
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and biological evaluation of novel neoflavonoid derivatives as potential antidiabetic agents†
Bing Wang‡
abcd,
Na Li‡abcd,
Teng Liuabcd,
Jie Sun *abcd and
Xiaojing Wang*abcd
*abcd and
Xiaojing Wang*abcd
aSchool of Medicine and Life Sciences, University of Jinan, Shandong Academy of Medical Sciences, Jinan 250200, Shandong, China. E-mail: sunjie310@126.com; xiaojing6@gmail.com
bInstitute of Materia Medica, Shandong Academy of Medical Sciences, Jinan 250062, Shandong, China
cKey Laboratory for Biotech-Drugs, Ministry of Health, Jinan 250062, Shandong, China
dKey Laboratory for Rare & Uncommon Diseases of Shandong Province, Jinan 250062, Shandong, China
First published on 7th July 2017
Abstract
Various substituted neoflavonoid derivatives were synthesized using sulfated montmorillonite K-10 as a catalyst. This method is environmental friendly, sustainable and economical, convenient in isolation and purification processes, with little byproducts, using earth-abundant catalysts and has relatively high yield. Those neoflavonoid derivatives were screened for antioxidant, a-glucosidase inhibitory, aldose reductase 2 (ALR2) inhibitory and advanced glycation end-product formation inhibitory effects. Most compounds exhibited significant antioxidant and advanced glycation end-product (AGE) formation inhibitory activities. It was interesting to note that out of thirty compounds, 8k and 8l were found to have greater ALR2 inhibitory activity than the standard drug quercetin. The pharmacological studies suggested neoflavonoid with adjacent 7,8-dihydroxy groups were more effective in inhibiting ALR2. Antidiabetic activity studies had shown that compounds 8l and 8m were equipotent to the standard drug glibenclamide in vivo. In summary, the target compound 8l provided a potential drug design concept for the development of therapeutic or prophylactic agents of diabetes and diabetes complications.
1. Introduction
Derivatives of neoflavonoids, also named 4-arylcoumarins, are widespread in nature and have possessed a broad spectrum of biological activities1 including antioxidant,2–4 antidiabetic,5 cytotoxic,6–8 antimicrobial,9–11 anti-inflammatory,12 antiprotozoal13–15 and estrogenic16 activities. Korec and co-workers17 reported that oral administration of an extract prepared from a commercial mixture of the stem barks of H. latiflora and E. caribaeum (CM) of uncertain origin decreased glucose levels in streptozotocin (STZ)-diabetic mice during short term experiments in 2000. Guerrero-Analco and co-workers18 found that the extract of H. latiflora and several 4-phenylcoumarins isolated from Rubiaceae were very effective in regulating blood glucose levels in hyperglycemia in 2007. The structure of the most effective compound (1,5-O-β-D-glucopyranosyl-7,3′,4′-trihydroxy-4-phenylcoumarin) was displayed in Fig. 1. | ||
| Fig. 1 Structure of compound 1. | ||
Owing to the potential biological activity and structural diversity of neoflavonoid, they have attracted an appreciable amount of attention in the synthetic community in recent decades.19 There are numerous reports of their synthesis in literature, but most of them have some disadvantages such as using expensive raw materials, delivering low regioselectivities and not economical or environmental friendly.20 Consequently, researchers are still looking for more environmental benign and economical synthetic processes. Lee and coworkers21 disclosed a new approach to the synthesis of neoflavonoid, based on a high yielding montmorillonite K-10 catalyzed lactone ring forming cyclization process (Fig. 2). In this article, we simplified and improved Lee's method by employing sulfated montmorillonite K-10 as catalyst (Fig. 2). In contrast to Lee's method, our substituted phenylpropiolic acids were not converted to acylate and the final product could be purified by recrystallization rather than chromatography on silica gel. In addition, we tried to use acetic acid as the solvent for this reaction, which was in accord with the concept of modern green chemistry.
 | ||
| Fig. 2 Methods to synthesize neoflavonoid. | ||
2. Results and discussion
2.1. Chemistry
The common synthetic strategies for the target compounds 4a–4p and 8a–8n are summarized in Scheme 1. The synthetic pathway was started from Knoevenagel condensation reaction of commercially available material 1a–1e and malonic acid to afford corresponding 2a–2e.22 Then under the catalysis of sulfated montmorillonite K-10, 4a–4p can be prepared by the esterification–cyclization reaction of 2a–2e and different phenols 3a–3e. In order to confirm the optimal reaction conditions, 4-methoxyphenylacrylic acid 2a with resorcinol 3a were chosen as model substrates. This paper screened different solvents such as nitrobenzene, chlorobenzene and acetic acid. It was found that this reaction had the highest yield of 79% in the nitrobenzene and a lower yield of 66% in the acetic acid (Table 1). The further studies found that the optimal H2SO4 concentration was 30% to treat montmorillonite K-10. This paper also examined the reuse of the catalyst on the tandem esterification alkylation of substituted cinnamic acids and resorcinol to get dihydrocoumarin 4a. The results showed that montmorillonite K-10 could be reused without extra treatment to catalyze the same reaction, and the yield of 4a didn't have significant decrease (Table 2). Montmorillonite K-10 also had such advantages as low cost, non-toxicity, and commercial availability, rendering the synthetic process more environmental friendly and economical. | ||
| Scheme 1 General synthetic route to neoflavonoid derivatives 4a–4p and 8a–8n. Reagents and conditions: (a) malonic acid, piperidine, pyridine, 90 °C; (b) nitrobenzene or acetic acid, montmorillonite K-10, 100 °C; (c) MeOH, SOCl2, reflux, 4 h; (d) Br2, CH2Cl2, 0 °C, 20 min; (e) KOH, EtOH, reflux, 6 h; (f) nitrobenzene, montmorillonite K-10, 100 °C; (g) I2, Al, acetonitrile, reflux, 5 h. | ||
| No. | Solvents | Concentration of H2SO4 (%) | Montmorillonite K-10 (g) | Reaction time (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 | Nitrobenzene | 0 | 2 | 27 | 5 |
| 2 | Nitrobenzene | 5 | 2 | 20 | 23 |
| 3 | Nitrobenzene | 10 | 2 | 16 | 41 |
| 4 | Nitrobenzene | 20 | 2 | 8 | 61 |
| 5 | Nitrobenzene | 30 | 2 | 3 | 79 |
| 6 | Nitrobenzene | 30 | 1 | 6 | 65 |
| 7 | Nitrobenzene | 30 | 4 | 3 | 79 |
| 8 | Nitrobenzene | 30 | 6 | 3 | 71 |
| 9 | Chlorobenzene | 30 | 2 | 5 | 45 |
| 10 | Acetic acid | 30 | 2 | 10 | 66 |
| Recycling times | Reaction time (h) | Yield (%) |
|---|---|---|
| 0 | 3 | 79 |
| 1 | 4.5 | 78 |
| 2 | 4.5 | 74 |
| 3 | 4.5 | 74 |
| 4 | 6 | 72 |
| 5 | 4 | 71 |
8a–8h were synthesized from different substituted cinnamic acids 2a–2c through four steps as outlined in Scheme 1. The key intermediates 4-methoxyphenylpropiolic acid (7a), 3,4-dimethoxyphenylpropiolic acid (7b) and 3,4,5-trimethoxyphenylpropiolic acid (7c) were prepared according to the routine reported previously by Sun's group.20 This procedure mainly included esterification, addition, elimination and acidification reactions. Then the neoflavonoids 8a–8h were successfully obtained via the reaction between key intermediates 7a–7c and different phenols 3a–3e in the presence of sulfated montmorillonite K-10 and nitrobenzene at 100 °C for 3–10 hours. The crude products were purified by recrystallization in pretty good yields (64–75%, Table 3). These neoflavonoids 8a–8h with 1, 2 or 3 methoxy group at the aryl in C4 could be easily converted to neoflavonoids 8i–8n with 1, 2 or 3 hydroxyl group at the aryl in C4 under the catalysis of I2 and Al in refluxing acetonitrile. Notably, the yield of this demethylating reaction was more than 90% without complex purification.
| Product | R1 | R2 | R3 | R4–7 | Yield (%) |
|---|---|---|---|---|---|
| 4a | H | OCH3 | H | 7-Hydroxy | 79 |
| 4b | OCH3 | OCH3 | H | 53 | |
| 4c | OCH3 | OH | H | 55 | |
| 4d | H | OCH3 | H | 6-Hydroxy | 52 |
| 4e | OCH3 | OCH3 | H | 53 | |
| 4f | H | OCH3 | H | 7,8-Dihydroxy | 79 |
| 4g | OCH3 | OCH3 | H | 83 | |
| 4h | OCH3 | OH | H | 53 | |
| 4i | OH | OCH3 | H | 66 | |
| 4j | H | OCH3 | H | 5,7-Dihydroxy | 72 |
| 4k | OCH3 | OCH3 | H | 62 | |
| 4l | OCH3 | OH | H | 53 | |
| 4m | OH | OCH3 | H | 64 | |
| 4n | H | OH | H | 62 | |
| 4o | OH | OH | H | 60 | |
| 4p | OCH3 | OCH3 | H | 6,8-Dihydroxy | 70 |
| 8a | H | OCH3 | H | 7-Hydroxy | 75 |
| 8b | OCH3 | OCH3 | H | 64 | |
| 8c | H | OCH3 | H | 7,8-Dihydroxy | 67 |
| 8d | OCH3 | OCH3 | H | 73 | |
| 8e | OCH3 | OCH3 | OCH3 | 70 | |
| 8f | H | OCH3 | H | 5,7-Dihydroxy | 65 |
| 8g | OCH3 | OCH3 | H | 70 | |
| 8h | OCH3 | OCH3 | H | 6,8-Dihydroxy | 72 |
| 8i | OH | OH | H | 7-Hydroxy | 93 |
| 8j | H | OH | H | 7,8-Dihydroxy | 93 |
| 8k | OH | OH | H | 97 | |
| 8l | OH | OH | OH | 90 | |
| 8m | OH | OH | H | 5,7-Dihydroxy | 93 |
| 8n | OH | OH | H | 6,8-Dihydroxy | 95 |
2.2. Biological evaluation
| Product | DPPH | OH | a-Glucosidase inhibitory activity | AGEs inhibitory activity | ALR2 inhibitory activity |
|---|---|---|---|---|---|
| IC50 value (μg mL−1) | |||||
| 4a | >1000 | >2000 | >1000 | >1000 | N/D |
| 4b | >1000 | >2000 | >1000 | 51.472 ± 1.364 | N/D |
| 4c | 25.146 ± 3.160 | >2000 | >1000 | 193.967 ± 15.232 | N/D |
| 4d | >1000 | >2000 | >1000 | >1000 | N/D |
| 4e | >1000 | >2000 | >1000 | >1000 | N/D |
| 4f | 9.655 ± 0.151 | 996 ± 39 | >1000 | 74.140 ± 0.420 | N/D |
| 4g | 7.828 ± 0.041 | 1061 ± 29 | >1000 | 6.730 ± 0.269 | N/D |
| 4h | 6.929 ± 0.016 | 908 ± 32 | >1000 | 121.279 ± 7.062 | N/D |
| 4i | 6.396 ± 0.067 | 982 ± 27 | >1000 | 29.706 ± 1.808 | N/D |
| 4j | 568.079 ± 4.537 | >2000 | >1000 | >1000 | N/D |
| 4k | 459.341 ± 21.791 | >2000 | >1000 | 7.717 ± 0.241 | N/D |
| 4l | 55.906 ± 1.589 | >2000 | >1000 | 5.572 ± 0.264 | N/D |
| 4m | 24.462 ± 1.160 | >2000 | >1000 | >1000 | N/D |
| 4n | 250.661 ± 3.690 | >2000 | >1000 | >1000 | N/D |
| 4o | 3.673 ± 0.029 | 721 ± 20 | >1000 | 4.712 ± 0.383 | N/D |
| 4p | 15.732 ± 0.465 | >2000 | >1000 | 6.454 ± 0.090 | N/D |
| 8a | >1000 | >2000 | >1000 | >1000 | N/D |
| 8b | >1000 | >2000 | >1000 | 1.605 ± 0.038 | N/D |
| 8c | 9.544 ± 0.139 | 599 ± 16 | >1000 | 2.257 ± 0.619 | N/D |
| 8d | 6.961 ± 0.175 | 573 ± 8 | >1000 | 4.356 ± 0.288 | N/D |
| 8e | 24.544 ± 0.275 | 938 ± 18 | >1000 | 6.329 ± 1.713 | N/D |
| 8f | 543.121 ± 23.144 | >2000 | 2.574 ± 0.496 | >1000 | N/D |
| 8g | 95.245 ± 17.881 | >2000 | >1000 | 5.467 ± 0.463 | N/D |
| 8h | 10.059 ± 0.204 | 410 ± 22 | 0.499 ± 0.164 | 33.418 ± 0.160 | N/D |
| 8i | 5.623 ± 0.024 | 486 ± 20 | >1000 | >1000 | N/D |
| 8j | 11.450 ± 0.185 | 619 ± 6 | >1000 | 24.200 ± 1.268 | N/D |
| 8k | 3.390 ± 0.006 | 341 ± 5 | 1.020 ± 0.062 | 0.617 ± 0.017 | 0.357 ± 0.015 |
| 8l | 3.583 ± 0.126 | 289 ± 0 | 0.250 ± 0.042 | 1.148 ± 0.125 | 0.203 ± 0.001 |
| 8m | 4.014 ± 0.032 | 338 ± 10 | 0.469 ± 0.042 | 2.493 ± 0.079 | 4.97 ± 0.052 |
| 8n | 3.891 ± 0.002 | 417 ± 21 | 0.400 ± 0.036 | 0.468 ± 0.028 | 4.815 ± 0.189 |
| Vitamin C | 6.509 ± 0.153 | 837 ± 24 | |||
| Acarbose | 0.032 ± 0.002 | ||||
| AG | 31.265 ± 0.942 | ||||
| Quercetin | 1.119 ± 0.088 | ||||
DPPH is a widely used method to evaluate antioxidant capacities of natural and synthetic products. Although the main skeleton for all compounds is same, the slight difference in their inhibitory potential might be due to the different substitution patterns on benzene ring. As shown in Table 4, the number and position of hydroxy groups were relatively correlated with DPPH radical scavenging activity. Generally, the increase of hydroxy groups and the decrease of methoxy groups were positive to this activity, while 3,4-dihydrogen structure was adverse to radical scavenging capacity. Moreover, compounds with adjacent 7,8-dihydroxy groups or 3′,4′-dihydroxy groups had great capacity to scavenge DPPH radical, especially compounds 4i, 4o, 8i, 8k, 8l, 8m and 8n were better than positive reference substance vitamin C (IC50 = 6.509 ± 0.153 μg mL−1). These conclusions were similar to ref. 25.
In addition, it can be found from the data that almost all compounds with two adjacent hydroxy groups exhibit great hydroxyl radical scavenging activity. Especially, the IC50 values of compounds 8h, 8k, 8l, 8m and 8n were from 289 ± 0 to 417 ± 21 μg mL−1, which were 2 to 3 folds of the value of vitamin C (IC50 = 837 ± 24 μg mL−1). The IC50 values, 4f > 8c and 4g > 8d, indicated that neoflavonoid with 3,4-dihydrogen had weaker hydroxyl radical scavenging activity.
| Test samples | Blood glucose concentration (mM) | ||||||
|---|---|---|---|---|---|---|---|
| Dose (per os) mg kg−1 of bw | 0 h | 1.5 h | 3 h | 5 h | 7 h | 9 h | |
| a *p < 0.05 significantly different ANOVA followed by Dunnett's t-test for comparison with respect to initial levels in each group. % variation of glycemia are in parentheses. | |||||||
| Control (vehicle) | — | 14.7 ± 4.3 | 14.8 ± 4.8 (0.68) | 12.3 ± 6.5 (−15.65) | 11.6 ± 8.4 (−21.09) | 9.2 ± 6.0 (−31.41) | 9.1 ± 4.8 (−31.41) |
| Glibenclamide | 10 | 16.1 ± 4.1 | 7.2 ± 1.2* (−55.28) | 6.9 ± 1.9 (−57.14) | 7.1 ± 1.6 (−55.90) | 6.6 ± 1.4 (−59.63) | 4.7 ± 1.1* (−70.81) |
| 8l | 10 | 18.7 ± 7.3 | 11.1 ± 4.3 (−40.64) | 9.9 ± 5.6 (−47.06) | 8.9 ± 4.4 (−52.94) | 7.9 ± 3.7 (−57.75) | 7.7 ± 3.3 (−59.36) |
| 8l | 30 | 17.5 ± 7.5 | 8.3 ± 2.6* (−51.43) | 5.7 ± 1.1* (−66.86) | 6.1 ± 0.9 (−65.14) | 4.0 ± 1.4* (−77.14) | 5.4 ± 1.9 (−69.14) |
| 8l | 100 | 18.0 ± 5.3 | 9.9 ± 3.2 (−45.00) | 8.7 ± 2.9 (−51.67) | 6.2 ± 1.6 (−65.56) | 5.2 ± 1.5 (−70.56) | 6.5 ± 1.3 (−63.89) |
| 8m | 10 | 14.4 ± 5.5 | 9.6 ± 2.7 (−33.30) | 8.8 ± 3.1 (−38.90) | 8.1 ± 2.2 (−43.05) | 6.5 ± 1.8 (−54.16) | 5.2 ± 1.2* (−63.19) |
| 8m | 30 | 15.0 ± 2.1 | 9.1 ± 1.6 (−39.33) | 8.3 ± 3.0 (−44.53) | 8.5 ± 2.5 (−43.33) | 6.7 ± 2.1 (−55.20) | 5.4 ± 1.3 (−64.20) |
| 8m | 100 | 13.6 ± 3.1 | 9.0 ± 1.9 (−33.82) | 8.3 ± 1.4 (−38.97) | 7.7 ± 1.2 (−45.59) | 6.1 ± 0.7 (−55.15) | 4.9 ± 0.6* (−63.97) |
| 8n | 10 | 17.6 ± 5.1 | 11.7 ± 5.0 (−33.52) | 11.2 ± 3.3 (−36.36) | 10.0 ± 3.0 (−43.75) | 9.4 ± 2.8 (−46.59) | 7.4 ± 2.1 (−57.39) |
| 8n | 30 | 18.8 ± 7.4 | 11.6 ± 4.0 (−38.30) | 10.5 ± 3.7 (−44.15) | 9.1 ± 2.9 (−51.60) | 8.7 ± 4.3 (−53.72) | 7.0 ± 2.9 (−62.77) |
| 8n | 100 | 17.5 ± 5.7 | 11.1 ± 5.9 (−36.57) | 9.9 ± 6.0 (−43.43) | 8.3 ± 1.7 (−52.57) | 8.1 ± 1.8 (−53.71) | 6.9 ± 1.4 (−60.57) |
| Test samples | Blood glucose concentration (mM) | ||||||
|---|---|---|---|---|---|---|---|
| Dose (per os) mg kg−1 of bw | 0 h | 1.5 h | 3 h | 5 h | 7 h | 9 h | |
| a *p < 0.05 significantly different ANOVA followed by Dunnett's t-test for comparison with respect to initial levels in each group. % variation of glycemia are in parentheses. | |||||||
| Control (vehicle) | — | 10.2 ± 1.0 | 8.1 ± 0.9 (−20.59) | 8.2 ± 1.0 (−18.82) | 8.4 ± 1.5 (−17.65) | 7.1 ± 1.1 (−29.41) | 6.4 ± 0.9 (−36.27) |
| Glibenclamide | 10 | 9.0 ± 1.9 | 6.0 ± 1.0* (−33.33) | 5.5 ± 0.8* (−38.89) | 6.0 ± 1.1* (−33.33) | 5.4 ± 1.1 (−40.00) | 5.5 ± 1.0 (−38.89) |
| 8l | 10 | 10.3 ± 0.6 | 8.1 ± 1.0 (−21.36) | 6.5 ± 0.7* (−36.89) | 7.3 ± 1.2 (−29.13) | 6.2 ± 0.9 (−39.81) | 6.6 ± 0.8 (−35.92) |
| 8l | 30 | 10.5 ± 1.3 | 6.6 ± 0.8 (−37.14) | 7.3 ± 0.7 (−30.48) | 6.5 ± 1.6 (−38.10) | 5.8 ± 1.4 (−44.76) | 7.1 ± 1.5 (−32.38) |
| 8l | 100 | 10.4 ± 0.8 | 8.0 ± 0.9 (−24.04) | 7.0 ± 0.8 (−32.69) | 7.2 ± 0.8 (−30.77) | 6.4 ± 0.7 (−39.42) | 7.2 ± 0.7 (−31.73) |
| 8m | 10 | 10.7 ± 1.1 | 9.4 ± 1.2 (−12.15) | 8.7 ± 1.3 (−18.69) | 9.3 ± 1.0 (−13.08) | 8.6 ± 1.4 (−20.56) | 7.8 ± 1.2 (−27.10) |
| 8m | 30 | 10.8 ± 1.3 | 9.0 ± 1.4 (−16.67) | 8.7 ± 1.3 (−19.44) | 9.3 ± 1.0 (−13.89) | 8.6 ± 1.5 (−20.37) | 7.9 ± 1.2 (−27.78) |
| 8m | 100 | 10.6 ± 1.1 | 9.2 ± 1.1 (−13.40) | 8.3 ± 1.0 (−21.70) | 8.1 ± 1.7 (−22.64) | 7.7 ± 1.4 (−27.36) | 7.8 ± 1.2 (−26.42) |
| 8n | 10 | 10.7 ± 1.1 | 9.6 ± 0.7 (−10.28) | 9.3 ± 0.9 (−13.08) | 8.8 ± 0.7 (−17.76) | 8.8 ± 0.9 (−17.76) | 8.2 ± 1.2 (−23.36) |
| 8n | 30 | 10.7 ± 1.0 | 9.7 ± 1.1 (−9.35) | 8.9 ± 1.0 (−16.82) | 8.9 ± 0.9 (−16.82) | 8.7 ± 1.1 (−18.69) | 7.9 ± 0.9 (−26.17) |
| 8n | 100 | 10.6 ± 1.1 | 9.5 ± 1.0 (−10.38) | 8.7 ± 0.9 (−17.92) | 8.4 ± 1.1 (−20.75) | 8.6 ± 0.9 (−18.87) | 8.1 ± 1.0 (−24.53) |
 | ||
| Fig. 3 Long-term effect of the compound 8l and 8m on blood glucose levels in STZ-diabetic mice. Each value is the mean ± SEM for six mice in each group. *p < 0.05 significantly different ANOVA followed by Dunnett's t-test for comparison with the diabetic control group at same time. | ||
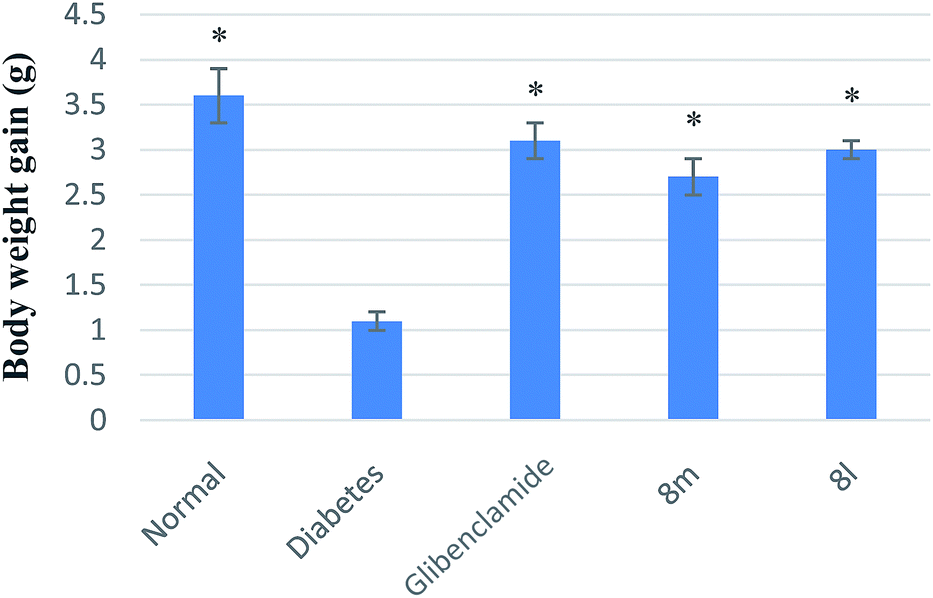 | ||
| Fig. 4 Body weight changes of daily treatment with compounds 8l and 8m in STZ-induced diabetic mice. Each value is the mean ± SEM for six mice in each group. *p < 0.05 significantly different ANOVA followed by Dunnett's t-test vs. the diabetic control group at same time. | ||
 | ||
| Fig. 5 Oral glucose tolerance test of compounds 8m and 8l on in STZ-induced diabetic mice. Each value is the mean ± SEM for six mice in each group. * p < 0.05 significantly different ANOVA followed by Dunnett's t-test vs. the diabetic control group at same time. | ||
3. Conclusions
Thirty neoflavonoid derivatives were synthesized by using sulfated montmorillonite K-10 as catalyst. Compared with traditional methods, this method is more environmental friendly, more sustainable and economical owning to the earth-abundant catalysts used, more convenient in isolation and purification processes due to less byproducts, and the yield is relatively higher than traditional methods. We then studied the pharmacological activity of the synthesized compounds. In the respect to its antioxidant activity, compounds 8k, 8l, 8m and 8n had strong hydroxyl radical scavenging activity. Compounds 8k and 8l showed significant effect on treatment of diabetic complications. Notably, 8l had relatively strong activity, which displayed little weaker capacity than acarbose. Oral toxicity tests indicated their innocuousness for mice. We selected compounds 8l, 8m and 8n to investigate their antidiabetic activity in vivo. The results showed the effect of the target compounds 8l and 8m were equipotent to that of the glibenclamide used as a positive control in vivo, in all cases. In conclusion, the target compound 8l offered a potential drug design concept for the development of therapeutic or preventive agents for diabetes and complications of diabetes.4. Experimental
4.1. Synthesis
Mass spectra were obtained with an Agilent Trap VL LC/MS spectrometer. The absorbance was recorded by a Hitachi U-3000 UV spectrophotometer. Column chromatography was performed on silica gel (200–300 mesh). Unless otherwise noted, all solvents and reagents were commercially available and used without further purification.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The suspension was directly filtered and a suitable amount of petroleum ether was added to the filtrate. Then cooled the mixture and stewing or stirred under low temperature for a certain length of time to promote crystallization of the product. The crude product was recrystallized from EtOAc–PE to afford 7-hydroxy-4-(4-methoxyphenyl)-3,4-dihydrocoumarin 4a. Compounds 4b–4p were obtained using the same procedures.
:1). The suspension was directly filtered and a suitable amount of petroleum ether was added to the filtrate. Then cooled the mixture and stewing or stirred under low temperature for a certain length of time to promote crystallization of the product. The crude product was recrystallized from EtOAc–PE to afford 7-hydroxy-4-(4-methoxyphenyl)-3,4-dihydrocoumarin 4a. Compounds 4b–4p were obtained using the same procedures.
4.1.3.1. 7-Hydroxy-4-(4-methoxyphenyl)-3,4-dihydrocoumarin (4a). White solid, yield: 79%, mp 177.2–178.3 °C, IR (KBr, ν, cm−1): 3332 (OH); 1736 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 3040, 1614, 1583, 1507, 1450, 1105, 1028, 984, 832 (Ar); 1236, 1146 (C–O); 2800 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.03 (m, 2H), 3.73 (s, 3H), 4.32 (t, J = 6.0 Hz, 1H), 6.53 (dd, J = 2.4 and 8.4 Hz, 1H), 6.55 (d, J = 2.4 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 6.90 and 7.06 (d, J = 1.8 Hz, 4H), 9.74 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 36.76, 37.80, 54.95, 103.17, 111.51, 114.03, 116.47, 128.23, 128.81, 133.53, 151.81, 157.43, 158.10 and 167.73. MS: m/z (%): 207.7 [M + 1]+, 162.6.
O); 3040, 1614, 1583, 1507, 1450, 1105, 1028, 984, 832 (Ar); 1236, 1146 (C–O); 2800 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.03 (m, 2H), 3.73 (s, 3H), 4.32 (t, J = 6.0 Hz, 1H), 6.53 (dd, J = 2.4 and 8.4 Hz, 1H), 6.55 (d, J = 2.4 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 6.90 and 7.06 (d, J = 1.8 Hz, 4H), 9.74 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 36.76, 37.80, 54.95, 103.17, 111.51, 114.03, 116.47, 128.23, 128.81, 133.53, 151.81, 157.43, 158.10 and 167.73. MS: m/z (%): 207.7 [M + 1]+, 162.6.
4.1.3.2. 7-Hydroxy-4-(3,4-dimethoxyphenyl)-3,4-dihydrocoumarin (4b). White solid, yield 53%, mp 144.8–146.1 °C. IR (KBr, ν, cm−1): 3433 (OH); 1767 (C
O); 3030, 1628, 1597, 1514, 1447, 1101, 1025, 848, 810 (Ar); 1267, 1142 (C–O); 2790 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.05 (m, 2H), 3.70 and 3.71 (s, 6H), 4.29 (t, J = 6.0 Hz, 1H), 6.52 (s, 1H), 6.53–6.56 (m, 2H), 6.83–6.89 (m, 3H), 9.74 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 37.27, 38.81, 55.93, 55.95, 103.72, 111.76, 112.09, 112.34, 119.54, 129.40, 149.37, 158.00 and 168.37. MS: m/z (%): 300.8 [M + 1]+, 258.7, 190.6, 162.7.
4.1.3.3. 7-Hydroxy-4-(3-methoxy-4-hydroxyphenyl)-3,4-dihydrocoumarin (4c). White solid, yield 55%, mp 194.3–196.3 °C. IR (KBr, ν, cm−1): 3415 (OH); 1724 (C
O); 2994, 1625, 1602, 1518, 1454, 1112, 1028, 841, 809 (Ar); 1259, 1152 (C–O); 2929 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.03 (m, 2H), 3.71 (s, 3H), 4.24 (t, J = 6.0 Hz, 1H), 6.44 (d, J = 7.8 Hz, 1H), 6.51 (s, 1H), 6.54 (d, J = 8.4 Hz, 1H), 6.70 (d, J = 7.8 Hz, 1H), 6.78 (s, 1H), 6.84 (d, J = 7.8 Hz, 1H), 8.93 and 9.72 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 37.35, 38.81, 56.05, 103.69, 112.05, 115.93, 117.23, 119.84, 129.42, 132.94, 146.01, 148.20, 152.37, 157.95 and 168.45. MS: m/z (%): 287.1 [M + 1]+, 162.8.
4.1.3.4. 6-Hydroxy-4-(4-methoxyphenyl)-3,4-dihydrocoumarin (4d). White solid, yield 52%, mp 167.7–169.7 °C. IR (KBr, ν, cm−1): 3332, (OH); 1728 (C
O); 3000, 1613, 1600, 1502, 1450, 1105, 1028, 831, 807 (Ar); 1236, 1146 (C–O), 2829 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.01 (m, 2H), 3.73 (s, 3H), 4.34 (t, J = 6.3 Hz, 1H), 6.37 (s, 1H), 6.68 (d, J = 8.6 Hz, 1H), 6.92 (d, J = 8.0 Hz, 2H), 6.96 (d, J = 8.6 Hz, 1H), 7.1 (d, J = 8.6 Hz, 2H), 9.34 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 36.8, 39.99, 55.55, 114.68, 115.23, 117.76, 128.05, 129.05, 133.34, 144.43, 154.37, 158.81 and 168.53. MS: m/z (%): 207.7 [M + 1]+, 162.6.
4.1.3.5. 6-Hydroxy-4-(3,4-dimethoxyphenyl)-3,4-dihydrocoumarin (4e). White solid, yield 53%, mp 166.7–168.5 °C, IR (KBr, ν, cm−1): 3259 (OH); 1709 (C
O); 3071, 1593, 1522, 1487, 1451, 1106, 1022, 856, 817 (Ar); 1267, 1142 (C–O); 2790 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.04 (m, 2H), 3.72 and 3.73 (s, 6H), 4.33 (t, J = 6.3 Hz, 1H), 6.37 (d, J = 2.6 Hz, 1H), 6.62 (dd, J = 1.6 and 8.2 Hz, 1H), 6.68 (dd, J = 2.7 and 8.7 Hz, 1H), 6.88 (d, J = 1.5 Hz, 1H), 6.91 (d, J = 8.3 Hz, 1H), 6.96 (d, J = 8.7 Hz, 1H), 9.32 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 36.70, 39.99, 55.90, 111.99, 112.43, 114.67, 115.22, 117.70, 119.88, 128.06, 133.72, 144.42, 148.42, 149.41, 154.35 and 168.59. MS: m/z (%): 301.0 [M + 1]+, 259.0, 162.9.
4.1.3.6. 7,8-Dihydroxy-4-(4-methoxyphenyl)-3,4-dihydrocoumarin (4f). White solid, yield 79%, mp 168.9–171.1 °C. IR (KBr, ν, cm−1): 3330 (OH); 1738 (C
O); 3050, 1634, 1612, 1512, 1458, 1109, 1030, 1002, 830, 802 (Ar); 1236, 1154 (C–O); 2805 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.01 (m, 2H), 3.72 (s, 3H), 4.28 (t, J = 6.8 Hz, 1H), 6.28 (d, J = 8.0 Hz, 1H), 6.53 (d, J = 8.0 Hz, 1H), 6.88 and 7.06 (d, J = 8.0 Hz, 4H), 8.91 and 9.29 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 37.35, 38.93, 55.53, 111.7, 114.9, 118.5, 119.1, 128.83, 133.74, 134.11, 140.99, 146.31, 158.66 and 168.25. MS: m/z (%): 286.8 [M + 1]+, 244.6, 178.6, 160.6.
4.1.3.7. 7,8-Dihydroxy-4-(3,4-dimethoxyphenyl)-3,4-dihydrocoumarin (4g). White solid, yield 83%, mp 173.7–174.6 °C. IR (KBr, ν, cm−1): 3423 (OH); 1754 (C
O); 2935, 1610, 1515, 1470, 1413, 1025, 807 (Ar); 1240, 1137 (C–O); 2829 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.04 (m, 2H), 3.70 and 3.71 (s, 6H), 4.27 (t, J = 6.3 Hz, 1H), 6.28, 6.53, 6.56 and 6.87 (d, J = 8.2 Hz, 4H), 6.84 (s, 1H), 8.90 and 9.28 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 37.25, 39.98, 55.96, 111.48, 111.87, 112.32, 117.61, 118.26, 119.62, 133.68, 134.56, 140.99, 146.28, 148.26, 149.32 and 168.34. MS: m/z (%): 316.8 [M + 1]+, 178.5.
4.1.3.8. 7,8-Dihydroxy-4-(3-methoxy-4-hydroxyphenyl)-3,4-dihydrocoumarin (4h). White solid, yield 53%, mp 171.7–173.1 °C. IR (KBr, ν, cm−1): 3373 (OH); 1743 (C
O); 3018, 1612, 1517, 1464, 1033, 812, 693 (Ar); 1274, 1067 (C–O); 2810 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.01 (m, 2H), 3.71 (s, 3H), 4.22 (t, J = 6.2 Hz, 1H), 6.29 (d, J = 8.3 Hz, 1H), 6.45, 6.52 and 6.69 (d, J = 8.1 Hz, 3H), 6.78 (d, J = 1.5 Hz, 1H), 8.88, 8.90 and 9.26 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 37.34, 39.98, 56.08, 111.45, 112.19, 115.88, 117.63, 118.45, 119.90, 132.95, 133.65, 140.96, 145.97, 146.21, 148.14 and 168.41. MS: m/z (%): 303.1 [M + 1]+, 283.7, 260.7, 178.7, 150.6.
4.1.3.9. 7,8-Dihydroxy-4-(3-hydroxy-4-methoxyphenyl)-3,4-dihydrocoumarin (4i). White solid, yield 66%, mp 185.0–187.1 °C. IR (KBr, ν, cm−1): 3499, 3420 (OH); 1752 (C
O); 3010, 1610, 1591, 1518, 1469, 1030, 786, 698 (Ar); 1277, 1068 (C–O); 2810 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 2.97 (m, 2H), 3.72 (s, 3H), 4.19 (t, J = 5.4 Hz, 1H), 6.32 (d, J = 7.8 Hz, 1H), 6.51, 6.53 and 6.54 (d, J = 6.0 Hz, 3H), 6.84 (d, J = 7.8 Hz, 1H), 8.90, 8.95 and 9.29 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 37.44, 39.11, 56.10, 111.47, 112.85, 114.92, 117.73, 118.25, 133.68, 134.92, 140.96, 146.23, 147.03 and 168.26. MS: m/z (%): 302.9 [M + 1]+, 283.7, 260.7, 178.6, 150.6.
4.1.3.10. 5,7-Dihydroxy-4-(4-methoxyphenyl)-3,4-dihydrocoumarin (4j). White solid, yield 72%, mp 148.4–149.9 °C. IR (KBr, ν, cm−1): 3510 (OH); 1759 (C
O); 3050, 1635, 1611, 1513, 1457, 1134, 1061, 1032, 826 (Ar); 1231, 1179 (C–O); 2805 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 2.97 (m, 2H), 3.69 (s, 3H), 4.38 (d, J = 6.5 Hz, 1H), 6.02 (d, J = 2.1, 1H), 6.17 (d, J = 2.2 Hz, 1H), 6.83 and 6.98 (d, J = 8.6 Hz, 4H), 9.55 and 9.73 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 33.40, 37.75, 61.07, 94.51, 95.15, 99.18, 103.88, 114.44, 128.13, 134.74, 153.38, 155.79, 158.28 and 168.41. MS: m/z (%): 286.9 [M + 1]+, 178.7, 110.8.
4.1.3.11. 5,7-Dihydroxy-4-(3,4-dimethoxyphenyl)-3,4-dihydrocoumarin (4k). White solid, yield 62%, mp 185.8–187.7 °C. IR (KBr, ν, cm−1): 3402, (OH); 1778 (C
O); 1632, 1610, 1500, 1460, 1012, 825 (Ar); 1237, 1148 (C–O). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.02 (m, 2H), 3.68 and 3.70 (s, 6H), 4.37 (d, J = 6.6 Hz, 1H), 6.01 (s, 1H), 6.17 (s, 1H), 6.40 (d, J = 8.4 Hz, 1H), 6.81 (d, J = 9.6 Hz, 2H), 9.56 and 9.74 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 33.76, 37.74, 55.87, 55.95, 95.11, 99.17, 103.74, 111.49, 112.22, 118.39, 135.29, 148.08, 153.45, 155.81, 158.27 and 168.47. MS: m/z (%): 316.9 [M + 1]+, 178.7, 164.7, 110.8.
4.1.3.12. 5,7-Dihydroxy-4-(3-methoxy-4-hydroxyphenyl)-3,4-dihydrocoumarin (4l). White solid, yield 53%, mp 173.0–174.0 °C. IR (KBr, ν, cm−1): 3404, (OH); 1767 (C
O); 3030, 1628, 1597, 1514, 1447, 1101, 1025, 848, 810 (Ar); 1267, 1142 (C–O), 2790 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.01 (m, 2H), 3.70 (s, 3H), 4.33 (d, J = 6.6 Hz, 1H), 6.00 (s, 1H), 6.16 (s, 1H), 6.30 (d, J = 7.8 Hz, 1H), 6.62 (d, J = 8.4 Hz, 1H), 6.75 (s, 1H), 8.91, 9.54 and 9.72 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 33.73, 37.86, 56.00, 95.09, 99.16, 103.95, 111.78, 115.72, 118.78, 133.68, 145.78, 148.05, 153.42, 155.78, 158.19 and 168.55. MS: m/z (%): 303.1 [M + 1]+, 261.1, 179.1, 151.1, 111.2.
4.1.3.13. 5,7-Dihydroxy-4-(3-hydroxy-4-methoxyphenyl)-3,4-dihydrocoumarin (4m). White solid, yield 64%, mp 228.3–230.3 °C. IR (KBr, ν, cm−1): 3332 (OH); 1751 (C
O); 3065, 1638, 1589, 1514, 1463, 1137, 1026, 841, 797 (Ar); 1285, 1166 (C–O), 2840 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 2.98 (m, 2H), 3.70 (s, 3H), 4.29 (d, J = 6.4 Hz, 1H), 6.01 (d, J = 2.2 Hz, 1H), 6.16 (d, J = 2.2 Hz, 1H), 6.55–6.39 (m, 2H), 6.80 (d, J = 8.3 Hz, 1H), 8.93 (s, 1H), 9.57 (s, 1H), 9.74 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 33.55, 37.84, 56.07, 95.08, 99.11, 103.92, 112.82, 114.49, 117.61, 135.46, 146.82, 146.85, 153.37, 155.80, 158.20 and 168.47. MS: m/z (%): 303.0 [M + 1]+, 260.8, 178.7, 150.7, 110.8.
4.1.3.14. 5,7-Dihydroxy-4-(4-hydroxyphenyl)-3,4-dihydrocoumarin (4n). White solid, yield 62%, mp 269.8–270.6 °C. IR (KBr, ν, cm−1): 3334 (OH); 1724 (C
O); 1616, 1513, 1464, 1413, 1132, 1062, 829 (Ar); 1288, 1190 (C–O). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 2.76 (dd, J = 15.8, 1.6 Hz, 1H), 3.13 (dd, J = 15.8, 6.9 Hz, 1H),4.32 (m, 1H), 6.01 (d, J = 2.2 Hz, 1H), 6.16 (d, J = 2.3 Hz, 1H), 6.65 (m, 2H), 6.85 (d, J = 8.5 Hz, 2H), 9.30 (s, 1H), 9.57 (s, 1H), 9.75 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 33.37, 37.87, 95.09, 98.63, 104.03, 116.19, 122.46, 128.15, 132.95, 153.36, 155.75, 156.51, 158.18, 159.37, and 168.53. MS: m/z (%): 272.7 [M + 1]+, 178.5.
4.1.3.15. 5,7-Dihydroxy-4-(3,4-dihydroxyphenyl)-3,4-dihydrocoumarin (4o). White solid, yield 60%, mp 214.7–216.4 °C. IR (KBr, ν, cm−1): 3272 (OH); 1751 (C
O); 1620, 1521, 1477, 1420, 1190, 1055, 799 (Ar); 1294, 1163 (C–O). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 2.72 (dd, J = 15.7, 1.6 Hz, 1H), 3.12 (m, 1H), 4.25 (m, 1H), 6.00 (d, J = 2.2 Hz, 1H), 6.16 (d, J = 2.3 Hz, 1H), 6.34 (dd, J = 8.2, 2.1 Hz, 1H), 6.42 (d, J = 2.1 Hz, 1H), 6.61 (d, J = 8.1 Hz, 1H), 8.75 (s, 1H), 8.84 (s, 1H), 9.55 (s, 1H), 9.72 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 33.56, 39.48, 94.53, 99.17, 104.17, 116.04, 117.85, 133.75, 144.42, 145.54, 153.36, 155.79, 158.13, 159.37, and 168.50. MS: m/z (%): 288.9 [M + 1]+, 178.7.
4.1.3.16. 6,8-Dihydroxy-4-(3,4-dimethoxyphenyl)-3,4-dihydrocoumarin (4p). White solid, yield 70%, mp 192.8–193.8 °C. IR (KBr, ν, cm−1): 3385 (OH); 1743 (C
O); 3058, 1638, 1608, 1518, 1450, 1311, 1106, 1024, 867 (Ar); 1267, 1171 (C–O), 2821 (–OCH3). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 2.99 (m, 2H), 3.72 (d, J = 4.9 Hz, 6H), 4.21 (t, J = 6.7 Hz, 1H), 6.34 (s, 1H), 6.52 (s, 1H), 6.59 (dd, J = 8.2, 1.9 Hz, 1H), 6.84 (d, J = 1.9 Hz, 1H), 6.89 (d, J = 8.3 Hz, 1H), 8.86 (s, 1H), 9.19 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ (ppm): 37.32, 39.07, 55.98, 55.99, 104.42, 111.92, 112.42, 114.65, 116.68, 119.79, 134.53, 142.44, 144.15, 145.57, 148.32, 149.38 and 168.69. MS: m/z (%): 317.0 [M + 1]+, 178.8.
SOCl2 (60 mmol) was added to a solution of 2a (40 mmol) in anhydrous methanol (100 mL). The solution was then mechanically stirred and refluxed for 2 h. Stop the reaction and cool for 10 minutes to concentrate, then ice water (50 mL) was added and followed by removal of the solvent in vacuo to obtain the crude product 4-methoxyphenyl ethyl acrylate 5a.
To a solution of 4-methoxyphenyl ethyl acrylate 5a in CH2Cl2 (80 mL) was introduced Br2 (2.1 mL, 41 mmol) dropwise into the solution at 0 °C for 20 min until the reaction solution does not fade. Continue stirring for one hour, the reaction solution was washed with 10% aqueous solution of sodium bisulfite to remove excess bromine and washed again with water. The organic layer was concentrated in vacuo and afforded the crude product 2,3-dibromo-4-methoxyphenylpropionate 6a. A mixture of 6a and KOH (6.8 g, 121 mmol) in ethyl alcohol (100 mL) was heated to reflux for 10 h. After the reaction stops, add 20 mL water to dissolve the salt, with concentrated hydrochloric acid to adjust the pH to strong acid, with dichloromethane to extract the reaction solution, then concentrating in vacuo to obtain the crude product. The crude product was subjected to column chromatography (silica, EtOAc–PE, 1:2) to obtain purified compound 4-methoxybenzene propionic acid 7a (4.6 g, in 65% yield).
To a solution of 4-methoxyphenylpropionic acid 7a (1.76 g, 10 mmol) and catechol (1.2 g, 11 mmol), in nitrobenzene (20 mL, anhyd) and the solution was stirred and heated to 100 °C. Then, 4 g acidified montmorillonite K-10 was added and the reaction was monitored by TLC (CH2Cl2:CH3OH, 10:1) and the reaction was completed after 5 hours. Hot filter, add 30 mL of petroleum ether in the filtrate and place the natural crystallization overnight. The solvent was removed by vacuum suction filtration, washed with 20 mL of petroleum ether and dried to obtain pure product 4-(4′-methoxyphenyl)-7-hydroxycoumarin 8a (2.0 g, in 75% yield). Compounds 8b–8h were obtained by using the same procedures.
4.1.4.1. 7-Hydroxy-4-(4-methoxyphenyl)-coumarin (8a). Pink solid, yield: 75%, mp 265.1–266.3 °C, IR (KBr, ν, cm−1): 3206 (OH); 1702 (C
O); 3006, 1617, 1608, 1511, 1443, 1122, 1036, 1002, 828 (Ar); 1251, 1184 (C–O); 2841 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.84 (s, 3H), 6.11 (s, 1H), 6.79 (d, J = 8.7 Hz, 2H), 7.11 (d, J = 7.6 Hz, 2H), 7.36 (d, J = 8.1 Hz, 1H), 7.48 (d, J = 7.6 Hz, 2H), 10.65 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 39.57, 55.81, 60.23, 103.16, 110.26, 111.25, 113.58, 114.75, 127.78, 128.66, 130.47, 155.57, 156.07, 160.71, 160.82 and 161.78. MS: m/z (%): 268.9 [M + 1]+, 162.7.
4.1.4.2. 7-Hydroxy-4-(3,4-dimethoxyphenyl)-coumarin (8b). Pink solid, yield: 64%, mp 239.4–240.5 °C, IR (KBr, ν, cm−1): 3355 (OH); 1732 (C
O); 3069, 1622, 1558, 1519, 1450, 1139, 1014, 860, 820 (Ar); 1253, 1178 (C–O); 2843 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.83 (d, J = 12.1 Hz, 6H), 6.16 (s, 1H), 6.79 (dd, J = 6.3, 2.3 Hz, 2H), 7.09 (ddd, J = 11.2, 10.1, 5.1 Hz, 3H), 7.41–7.47 (m, 1H), 10.62 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 39.57, 49.07, 56.09, 56.09, 103.12, 110.32, 111.26, 112.18, 112.56, 113.60, 121.62, 127.94, 128.82, 149.28, 150.39, 155.75, 156.08, 160.75 and 161.75. MS: m/z (%): 299.0 [M + 1]+, 282.9, 256.8, 164.7.
4.1.4.3. 7,8-Dihydroxy-4-(4-methoxyphenyl)-coumarin (8c). Red solid, yield: 67%, mp 108–110 °C, IR (KBr, ν, cm−1): 3480 (OH); 1685 (C
O); 3165, 1601, 1560, 1508, 1442, 1177, 1017, 856, 819 (Ar); 1297, 1177 (C–O); 2841 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.84 (s, 3H), 6.09 (s, 1H), 6.82 (dd, J = 29.0, 8.7 Hz, 2H), 7.10 (d, J = 8.7 Hz, 2H), 7.47 (d, J = 8.6 Hz, 2H), 9.42 (s, 1H), 10.16 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 39.56, 39.70, 55.79, 110.16, 112.06, 112.72, 114.65, 117.83, 128.07, 130.48, 133.08, 144.42, 150.00, 156.11, 160.67 and 161.73. MS: m/z (%): 284.8 [M + 1]+, 256.7, 152.6.
4.1.4.4. 7,8-Dihydroxy-4-(3,4-dimethoxyphenyl)-coumarin (8d). Pink solid, yield: 73%, mp 126.1–127.3 °C, IR (KBr, ν, cm−1): 3322 (OH); 1700 (C
O); 2954, 1604, 1519, 1505, 1440, 1020, 817 (Ar); 1257, 1142 (C–O); 2834 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.82 (s, 3H), 3.84 (s, 3H), 6.15 (s, 1H), 6.80 (d, J = 8.7 Hz, 1H), 6.93 (d, J = 8.7 Hz, 1H), 7.06 (d, J = 8.2 Hz, 1H), 7.10 (dd, J = 8.8, 4.8 Hz, 2H), 9.38 (s, 1H),10.17 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 39.56, 56.08, 110.23, 112.05, 112.11, 112.61, 112.73, 117.97, 121.64, 128.23, 133.06, 144.45, 149.20, 149.97, 150.31, 156.27 and 160.72. MS: m/z (%): 314.9 [M + 1]+, 299.8, 164.6.
4.1.4.5. 7,8-Dihydroxy-4-(3,4,5-trimethoxyphenyl)-coumarin (8e). Pink solid, yield: 70%, mp 281.3–281.5 °C, IR (KBr, ν, cm−1): 3266 (OH); 1702 (C
O); 2944, 1624, 1519, 1504, 1446, 1037, 867 (Ar); 1255, 1171 (C–O); 2847 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.74 (s, 3H), 3.83 (s, 6H), 6.21 (s, 1H), 6.81 (d, J = 10.3 Hz, 3H), 6.95 (d, J = 8.7 Hz, 1H), 9.43 (s, 1H), 10.24 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 39.49, 49.07, 56.49, 60.54, 106.42, 110.54, 111.91, 112.83, 118.02, 131.35, 133.03, 138.60, 144.34, 150.03, 153.39, 156.41 and 160.68. MS: m/z (%): 345.0 [M + 1]+, 318.9, 299.0.
4.1.4.6. 5,7-Dihydroxy-4-(4-methoxyphenyl)-coumarin (8f). Red solid, yield: 65%, mp 256.6–258.2 °C, IR (KBr, ν, cm−1): 3208 (OH); 1696 (C
O); 1635, 1514, 1468, 1023, 825 (Ar); 1267, 1182 (C–O); 2868 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.79 (s, 3H), 5.74 (s, 1H), 6.22 (dd, J = 48.3, 2.3 Hz, 2H), 6.93 (m, 2H), 7.29 (m, 2H), 10.15 (s, 1H), 10.42 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 49.07, 55.57, 95.10, 99.61, 101.11, 110.45, 111.64, 113.10, 129.52, 130.96, 140.06, 156.29, 157.60, 159.64, 160.46 and 162.03. MS: m/z (%): 285.0 [M + 1]+, 252.8, 152.6.
4.1.4.7. 5,7-Dihydroxy-4-(3,4-methoxyphenyl)-coumarin (8g). Red solid, yield: 70%, mp 183.6–184.8 °C, IR (KBr, ν, cm−1): 3468 (OH); 1675 (C
O); 3051, 1595, 1516, 1458, 1079, 840 (Ar); 1246, 1171 (C–O); 2841 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.75 (s, 3H), 3.79 (s, 3H), 5.79 (s, 1H),6.19 (d, J = 2.3 Hz, 1H), 6.26 (d, J = 2.3 Hz, 1H), 6.88 (dd, J = 8.2, 2.0 Hz, 1H), 6.95 (dd, J = 5.1, 3.1 Hz, 2H), 10.14 (s, 1H), 10.42 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 39.5, 55.95, 95.09, 99.69, 101.15, 110.49, 111.01, 112.40, 120.45, 132.32, 147.94, 149.18, 156.36, 157.30, 157.61, 160.49 and 162.02. MS: m/z (%): 315.0 [M + 1]+, 164.7.
4.1.4.8. 6,8-Dihydroxy-4-(3,4-dimethoxyphenyl)-coumarin (8h). Red solid, yield: 72%, mp 311.6–313.7 °C. IR (KBr, ν, cm−1): 3230 (OH); 1669 (C
O); 3003, 1598, 1516, 1464, 1023, 847 (Ar); 1262, 1170 (C–O); 2831 (–OCH3). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 3.84 (d, J = 7.3 Hz, 6H), 6.14 (s, 1H), 6.82 (s, 1H), 6.97 (s, 1H), 7.10 (m, 3H), 9.48 (s, 1H), 10.25 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 39.50, 56.09, 103.64, 110.41, 110.68, 111.38, 112.11, 112.45, 121.47, 128.23, 143.27, 148.98, 149.16, 150.24, 150.84, 155.57 and 162.02. MS: m/z (%): 315.0 [M + 1]+, 164.7.
:CH3OH, 6:1) until the starting material disappeared completely. The reaction solution was concentrated, and 5% sodium bisulfite was added to remove excess iodine. After the hydrochloric acid was acidified, the mixture was extracted with ethyl acetate. The organic layer was concentrated to obtain 4-(4′-hydroxyphenyl)-7-hydroxycoumarin 8i (0.46 g, in 92% yield). Compounds 8j–8n were obtained by using the same procedures.
4.1.5.1. 7-Hydroxy-4-(3,4-dihydroxyphenyl)-coumarin (8i). Yellow solid, yield: 92%, mp 265.5–267.9 °C, IR (KBr, ν, cm−1): 3430 (OH); 1667 (C
O); 1627, 1542, 1513, 1444, 1116, 1007, 839 (Ar); 1263, 1194 (C–O). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 6.04 (s, 1H), 6.81 (dt, J = 10.3, 1.9 Hz, 3H), 6.90 (dd, J = 5.1, 2.9 Hz, 2H), 7.46 (d, J = 8.5 Hz, 1H), 9.37 (s, 1H), 9.52 (s, 1H), 10.66 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 49.07, 103.10, 109.61, 111.24, 113.47, 116.31, 120.45, 126.55, 128.87, 145.89, 147.55, 156.06, 160.82 and 161.67. MS: m/z (%): 270.9 [M + 1]+, 242.2.
4.1.5.2. 7,8-Dihydroxy-4-(4-hydroxyphenyl)-coumarin (8j). Yellow solid, yield: 93%, mp 272.0–273.1 °C, IR (KBr, ν, cm−1): 3352 (OH); 1685 (C
O); 2959, 1610, 1586, 1514, 1454, 1111, 1042, 838 (Ar); 1229, 1066 (C–O). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 6.06 (s, 1H), 6.80 (d, J = 8.8 Hz, 1H), 6.90 (dd, J = 18.2, 8.6 Hz, 3H), 7.36 (d, J = 8.5 Hz, 2H), 9.93 (s, 1H), 9.37 (s, 1H), 10.14 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 39.56, 60.23, 109.76, 112.09, 112.66, 115.98, 117.93, 126.43, 130.54, 133.04, 144.45, 149.91, 156.46, 159.25 and 160.74. MS: m/z (%): 270.8 [M + 1]+, 242.7, 152.5.
4.1.5.3. 7,8-Dihydroxy-4-(3,4-dihydroxyphenyl)-coumarin (8k). Yellow solid, yield: 97%, mp 182.0–183.1 °C, IR (KBr, ν, cm−1): 3265 (OH); 1666 (C
O); 2918, 1597, 1531, 1449, 1343, 1119, 1043, 847 (Ar); 1243, 1043 (C–O). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 6.02 (s, 1H), 6.80 (s, 1H), 6.81 (d, J = 2.0 Hz, 1H), 6.89 (m, 2H), 6.95 (d, J = 8.8 Hz, 1H), 9.35 (s, 1H), 9.41 (s, 1H), 9.48 (s, 1H), 10.20 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 49.06, 109.54, 112.60, 116.31, 118.02, 120.45, 126.88, 133.01, 144.44, 145.79, 147.43, 149.87, 156.62, 160.77 and 172.51. MS: m/z (%): 286.9 [M + 1]+, 240.8, 152.6.
4.1.5.4. 7,8-Dihydroxy-4-(3,4,5-trihydroxyphenyl)-coumarin (8l). Light yellow solid, yield: 90%, mp 312.5–313.5 °C, IR (KBr, ν, cm−1): 3386 (OH); 1704 (C
O); 2851, 1600, 1539, 1452, 1317, 1038, 833 (Ar); 1226, 1077 (C–O). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 5.98 (s, 1H), 6.42 (s, 2H), 6.80 (d, J = 8.8 Hz, 1H), 6.99 (d, J = 8.8 Hz, 1H), 8.60 (s, 1H), 9.22 (s, 2H), 9.36 (s, 1H), 10.13 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 56.54, 60.23, 107.99, 109.35, 112.06, 112.55, 118.11, 125.98, 132.98, 135.19, 144.43, 146.60, 149.86, 156.95 and 160.76. MS: m/z (%): 302.9 [M + 1]+, 256.8, 152.7.
4.1.5.5. 5,7-Dihydroxy-4-(3,4-dihydroxyphenyl)-coumarin (8m). Light yellow solid, yield: 93%, mp 261.5–262.5 °C, IR (KBr, ν, cm−1): 3305 (OH); 1698 (C
O); 1597, 1522, 1442, 1349, 1033, 845 (Ar); 1265, 1083 (C–O). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 5.69 (s, 1H), 6.18 (d, J = 2.4 Hz, 1H), 6.25 (d, J = 2.3 Hz, 1H), 6.62 (dd, J = 8.1, 2.1 Hz, 1H), 6.72 (m, 2H), 9.01 (s, 1H), 9.11 (s, 1H), 10.11 (s, 1H), 10.40 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 49.07, 60.26, 95.02, 99.61, 101.19, 110.04, 115.94, 119.26, 130.93, 144.50, 146.03, 156.85, 157.65, 160.55 and 161.90. MS: m/z (%): 286.9 [M + 1]+, 152.6.
4.1.5.6. 6,8-Dihydroxy-4-(3,4-dihydroxyphenyl)-coumarin (8n). Light yellow solid, yield: 95%, mp 301.5–303.5 °C, IR (KBr, ν, cm−1): 3365 (OH); 1683 (C
O); 1607, 1520, 1446, 1395, 861 (Ar); 1276 (C–O). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 5.99 (s, 1H), 6.80 (dd, J = 7.9, 2.3 Hz, 2H), 6.88 (d, J = 2.1 Hz, 1H), 6.90 (d, J = 8.1 Hz, 1H), 6.98 (s, 1H), 9.31 (s, 1H), 9.40 (s, 1H), 9.44 (s, 1H), 10.20 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 56.19, 103.61, 109.82, 110.78, 111.58, 116.30, 120.27, 126.95, 143.27, 145.84, 147.39, 148.97, 150.85, 155.98 and 161.15. MS: m/z (%): 287.1 [M + 1]+, 152.8.
4.2. Biological activity
| ·OH scavenging effect (%) = [A0 − (A1 − A2)]/A0 × 100% |
The effect of target compounds and vitamin C to scavenge the DPPH free radical was evaluated with method described by Villaño and coworkers37 with slight modifications.
2.0 mL of different concentrations of the sample solution was added to 2.0 mL of DPPH solution (0.04 mg mL−1), shaken and allowed to react at room temperature for 30 min. The absorbance value was measured at 517 nm with an ultraviolet-visible spectrophotometer. The blank reference cuvette contained absolute ethanol. All measurements were performed in triplicate. IC50 values were calculated. The percentage DPPH free radical scavenging rate was determined as
| DPPH free radical scavenging effect (%) = [A0 − (A1 − A2)]/A0 × 100% |
| Inhibition rate% = {[(AC − AB) − (AS − ASB)]/(AC − AB)} × 100% |
| Reagents | Volume (μL) | |||
|---|---|---|---|---|
| Blank group | Control group | Sample blank group | Sample group | |
| PBS | 20 | 10 | 20 | 10 |
| Compounds/inhibitors | 0 | 0 | 10 | 10 |
| PNPG | 20 | 20 | 20 | 20 |
| Water | 10 | 10 | 0 | 0 |
| Mix well and incubate at 37 °C for 10 minutes | ||||
| α-Glucosidase | 0 | 10 | 0 | 10 |
| Mix well and react at 37 °C for 20 minutes | ||||
| Na2CO3 | 70 | 70 | 70 | 70 |
| Inhibition rate% = {[(AC − AB) − (AS − ASB)]/(AC − AB)} × 100% |
STZ-Induced diabetic mice and normal mice were placed in single cages with wire-net floors and deprived of food for 12 h before experimentation but allowed free access to tap water throughout. The compounds (at the doses of 10, 30 and 100 mg kg−1 of bw) were suspended in 0.05% Tween-80 in saline solution. Glibenclamide (10 mg kg−1 of bw) was suspended in the same vehicle. The target compounds were freshly prepared immediately before experimentation and administered by the intragastrical route at the doses of 10 mL kg−1 of bw. Control mice received only the vehicle (0.05% Tween-80 in saline solution) by the same route. Blood glucose levels were measured at 0, 1.5, 3, 5, 7, 9 h after drugs administration.
| % variation of glycemia = [(Gi − Gt)/Gi] × 100% |
Conflict of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are grateful to support from the Project of Shandong Province Higher Educational Science and Technology Program (J14M02), the Science and Technology Research Program of Shandong Academy of Medical Sciences (2014-4), Shandong Provincial Natural Science Foundation (ZR2015YL041) and the Innovation Project of Shandong Academy of Medical Sciences.References
- M. M. Garazd, Y. L. Garazd and V. P. Khilya, Chem. Nat. Compd., 2003, 39, 54–121 CrossRef CAS
.
- M. H. Lin, Y. S. Chou, Y. J. Tsai and D. S. Chou, J. Exp. Clin. Med., 2011, 3, 126–131 CrossRef CAS
- K. Zhang, W. Ding, J. Sun, B. Zhang, F. Lu, R. Lai, Y. Zou and G. Yedid, Chem. Nat. Compd., 2012, 107, 203–210 Search PubMed
- F. Pérez-Cruz, F. A. Villamena, G. Zapata-Torres, A. Das, C. A. Headley, E. Quezada, C. Lopez-Alarcon and C. Olea-Azar, J. Phys. Org. Chem., 2014, 26, 773–783 CrossRef
- R. Korec, M. Korecová, K. H. Sensch and T. Zoukas, Diabetes Res. Clin. Pract., 2000, 50, 42 CrossRef
- E. Rizzi, S. Dallavalle, L. Merlini, G. L. Beretta, G. Pratesi and F. Zunino, Bioorg. Med. Chem. Lett., 2005, 15, 4313 CrossRef CAS PubMed
- C. Billard, F. Menasria, C. Quiney, A. M. Faussat, J. P. Finet, S. Combes and J. P. Kolb, Exp. Hematol., 2008, 36, 1625 CrossRef CAS PubMed
- F. Menasria, A. G. B. Azebaze, C. Billard, A. M. Faussat, A. E. Nkengfack, M. Meyer and J. P. Kolb, Leuk. Res., 2008, 32, 1914–1926 CrossRef CAS PubMed
- Y. Kong, Y. J. Fu, Y. G. Zu, F. R. Chang, Y. H. Chen, X. L. Liu, J. Stelten and H. M. Schiebel, Food Chem., 2010, 121, 1150–1155 CrossRef CAS
- S. K. Roy, N. Kumari, S. Pahwa, U. C. Agrahari, K. K. Bhutani, S. M. Jachak and H. Nandanwar, Fitoterapia, 2013, 90, 140 CrossRef CAS PubMed
- C. Canning, S. Sun, X. Ji, S. Gupta and K. Zhou, J. Ethnopharmacol., 2013, 147, 259–262 CrossRef CAS PubMed
- T. Taechowisan, C. Lu, Y. Shen and S. Lumyong, Microbiology, 2005, 151, 1691 CrossRef CAS PubMed
- R. Argotteramos, G. Ramírezavila, M. C. Rodríguezgutiérrez, M. Ovillamuñoz, H. Lanzmendoza, M. H. Rodríguez, M. Gonzalezcortazar and L. Alvarez, J. Nat. Prod., 2006, 69, 1442–1444 CrossRef CAS PubMed
- J. T. Pierson, A. Dumètre, S. Hutter, F. Delmas, M. Laget, J. P. Finet, N. Azas and S. Combes, Eur. J. Med. Chem., 2010, 45, 864–869 CrossRef CAS PubMed
- J. V. Richard, K. A. Werbovetz, M. Gelb and A. Whitty, Curr. Opin. Chem. Biol., 2010, 14, 447 CrossRef CAS PubMed
- S. Nishimura, M. Taki, S. Takaishi, Y. Iijima and T. Akiyama, ChemInform, 2000, 31, 505–508 Search PubMed
- R. Korec, S. K. Heinz and T. Zoukas, Arzneim. Forsch., 2000, 50, 122–128 CAS
- J. Guerrero-Analco, O. Medina-Campos, F. Brindis, R. Bye, J. Pedraza-Chaverri, A. Navarrete and R. Mata, Phytochemistry, 2007, 68, 2087–2095 CrossRef CAS PubMed
- M. M. Garazd, Y. L. Garazd and V. P. Khilya, Chem. Nat. Compd., 2005, 41, 245–271 CrossRef CAS
- J. Sun, X. W. Ding, K. Y. Zhang and Y. Zou, Chin. Chem. Lett., 2011, 22, 667–670 CrossRef CAS
- L. Jian-Ming, T. Tsui-Hwa and L. Yean-Jang, Synthesis, 2001, 2001, 2247–2254 CrossRef
- J. Sun, X. W. Ding, X. P. Hong and K. Y. Zhang, Chem. Nat. Compd., 2012, 48, 16–22 CrossRef CAS
- J. B. Veselinović, A. M. Veselinović and Ž. J. Vitnik, et al., Antioxidant properties of selected 4-phenyl hydroxycoumarins: integrated in vitro and computational studies, Chem.–Biol. Interact., 2014, 214(1), 49 CrossRef PubMed
- L. Ismaili, A. Nadaradjane, L. Nicod, C. Guyon, A. Xicluna, J. F. Robert and B. Refouvelet, Eur. J. Med. Chem., 2008, 43, 1270–1275 CrossRef CAS PubMed
- K. Y. Zhang, W. X. Ding, J. Sun, B. Zhang, F. J. Lu, R. Lai, Y. Zou and G. Yedid, Biochimie, 2014, 48(1), 16–22 Search PubMed
- R. Mata, S. Cristians, S. Escandónrivera, K. Juárezreyes and I. Riverocruz, J. Nat. Prod., 2013, 76, 468–483 CrossRef CAS PubMed
- J. Riverachávez, M. Figureroa, M. C. González, A. E. Glenn and R. Mata, J. Nat. Prod., 2015, 78, 730 CrossRef PubMed
- D. Liu, W. He, Z. Wang, L. Liu, C. Wang, C. Zhang, C. Wang, Y. Wang, G. Tanabe and O. Muraoka, Eur. J. Med. Chem., 2016, 110, 224 CrossRef CAS PubMed
- Z. Ni, Z. Zhuge, W. Li, H. Xu, Z. Zhang and H. Dai, Biol. Pharm. Bull., 2012, 35, 2050–2053 CAS
- X. Q. Zou, S. C. Peng, L. F. Tan, Q. Yuan, H. W. Deng and Y. J. Li, Bioorg. Med. Chem., 2010, 18, 3020–3025 CrossRef CAS PubMed
- X. Qin, X. Hao, H. Han, S. Zhu, Y. Yang, B. Wu, S. Hussain, S. Parveen, C. Jing and B. Ma, J. Med. Chem., 2015, 58, 1254–1267 CrossRef CAS PubMed
- Y. C. Hwang, M. Kaneko, S. Bakr, H. Liao, Y. Lu, E. R. Lewis, S. Yan, S. Ii, M. Itakura and L. Rui, FASEB J., 2004, 18, 1192–1199 CrossRef CAS PubMed
- D. Lorke, Arch. Toxicol., 1983, 54(4), 275–287 CrossRef CAS PubMed
- S. Cristians, R. Bye, A. Navarrete and R. Mata, J. Ethnopharmacol., 2013, 145, 530–535 CrossRef CAS PubMed
- S. Cristians, R. Mata and R. Bye, J. Ethnopharmacol., 2014, 152, 308–313 CrossRef CAS PubMed
- C. N. Rhodes and D. R. Brown, Catal. Lett., 1994, 24, 285–291 CrossRef CAS
- D. Villaño, M. S. Fernándezpachón, M. L. Moyá, A. M. Troncoso and M. C. Garcíaparrilla, Talanta, 2007, 71, 230 CrossRef PubMed
- S. Chethan, S. M. Dharmesh and N. G. Malleshi, Bioorg. Med. Chem., 2008, 16, 10085–10090 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06457h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |