
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Studying a novel AIE coating and its handling process via fluorescence spectrum†
Weili Li *a,
Wei Yaoa,
Jun Wanga,
Zhenyu Qiua,
Jijun Tanga,
Shengyuan Yangb,
Meifang Zhu*b,
Zexiao Xuc,
Rong Hud,
Anjun Qind and
Ben Zhong Tang*de
*a,
Wei Yaoa,
Jun Wanga,
Zhenyu Qiua,
Jijun Tanga,
Shengyuan Yangb,
Meifang Zhu*b,
Zexiao Xuc,
Rong Hud,
Anjun Qind and
Ben Zhong Tang*de
aSchool of Material Science and Engineering, National Demonstration Center for Experimental Materials Science and Engineering Education, Jiangsu University of Science and Technology, Zhenjiang 212003, China. E-mail: just_liweili@163.com
bState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, Donghua University, Shanghai, 201620, China. E-mail: zmf@dhu.edu.cn
cSuzhou Jiren Hi-Tech Material Co., Ltd, Suzhou 215143, China
dGuangdong Innovative Research Team, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China
eDepartment of Chemistry, Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction, The Hong Kong University of Science & Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: tangbenz@ust.hk
First published on 23rd August 2017
Abstract
For polymer materials, both their compositions and preparation process greatly influence their service performance. Thus, the sound understanding of the relationship between materials' preparation processes and their properties is paramount. However, current research methods are partially limited due to the absence of a direct testing method to track the entire process, e.g. synthesizing, curing, ageing, and so on. With the ability for real-time sensitive characterization, fluorescence spectroscopy may be applied in testing polymer materials' performance. Here, we synthesized a novel aggregation induced emission (AIE) resin named TPE–EPOXY resin and prepared an AIE coating based on it. According to restriction of intramolecular rotation (RIR) mechanism, the preparation, curing, and aging processes for the AIE polymer resins & coatings could be studied with real-time observation. In addition, their properties could also be studied systematically. The results in this paper pave a good way to understand the relationship between the internal structure and the properties of polymer materials. Moreover, the prepared AIE polymer resins has a potential to expand the application fields of the AIE mechanism.
Introduction
Polymer resins such as epoxy resin, polyurethane resin, and acrylic resin are now indispensable to be applied in areas such as coatings, adhesives, engineering plastics, fibers and so on.1–7 Besides being applied as building block materials, polymer materials can also be designed and synthesized to be intelligent. However, to achieve this goal, better understanding of the relationship between the polymer structure and their properties is a requirement. Due to their high molecular weight and complex structure, it is hard to intimately correlate polymer materials' structures with their properties. To understand the microstructure information of polymer materials, traditional characterization methods, such as FT-IR, XRD, SEM, TEM, DSC, TGA are traditionally used.8 However, they are not good enough due to the fact that the internal interaction and mutual movement between the polymer chains are also important, because these features are the bridges to understanding the relationship between the microstructure and macro performance of polymer materials.9,10Fluorescence spectroscopy (also known as fluorometry or spectrofluorometry) is a type of electromagnetic spectroscopy that analyzes fluorescence from a sample. It involves using a beam of light, usually ultraviolet light, that excites the electrons in molecules of certain compounds and causes them to emit light; typically, but not necessarily, visible light. With real-time feature and sensitivity, it may be applied in studying the relationship between the structure and the properties of the polymer materials. Fluorescent organic molecules have been vigorously investigated due to the interesting photophysical properties accompanied by their intrinsic softness and lightness. However, they usually present prominent fluorescence in a solution, not in a solid state due to aggregation-caused quenching (ACQ) phenomenon,11,12 which limits its application in solid polymer materials. Aggregation-induced emission (AIE), such an abnormal emission behavior has drawn great research interest in the recent years, for it is exactly the opposite to the common belief that the emission of chromophores decreases in the aggregated state.13–16 For various AIEgens, tetraphenylethene (TPE) is one of the typical fluorescent molecule found to be non-emissive in dilute solution but highly luminescent when its molecules are aggregated in concentrated solutions or cast in solid films.17 In detail, in a dilute solution, TPE undergoes dynamic intramolecular rotations against its double bond and renders its molecule non-luminescent. On the other hand, in the aggregate state, the molecules cannot pack through a π–π stacking process due to its propeller shape, while the intramolecular rotations of its aryl rotors are greatly restricted owing to the physical constraint. This restriction blocks the non-radiative pathway and opens up the radiative channel.18,19 Besides when in aggregate state, AIEgens can also emit fluorescence when they are restricted with the help of surrounding polymer chains by covalent bond. Our group previously reported a facile approach to synthesize stimuli-responsive fluorescent elastomer by linking tetraphenylethylene (TPE) derivant to flexible polydimethylsiloxane (PDMS) polymer chains with covalent-bond. Beyond this, we also used TPE derivative as the fluorescent marker to study emulsion polymerization process and the properties of fluorescent emulsions.20,21
In the recent years, TPE derivative was frequently used as fluorescent marker to study the properties of the polymer based materials and various other applications in polymer science.1,22–29 In this line, Guan et al. designed and synthesized a unique fluorescent surfactant (TPE–DTAB), which combined both the properties of the aggregation-induced emission (AIE) and amphilicity which together were used to image the macrodispersion of the layered double hydroxide fillers (montmorillonite) in the polymer matrix.30 Wang's group also visualized the entire gelation process of chitosan (CS) by the synthesized AIE fluorogenic probe, that is, tetraphenylethene-labelled chitosan (TPE–CS).31
Inspired by these research findings, we synthesized and studied a new kind of fluorescent resin by modifying epoxy with AIEgen named dibenzyl amine tetraphenylethene (2CH2NH2–TPE). The preparation, curing, and aging process of fluorescent resin were trailed according to fluorescence spectrum. In addition, the interaction between polymer chains and knitted AIEgen was investigated systematically by combining the results of fluorescence spectrum with other characterization methods. Due to the labelling effect of AIEgen, the relationship between polymer materials structures and their properties could be studied systematically.
Results and discussions
Synthesis of p-dimethylamino tetraphenylethylene (2CH2NH2–TPE)
The molecular structure of 2CH2NH2–TPE is shown in Scheme 1 which is a TPE derivative with dimethylamino.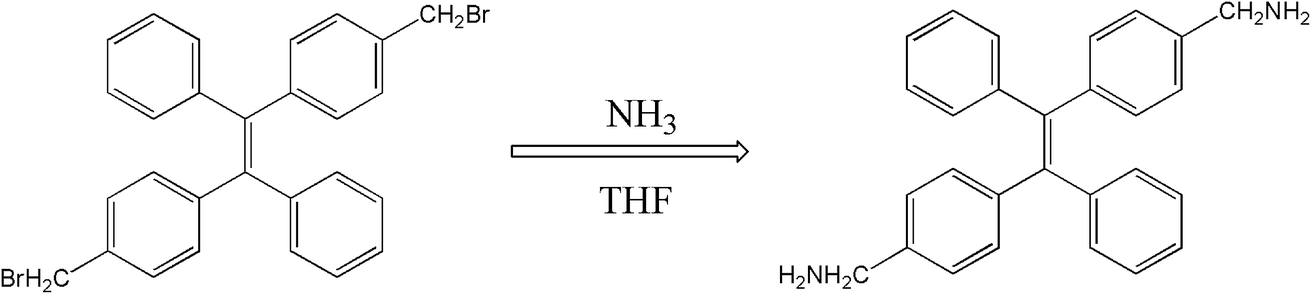 | ||
| Scheme 1 The synthesis process of 2CH2NH2–TPE. | ||
At first, p-bromide tetraphenylethylene (2CH2Br–TPE) was synthesized according to the reference in our lab.20 Then p-dimethylamino tetraphenylethylene (2CH2NH2–TPE) was prepared as follows, which was presented as Scheme 1. In a typical experiment, 40 ml of THF and 80 ml of NH3·H2O were added into a 250 ml two-necked round bottom flask, then 2.07 g 2CH2Br–TPE (0.004 mol) was dissolved in THF and added into the system drop-wise over a period of 4 h, and the mixture was then continued under stirring overnight. After that, THF was removed by rotoevaporation, and the product was extracted out with dichloromethane. 2CH2Br–TPE was crystallized out when its dichloromethane solution was dropped in petroleum ether. The solid was isolated by filtration, and then redissolved in dichloromethane. The process of dissolving and crystallization was repeated for 3–4 times. Pure 2CH2NH2–TPE was obtained (53% yield). 1H-NMR (400 MHz): δ 8.61 (m, 4H, NH2), 7.16–7.49 (m, 18H, Ar), 4.31 (s, 4H, CH2) (Fig. 1).
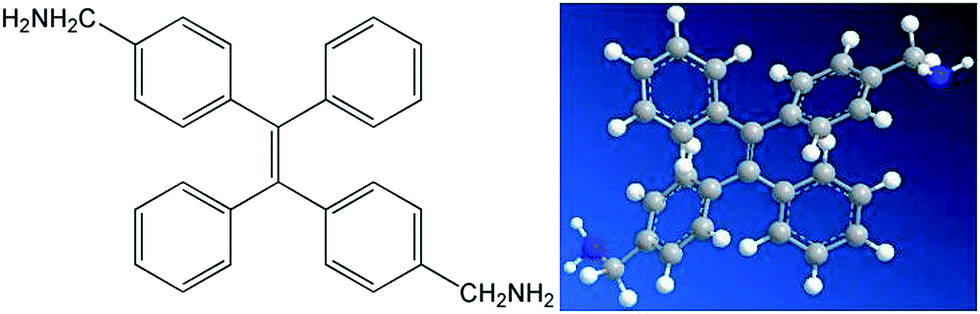 | ||
| Fig. 1 Chemical structure and ball-rod modelling of 2CH2NH2–TPE. | ||
Synthesis and curing of TPE–EPOXY resin
According to electrophilic addition mechanism, epoxy functional groups can be opened by benzylamino of 2CH2NH2–TPE. In a typical experiment, a certain amount of epoxy resin was dissolved in THF solution, then 2CH2NH2–TPE was dissolved in THF and added into the epoxy/THF solution drop by drop at 60 °C. The concentration of reactant mixture was set at 0.2 g ml−1. After the two reactants were mixed evenly, the reaction was carried on at 70 °C for about 8 h. Upon cooling to room temperature, the mixture was added dropwise into the adequately cooled methanol. The obtained gel with high viscosity was isolated by filtration, rinsed with methanol for 3 times, and dried in vacuum at 40 °C overnight to afford a pale yellow transparent gel, named TPE–EPOXY (as is presented in Fig. 2). To study the degree of labelling on the properties of prepared TPE–EPOXY resin, five samples were prepared as summarized in Table 1.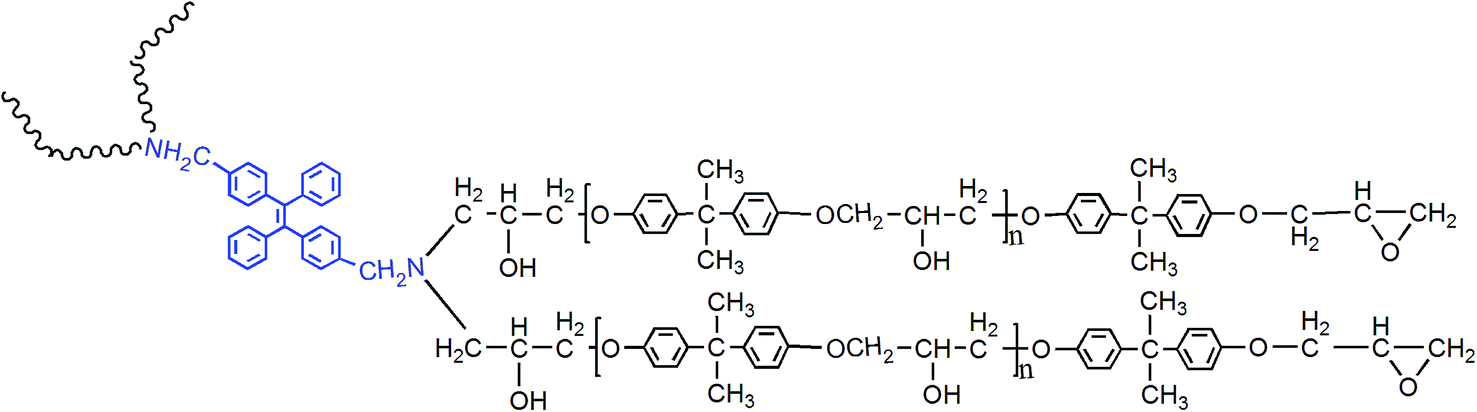 | ||
| Fig. 2 Chemical structure of TPE–EPOXY resin. | ||
| Sample | Reaction materials | Degree of labeling |
|---|---|---|
| TPE–EPOXY-1 | E-44, 2CH2NH2–TPE | 0.1 |
| TPE–EPOXY-2 | E-44, 2CH2NH2–TPE | 0.02 |
| TPE–EPOXY-3 | E-44, 2CH2NH2–TPE | 0.01 |
| TPE–EPOXY-4 | E-44, 2CH2NH2–TPE | 0.002 |
| TPE–EPOXY-5 | E-44, 2CH2NH2–TPE | 0.001 |
Comparing with 1HNMR spectra of TPE–EPOXY-1 resin and original epoxy resin (CDCl3 is used as the solution), the present peaks at around 7.000 (m, H-benzene) are attributed to the benzene ring of TPE, as observed in Fig. 1S.† The data indicate that the reaction successfully proceeded without damaging the structures, and AIEgen was linked onto the polymer chains of the epoxy resin.
2CH2NH2–TPE can be also be a cross-linking agent, the content of which may influence the property of the prepared TPE–EPOXY resin. The molecular weight of TPE–EPOXY resins varied with the content of 2CH2NH2–TPE which was characterized and presented in Table 2. With the relative content of 2CH2NH2–TPE increasing, the value of Mn and Mw of the prepared TPE–EPOXY increased gradually, which interfered with the property of the cured fluorescent coating. Fortunately, the results show that no gel was formed due to the controlled content of 2CH2NH2–TPE.
| Sample | Mn | Mw | ρ |
|---|---|---|---|
| TPE–EPOXY-1 | 3168 | 5733 | 1.810 |
| TPE–EPOXY-2 | 1247 | 1303 | 1.045 |
| TPE–EPOXY-3 | 851 | 854 | 1.0043 |
| TPE–EPOXY-4 | 676 | 678 | 1.0003 |
| TPE–EPOXY-5 | 468 | 484 | 1.034 |
To cure the fluorescent resin, the curing agent diethylenetriamine (DETA), was added into the TPE–EPOXY resin, and the molar ratio between resin and curing agent was set at 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.4. After being laid aside for about 1 h, the pre-treated glass plates were dipped into the blending solution at a speed of 200 mm min−1 by using a specially designed apparatus. According to FTIR spectra (Fig. 2S†), the absorption peak for epoxy group, which is located at 915 cm−1, weakens obviously due to the cured AIE coating because it was consumed during the curing process. Fig. 6 presents the surface of the cured AIE coating based on TPE–EPOXY-1 resin observed using SEM measurement. The AIEgen was uniformly dispersed in the EPOXY resin matrix without the formation of large aggregates. A slight wrinkle could have been caused by the shrinkage of polymer chains during the curing process.
:2.4. After being laid aside for about 1 h, the pre-treated glass plates were dipped into the blending solution at a speed of 200 mm min−1 by using a specially designed apparatus. According to FTIR spectra (Fig. 2S†), the absorption peak for epoxy group, which is located at 915 cm−1, weakens obviously due to the cured AIE coating because it was consumed during the curing process. Fig. 6 presents the surface of the cured AIE coating based on TPE–EPOXY-1 resin observed using SEM measurement. The AIEgen was uniformly dispersed in the EPOXY resin matrix without the formation of large aggregates. A slight wrinkle could have been caused by the shrinkage of polymer chains during the curing process.
The structure and thermodynamics properties of TPE–EPOXY resin
The structure and thermodynamic property of the prepared fluorescent resin was also investigated. Fig. 3 shows the X-ray diffractometry (XRD) results of TPE–EPOXY-1 resin and epoxy resin (E-44). No diffraction peaks of 2CH2NH2–TPE were detected from TPE–EPOXY-1 resin. This suggested that 2CH2NH2–TPE was uniformly dispersed in the epoxy resin without the formation of large aggregates probably due to its connection onto polymer chains of the epoxy resin with covalent bonds.32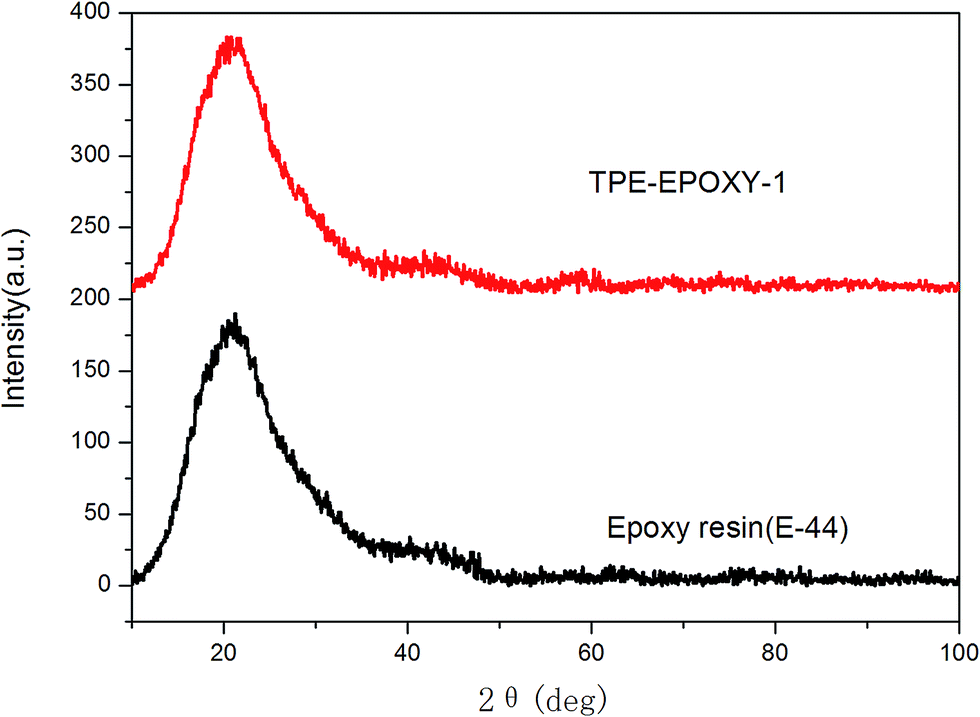 | ||
| Fig. 3 XRD curves of TPE–EPOXY-1 resin and epoxy resin (E-44). | ||
When the fluorescent resin was cured by DETA, the obtained solid fluorescent coating was tested using DSC measurement. TPE–EPOXY-1 resin was taken as the specimen. As observed from Fig. 4, the material presented an amorphous state with Tg value at around 55 °C. In addition, no melting peaks attributed to AIEgens means that it was dispersed in the epoxy resin effectively by chemical bond. This result is consistent with the XRD measurement.
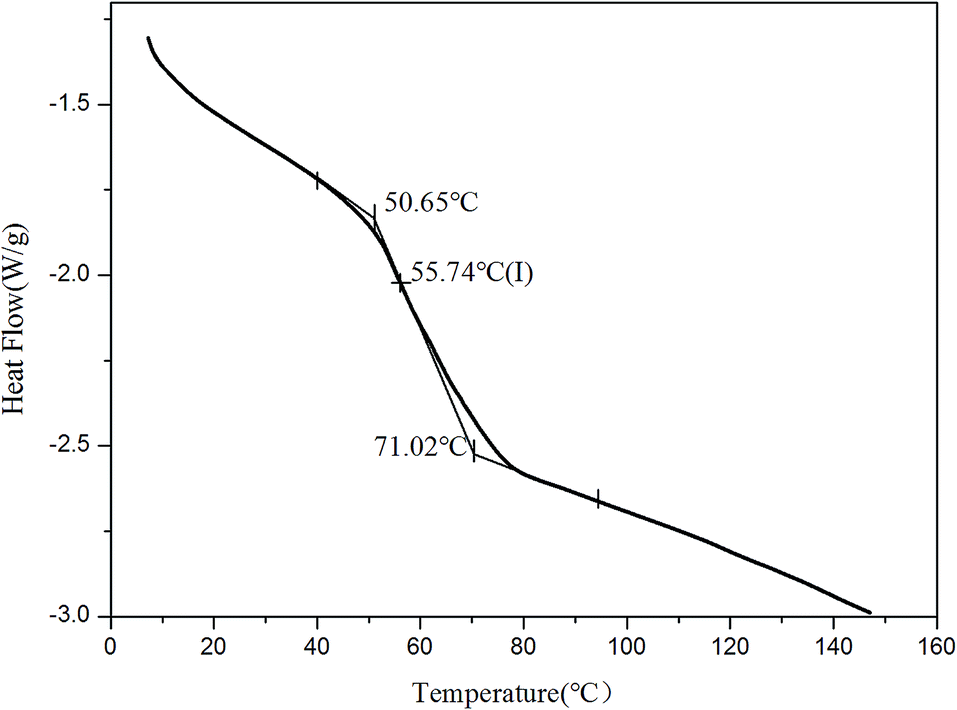 | ||
| Fig. 4 DSC charts of fluorescent coating based on TPE–EPOXY-1 resin. | ||
Thermotolerance property is one of the important indexes for evaluating the performance of polymeric materials. Fig. 5 presents thermogravimetric results of pure epoxy resin, the fluorescent resin, and the cured fluorescent coating, respectively. This test was used to study the effect of knitted TPE on thermotolerance of resin and the cured coating. From the figure, TPE–EPOXY-1 resin starts to lose weight at 292 °C, which is higher than that of pure epoxy resin (271 °C). After being cured by DETA, the fluorescent coating starts to decompose at 316 °C although the unreacted small organic molecules constantly evaporate during being heated. The knitted AIEgen does not negatively effect the heat resistance of epoxy resin. On the contrary, slightly crosslinked structure is capable of decomposing TPE–EPOXY at a higher temperature.
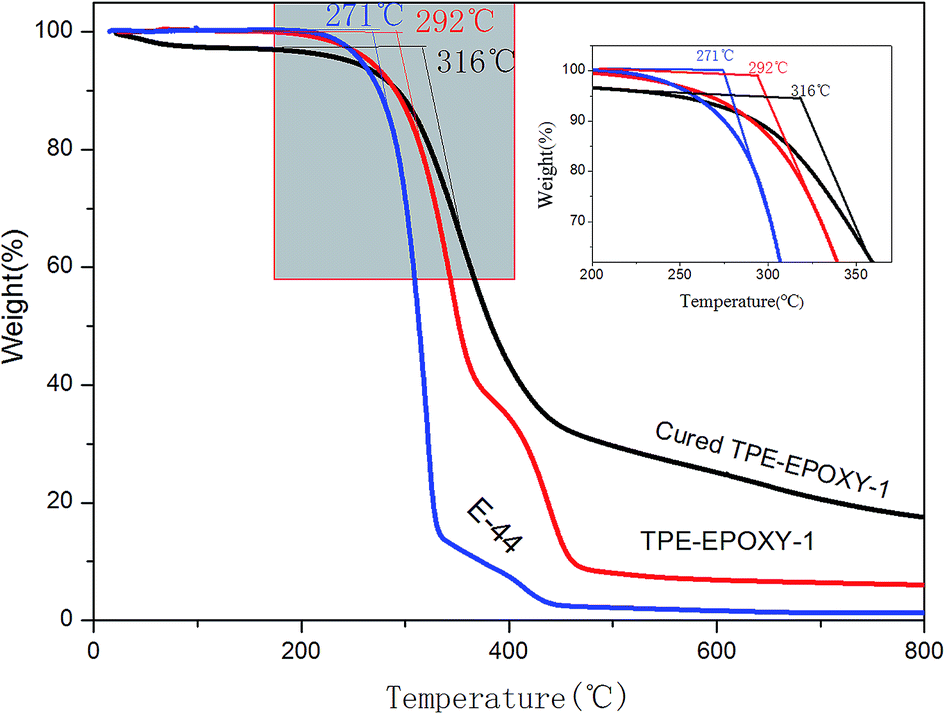 | ||
| Fig. 5 Thermogravimetric result of epoxy resin (E-44), TPE–EPOXY-1 resin, and the cured fluorescent coating based on TPE–EPOXY-1. | ||
Study the dissolution behavior of TPE–EPOXY resin according to PL spectra
The knitted AIEgen (2CH2NH2–TPE) was used as the fluorescent marker, and the dissolution behaviors of TPE–EPOXY resins were studied using the PL spectra.At first, the impact of interaction between AIEgen and polymer chains on the photophysical property of the fluorescent resin was studied. The results are presented in Fig. 6. Comparing with the two samples, when the epoxy resin was blended with the AIEgen prior to dissolving in the THF, the blending showed a very weak fluorescence. However, when 2CH2NH2–TPE and epoxy were knitted together by a covalent bond, such as the sample TPE–EPOXY-1 resin, it showed a very strong fluorescence even when dissolved in the solution. We propose that the restriction of the intramolecular rotation (RIR) was the main cause for the AIE phenomenon.33,34 The linked polymer chains tended to restrict the intramolecular rotation of TPE molecular, and thus the absorbed UV light was released by fluorescent luminescence behavior for TPE–EPOXY samples.
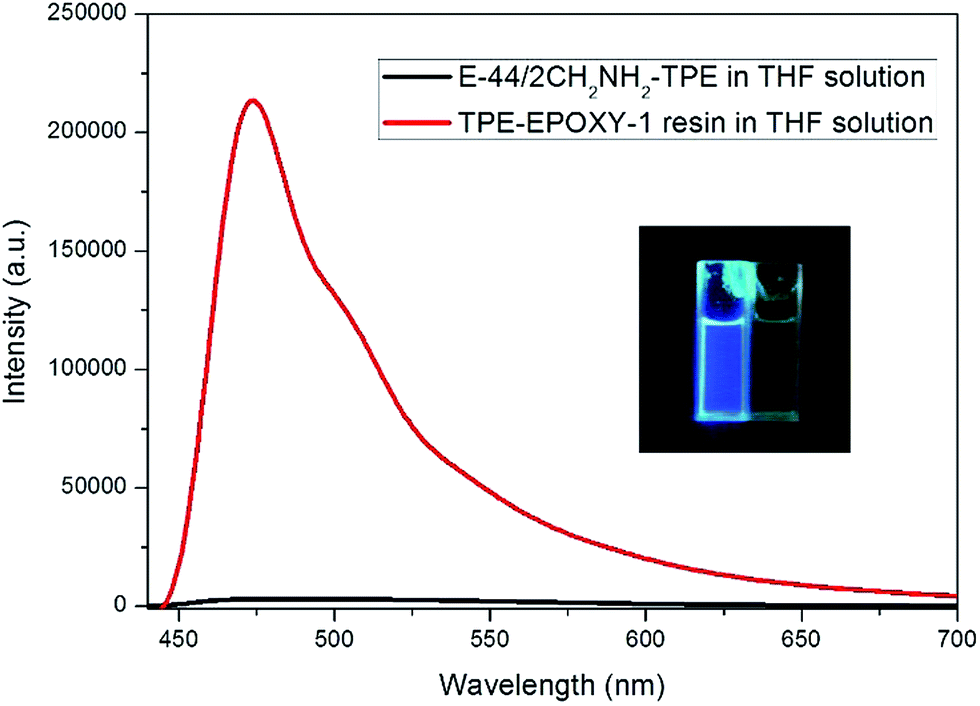 | ||
| Fig. 6 PL-spectra of the epoxy resin blended with 2CH2NH2–TPE (100:1) in THF solution TPE–EPOXY-1 resin (0.01 g ml−1). | ||
Various TPE–EPOXY resins were dissolved in THF at 0.01 g ml−1 in order to study the effect of DL on the fluorescence property of the synthesized TPE–EPOXY resin. Photoluminescence (PL) spectra of TPE–EPOXY resins presented a strong fluorescent property and concentration which was dependent on the intensity increase. We proposed that the restriction of intramolecular rotation (RIR) was the primary cause for the AIE phenomenon.35,36 When TPE deviant and polymer chains were knitted together by covalent bonds, the RIR process partially activated and thus made the resin somewhat emissive. According to Fig. 7, the intensity of PL spectra increased linearly with the concentration of TPE deviant. With the connection effect of covalent bonds and similar chemical environment, the intensity of PL spectra did not present an abrupt change. These data demonstrated that TPE deviant homogeneously dispersed in the elastomeric samples without forming macroscopic aggregates or domains.
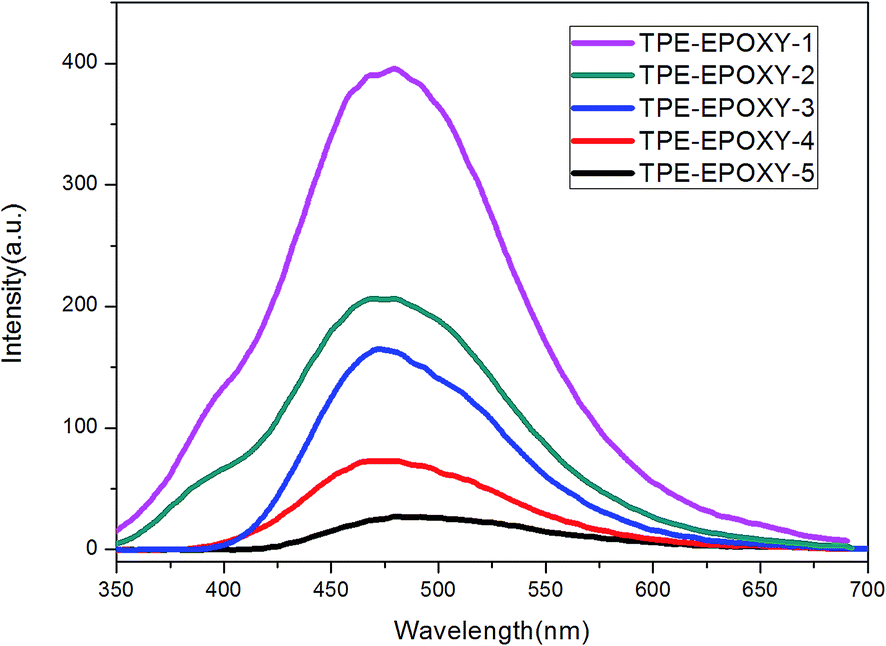 | ||
| Fig. 7 PL spectra of TPE–EPOXY resins varied with DL of TPE molecular in THF solution (0.01 g ml−1). | ||
In the next, TPE–EPOXY-1 resin was selected as example to study the resin's dissolution behavior. At first, it was dissolved into THF-water mixed solvents to study its dissolution behavior in good and poor solvents (Fig. 8). As discussed above, when AIEgen is knitted onto polymer chains, the synthesized TPE–EPOXY resin could still emit fluorescence (the emission peak emerged at round 480 nm) under UV light even when they are dissolved in good solution (THF) due to RIR effect. In the experiment, the emission became stronger progressively with increasing the content of water in the mixed solvents. When a large amount of water (60%) was added into the THF solution, an emission peak emerged at around 480 nm, demonstrating a typical AEE (aggregation enhanced emission) phenomenon. During the process, the solubility of TPE–EPOXY-1 resin got worse and the polymer chains changed from stretched conformation to curled conformations, which as the result enhance RIR effect of the knitted AIEgens.
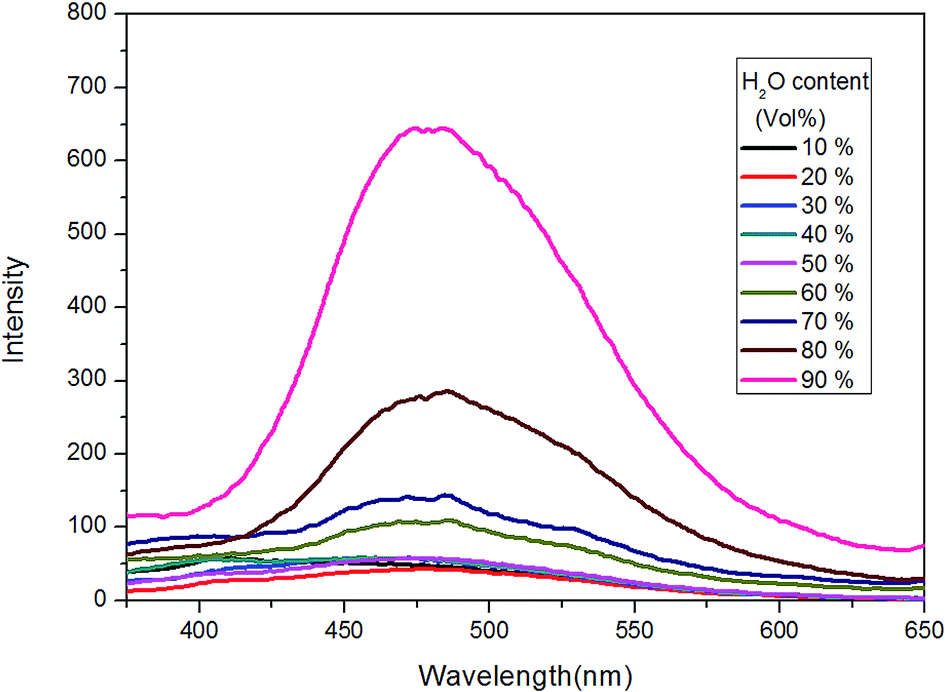 | ||
| Fig. 8 PL spectra of TPE–EPOXY-1 resin in THF/H2O mixed solution (0.01 g ml−1) varied with the content of H2O. | ||
Inspired by this result, TPE–EPOXY-1 resin was then dissolved in different solutions such as butanol, ethyl acetate (EA), THF, and toluene, to study their dissolution behavior according to PL spectra. Traditionally, this property is tracked by comparing the solubility parameter between the dissolved polymers and the solutions, which is presented in Table 3.
| Sample | Solutions | Polymer | |||
|---|---|---|---|---|---|
| Toluene | Butanol | EA | THF | Epoxy resin | |
| Solubility parameter | 8.9 | 23.32 | 9.1 | 9.2 | 8–13 |
For TPE–EPOXY-1 solutions with different solvents (Fig. 9), the emission peaks of PL spectra appeared in the 470 nm–480 nm range. However, their intensities varied with the dissolving solutions. For good solutions, such as toluene, ethyl acetate (EA), and THF, their solubility parameter are close to that of epoxy resin. Full stretching of polymer chains in these solutions are believed to have faded the bonding effect of the AIEgens. This lead to the decrease in the fluorescence intensity. However, when epoxy resin was dissolved in butanol, large differences of solubility parameter between them made the polymer chains of fluorescent resin to shrink and agglomerate in the solution. This restricted the intramolecular rotation of the knitted AIEgens. So the fluorescence intensity increased significantly (about four to five fold).
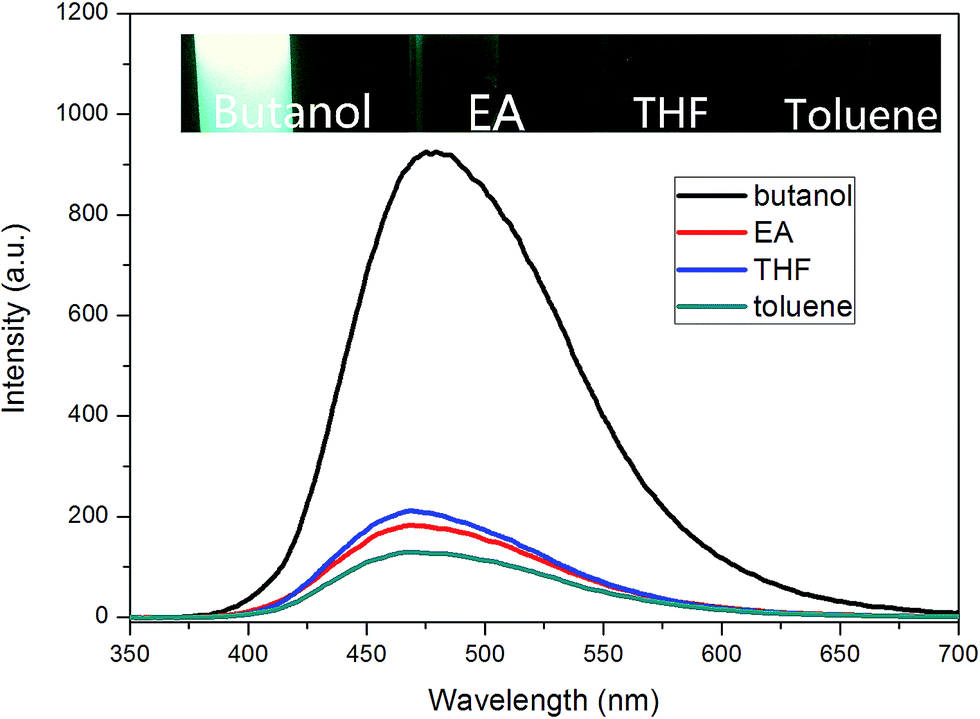 | ||
| Fig. 9 PL spectra of EA–TPE-1 reins dissolved in different solvent (0.01 g ml−1). | ||
EA–TPE-1 resin was then dissolved in THF under varying concentrations in order to study the fluorescent resin dissolution behavior in its good solution. According to Fig. 10, the polymer solution did not present an obvious fluorescence when EA–TPE-1 resin concentration was extremely low (<1 × 10−4 g ml−1). At this state, the polymer chain segments stretched freely in the solvent and there existed little tangles between them. Therefore, the intramolecular rotation of the knitted AIEgens was not under restriction, and the absorbed energy from UV light was possibly consumed mostly by random molecular motion of AIEgens. The degree of interaction between polymer chains in solution increased linearly with the increase in the polymer concentration, which resulted in the restriction of intramolecular rotation of AIEgens.
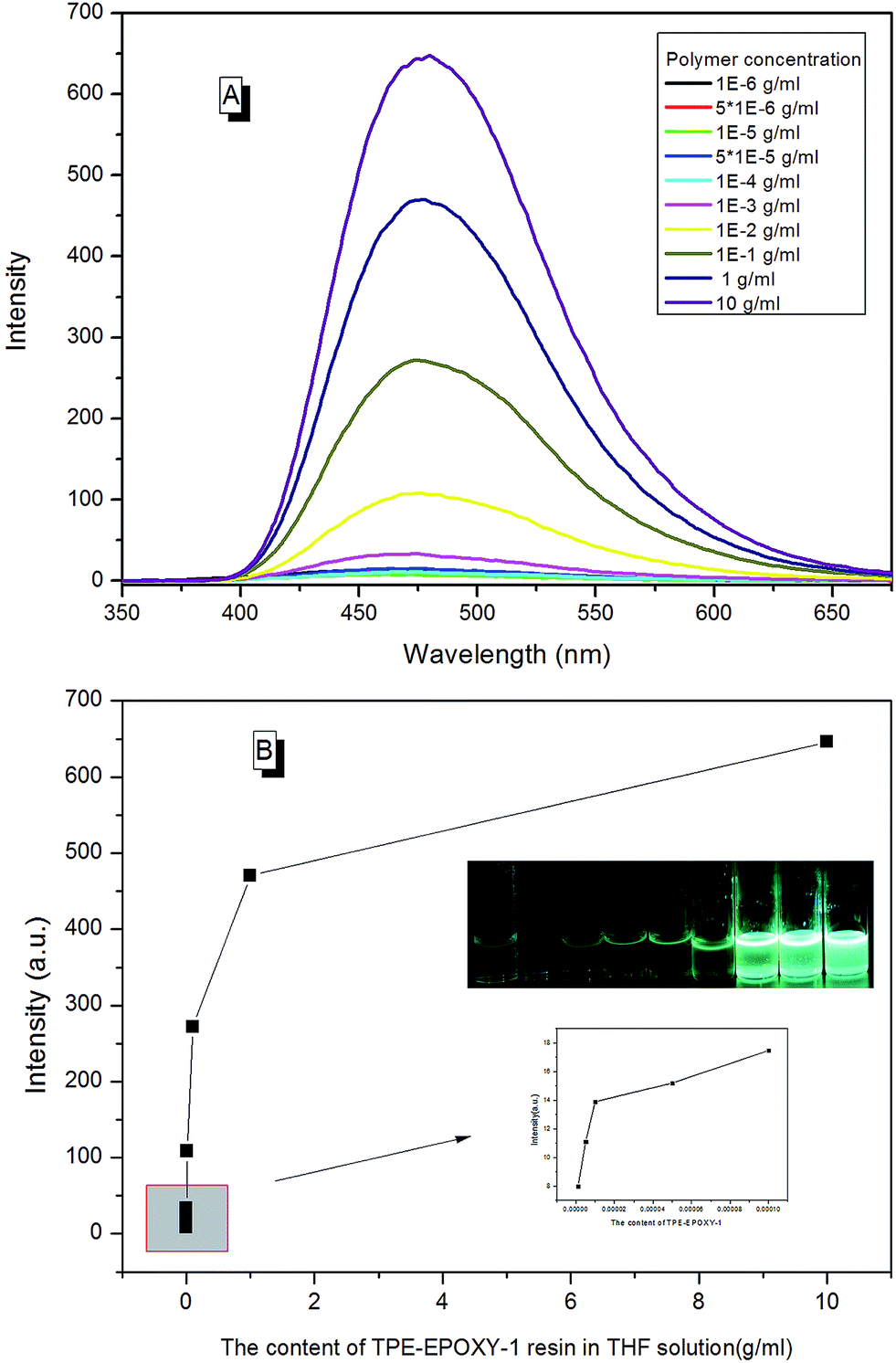 | ||
| Fig. 10 (A) PL spectra of EA–TPE-1 resin in THF varied with its concentration, (B) PL intensity of EA–TPE-1 in THF at 480 nm varied with its concentration. | ||
According to the taken pictures and the results of PL spectra, the interaction between polymer chains in solution varied with the type of solvents. Still, their concentrations could be trailed lively and accurately, which totally agreed with the perception of the past findings.
Tracking the handling process for the fluorescent resin according to PL spectra
With real-time capability and sensitivity, PL spectra could be used to track the preparation process, curing process, and accelerated aging process of the fluorescent resin. Theoretically, the luminescence behavior of AIEgen are related to the segmental motion of the knitted polymer chains when together. When 2CH2NH2–TPE was reacted with epoxy resin by condensation polymerization, the molecular weight of the reaction product increased gradually with the reaction time. During the reaction, a small amount of reaction mixtures was taken out and measured at the same temperature every once in a while. As presented in Fig. 11, the fluorescence intensity of the reaction mixture increased linearly with the reaction time, with the peak positions locking at around 475 nm. When more AIEgen were knitted onto polymer chains, the bondage of polymer chains restricted their intramolecular rotation, and thus the absorbed energy was released as fluorescent light.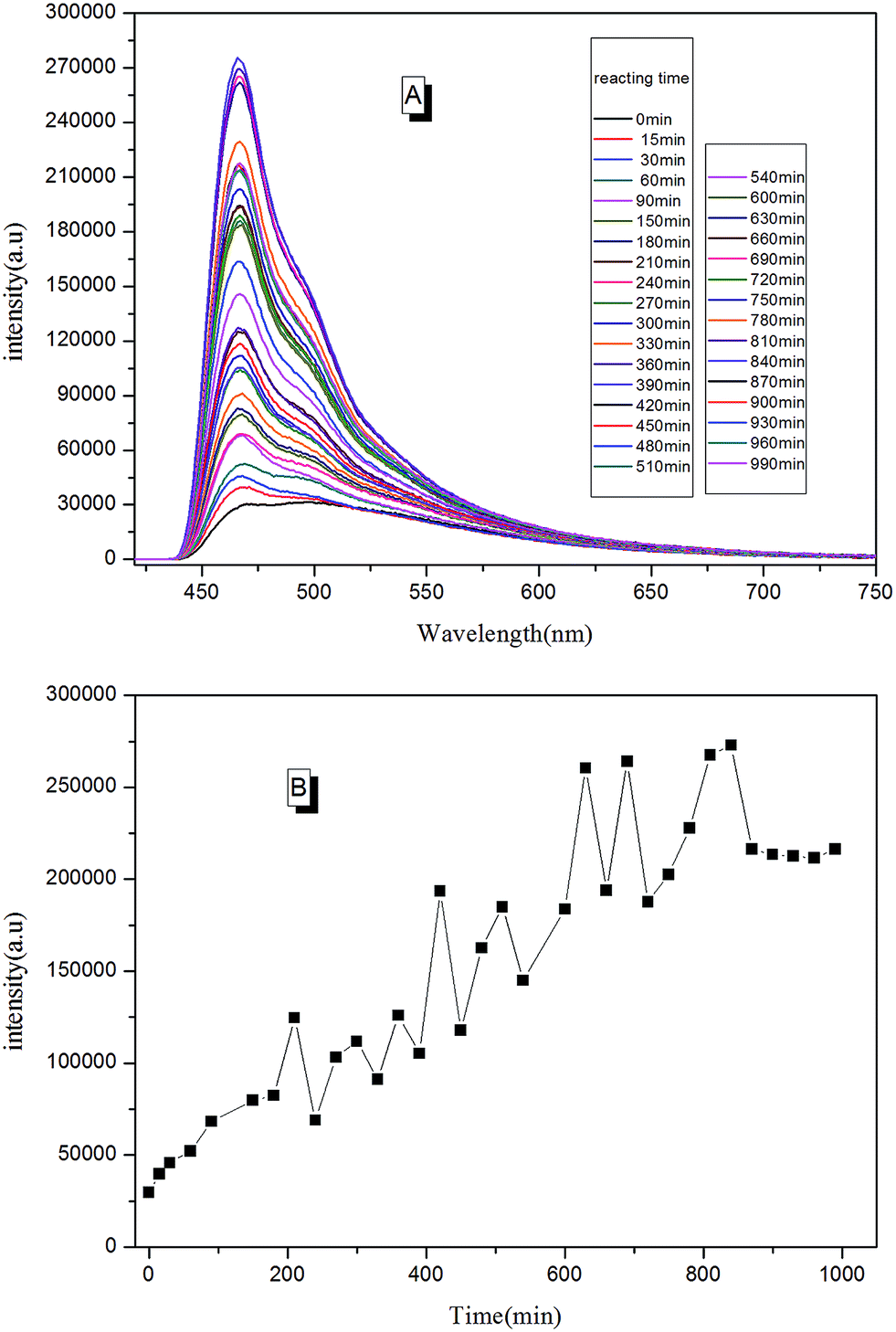 | ||
| Fig. 11 (A) The trend of PL spectra for the process of preparing fluorescent resin (TPE–EPOXY-1) varied with the curing time; (B) the trend of PL intensity for the process of preparing fluorescent resin varied with the reacting time. | ||
When the prepared fluorescent resin was cured with the help of the hardening agent, it formed a reticular conformation. Polymer chains were fixed to the hardening agent via covalent bonding. As AIEgens were knitted onto the polymer chains, the crosslinked polymer system would further restrict the intramolecular rotation of AIEgens, which led to the enhancement of PL intensity. According to Fig. 12, the peak positions locked were at around 475 nm during the curing process. There was a large increase in the PL intensity before 50 min. This is attributed to the simultaneously volatilizing solution at the beginning of the curing process. These two effects both limited the segmental motion, which made PL intensity to increase rapidly. However, when the solution was completely evaporated, there was only one way to further restrict the segmental motion. When the curing process completed, PL intensity showed less changes with the curing time.
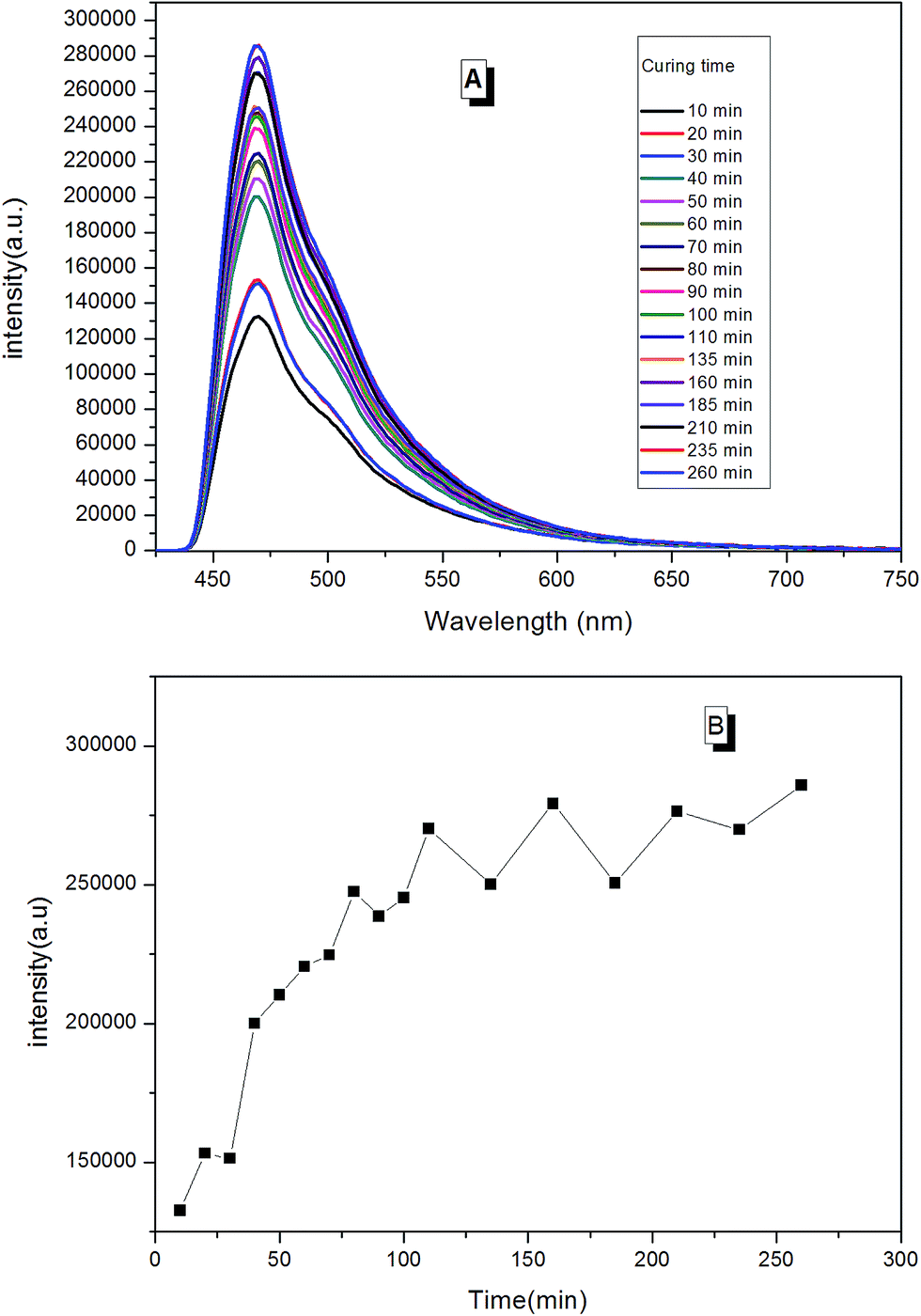 | ||
| Fig. 12 (A) The trend of PL spectra for the curing process of fluorescent resin (TPE–EPOXY-1) varied with the curing time; (B) the trend of PL intensity for the curing process of fluorescent resin varied with the curing time. | ||
Due to the molecular structure characteristics of epoxy resin, its anti-UV property is relatively poor. Previously, its weatherability property was checked using visual inspection. However, the method is known to be relative rough with relatively non precise judgment. In this paper, we tried to tell the whole aging process of epoxy resin according to PL spectra. In a typical testing procedure, the cured TPE–EPOXY-1 coating was placed under UV radiation (313 nm, 1.25 kW) constantly. Then the tested coating was taken out and checked by PL spectra at every 0.5 h. In contrast to the curing process, polymer chains of the cured fluorescent resin fractured constantly during the aging process, which is believed to have weakened the binding effect of AIEgens and lowered the PL intensity of the tested coatings. According to Fig. 13, the peak positions were locked at around 475 nm. However, the peak intensity changed with the aging time. From 0 min to 2000 min, it fluctuated to a certain range. With the extended aging time, there was a sudden weakening process for the peak intensity. This meant that the AIEgens started carrying the intramolecular rotation because of the loosening of the linked polymer chain segments. PL intensity weakened constantly as time went on.
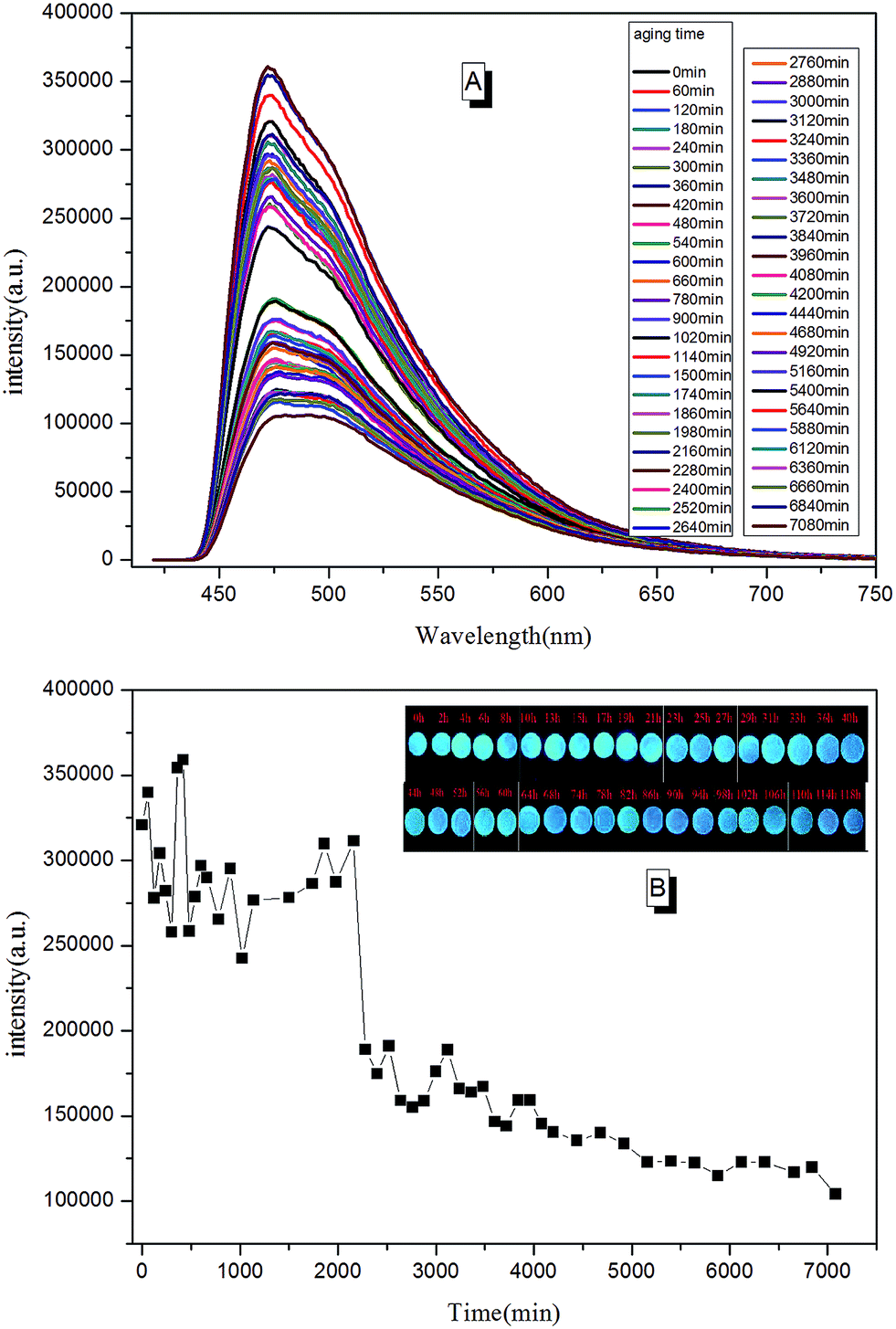 | ||
| Fig. 13 (A) The trend of PL spectra for the aging process of cured fluorescent coating (TPE–EPOXY-1) varied with the aging time; (B) the trend of PL intensity for the aging process of fluorescent coating (TPE–EPOXY-1) varied with the aging time. | ||
Conclusions
In this paper, a novel fluorescent resin was prepared by modifying epoxy resin with AIEgen (2CH2NH2–TPE). The intramolecular motions (i.e. rotation, vibration, bending, and so on) of the phenyl rings of TPE are restricted, giving rise to an intense fluorescence emission. According to this specialty, the dissolution behavior of fluorescent resin was studied systematically. In addition, according to the variation tendency of PL spectra, the fabrication process, the curing process, and the aging process of fluorescent resin were tracked in real time. This study thus opens up a new avenue for the research of fluorescent resin and its application in other various fields.Experimental section
Materials
Bisphenol-A epoxy resin (E-44), the typical structure in Fig. 14, and the curing agent diethylenetriamine (DETA) were purchased from Sanmu Group Co., China and were used as received. E-44 held a number-average molecular weight (Mn) of ∼450.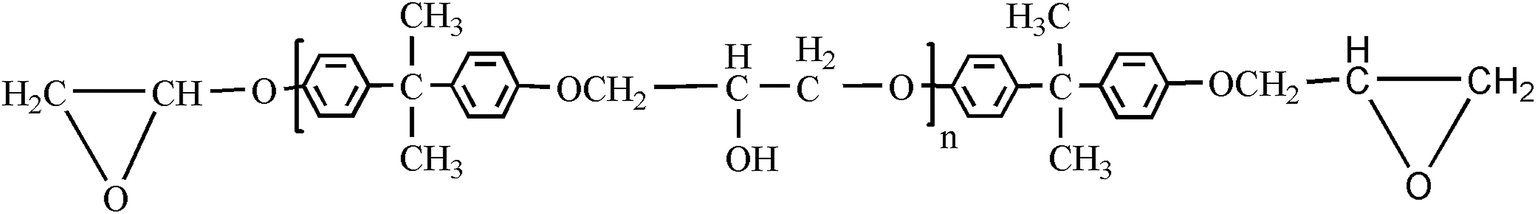 | ||
| Fig. 14 General molecular structure of bisphenol-A epoxy resin. | ||
p-Methyl benzophenone, N-bromobutanimide, zinc powder and titanium(IV) chloride (TiCl4) were purchased from Aladdin Reagent Co., Ltd. (Shanghai, China). Ammonia and tetrahydrofuran (THF) were purchased from Sinopharm Group Chemical Reagent Co., Ltd. (Shanghai, China). THF was distilled under normal pressure from sodium benzophenone ketyl under nitrogen, in addition, THF used in fluorescence spectra measurements was spectral purity grade.
Instruments
1HNMR spectra were measured on Bruker ARX 400 NMR spectrometers using CDCl3 as the deuterated solvent and tetramethylsilane (TMS; δ = 0 ppm) as the internal standard. FTIR spectra were recorded on a Perkin-Elmer 16 PC FT-IR spectrophotometer. Images of scanning electron microscopy (SEM) were taken on a JSM-6700F electron microscope. Thermogravimetric analysis (TGA) measurements were carried out under nitrogen or in air on a Perkin-Elmer TGA 7 analyzer at a heating rate of 10 °C min−1. DSC measurements were carried out using DSC Q100 (TA Instruments, USA) over the temperature of 0 °C to 150 °C at a scan rate of 10 °C min−1. All the thermograms were base line corrected and calibrated using Indium metal. The experimental specimens (8–10 mg) were dried at 60 °C under vacuum for 24 h, before being measured. All the samples were firstly annealed at 120 °C for 3 min, and cooled to 0 °C by using liquid nitrogen and then scanned for the measurement. X-ray diffraction (XRD) measurements were performed using an XRD diffractometer (D-MAX 2200 VPC) equipped with Ni-filtered Cu Kα radiation, having a wavelength of 0.154 nm. UV-vis absorption and light transmission spectra were measured on a Milton Roy Spectronic 3000 array spectrophotometer. Photoluminescence (PL) spectra were recorded on a Perkin Elmer LS 55 spectrofluoromerter.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by the National Natural Science Foundation of China (Grant No. 51673088), 60th Postdoctoral Foundation of China (Grant No. 2016M601750), State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, Donghua University, NSFC (LK1509), JSTD Industry-University Research Cooperation Project (BY2016073-02).References
- B. Zimmer, C. Nies, C. Schmitt and W. Possart, Polymer, 2017, 115, 77–95 CrossRef CAS
.
- X. Chen, W. Wang and C. Jiao, J. Hazard. Mater., 2017, 331, 257–264 CrossRef CAS PubMed
- H. Gu, S. Tadakamalla, X. Zhang, Y. Huang, Y. Jiang, H. A. Colorado, Z. Luo, S. Wei and Z. Guo, J. Mater. Chem. C, 2013, 1, 729–743 RSC
- F. Meng, S. Zheng, W. Zhang, H. Li and Q. Liang, Macromolecules, 2006, 39, 711–719 CrossRef CAS
- W. Li, Y. Xing, Y. Wu, J. Wang, L. Chen, G. Yang and B. Tang, Electrochim. Acta, 2015, 151, 289–296 CrossRef CAS
- D. Huang, J. Wang, J. Tang, W. Shen, W. Li, J. Zhang, J. Zhang and Z. Xu, J. Coat. Technol. Res., 2016, 13, 691–701 CrossRef CAS
- W. Li, Y. Xing, X. Xing, Y. Li, G. Yang and L. Xu, Electrochim. Acta, 2013, 112, 183–190 CrossRef CAS
- M. Abadi, M. F. Serag and S. Habuchi, Macromolecules, 2015, 48, 6263–6271 CrossRef CAS
- A. M. Aerdts, A. M. van Herk, B. Klumperman, J. Kurja and A. L. German, in Synthesis of Polymers, Wiley-VCH Verlag GmbH, 2008, pp. 268–316, DOI:10.1002/9783527619313.ch9
- A. P. Richez, H. N. Yow, S. Biggs and O. J. Cayre, Prog. Polym. Sci., 2013, 38, 897–931 CrossRef CAS
- J. R. Lakowicz, in Principles of Fluorescence Spectroscopy, Springer, US, 2006, DOI:10.1007/978-0-387-46312-4
- Z. Qiu, T. Han, R. T. Kwok, J. W. Lam and B. Z. Tang, Macromolecules, 2016, 49, 8888–8898 CrossRef CAS
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 1740–1741 RSC
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC
- J. Mei, Y. Hong, J. W. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed
- R. Hu, N. L. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC
- Z. Chang, Y. Jiang, B. He, J. Chen, Z. Yang, P. Lu, H. S. Kwok, Z. Zhao, H. Qiu and B. Z. Tang, Chem. Commun., 2013, 49, 594–596 RSC
- J. Yang, N. Sun, J. Huang, Q. Li, Q. Peng, X. Tang, Y. Dong, D. Ma and Z. Li, J. Mater. Chem. C, 2015, 3, 2624–2631 RSC
- J. Shi, N. Chang, C. Li, J. Mei, C. Deng, X. Luo, Z. Liu, Z. Bo, Y. Q. Dong and B. Z. Tang, Chem. Commun., 2012, 48, 10675–10677 RSC
- W. Li, D. Huang, J. Wang, W. Shen, L. Chen, S. Yang, M. Zhu, B. Tang, G. Liang and Z. Xu, Polym. Chem., 2015, 6, 8194–8202 RSC
- S. Yang, W. Shen, W. Li, J. Tang, W. Yao, J. Wang, M. F. Zhu, B. Z. Tang, G. Liang and Z. Xu, RSC Adv., 2016, 6, 74225–74233 RSC
- R. Taniguchi, T. Yamada, K. Sada and K. Kokado, Macromolecules, 2014, 47, 6382–6388 CrossRef CAS
- S. Bao, Q. Wu, W. Qin, Q. Yu, J. Wang, G. Liang and B. Z. Tang, Polym. Chem., 2015, 6, 3537–3542 RSC
- S. Jiang, T. Y. Huang, K. M. Wang, B. Z. Tang and Q. Yu, Molecules, 2016, 22, 54 CrossRef PubMed
- X. Guan, D. Zhang, T. Jia, Y. Zhang, L. Meng, Q. Jin, H. Ma, D. Lu, S. Lai and Z. Lei, RSC Adv., 2016, 6, 107622–107627 RSC
- X. Zhang, Z. Chi, H. Li, B. Xu, X. Li, W. Zhou, S. Liu, Y. Zhang and J. Xu, Chem.–Asian J., 2011, 6, 808–811 CrossRef CAS PubMed
- X. Zhang, Z. Chi, B. Xu, C. Chen, X. Zhou, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem., 2012, 22, 18505–18513 RSC
- Z. Zhao, S. Chen, J. W. Lam, P. Lu, Y. Zhong, K. S. Wong, H. S. Kwok and B. Z. Tang, Chem. Commun., 2010, 46, 2221–2223 RSC
- X. Zhang, Z. Chi, H. Li, B. Xu, X. Li, S. Liu, Y. Zhang and J. Xu, J. Mater. Chem., 2011, 21, 1788–1796 RSC
- W. Guan, S. Wang, C. Lu and B. Z. Tang, Nat. Commun., 2016, 7, 11811 CrossRef PubMed
- Z. Wang, J. Nie, W. Qin, Q. Hu and B. Z. Tang, Nat. Commun., 2016, 7, 12033 CrossRef PubMed
- W. L. Li, Y. M. Gao and S. M. Wang, J. Appl. Polym. Sci., 2012, 125, 1027–1032 CrossRef CAS
- Q. Liu, X. Wang, H. Yan, Y. Wu, Z. Li, S. Gong, P. Liu and Z. Liu, J. Mater. Chem. C, 2015, 3, 2953–2959 RSC
- A. Qin, J. W. Lam and B. Z. Tang, Prog. Polym. Sci., 2012, 37, 182–209 CrossRef CAS
- W. Rettig, Angew. Chem., Int. Ed., 1986, 25, 971–988 CrossRef
- P. F. Barbara, T. J. Meyer and M. A. Ratner, J. Phys. Chem., 1996, 100, 13148–13168 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06527b |
| This journal is © The Royal Society of Chemistry 2017 |