.5°, 29.5°, 31.3° and 41.8°, which could be indexed to the (100), (101), (110), (200), (102) and (211) planes of h-BiPO4, respectively. No impurity peaks are found, indicating high purity of the sample S-MH. After calcination at high temperature, the crystalline phase of S-MHT is converted into m-BiPO4, and all the diffraction peaks of S-MHT are in good agreement with m-BiPO4. However, the sample S-OH synthesized by a hydrothermal method is a mixture of h-BiPO4 and n-BiPO4. The peaks at 14.6° and 20.1° belong to (100) and (101) planes of h-BiPO4 can be observed obviously. It is clear that the XRD pattern of S-OHT corresponds to n-BiPO4 can be obtained by calcination of S-OH at 600 °C, and no impurity peaks are found. It is interesting to find that the h-BiPO4 (S-MH) is easily transformed into m-BiPO4 (S-MHT). But if it is a mixture of h-BiPO4 and n-BiPO4 (S-OH), it would be converted into n-BiPO4 (S-OHT). The existence of n-BiPO4 would play the part of seed crystal. Moreover, the sharp and intense XRD peaks suggest that the as-prepared samples S-MH, S-MHT and S-OHT are well-crystallized.
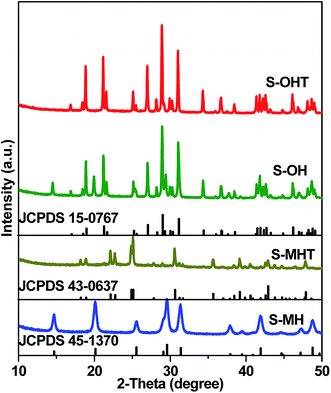 |
| | Fig. 1 XRD patterns of the as-synthesized S-MH, S-MHT, S-OH, S-OHT, together with the standard patterns of h-BiPO4 (JCPDS 45-1370), m-BiPO4 (JCPDS 43-0637) and n-BiPO4 (JCPDS 15-0767). | |
The morphology, particle size and micro-structure of the as-prepared samples were investigated by SEM and TEM (Fig. 2). As shown in Fig. 2a and e, the morphology of S-MH is the solid sphere with the diameter of 1–3 μm, which is further composed of tiny nanoparticles. After heat treatment of S-MH, S-MHT is also kept sphere shape, while its surface is rugged (Fig. 2b and f). This may be caused by sintering of tiny S-MH nanoparticles under high temperature calcination. HRTEM images of S-MH (Fig. 2i) and S-MHT (Fig. 2j) display the resolved lattice spacings of 0.28 nm and 0.32 nm, which corresponding to the d-spacing values for (102) plane of h-BiPO4 and (101) plane of m-BiPO4, respectively. It is in good agreement with the XRD results. For S-OH, its morphology is very interesting. As shown in Fig. 2c and g, the shape of S-OH is like sea urchin. It shows spherically hierarchical micro-structure, which is built by some connected nanoparticles, some nanorods like needle inserted into the sphere partly and pores. While for S-OHT (Fig. 2d and h), the sphere is different from S-OH. It is composed of nanorods not just in the surface, but some shortly scattered nanorods inside sphere, and some spheres are broken. From Fig. 2k and l, it can be observed that the fringe spacings detected at nanorods of S-OH and S-OHT are about 0.32 nm and 0.47 nm, corresponding to the (200) and (011) lattice planes of n-BiPO4, respectively. In addition, the lattice fringes of S-OHT are very clear, indicating high degree of crystallinity. It is in good agreement with the results of the XRD analysis. Based on XRD and the morphology transformation analysis between S-OH and S-OHT, it can be concluded that the nanoparticles with hexagonal phase in S-OH would be transformed into nanorods with monoclinic phase after calcination at 600 °C. S-OHT is the pure monoclinic phase of BiPO4 with P21/n space group, lattice constants a = 6.752, b = 6.933 and c = 6.468 Å, and the spherically hierarchical micro-structure is composed of some shortly scattered nanorods. S-MH and S-MHT both exhibit the solid sphere shape, which is constituted by tiny and sintered nanoparticles, respectively. While S-MH is the pure hexagonal phase of BiPO4 with P3121 space group and lattice constants a = 6.986, b = 6.986 and c = 6.475 Å; S-MHT is the pure monoclinic phase of BiPO4 with P21/m space group and lattice constants a = 4.883, b = 7.069 and c = 4.704 Å.
 |
| | Fig. 2 (a–d) SEM, (e–h) TEM and (i–l) HRTEM images of the as-synthesized (a, e, i) S-MH, (b, f, j) S-MHT, (c, g, k) S-OH and (d, h, l) S-OHT. | |
Fig. 3 shows the UV-vis DRS of S-MH, S-MHT and S-OHT. The absorption edges of these three samples all occur at about 275 nm. The band gap energy (Eg) is calculated via the Tauc equation,28–30 (αhν)n = A(hν − Eg), where A, α, h and ν are proportionality constant, absorption coefficient, Planck constant and light frequency, respectively. The value of the exponent n is decided from the properties of the transition in a semiconductor.21,30 Monoclinic BiPO4 (S-MHT and S-OHT) is an indirect band semiconductor, the value of n is 0.5. While hexagonal BiPO4 (S-MH) is direct, the value of n is 2. As shown in the inset of Fig. 3, the band gaps of S-MH, S-MHT and S-OHT are about 4.65, 4.54 and 4.49 eV, respectively. The difference among the band gaps of these three samples may be caused by their electronic structural difference limited by the crystalline phase, as also reported by Zhu's group21 and observed in anatase and brookite titanium dioxide.31
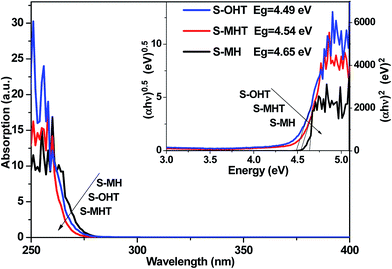 |
| | Fig. 3 UV-vis DRS spectra and bandgap energies (the inset) of the as-synthesized S-MH, S-MHT, S-OH and S-OHT. | |
3.2. Photocatalytic properties
The photocatalytic activities of the as-prepared S-MH, S-MHT and S-OHT were tested by photocatalytic degradation of BPA, phenol and 4-CP under UV light irradiation. To investigate the effect of the amount of photocatalyst on the photodegradation efficiency of BPA, experiments performed (Fig. S1†) with different amount of n-BiPO4 exhibited that the photocatalytic efficiency increases with an increase in n-BiPO4 amount up to 0.15 g, and is then decreased. A similar phenomenon had been reported by others when studying photocatalytic degradation of dyes,32 and it can be explained in terms of availability of active sites on the catalyst surface and the penetration of light into the suspension.32 To further study the activity of n-BiPO4 for photocatalytic degradation of low concentration of BPA, the influence of the BPA concentration was investigated and shown in Fig. S2.† The results showed that the as-prepared n-BiPO4 could effectively photocatalytic degrade different concentrations of BPA (5, 10, 15 and 20 mg L−1). Additionally, 150 mL BPA with concentration of 5 mg L−1 could be eliminated completely over n-BiPO4 (0.1 g) within irradiation for 30 min. Fig. 4a displays the degradation profiles of BPA with BiPO4 samples. After 120 min of irradiation, 55.5%, 87.9% and 94.6% of BPA are degraded by the S-MH, S-MHT and S-OHT photocatalysts, respectively. The outstanding activity of the BiPO4 is further detected by comparing the apparent reaction rate constants (k). As shown in Fig. 4b, the degradation of BPA can be described by the pseudo first-order reaction kinetics, ln(C/C0) = −kt, where C0 and C are the initial concentration and the concentration at reaction time t, respectively. The ks for the S-MH, S-MHT and S-OHT are estimated to be about 0.42, 0.99 and 1.32 h−1, respectively. The k of S-OHT is 1.33-fold and 3.14-fold higher than that of S-MHT and S-MH. As we all known, degradation does not mean mineralization, because the original organic pollutants would be degraded to small organic molecules. Therefore, total organic carbon (TOC) technique was applied to further measure the mineralization of the solution, and the results are shown in Fig. 4c. It is clear that S-OHT exhibits the higher capacity (76.8%) of mineralization for BPA than S-MHT (65.2%) and S-MH (25.1%). It means that the reactive species produced on n-BiPO4 photocatalyst not only can degrade BPA, but also can efficient decompose BPA into CO2. Moreover, the as-prepared S-OHT is a stable photocatalyst in the process of photocatalytic degradation of BPA in water. After 3 cycles of the photocatalytic reaction, the photodegradation efficiency of BPA over the used S-OHT is similar to that over the fresh sample (Fig. 4d). The similar result was also reported by Zhu's group that n-BiPO4 is stable during the photocatalytic degradation of methylene blue.13
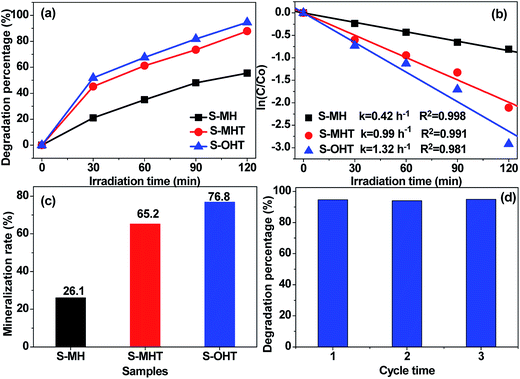 |
| | Fig. 4 (a) Photocatalytic degradation curves of BPA (20 mg L−1) for different samples (0.1 g) under UV light irradiation; (b) kinetic plots of ln(C/C0) vs. irradiation time; (c) mineralization rate of BPA by different samples under UV light irradiation for 2 h; (d) cycling runs of S-OHT for BPA degradation under UV light irradiation. | |
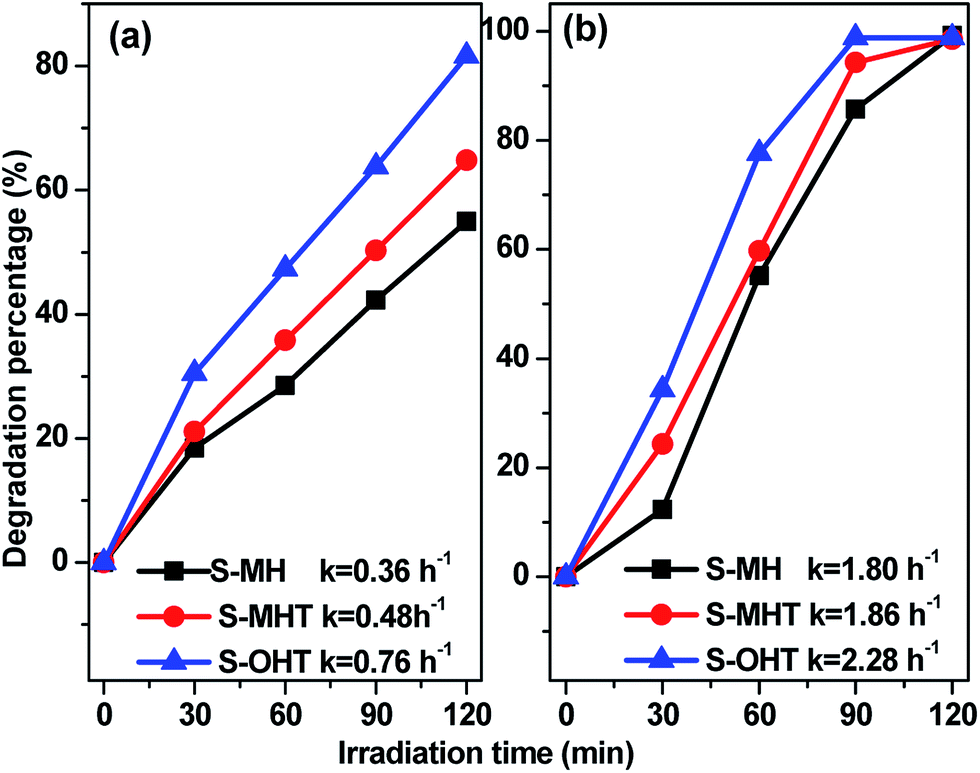
To study the broad-spectrum activity of BiPO4 for other phenols, phenol and 4-chlorophenol (4-CP) were further selected as probe molecules to investigate the photocatalytic activities of the as-prepared BiPO4 samples. As shown in Fig. 5, whether the degradation of phenol or 4-CP over BiPO4 samples follows pseudo-first-order kinetics and the corresponding kinetic constants are showed in Fig. 5. Moreover, the photocatalytic activity for phenol and 4-CP degradation decreases in the order S-OHT > S-MHT > S-MH. The sequence is consistent with their performance for BPA degradation. In order to further illustrate the photocatalytic activity of the photocatalysts, the dark absorption test and the blank test were carried out. As shown in Fig. S3a,† the dark adsorption test in the absence of irradiation but with the catalysts showed that no significant change in the substrate concentration is found, and an adsorption–desorption equilibrium had been established after stir in dark for 30 min. On the other hand, the blank tests (Fig. S3b†) showed that 11.7% of BPA, 16.6% of phenol and 24.3% of 4-CP were removed after irradiation for 2 h due to photo-induced self-photodegradation. However, compared with the distinctively photocatalytic activities of n-BiPO4 (94.6% of BPA, 81.6% of phenol and 100% of 4-CP were eliminated after irradiation for 2 h), the presence of both photocatalyst and illumination is necessary for the effectively photocatalytic degradation of refractory phenols.
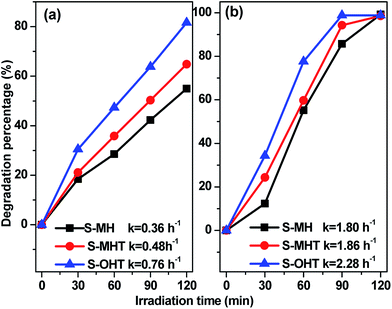 |
| | Fig. 5 Photocatalytic activities of the samples (0.1 g) for (a) phenol and (b) 4-CP degradation under UV light irradiation. | |
The photocatalytic degradation of phenols over S-OHT indicates that S-OHT has a great capability to decompose the benzene ring in the phenols. However, the degradation of benzene in the gas phase may be different to that in a liquid phase for the deficiency of water, which is crucial for the elimination of volatile organic compounds. To verify the validity of S-OHT on the photocatalytic decomposition of the benzene ring, the photocatalytic degradation behaviors of gaseous benzene by BiPO4 samples were carried out with the dry O2 atmosphere under the UV-light irradiation. As shown in Fig. 6, excluding S-MH, the degradation of benzene can be achieved on the S-MHT and S-OHT. The initial conversion of benzene on S-MHT sample was about 80%, with 500 ppm CO2 produced. However, after 10 h reaction time, the amount of CO2 gradually dropped to 105 ppm, and the conversion decreased to 13%. Compared with S-MHT, the conversion of benzene over S-OHT sample was about 85% with 800 ppm CO2 produced during the first half hour. After irradiation for 7 h, the benzene conversion remained 67% with more than 675 ppm CO2 generated, and the average mineralization ratio of benzene was about 55%, suggesting that most of benzene was mineralized to CO2. In the following 3 h, the S-OHT sample displayed no obvious inactivation. Moreover, benzene could not be decomposed under UV-light irradiation, and the S-OHT sample didn't produce CO2 in the dark reaction. The results proved that the benzene ring was indeed decomposed by the S-OHT.
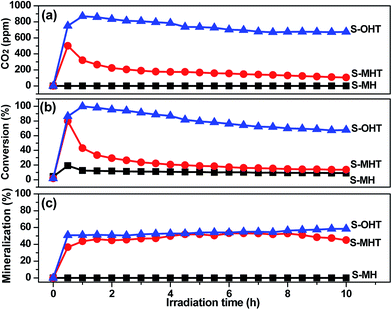 |
| | Fig. 6 (a) The production of CO2 and (b) the conversion and (c) mineralization efficiencies of C6H6 over prepared S-MH, S-MHT and S-OHT as a function of irradiation time. | |
3.3. Mechanism of photocatalysis
In general, there are many factors influencing the photocatalytic activity of photocatalyst, such as the phase structure, light absorption, surface area, and the separation efficiency of photogenerated electron–hole pairs.1 It should be noted that the light source used in this work is monochromatic light with wavelength of 254 nm. The light absorption of these three samples is all around 275 nm, as shown in Fig. 3. Moreover, the BET surface area is all negligible (<1 m2 g−1). Therefore, it may be due to other factors that influence the activities of S-MH, S-MHT and S-OHT. In the previous report, Zhu's group21 found that the highest photoactivity of n-BiPO4 originates from the most distorted PO4 tetrahedron, not other factors such as photo absorption, BET surface area and the oxidation potential of photoexcited holes. The distorted PO4 tetrahedron facilitates the separation of photoexcited electron–hole pairs. Therefore, the photocatalytic activity of BiPO4 is deeply relying on the separation efficiency of photogenerated electrons and holes. To further confirm this summing-up, electrochemical impedance spectroscopy (EIS) and photoluminescence test (PL) were carried out in our experiments to investigate the differences of separation, transfer and recombination of the photoexcited electrons and holes between these samples. As previous studies,33,34 the diameters of the EIS plots under light irradiation are supposed to manifest the charge separation and transfer resistance across the solid–liquid junction in the electrode–electrolyte interface region. Besides, the smallest arc radius indicates most effective separation of the photoexcited electrons and holes, and vice versa. The typical EIS responses of BiPO4 with different crystal phase are presented in Fig. 7. The arc radiuses of prepared BiPO4 samples are different from each other. EIS data was analyzed in terms of an equivalent circuit model and the values of charge transfer resistance were about 330, 435 and 579 Ω cm−2 for n-BiPO4, m-BiPO4 and h-BiPO4, respectively. It indicates that S-OHT with n-BiPO4 phase possesses the highest efficiency of electron–hole separation among these three BiPO4 samples. This is also supported by the results of PL spectra, which can reflect the recombination rate of photoexcited electros and holes.
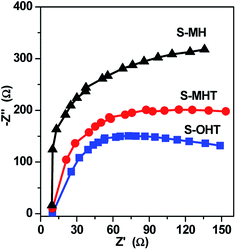 |
| | Fig. 7 EIS plots for the as-synthesized samples under UV-light irradiation. | |
As well known, there are certain intrinsic relationships between the PL spectrum and photocatalytic activity of a semiconductor material according to the mechanisms of PL and photocatalysis.35 The lower the PL intensity, the higher the separation efficiency of photoexcited carriers, and thus the higher the photocatalytic activity.29,35 Fig. 8 presents the PL spectra excited at 255 nm wavelength at room temperature. A main emission peak centered at about 301 nm are observed for all samples. A weaker PL emission intensity than that of S-MH and S-MHT is produced on S-OHT, indicating that the recombination of the photoexcited electrons and holes is efficiently hampered on S-OHT. The result is in good agreement with the EIS. According to the above analysis, the photocatalytic activity difference between h-BiPO4, m-BiPO4 and n-BiPO4 is deeply relying on the separation efficiency of photogenerated electrons and holes. The results further confirm the previous report.21
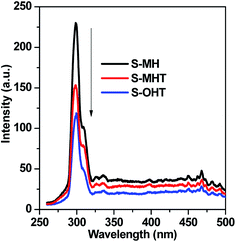 |
| | Fig. 8 Photoluminescence spectra of the as-synthesized samples (excitation wavelength was 256 nm). | |
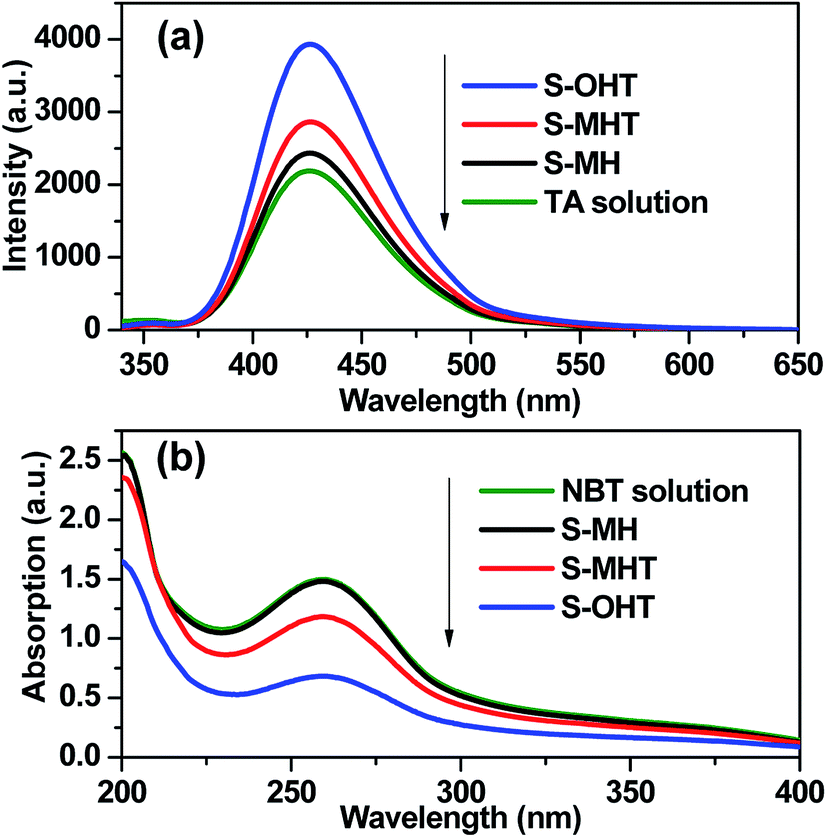
It is well known that the photoexcited electrons and holes are the cornerstone of the photocatalysis.1 The efficient separation of photoexcited electrons (e−) and holes (h+) favors the production of hydroxyl radicals (˙OH) and superoxide radicals (˙O2−), which are the primary and straightforward reactive species for the photocatalytic degradation of organic pollutants in the water. The photoexcited electrons and holes at the conduction band and valance band of BiPO4 can reduce O2 and oxidize OH− to form ˙O2− and ˙OH, respectively.15 Moreover, ˙OH may come from ˙O2− via chain reactions, and H2O2 is a significant intermediate species in the process. In order to confirm the presence of the photoactive radicals in the reaction system, following experiments were carried out. Terephthalic acid-photoluminescence (TA-PL), nitroblue tetrazolium (NBT) and N,N-diethyl-p-phenylenediamine (DPD) methods were used to determine the photogenerated radical species ˙OH, ˙O2− and H2O2 over BiPO4 upon the irradiation of light, respectively.36–39
TA used as the indicator can readily react with the generated ˙OH producing 2-hydroxyterephthalic acid (HOA), which has a good fluorescence. The intensity of PL implies the amount of generated ˙OH in the reaction system.36,37 As shown in Fig. 9a, it is clear that the PL peaks located at about 426 nm for these three samples are observed, indicating that ˙OH is produced in the photocatalytic reaction process. Moreover, S-OHT displays the higher formation efficiency of ˙OH than S-MHT and S-MH. The formation of ˙OH order is S-OHT > S-MHT > S-MH, which is in accordance with the results of the photocatalytic activity and separation of photoexcited carriers tests. Nitroblue tetrazolium, exhibiting an absorption maximum at 259 nm, was used for the analysis of ˙O2−. NBT can react with the formed ˙O2− in the water solution, and then its absorption peak would be reduced.38 Therefore, NBT method used as a simple approach for detection of ˙O2− has been widely adopted. As shown in Fig. 9b, the intensity of NBT at about 259 nm decreases gradually with the order: S-MH > S-MHT > S-OHT. It indicates that the concentration of NBT in the presence of S-OHT is reduced greatly. That is more ˙O2− is produced over S-OHT in the photocatalytic reaction process. Furthermore, the DPD method was employed for detecting the existence of H2O2.39 As shown in Fig. 10, in the presence of either S-MH, S-MHT or S-OHT under UV-light irradiation, two clearly characteristic peaks of existence of H2O2 with absorption maxima at 510 nm and 550 nm are found. Moreover, the production of H2O2 decreases in the order S-OHT > S-MHT > S-MH. It is in line with the results of TA-PL experiment and the photocatalytic activity test.
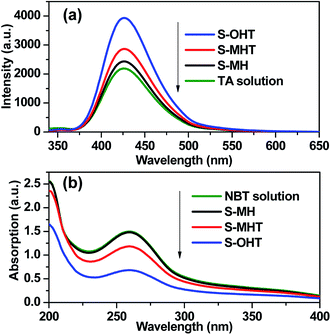 |
| | Fig. 9 (a) The photoluminescence emission spectra of TA solution and (b) the absorption of NBT solution in the presence of different samples under UV light irradiation. | |
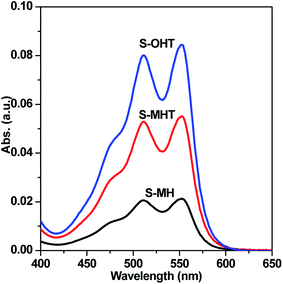 |
| | Fig. 10 H2O2 was detected by addition of N,N-diethyl-p-phenylenediamine and horseradish peroxidase to S-MH, S-MHT and S-OHT aqueous solution after UV light irradiation for 20 min, respectively. | |
In order to further understand the roles of the main oxidative species involved in the decomposition of BPA over BiPO4 photocatalyst, the trapping experiments of active species was performed, using isopropanol (IPA) as ˙OH scavenger,40 ammonium oxalate (AO) as h+ capturer,41 and benzoquinone (BQ) as ˙O2− scavenger.42 As shown in Fig. 11, the photocatalytic activity of S-OHT is obviously suppressed by adding AO or BQ, whereas slightly suppressed by ˙OH scavenger. It indicates that h+, ˙O2− and ˙OH are all the oxidative species for the photocatalytic degradation of BPA over n-BiPO4 photocatalyst, and the effect to BPA degradation from strong to weak follows the order of ˙O2−, h+ and ˙OH. Therefore, on the basis of the discussion and results above, a possible mechanism for the photocatalytic decomposition of phenols and benzene over n-BiPO4 is proposed in Fig. S4.†
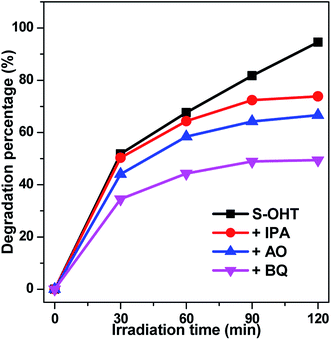 |
| | Fig. 11 Plots of photogenerated carriers trapping in the system of photocatalytic degradation of BPA on S-OHT. | |
4. Conclusions
Sphere-like BiPO4 photocatalysts with h-BiPO4, m-BiPO4 and n-BiPO4 crystal phases were controllably synthesized through a simple hydrothermal-calcination method, respectively. The n-BiPO4 exhibited much higher photocatalytic activity towards degradation of BPA, phenol, 4-CP and benzene than h-BiPO4 and m-BiPO4. The BPA degradation and mineralization rates could reach up to 94.6% and 76.8% under UV light irradiation for 2 h, respectively. And the degradation efficiency is maintained at over 94% after three cycles. The excellent photocatalytic activity could be attributed to the distortion of PO4 tetrahedron, which improves the separation efficiency of the photoexcited electron–hole pairs and produces more oxidative species. During the photocatalytic process, effect of the oxidative specie to BPA degradation follows the order of ˙O2− > h+ > ˙OH.
Acknowledgements
This work was supported by the Natural Science Foundation of Anhui Province (1608085QB37), and the Collaborative Innovation Center of Advanced Functional Materials (XTZX103732016001).
References
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- Y. Sun, S. Gao, F. Lei and Y. Xie, Chem. Soc. Rev., 2015, 44, 623–636 RSC.
- E. L. Cates, Environ. Sci. Technol., 2017, 51, 757–758 CrossRef CAS PubMed.
- N. Barbero and D. Vione, Environ. Sci. Technol., 2016, 50, 2130–2131 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- J. H. Carey, J. Lawrence and H. M. Tosine, Bull. Environ. Contam. Toxicol., 1976, 16, 697–701 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- R. Daghrir, P. Drogui and D. Robert, Ind. Eng. Chem. Res., 2013, 52, 3581–3599 CrossRef CAS.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton and J. A. Byrne, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Z. G. Yi, J. H. Ye, N. kikugawa, T. Kako, S. Ouyang, H. Stuart-williams, H. Yang, J. Y. Cao, W. J. Luo, Z. S. Li, Y. Liu and R. L. Withers, Nat. Mater., 2010, 9, 559–564 CrossRef CAS PubMed.
- X. Fu, J. Wang, D. Huang, S. Meng, Z. Zhang, L. Li, T. Miao and S. Chen, ACS Catal., 2016, 6, 957–968 CrossRef CAS.
- C. Pan and Y. Zhu, Environ. Sci. Technol., 2010, 44, 5570–5574 CrossRef CAS PubMed.
- C. Pan and Y. Zhu, J. Mater. Chem., 2011, 21, 4235–4241 RSC.
- Y. Zhu, Y. Liu, Y. Lv, Q. Lin, D. Liu and Y. Zhu, J. Mater. Chem. A, 2014, 2, 13041–13048 CAS.
- Q. Zhang, H. Tian, N. Li, M. Chen and F. Teng, CrystEngComm, 2014, 16, 8334–8339 RSC.
-
(a) B. Lu, X. Ma, C. Pan and Y. Zhu, Appl. Catal., A, 2012, 435, 93–98 CrossRef;
(b) C. Pan, J. Xu, Y. Wang, D. Li and Y. Zhu, Adv. Funct. Mater., 2012, 22, 1518–1524 CrossRef CAS;
(c) Y. Liu, P. Zhang, H. Lv, J. Guang, S. Li and J. Jiang, RSC Adv., 2015, 5, 83764–83772 RSC.
- Y. Wang, X. Guan, L. Li and G. Li, CrystEngComm, 2012, 14, 7907–7914 RSC.
- M. Zhao, L. Li, L. Yang, J. Zheng and G. Li, CrystEngComm, 2013, 15, 609–615 RSC.
- C. Pan and Y. Zhu, Catal. Sci. Technol., 2015, 5, 3071–3083 CAS.
-
(a) C. Pan, D. Li, X. Ma, Y. Chen and Y. Zhu, Catal. Sci. Technol., 2011, 1, 1399–1405 RSC;
(b) Y. Liu, Y. Zhu, J. Xu, X. Bai, R. Zong and Y. Zhu, Appl. Catal., B, 2013, 142, 561–567 CrossRef.
- P. Shan, C. Niu, D. Huang, G. Zeng and H. Zhang, RSC Adv., 2015, 5, 89105–89112 RSC.
-
(a) C. Pan, J. Xu, Y. Chen and Y. Zhu, Appl. Catal., B, 2012, 115, 314–319 CrossRef;
(b) H. Lv, J. Guang, Y. Liu, H. Tang, P. Zhang, Y. Lu and J. Wang, RSC Adv., 2015, 5, 100625–100632 RSC.
-
(a) B. Long, J. Huang and X. Wang, Prog. Nat. Sci.: Mater. Int., 2012, 22, 644–653 CrossRef;
(b) X. Zou, C. Ran, Y. Dong, Z. Chen, D. Dong, D. Hu, X. Li and Y. Cui, RSC Adv., 2016, 6, 20664–20670 RSC.
- J. G. Hengstler, H. Foth, T. Gebel, P. J. Kramer, W. Lilienblum, H. Schweinfurth, W. Volkel, K. M. Wollin and U. Gundert-Remy, Crit. Rev. Toxicol., 2011, 41, 263–291 CrossRef CAS PubMed.
- L. N. Vandenberg, I. Chahoud, J. J. Heindel, V. Padmanabhan, F. J. R. Paumgartten and G. Schoenfelder, Environ. Health Perspect., 2010, 118, 1055–1070 CrossRef CAS PubMed.
-
(a) C. Chang, Y. Fu, M. Hu, C. Wang, G. Shan and L. Zhu, Appl. Catal., B, 2013, 142, 553–560 CrossRef;
(b) H. Katsumata, M. Taniguchi, S. Kaneco and T. Suzuki, Catal. Commun., 2013, 34, 30–34 CrossRef CAS;
(c) A. Omoike, T. Wacker and M. Navidonski, Chemosphere, 2013, 91, 1643–1647 CrossRef CAS PubMed;
(d) J. Xu, L. Wang and Y. Zhu, Langmuir, 2012, 28, 8418–8425 CrossRef CAS PubMed.
- M. A. Butler, J. Appl. Phys., 1977, 48, 1914–1920 CrossRef CAS.
- S. Meng, X. Ning, T. Zhang, S. Chen and X. Fu, Phys. Chem. Chem. Phys., 2015, 17, 11577–11585 RSC.
- Y. F. Liu, Y. H. Lv, Y. Y. Zhu, D. Liu, R. L. Zong and Y. F. Zhu, Appl. Catal., B, 2014, 147, 851–857 CrossRef CAS.
- T. A. Kandiel, A. Feldhoff, L. Robben, R. Dillert and D. W. Bahnemann, Chem. Mater., 2010, 22, 2050–2060 CrossRef CAS.
-
(a) N. Naneshvar, D. Salari and A. R. Khataee, J. Photochem. Photobiol., A, 2004, 162, 317–322 CrossRef;
(b) M. S. T. Goncalves, A. M. F. Oliveira-Campos, E. M. M. S. Pinto, P. M. S. Plasência and M. J. R. P. Queiroz, Chemosphere, 1999, 39, 781–786 CrossRef CAS.
- B. L. He, B. Dong and H. L. Li, Electrochem. Commun., 2007, 9, 425–430 CrossRef CAS.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS PubMed.
- L. Jing, Y. Qu, B. Wang, S. Li, B. Jiang, L. Yang, W. Fu, H. Fu and J. Sun, Sol. Energy Mater. Sol. Cells, 2006, 90, 1773–1787 CrossRef CAS.
- J. C. Barreto, G. S. Smith, N. H. P. Strobel, P. A. McQuillin and T. A. Miller, Life Sci., 1994, 56, 89–96 CrossRef.
- K. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimato, J. Photochem. Photobiol., A, 2000, 134, 139–142 CrossRef CAS.
- B. H. J. Bielski and H. W. Richter, J. Am. Chem. Soc., 1977, 99, 3019–3023 CrossRef CAS.
- S. Meng, D. Li, X. Fu and X. Fu, J. Mater. Chem. A, 2015, 3, 23501–23511 CAS.
- M. Sadakane, K. Sadaki, H. Kunioku, B. Ohtani, W. Ueda and R. Abe, Chem. Commun., 2008, 48, 6552–6554 RSC.
- H. Kominami, A. Furusho, S. Murakami, H. Inoue, Y. Kera and B. Ohtani, Catal. Lett., 2001, 76, 31–34 CrossRef CAS.
- R. Palominos, J. Freer, M. A. Mondaca and H. D. Mansilla, J. Photochem. Photobiol., A, 2008, 193, 139–145 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06560d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Tong Xub,
Yunjian Wangb and
Sugang Meng
a,
Tong Xub,
Yunjian Wangb and
Sugang Meng