
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHemin-bound cysteinyl bolaamphiphile self-assembly as a horseradish peroxidase-mimetic catalyst
Chaemyeong Lee and
Sang-Yup Lee *
*
Department of Chemical and Biomolecular Engineering, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul, 03722, South Korea. E-mail: leessy@yonsei.ac.kr; Fax: +822-312-6401; Tel: +822-2123-5758
First published on 9th August 2017
Abstract
Horseradish peroxidase (HRP) is an important oxidative enzyme with a heme cofactor whose Fe centre acts as the active site. Its catalytic activity is expressed upon association with the heme cofactor through coordination between Fe and histidine imidazole. In this study, a HRP-mimetic catalyst was prepared by binding hemin to the self-assembled suprastructure of cysteinyl bolaamphiphiles through an Fe–thiol bond, and its oxidative catalytic activity was investigated. The cysteinyl bolaamphiphile self-assembly is rich with surface-exposed cysteine thiols, which served as binding sites. The activity of the prepared HRP-mimetic catalyst was pH-dependent and increased with increasing temperature, with an activation energy of 31.7 kJ mol−1. The kinetic parameters obtained by varying the substrate concentrations suggested a ping-pong mechanism where H2O2 and substrate bind sequentially to the active centre. The cysteinyl bolaamphiphile self-assembly provides a biomimetic support on which various metallic cofactors can bind to induce biochemical activity.
Introduction
Horseradish peroxidase (HRP) is a widely studied oxidative enzyme for H2O2 activation found in the roots of horseradish. It has long been used for biological and biochemical processes such as phagocytosis and ELISA experiments requiring selective oxidation of target substrates.1,2 HRP is a holoenzyme with a heme cofactor whose Fe centre acts as the active site. Structural studies revealed that the heme Fe ion coordinates with the proximal His170 ligand while the neighbouring Asp247 residue is supposed to impart anionic character to His170 imidazole by forming a hydrogen bond (Scheme 1).3,4 The versatile applicability and structural characteristics of HRP, particularly its catalytically active metalloporphyrin, prompted many researchers to devise HRP mimics. For example, hematine5,6 and other metalloporphyrins7 exhibit peroxidase-like activity originating from the central transition metal ion, e.g. Fe. Various materials, such as histidine-decorated solid electrodes,8 hydrogels,9 and activated carbon fibres,10 also have been exploited as supports of hemin to mimic the HRP structure. Recently, hemin alone was examined as an alternative oxidation catalyst to HRP,11 but was found to be less active than the hemins bound on a support.9 Notably, although the coordination of the central Fe ion is crucial to achieve a high activity, the coordination (or binding) method of hemin has not been considered in previous studies of HRP mimics. In the presence of a suitable hemin-binding ligand, an alternative of the His170/Asp247 combination, hemin will yield high oxidative catalytic activity upon binding.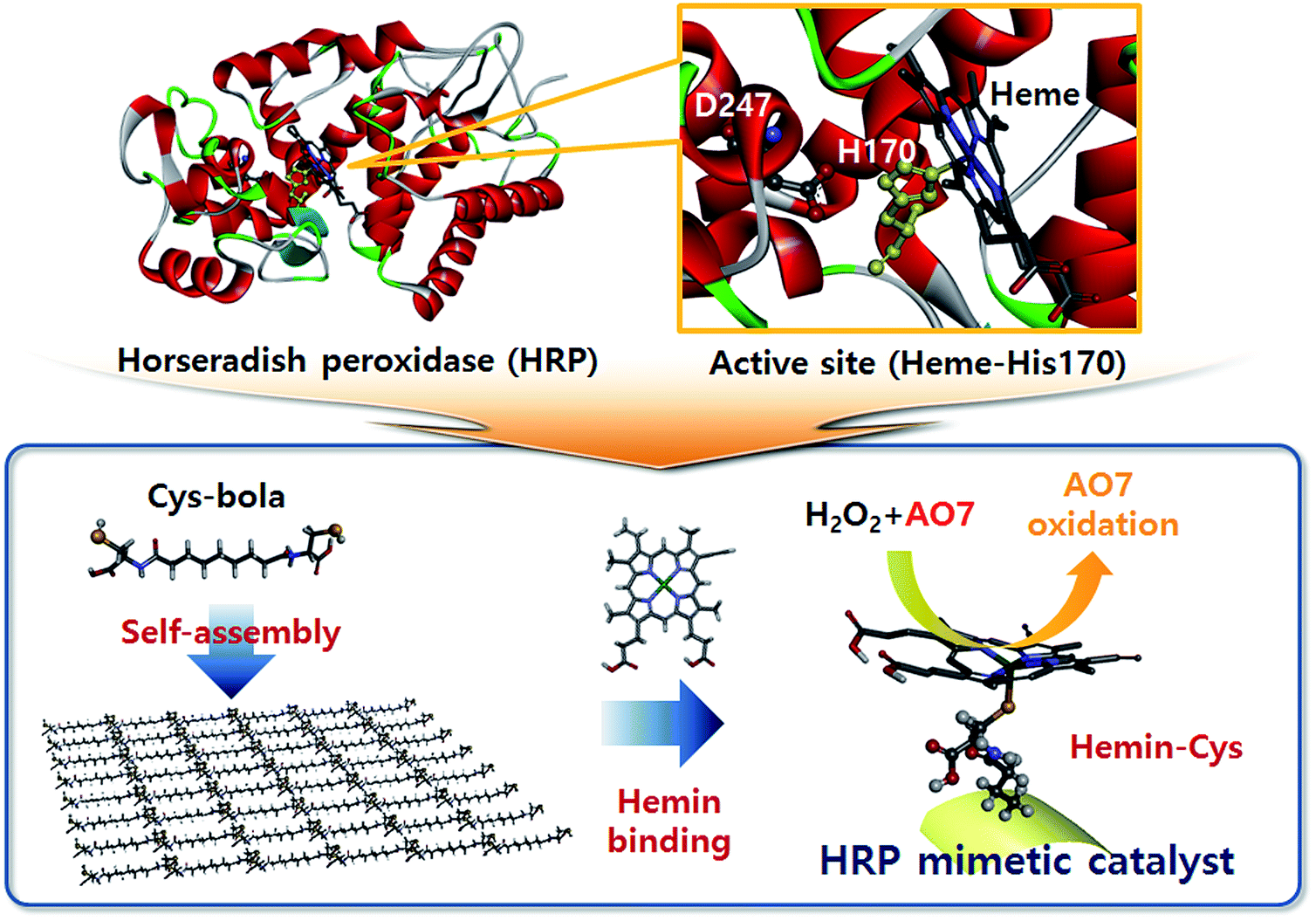 | ||
| Scheme 1 HRP structure and HRP-mimetic catalyst prepared from cysteinyl bolaamphiphile self-assemblies. At the active site of HRP, the heme cofactor is linked to His170. The cysteinyl bolaamphiphile self-assembled structure contains numerous cysteine sulfhydryl groups on the surface. Upon binding of hemin with the surface-exposed sulfhydryl groups, a catalytically active HRP mimic was obtained. | ||
Cysteine thiol is a possible binding ligand that can provide hemin with the enhanced catalytic activity and considerable binding strength. The cysteine thiol–heme Fe ion tethering is observed in natural enzymes. For example, cytochrome P450 (ref. 12) and chloroperoxidase13 have an active site of thiol-bound heme at which the enzymatic oxidation reaction is carried out. In addition, thiol–iron ion binding is advantageous to tether strongly the hemin on the substrate in the absence of additional binding motifs like the heme pocket.14 Considering these functions of thiol–Fe binding, the cysteine-decorated matrices are promising supports for hemin immobilization. Recently, we reported that the self-assembly of cysteinyl bolaamphiphiles (Cys-C7 hereafter) can produce a soft, biomimetic matrix with abundant surface-exposed thiols and disulfides.15 The Cys-C7 consists of a central hydrophobic heptyl chain conjugated to hydrophilic cysteine motifs through peptide bonds. Because of the amphiphilic character, the Cys-C7 molecules self-assemble to form suprastructures stabilized by hydrogen bonding. The self-assembly process results in an ordered arrangement of the bolaamphiphiles at the molecular level with exposure of thiol (and disulfide) groups to which hemin can bind. The hemin-decorated self-assembled structure showed good structural stability under chemical and thermal stresses,15 and can therefore be exploited as a solid HRP-mimetic catalyst, which undergoes a change in chemical composition during the oxidation reaction, as illustrated in Scheme 1.
Here, the effect of experimental conditions such as pH and temperature on the oxidative catalytic activity of the hemin-decorated self-assembled Cys-C7 was investigated. The orange dye Acid Orange 7 (AO7), known to be a serious water pollutant, was tested as a substrate for degradation by catalytic oxidation in the presence of H2O2. Decomposition of the dye was monitored to evaluate the catalytic activity of the HRP-mimetic catalyst, and the activation energy was determined from the reaction rates at various temperatures. The mechanism of the catalytic oxidation was deduced from kinetic studies. Our results revealed that the bolaamphiphile assembly was a reliable biomimetic support with Fe binding sites and a protein-like environment, which expressed the oxidative catalytic activity from hemin presumably by facilitating Fe(III)/Fe(IV) transition during the catalysis.
Experimental section
For the preparation of the cysteine-rich matrix, Cys-C7 was synthesized from L-cysteine and azelaic acid according to a previously reported procedure.15 Briefly, the cysteine benzyl ester p-toluene sulfonate was synthesized and was conjugated with azelaic acid through the carbodimide chemistry. After the hydrolysis of the intermediate product and subsequent crystallization, purified Cys-C7 powder was obtained. The pure Cys-C7 powder was dissolved in HEPES buffer (10 mM, pH 7.0) at a concentration of 7 mg mL−1 and left for one day in the buffer solution at room temperature for self-assembly. Bovine hemin (>90%, Sigma-Aldrich, St. Louis, MO) was dissolved in the prepared Cys-C7 assembly suspension at a concentration of 0.1 mM, and left for one day to bind to the surface of the Cys-C7 assemblies. The hemin-bound Cys-C7 assemblies were applied as the HRP-mimetic catalysts.When preparing the Cys-C7 assemblies, the reducing agent β-mercaptoethanol (β-ME) was added to the Cys-C7 solution at a concentration of 10 mM to reduce the disulfide bonds. In the absence of β-ME, cysteine thiols are oxidized to form disulphide bonds during the self-assembly. To evaluate the effect of cysteine thiol oxidation on the catalytic activity, Cys-C7 assemblies prepared in the presence/absence β-ME were used as supports, respectively, in preparing the HRP-mimetic catalysts by addition of hemin.
The catalytic activity was tested by adding H2O2 and AO7 (dye content ≥85%, Sigma-Aldrich) substrate solutions to the prepared HRP-mimetic catalyst suspension. Stock solutions of H2O2 and AO7 were prepared in HEPES buffer (10 mM, pH 7.0) at concentrations of 10 mM and 0.05 mM, respectively. To the 1 mL of the HRP-mimetic catalyst suspension, 1 mL of AO7 solution and 0.1 mL of H2O2 solution were added in sequence to initiate the catalytic reaction. After mixing the substrate solutions, the concentrations of the reaction mixture were as follows: Cys-C7 3.3 mg mL−1, hemin 0.048 mM, AO7 0.024 mM, and H2O2 0.48 mM. The progress of the catalytic reaction was monitored by following the disappearance of the characteristic AO7 absorption band at 484 nm by UV-vis spectroscopy (S-3100, Scinco).16
Since hemin binding to the support is a decisive factor of the catalytic activity, concentrations of Cys-C7 (0.48–6.67 mg mL−1) and hemin (0.005–0.048 mM) were controlled to observe the effect of each component on the catalysis. To observe the effect of pH, HEPES buffer solutions at various pH (pH = 3.5–8.2) were used whose pH was controlled with 0.1 M HCl and 0.1 M NaOH. Activation energy of the catalyst was determined from the reaction rate at various temperatures (10–50 °C). From the slope of the AO7 oxidation profiles at the beginning of the reaction, the reaction rate was determined and the reaction rate constants were determined assuming a first order reaction. The reusability of the catalyst was tested by repeating the AO7 oxidation reaction. After 10 min reaction time, the remaining AO7 concentration was measured, and the catalytic activity was determined. After each use, the catalysts were collected, washed with deionized water, and re-dispersed in the AO7 solution for repeated use.
Control experiments using the natural HRP (lyophilized, 148 U mg−1, Sigma) were performed for the activity comparison to the mimetic catalyst. Oxidation of AO7 was carried out in various HRP concentrations in HEPES buffer solution (10 mM, pH 7.0) in the presence of AO7 and H2O2 substrates.
Results and discussion
The catalytic oxidation of AO7 was represented by the degradation ratio that was calculated by the following equation:
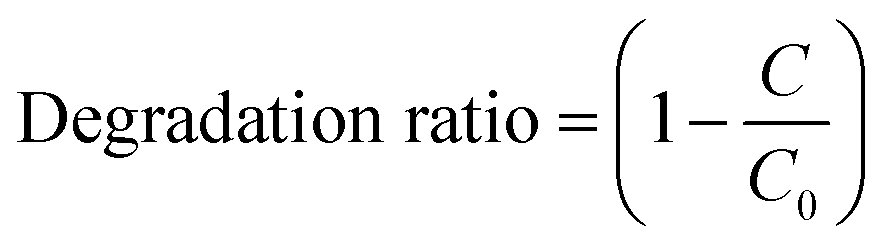 | (1) |
 | ||
| Fig. 1 Catalytic oxidation of AO7 by the HRP-mimetic catalyst and the individual components used for its preparation. (a) Normalized AO7 concentration representing the activity of the catalyst and its individual components, (b) AO7 degradation ratio by the catalyst and its individual components (error bar represents the standard deviation, n = 3), (c) oxidation profiles of AO7 by HRP and the HRP-mimetic catalyst at various concentrations. | ||
The activity of the HRP-mimetic catalyst was compared with that of HRP. As a control, HRP was dissolved in HEPES buffer (10 mM, pH 7) at a concentration of 0.35 mg mL−1. To this HRP solution, AO7 and H2O2 substrates were added at designed concentrations (0.024 and 0.48 mM, respectively) to initiate the oxidative catalysis. The oxidation profiles of AO7 by HRP and by the HRP-mimetic catalyst at various concentrations are shown in Fig. 1c. The oxidation profiles at the same concentration of 0.35 mg mL−1 indicate that the activity of the HRP-mimetic catalyst was a little lower than that of HRP. The activity of the HRP-mimetic catalyst was estimated to be between 110 and 130 U mg−1 from other oxidation profiles. This suggests that the prepared mimetic catalyst has considerable activity although it is not as active as HRP.
For the peroxidase, the oxidative catalytic activity originates from a reactive Fe(IV)-oxo radical intermediate in the centre of hemin,17–19 which is formed by heterolytic cleavage of H2O2 and binding of oxene on Fe ion that is coordinated with a chelating porphyrin ligand.8,20 The observed catalytic activity suggests that cysteine thiol of Cys-C7 assembly was an effective binding ligand for generating the active Fe(IV)-oxo intermediate species of hemin. The anionic cysteinate is likely to stabilize Fe(III) leading to formation of Fe(IV)-oxo intermediate when H2O2 is associated with.21 Compared to building His/Asp combination in a well-controlled conformation, usage of cysteine is advantageous to prepare a HRP mimic because of its simplicity and high binding affinity to Fe, which allows the solid immobilization of hemin on the surface of the support.
Because the catalytic activity stems from the binding of hemin to Cys-C7 assembly, the effect of each component of the HRP-mimetic catalyst was investigated. First, the catalytic activity was examined at different hemin concentrations. AO7 degradation increased with increasing hemin concentration at fixed concentrations of Cys-C7 (3.3 mg mL−1) and H2O2 (0.47 mM). Specifically, the degradation rate of AO7 was determined for hemin concentrations in the range of 0.005–0.04 mM from the slope of the degradation plot at the early stage (t < 400 s) of the reaction (Fig. 2a and b). The increasing degradation rate implies the formation of more active sites with increasing addition of hemin to the Cys-C7 assemblies, leading to faster degradation. At hemin concentrations above 0.04 mM, the binding sites of the Cys-C7 assembly seemed to be saturated, and no further increase in degradation rate was observed. When 0.048 mM hemin was used, the AO7 degradation ratio did not exceed 0.8 (C/C0 = 0.2), as shown in Fig. 2a, suggesting that complete degradation of AO7 could not be achieved, presumably because of inactivation of the catalyst. Inactivation of the active site of hemin may result from the interaction of the phenoxy radicals with the catalytic centre.22
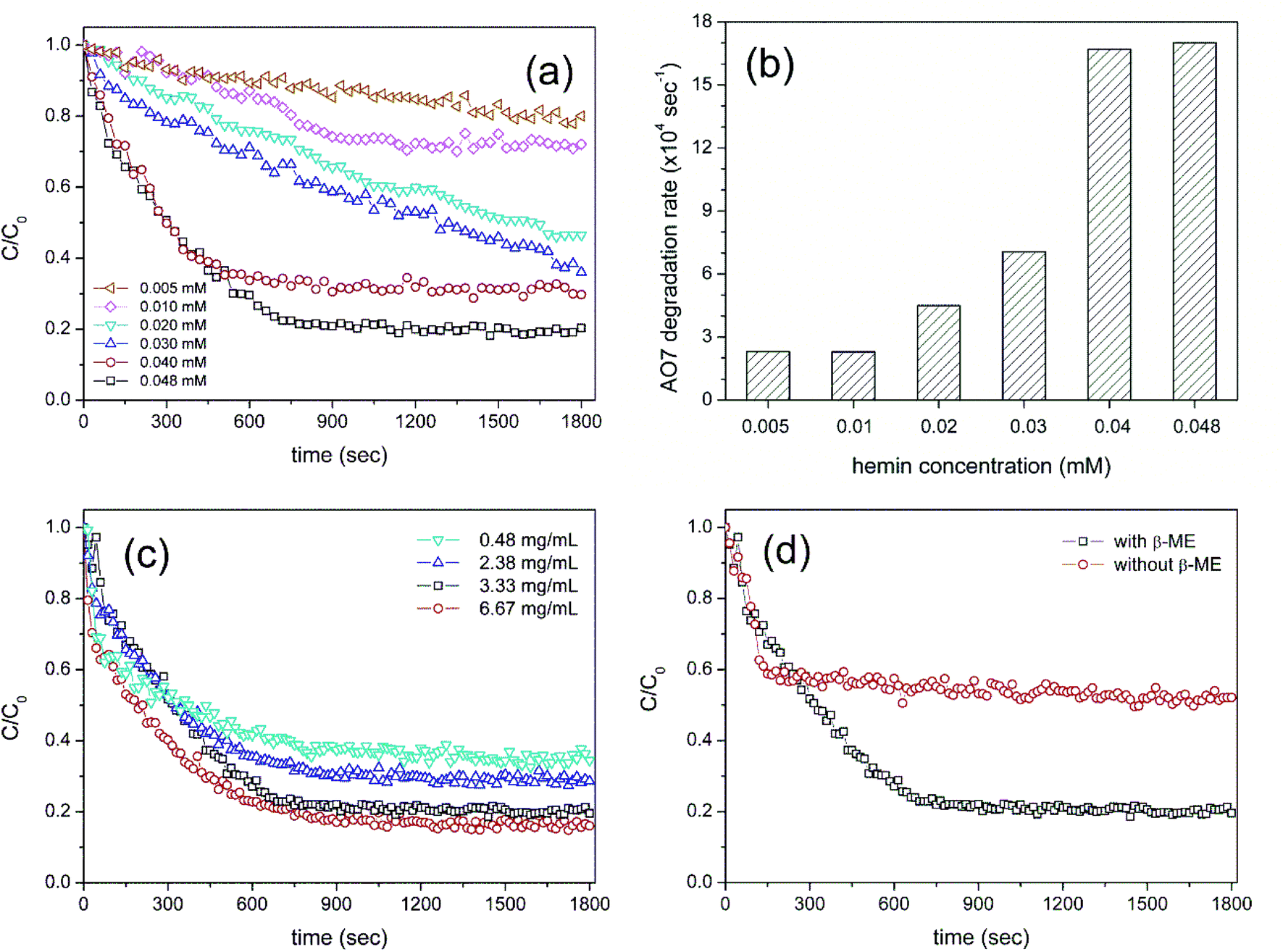 | ||
| Fig. 2 Effect of hemin and Cys-C7 assembly on the catalytic activity. (a) Normalized AO7 concentration and (b) corresponding initial oxidation reaction rate at different hemin concentrations (0.005–0.048 mM) and constant Cys-C7 concentration (3.3 mg mL−1). (c) Normalized AO7 concentration at different concentration of Cys-C7 (0.48–6.67 mg mL−1) and constant hemin concentration (0.048 mM). (d) Change in catalytic activity upon addition of β-mercaptoethanol, which reduces the disulfide bonds in the Cys-C7 assembly. | ||
The effect of hemin concentration suggests that the activity of the HRP-mimetic catalyst can be influenced by the amount of Cys-C7 assembly. Thus, we examined the catalytic activity at various Cys-C7 concentrations in the range of 0.48–6.67 mg mL−1 while keeping the hemin concentration at 0.048 mM. No significant differences in the initial degradation rate were observed, indicating that the initial reaction rate was mainly determined by the amount of hemin (Fig. 2c). However, overall, the degradation ratio increased proportionally with the amount of Cys-C7. It can be inferred that higher Cys-C7 concentrations provided more cysteine thiol binding sites for hemin resulting in the formation of more catalytically active sites and therefore higher degradation ratios.
The binding of hemin to the Cys-C7 assembly was mainly governed by the surface cysteine sulfhydryl groups. During the self-assembly process, the cysteine thiols were oxidized to form disulfide bonds. Addition of β-ME to the Cys-C7 assembly suspension would reduce the disulfide bonds to thiol groups providing more binding sites for hemin, thereby resulting in the formation of more active sites. Fig. 2d shows the AO7 degradation profiles before and after reduction of the disulfide bonds on the Cys-C7 assembly. Without β-ME, less hemin molecules could bind to the Cys-C7 assembly resulting in lower catalytic activity, whereas after reduction of the disulfide bonds, the degree of AO7 degradation increased. This result demonstrates that the binding between cysteine thiol and hemin Fe ion is the key to the catalytic activity. Formation of disulfide bonds during self-assembly is inevitable because of the vicinity of the cysteine thiols.
The catalytic reaction was also strongly affected by physicochemical conditions such as pH and temperature. Similar to the natural enzyme, the HRP-mimetic catalyst showed an optimal pH. Fig. 3a shows the AO7 concentration profiles during the catalytic oxidation at different pH values. The catalytic AO7 degradation was accelerated by increasing the solution pH from 3.5 to 7.0, at which the maximum catalytic performance was achieved, and was retarded at a basic condition of pH 8.2. The maximum performance at pH 7.0 is indicative of the biological character of the HRP-mimetic catalyst. We considered the pH effect on the individual components of the catalyst and on the substrate. In the absence of the catalyst, the degradation of AO7 is promoted in acidic solution due to the neutralization of the SO3− group of AO7 and formation of HOO˙ radicals from H2O2 dissociation.23 In addition, hemin alone shows maximum oxidation activity at pH 3.2.10 Thus, the maximum catalytic activity at pH 7.0 suggests that the optimal performance was not due to an enhanced substrate activity or hemin activation, but is indicative of the HRP-like character of the prepared catalyst, which is consistent with the optimal pH of HRP (6.0–7.2).24–26 The pH-dependent activity of the HRP-mimetic catalyst suggests the anionic cysteinate coordination to Fe(III) of hemin, which is common for many metalloproteins.21 Under acidic conditions, the cysteinate is protonated to form neutral cysteine that weakly binds Fe(III). Thus, less hemin binding binds on the Cys-C7 assembly resulting in decrease of catalytic activity. At the basic condition, promotion of the ionization of SO3− group of AO7 is likely to retard the catalysis.
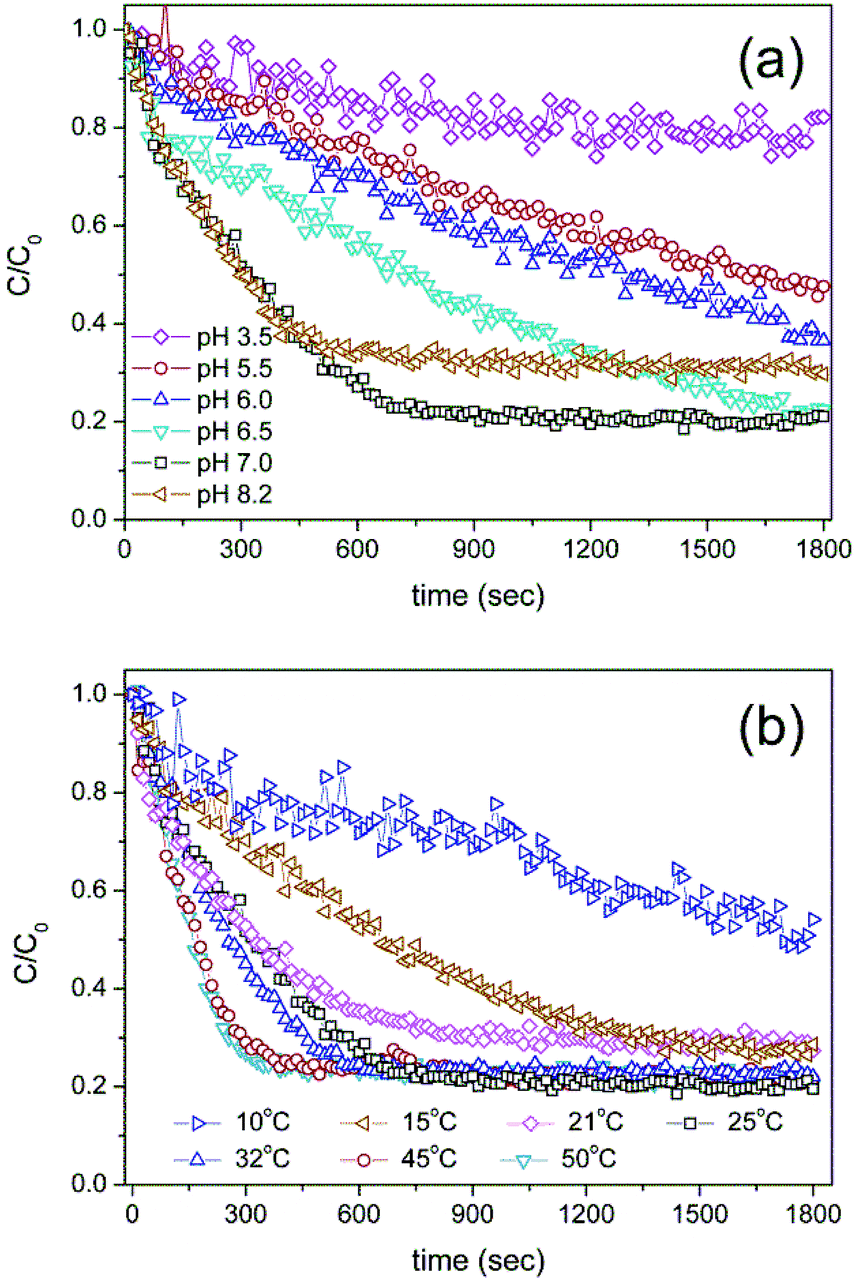 | ||
| Fig. 3 AO7 concentration profiles during the catalytic oxidation at different (a) pH values and (b) temperature. | ||
Interestingly, the catalytic activity of the HRP-mimetic catalyst continuously increased with increasing temperature up to 45 °C (Fig. 3b), in contrast with the natural HRP activity, which decreases at temperatures above 38 °C.26–28 The temperature dependency is a non-biological character of the HRP-mimetic catalyst, although the activity did not increase further at temperatures above 45 °C. This non-biological behaviour is presumably due to the thermal stability of the Cys-C7 assembly structure and solid hemin binding through the cysteinate-Fe bonding. On the other hand, the structure of HRP is destabilized above ∼40 °C, resulting in reduced catalytic activity.29,30 The molecular interactions responsible for the assembly of Cys-C7 molecules, including hydrophobic interactions, hydrogen bonding, and disulfide bondings, were weakened by increasing temperature, but were still strong enough to maintain the Cys-C7 assembly stability.
The activation energy of the HRP-mimetic-catalysed oxidation was evaluated from the reaction rate constants at different temperatures. To estimate the apparent rate constant, kobs, the reaction was treated as a pseudo-first-order reaction dependent only on the AO7 concentration. Because the concentration of H2O2 was about 20 times that of AO7, the effect of the H2O2 concentration was negligible. This pseudo-first-order reaction rate can be simply calculated by eqn (2):
| −rAO7 = kobsCAO7 | (2) |
The logarithm of the reaction rate constant, kobs, was plotted against 1/T according to the Arrhenius equation, and the activation energy was calculated from the slope of the linear regression (Fig. 4). The activation energy of AO7 oxidation catalysed by the HRP-mimetic catalyst was 31.7 kJ mol−1, which was higher than that of the reaction catalysed by HRP (15.6 kJ mol−1)31 or hemin–graphene composite (20.37 kJ mol−1),32 but lower than that of the oxidation promoted by hemin alone (37.0 kJ mol−1).11 The magnitude of the activation energy suggests that the interaction between the Cys-C7 assembly and the AO7 substrate was not sufficiently strong to accelerate the catalysis. As reported for other oxidation catalysts, the affinity of the catalyst support to the substrate influenced the reaction rate.33,34 The activation energy would increase with increasing inertness of the support to the target substrate. It is likely that the attractive interaction between AO7 and the Cys-C7 assembly was not significant because the aromatic nature of AO7 was repulsive to the surface carboxyl and sulfhydryl groups of the Cys-C7 assembly.
 | ||
| Fig. 4 Determination of the activation energy of AO7 oxidation catalysed by the HRP-mimetic catalyst from the Arrhenius plot. Assuming pseudo-first-order reaction kinetics, the apparent rate constant (kobs) was determined from the onset reaction rate at each temperature. | ||
Absence of distal H2O2 binding site is another factor for the high activation energy. Distal ligands of His42 and Arg38 work as acid/base to facilitate the heterolysis of H2O2 resulting in the promotion of the overall catalysis.35 It is natural that the activation energy (and rate) of the reaction is lowered in the absence of such assisting ligands.
The oxidation catalysed by the HRP-mimetic catalyst is a bi-substrate reaction with kinetics governed by both substrates. Detailed analysis of kinetics and mechanism was performed based on the reaction rates at different substrate concentrations. The catalytic reaction rate can be described by the bi-substrate Michaelis–Menten (MM) eqn (3):36
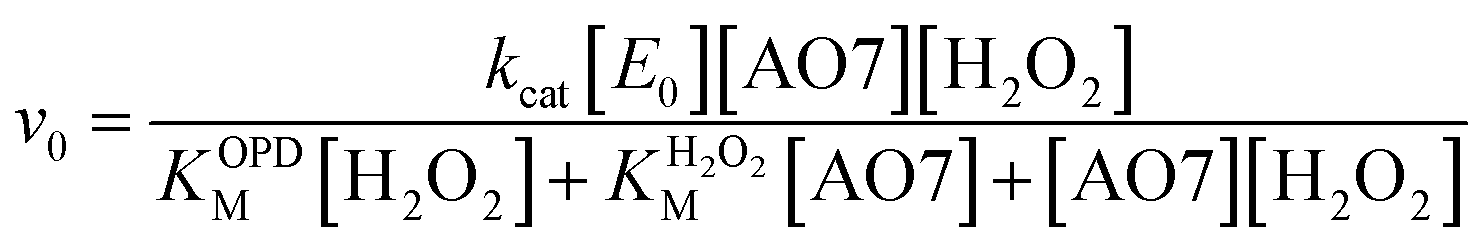 | (3) |
Fig. 5a and b show the MM kinetic and double-reciprocal plots, respectively, obtained using the reaction rates determined at fixed AO7 concentrations while varying the H2O2 concentrations. As can be seen from the MM kinetic plots, the substrate-dependent reaction rates indicated the enzyme-like behaviour of the HRP-mimetic catalyst. Similar concentration-dependent MM kinetic plots were obtained at fixed H2O2 concentrations (Fig. 5c and d). The kinetic parameters, namely, the Michaelis constants (KM) and turnover numbers (kcat), were determined from the double-reciprocal plots assuming a pseudo-first-order reaction, and the catalytic efficiency (kcat/KM) was calculated. The catalytic efficiencies and kinetic parameters are listed in Table 1. The substrates showed different affinities for the HRP-mimetic catalyst. The Michaelis constant (KAO7M) increased with increasing AO7 concentration (Fig. 5a and b), indicating a weak attractive interaction between AO7 and the HRP-mimetic catalyst. For substrates with good affinity for the catalyst, the KM decreases with increasing substrate concentration.37 The weak interaction between AO7 and the HRP-mimetic catalyst is consistent with previous results on the activation energy. The turnover number (kAO7cat) also increased with AO7 concentration, but not significantly. Thus, the catalytic efficiency (kAO7cat/KAO7M) decreased with AO7 concentration. The slight increase in turnover number was presumably due to the high AO7 content in the reaction mixture, which enhanced the chances of collision for reaction at the active sites.
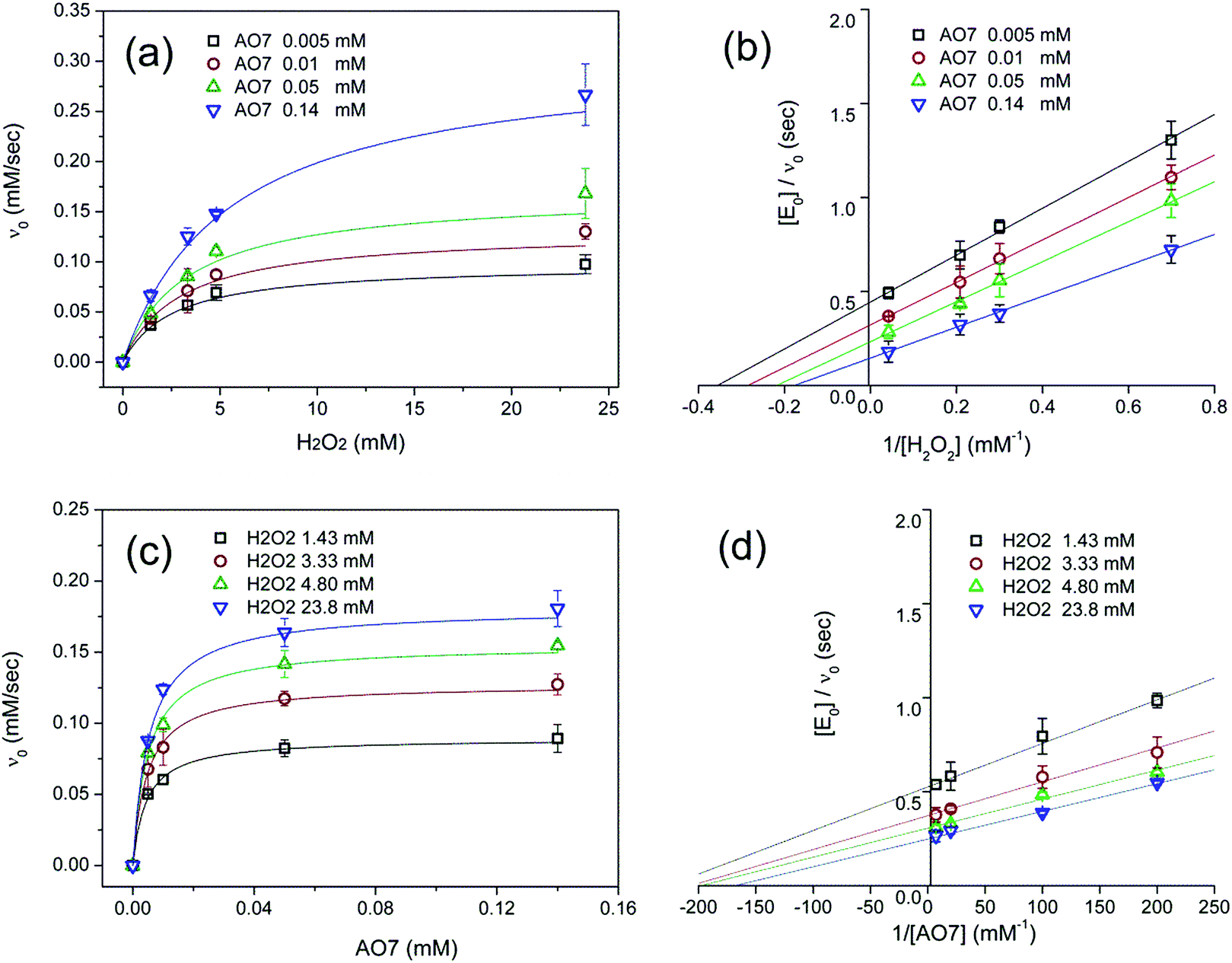 | ||
| Fig. 5 Steady-state kinetic assay of the HRP-mimetic catalysts. The apparent reaction rates obeyed the Michaelis–Menten kinetic model with linear double-reciprocal plots, which were obtained (a, b) for various H2O2 concentrations at a AO7 fixed concentration (0.005, 0.01, 0.05, and 0.14 mM) and (c, d) for various AO7 concentrations at a fixed H2O2 concentration (1.43, 3.33, 4.80, and 23.8 mM), respectively. The reaction rates were determined at pH 7.0 using the HRP-mimetic catalysts prepared from 0.048 mM hemin and 3.3 mg mL−1 Cys-C7. Error bars represent the standard deviation (n = 3). | ||
| AO7 conc. (mM) | kAO7cat (s−1) | KAO7M (mM) | kAO7cat/KAO7M (s−1 mM−1) |
|---|---|---|---|
| 0.005 | 35.9 ± 2.4 | 0.07 ± 0.01 | 536.1 ± 5.0 |
| 0.01 | 39.1 ± 1.6 | 0.1 ± 0.02 | 317.2 ± 4.8 |
| 0.05 | 44.2 ± 7.6 | 0.4 ± 0.05 | 112.6 ± 5.3 |
| 0.14 | 44.4 ± 9.1 | 1.4 ± 0.56 | 33.0 ± 9.9 |
| H2O2 conc. (mM) |
|
|
|
|---|---|---|---|
| 1.34 | 64.2 ± 6.4 | 45.7 ± 1.9 | 1.4 ± 0.1 |
| 3.33 | 84.5 ± 3.8 | 105.0 ± 4.3 | 0.8 ± 0.0 |
| 4.80 | 99.8 ± 4.2 | 147.4 ± 4.2 | 0.7 ± 0.1 |
| 23.8 | 104.5 ± 3.7 | 615.9 ± 2.8 | 0.2 ± 0.1 |
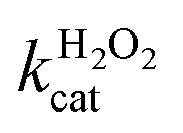
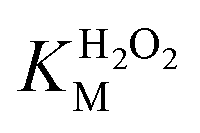
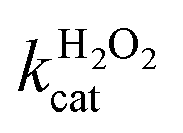
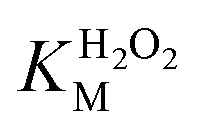
A similar change in the kinetic parameters was observed when varying the H2O2 concentration (Fig. 5c and d). With increasing H2O2 concentration, 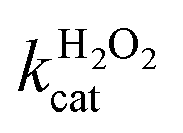 /
/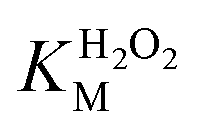 decreased because of a drastic increase of
decreased because of a drastic increase of 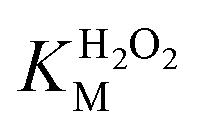 accompanied by a mild increase of
accompanied by a mild increase of 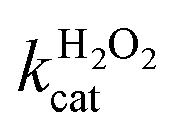 . The
. The 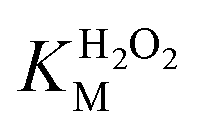 increase suggests that the driving force for H2O2 binding to the Fe centre of hemin is not significant, either. In natural HRP, the His 42 and Arg38 in distal helix promotes the binding of H2O2 to the Fe centre.38 Furthermore, these distal ligands are responsible for the high reaction rate of HRP.39 However, the lack of this assisting amino acid in the HRP-mimetic catalyst resulted in a low affinity for H2O2.
increase suggests that the driving force for H2O2 binding to the Fe centre of hemin is not significant, either. In natural HRP, the His 42 and Arg38 in distal helix promotes the binding of H2O2 to the Fe centre.38 Furthermore, these distal ligands are responsible for the high reaction rate of HRP.39 However, the lack of this assisting amino acid in the HRP-mimetic catalyst resulted in a low affinity for H2O2.
The reaction mechanism of the HRP-mimetic catalyst can be inferred from the parallel double-reciprocal plots, which suggested a ping-pong mechanism where an intermediate complex is formed by association of one substrate to the active site followed by reaction with the other substrate.31 This agrees with the generally accepted mechanism of HRP where the association of H2O2 with the Fe active centre of hemin generates the active Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O intermediate leading to subsequent oxidation of the other substrate. This ping-pong mechanism based on the formation of active intermediates with H2O2 was also observed in many other peroxidase-mimetic catalysts with active sites of biological and organic compounds.40–42
O intermediate leading to subsequent oxidation of the other substrate. This ping-pong mechanism based on the formation of active intermediates with H2O2 was also observed in many other peroxidase-mimetic catalysts with active sites of biological and organic compounds.40–42
To evaluate the practical applicability of the HRP-mimetic catalyst, its reusability was investigated. The oxidation of AO7 was monitored with repeated use. The catalytic activity gradually decreased (Fig. 6a), presumably because of hemin degradation by repeated exposure to H2O2. Heme was reported to be degraded by H2O2 leading to inactive species, which were produced by the formation of Fe(II)-oxy species or by binding of O2˙ radical.43 Fig. 6b shows the AO7 oxidation profiles with repeated use of the catalyst. The onset rate (corresponding to the initial slopes) of the normalized AO7 concentration profiles decreased with reuse of the catalyst, and the reaction reached a plateau at a lower degree of AO7 oxidation. These results confirm the degradation of the catalyst, indicating that the decrease in catalytic activity was not caused by the loss of catalyst or detachment of hemin during the collection and washing steps. Because hemin degradation is also observed even in natural enzymes with heme cofactors, the inherent vulnerability of hemin was considered responsible for the degradation of the catalyst.
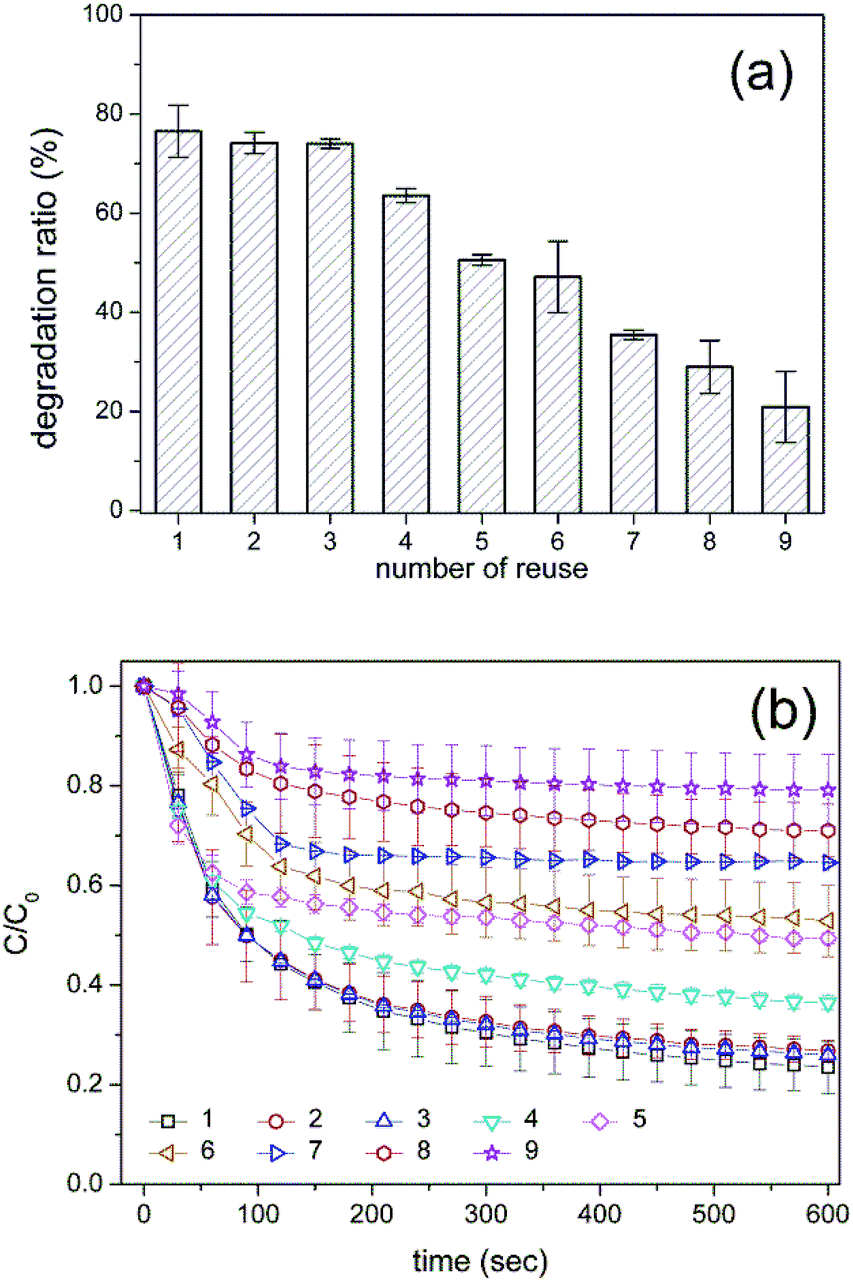 | ||
| Fig. 6 Reusability of the HRP-mimetic catalyst. (a) Changes in the degradation ratio and (b) AO7 degradation profiles with repeated use of the catalyst (error bar represents the standard deviation, n = 3). | ||
Finally, the HRP-mimetic catalyst was applied for the oxidation of other organic substrates. Two organic compounds of methylene blue (MB) and o-phenylenediamine (OPD) were tested as substrates. Since catalytic oxidation was mainly induced by the active Fe(IV)-oxo radical intermediate at the hemin centre, these organic compounds are readily oxidized in the presence of the HRP-mimetic catalyst. Degradation ratios of these two compounds are shown in Fig. 7. High degradation ratio of MB and OPD indicates that the oxidative catalysis mechanism works for other organic substrates as well as AO7. Except MB and OPD, other organic compounds (3,3′,5,5′-tetramethylbenzidine and pyrocatechol) were also oxidized by the catalyst (data not shown). Thus, the HRP-mimetic catalyst is applicable for the oxidation of various organic substrates without substrate specificity. The absence of the specific binding site for a target substrate in the HRP-mimetic catalyst accounts for the absence of substrate specificity.
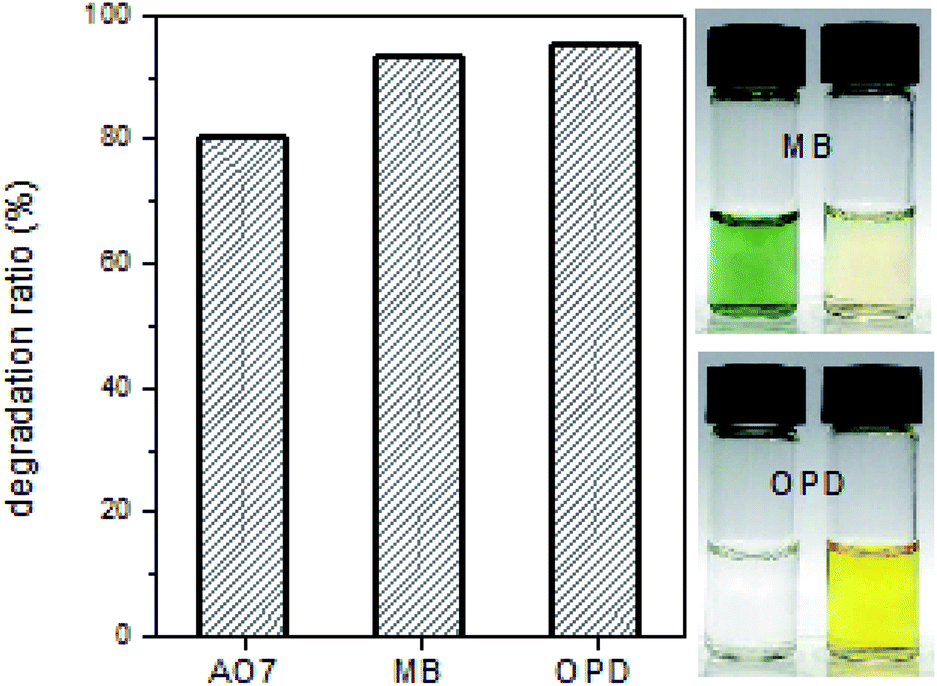 | ||
| Fig. 7 Catalytic oxidation of methylene blue (MB) and o-phenylenediamine (OPD) by the HRP-mimetic catalyst (left: degradation ratio of each component, right: photographic images of substrate solutions before and after the catalytic oxidation). | ||
Conclusions
In summary, experimental data demonstrated that hemin bound to Cys-C7 assemblies through a cysteine thiol–Fe bond showed oxidative catalytic activity. The Cys-C7 assembly served as a biomimetic matrix whose surface-exposed thiol groups bound Fe of hemin to express the oxidative catalytic activity by facilitating the formation of the active Fe(IV)-oxo radical intermediate during catalysis. The prepared HRP-mimetic catalyst displayed a lower activation energy than hemin alone and enhanced stability at temperatures above 40 °C. Compared to other polymeric and biomolecular supports using hemin as the active site, the Cys-C7 assembly provided an obvious binding site for hemin while the catalytic oxidation proceeded by the ping-pong mechanism. Expression of the catalytic activity of hemin after binding to the thiol-rich support confirmed that coordination of the central Fe ion was the key factor to form the active catalyst intermediate. The synergistic combination of natural cofactors and biomimetic supports prepared by the assembly of engineered bolaamphiphiles is an appealing method to develop enzyme-mimetic catalysts with designed activity.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by a grant from the Korean Research Foundation funded by the Korean Government (NRF-2016R1D1A1A09917929) and by the Human Resources Program in Energy Technology of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) granted financial resource from the Ministry of Trade, Industry & Energy, Republic of Korea (No. 20154010200810).References
- H. Bos and W. de Souza, J. Immunol. Methods, 2000, 238, 29–43 CrossRef CAS PubMed.
- A. M. Azevedo, V. C. Martins, D. M. F. Prazeres, V. Vojinovic, J. M. S. Cabral and L. P. Fonseca, Biotechnol. Annu. Rev., 2003, 9, 199–247 CAS.
- M. Gajhede, D. J. Schuller, A. Henriksen, A. T. Smith and T. L. Poulos, Nat. Struct. Mol. Biol., 1997, 4, 1032–1038 CAS.
- Q. Huang, K. Szigeti, J. Fidy and R. Schweitzer-Stenner, J. Phys. Chem. B, 2003, 107, 2822–2830 CrossRef CAS.
- A. Cordoba, I. Magario and M. L. Ferreira, Int. Biodeterior. Biodegrad., 2012, 73, 60–72 CrossRef CAS.
- S. Pirillo, F. S. G. Einschlag, E. H. Rueda and M. L. Ferreira, Ind. Eng. Chem. Res., 2010, 49, 6745–6752 CrossRef CAS.
- P. Zucca, C. M. B. Neves, M. M. Q. Simoes, M. G. P. M. S. Neves, G. Cocco and E. Sanjust, Molecules, 2016, 21, 964 CrossRef PubMed.
- G.-X. Wang, Y. Zhou, M. Wang, W.-J. Bao, K. Wang and X.-H. Xia, Chem. Commun., 2015, 51, 689–692 RSC.
- Q. Wang, Z. Yang, X. Zhang, X. Xiao, C. K. Chang and B. Xu, Angew. Chem., Int. Ed., 2007, 46, 4285–4289 CrossRef CAS PubMed.
- Y. Yao, Y. Mao, Q. Huang, L. Wang, Z. Huang, W. Lu and W. Chen, J. Hazard. Mater., 2014, 264, 323–331 CrossRef CAS PubMed.
- M. Yan, H. Xie, Q. Zhang, H. Qu, J. Shen and J. Kong, J. Mater. Sci. Chem. Eng., 2016, 4, 26–34 CAS.
- C. A. Hasemann, R. G. Kurumbail, S. S. Boddupalli, J. A. Peterson and J. Deisenhofer, Structure, 1995, 3, 41–62 CrossRef CAS PubMed.
- S. Colonna, N. Gaggero, C. Richelmi and P. Pasta, Trends Biotechnol., 1999, 17, 163–168 CrossRef CAS PubMed.
- D. J. Schuller, N. Ban, R. B. van Huystee, A. McPherson and T. L. Poulos, Structure, 1996, 4, 311–321 CrossRef CAS PubMed.
- C. Lee, M.-C. Kim and S.-Y. Lee, Colloids Surf., B, 2016, 142, 360–366 CrossRef CAS PubMed.
- Y.-Y. Lau, Y.-S. Wong, T.-T. Teng, N. Morad, M. Rafatullah and S.-A. Ong, Chem. Eng. J., 2014, 246, 383–390 CrossRef CAS.
- X. Yang, C. Fang, H. Mei, T. Chang, Z. Cao and D. Shangguan, Chem.–Eur. J., 2011, 17, 14475–14484 CrossRef CAS PubMed.
- K. D. Karlin, Nature, 2010, 463, 168–169 CrossRef CAS PubMed.
- J. G. Liu, T. Ohta, S. Yamaguchi, T. Ogura, S. Sakamoto, Y. Maeda and Y. Naruta, Angew. Chem., Int. Ed., 2009, 48, 9262–9267 CrossRef CAS PubMed.
- P. Campomanes, U. Rothlisberger, M. Alfonso-Prieto and C. Rovira, J. Am. Chem. Soc., 2015, 137, 11170–11178 CrossRef CAS PubMed.
- R. Perera, M. Sono, J. A. Sigman, T. D. Pfister, Y. Li and J. H. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3641–3646 CrossRef CAS PubMed.
- F. Gholami-Borujeni, A. H. Mahvi, S. Nasseri, M. A. Faramarzi, R. Nabizadeh and M. Alimohammadi, Appl. Biochem. Biotechnol., 2011, 165, 1274–1284 CrossRef CAS PubMed.
- N. H. Ince and G. Tezcaanli-Güyer, Ultrasonics, 2004, 42, 591–596 CrossRef CAS PubMed.
- S. Wang, H. Fang, Y. Wen, M. Cai, W. Liu, S. He and X. Xu, RSC Adv., 2015, 5, 57286–57292 RSC.
- Q. Chang and H. Tang, Molecules, 2014, 19, 15768–15782 CrossRef CAS PubMed.
- N. Gupta, A. Shrivastava and R. K. Sharma, Int. J. Nanomed., 2012, 7, 5491–5500 Search PubMed.
- Z. Yincan, L. Yan, G. Xueyong, W. Qiao and X. Xiaoping, RSC Adv., 2017, 7, 18976–18986 RSC.
- Y. Yang, D. Shen, Y. Long, Z. Xie and H. Zheng, Sci. Rep., 2017, 7, 43141 CrossRef CAS PubMed.
- D. G. Pina, A. V. Shnyrova, F. Gavilanes, A. Rodriguez, F. Leal, M. G. Roig, I. Y. Sakharov, G. G. Zhadan, E. Villar and V. L. Shnyrov, FEBS J., 2001, 268, 120–126 CrossRef CAS.
- C. Li, L. Zhou, C. Wang, X. Liu and K. Liao, RSC Adv., 2015, 5, 41994–41998 RSC.
- J. Terres, R. Battisti, J. Andreaus and P. C. De Jesus, Biocatal. Biotransform., 2014, 32, 64–73 CrossRef CAS.
- X. Lv and J. Weng, Sci. Rep., 2013, 3, 3285 CrossRef PubMed.
- T. Mallat and A. Baiker, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 11–28 CrossRef CAS PubMed.
- P. Sudarsanam, A. Rangaswamy and B. M. Reddy, RSC Adv., 2014, 4, 46378–46382 RSC.
- T. L. Poulos, in Biological Inorganic Chemistry: Structure and Reactivity, ed. I. Bertini, H. B. Gray, E. I. Stiefel and J. S. Valentine, University Science Books, Sausalito, California, 2007, XI.3, pp. 343–352 Search PubMed.
- M.-C. Kim and S.-Y. Lee, Nanoscale, 2015, 7, 17063–17070 RSC.
- M.-C. Kim, D. Lee, S. H. Jeong, S.-Y. Lee and E. Kang, ACS Appl. Mater. Interfaces, 2016, 8, 34317–34326 CAS.
- J. N. Rodriguez-Lopez, A. T. Smith and R. N. F. Thorneley, J. Biol. Chem., 1996, 271, 4023–4030 CrossRef CAS PubMed.
- K. Choudhury, M. Sundaramoorthy, A. Hickman, T. Yonetani, E. Woehl, M. F. Dunn and T. L. Poulos, J. Biol. Chem., 1994, 269, 20239–20249 CAS.
- X. H. Wang, K. G. Qu, B. L. Xu, J. S. Ren and X. G. Qu, Nano Res., 2011, 4, 908–920 CrossRef CAS.
- C. Hou, Q. Luo, J. Liu, L. Miao, C. Zhang, Y. Gao, X. Zhang, J. Xu, Z. Dong and J. Liu, ACS Nano, 2012, 6, 8692–8701 CrossRef CAS PubMed.
- L. Lai and A. S. Barnard, Nanoscale, 2014, 6, 14185–14189 RSC.
- M. Mylrajan, K. Valli, H. Wariishi, M. H. Gold and T. M. Loehr, Biochemistry, 1990, 29, 9617–9623 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |