DOI:
10.1039/C7RA06450K
(Paper)
RSC Adv., 2017,
7, 39962-39969
In vitro and in vivo investigation of chitosan–polylysine polymeric nanoparticles for ovalbumin and CpG co-delivery
Received
9th June 2017
, Accepted 4th August 2017
First published on 16th August 2017
Abstract
In this work, a simple and powerful vaccine delivery system was developed by the electrostatic binding of chitosan-based polycation methoxy poly(ethylene glycol)–chitosan–poly(L-lysine) (mPEG–CS–PLL) with ovalbumin (OVA) and unmethylated cytosine–phosphate–guanine (CpG). OVA was used as the model antigen and co-loaded with the adjuvant CpG in the nanoparticles (NPs) to increase the vaccine efficacy. This vaccine delivery system could induce enhanced immune responses by simultaneously delivering the immunostimulating adjuvant CpG with OVA antigen. The vaccine delivery system was found to promote the cell uptake of antigens into bone-marrow-derived dendritic cells (BMDCs). Moreover, the OVA and CpG co-loaded NPs showed much higher efficiency to stimulate BMDCs maturation and proinflammatory cytokine secretion compared with OVA alone and the OVA-loaded NPs. The cell viability assay in BMDCs verified that the chitosan-based NPs didn't hamper the cell viabilities. Finally, the OVA and CpG co-loaded NPs were confirmed to induce improved OVA-specific antibody production and increased IFN-γ producing T cells, suggesting that this simple vaccine delivery system had the ability to powerfully induce both humoral and cellular immune responses. This study clearly demonstrated that the chitosan-based NPs had great promise to be applied in vaccine delivery for the treatment of infectious diseases and cancers in the future.
1. Introduction
Nanoparticles (NPs) are widely used as efficient delivery systems to administer a broad range of bioactivators such as drug molecules, proteins, peptides and DNAs.1–3 NPs also have shown many advantages in the areas of vaccine delivery, such as reliable antigen protection, improved transportation and enhanced vaccine efficiency.4–6 Ideal NP-based vaccine delivery systems should provide practical advantages including simplicity, low cost for manufacturing and potent vaccine delivery efficiency. And recently, designing and producing effective vaccine delivery systems has become a research hotspot. Therefore, various advanced and practical NPs have been, and continue to be extensively designed and applied in vaccine delivery.
Chitosan is a natural linear polysaccharide composed by glucosamine and N-acetyl-D-glucosamine and derived by the deacetylation of chitin, it has been extensively researched as a practical biomaterial in many fields because of its biodegradable, nontoxic, non-immunogenic and non-carcinogenic properties.7–12 Chitosan contains many free amino groups which makes it be the only positively charged natural polysaccharide, so it can be used as the nanocarriers to load negatively charged substances such as proteins, peptides and genes.13 Many studies have shown that the chitosan-based NPs had excellent ability to enhance both systemic and local immune responses when used as vaccine carriers.14 For example, Yong Taik Lim et al. used chitosan containing bioderived polyelectrolyte NPs to load antigens, robust loading efficiency and enhanced immune responses were achieved.15 And Hee Dong Han et al. studied the in vivo stepwise immunomodulation by using chitosan NPs as a platform nanotechnology for cancer immunotherapy.16 Other researches about chitosan-based NPs were also widely studied for vaccine delivery, and the advantages of the chitosan-based NPs were highly demonstrated. However, chitosan has very poor water solubility, which has limited its applications in biomedical field.17,18 To improve this drawback, in our previous work, we have designed and synthesized the methoxy poly(ethylene glycol)–chitosan–poly(L-lysine) (mPEG–CS–PLL), this polycation showed very high encapsulation efficiency and loading capacity for model protein bovine serum albumin (BSA).19 The BSA-loaded NPs showed spherical and homogeneous size below 300 nm with positive charges. Moreover, the cytotoxicity of the polymer was very low, which was favorable to practical applications. It was suggested from our previous work that the mPEG–CS–PLL can be used as a safe and effective carrier for protein delivery.
Herein, we used the mPEG–CS–PLL (PCL) polycation to develop a simple and powerful antigen delivery system by electrostatic binding of PCL with a commonly used model antigen ovalbumin (OVA) and immunoadjuvant unmethylated cytosine-phosphate-guanine (CpG). The unmethylated CpG oligodeoxynucleotide (ODN) is a Toll-like receptor 9 (TLR9) ligand,20,21 it can efficiently drive dendritic cells (DCs) maturation and induce proinflammatory cytokine secretion from mammalian immune cells.22,23 Therefore, we selected CpG as a potent adjuvant to co-deliver with the antigen OVA to efficiently increase the antigen-specific immune responses. The OVA and CpG co-loaded NPs were easily prepared, the zeta potential, particle size, morphology, stability and in vitro OVA and CpG release were characterized in detail. The cell uptake and internalization of the NPs in bone-marrow-derived dendritic cells (BMDCs) were evaluated. The capability of OVA and CpG co-loaded NPs to stimulate BMDCs maturation was measured by detecting the expression of co-stimulatory molecules and proinflammatory cytokine secretions. The cell viability assay in BMDCs was conducted to verify the biocompatibility of the chitosan-based NPs. Moreover, the OVA and CpG co-loaded NPs were subcutaneously injected into the C57BL/6 mice, the OVA-specific antibody production and IFN-γ producing T-cells were detected to confirm whether this simple vaccine delivery system had the ability to raise humoral and cellular immune responses. We believed that this study might provide a simple but practical strategy for OVA and CpG co-delivery. The chitosan-based NPs were also expected to have the potentials to be widely applied for vaccine delivery to treat infectious diseases and cancers in biomedical field in the future.
2. Experimental section
2.1. Materials
The mPEG–CS–PLL (PCL) polycation was synthesized according to our previous study, the chemical structure, preparation method and the detailed characterizations were shown in detail.19 OVA was purchased from Sigma-Aldrich (St. Louis, MO, USA). CpG ODN 1826, 5′-TCCATGACGTTCCTGACGTT-3′, was synthesized by Sangon (Shanghai, China). C57BL/6 mice (female, 6–8 weeks old) were purchased from Beijing Huafukang Biological Technology Co. Ltd. (HFK Bioscience, Beijing, China). APC-conjugated anti-CD11c, PE-conjugated anti-CD80, PE-conjugated anti-CD86 were purchased from eBioscience (San Diego, CA). Mouse TNF-α and IL-6 ELISA kit and IFN-γ ELISPOT kit were purchased from BD Biosciences (San Diego, CA, USA). RPMI 1640 and fetal bovine serum (FBS) were purchased from Gibco (Grand Island, USA). Granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin-4 (IL-4) were purchased from R&D Systems (MN, USA). All other materials were analytical or pharmaceutical grade. Ultrapure water from Milli-Q water system was used to prepare the aqueous solutions.
2.2. Preparation of the NPs
PCL/OVA and PCL/CpG/OVA NPs were prepared through electrostatic binding and the preparation method was described in the following steps. Briefly, PCL, CpG and OVA were respectively dissolved in PBS buffer at 1 mg mL−1 before NPs preparation. PCL/OVA NPs (mass ratio, 1/1) were prepared simply by mixing 10 μL PCL solution with 10 μL OVA solution, and after 15 s vortex, the solution was incubated at room temperature for 20 min to form stable PCL/OVA NPs. PCL/CpG/OVA NPs (mass ratio, 1/0.5/1) were prepared via the similar method of mixing 10 μL PCL, 10 μL OVA and 5 μL CpG together. After 15 s vortex, the solution was incubated at room temperature for 20 min to form stable PCL/CpG/OVA NPs. The NPs should be freshly prepared before use.
2.3. Characterizations of the NPs
The zeta potential and particle size of the free OVA, PCL/OVA and PCL/CpG/OVA NPs were analyzed by a zeta potential/BI-90Plus particle size analyzer (Brookhaven, USA) at room temperature. Data were shown as mean ± standard deviation (SD) based on triplicate independent experiments. The transmission electron microscopy (TEM) (H-6009IV; Hitachi, Tokyo, Japan) was used to test the morphology of the PCL/OVA and PCL/CpG/OVA NPs. The NPs were prepared and placed on a copper grid covered with nitrocellulose. The sample was negatively stained with phosphotungstic acid and dried at room temperature before observation. To test the in vitro stability of the NPs, the PCL/OVA NPs and the PCL/CpG/OVA NPs were freshly prepared and incubated in PBS (pH 7.4) at 37 °C under gentle stirring. The particle size changes of the NPs were measured at different time points by the zeta potential/BI-90Plus particle size analyzer.
2.4. In vitro release of OVA and CpG
The in vitro release of OVA and CpG was further evaluated. FITC-OVA and Cy5-CpG were used to prepare the PCL/OVA or PCL/CpG/OVA NPs. The freshly prepared NPs were suspended in PBS (pH 7.4) and incubated at 37 °C under continuous shaking at 50 rpm in the dark. At fixed time intervals, the samples were centrifuged (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 10 min) and 1 mL of the supernatant was collected for analysis. And 1 mL of fresh PBS was added into the samples to maintain a constant volume. The cumulative release of FITC-OVA and Cy5-CpG was detected by fluorescence spectra and calculated based on the standard curve.
000g, 10 min) and 1 mL of the supernatant was collected for analysis. And 1 mL of fresh PBS was added into the samples to maintain a constant volume. The cumulative release of FITC-OVA and Cy5-CpG was detected by fluorescence spectra and calculated based on the standard curve.
2.5. Generation of BMDCs from mouse bone marrow cells
Bone-marrow-derived dendritic cells (BMDCs) were harvested from bone marrow of C57BL/6 mice. Both the femurs and tibiae were collected and the intact bones were soaked in 75% (v/v) ethanol for 1 min for disinfection. Next, both ends of the bones were cut with scissors, and the marrow was flushed with RPMI 1640 medium using a 1 mL syringe. Clusters within the marrow suspension were disintegrated by vigorous pipetting. The red blood cells were depleted with 0.83 M NH4Cl buffer. The bone marrow cells were collected and cultured in 10 mL of RPMI 1640 medium supplemented with 10% FBS, 50 IU mL−1 penicillin and streptomycin, and 20 ng mL−1 mouse recombinant granulocyte macrophage colony-stimulating factor (GM-CSF) and 10 ng mL−1 interleukin-4 (IL-4) at 37 °C in 5% CO2. After 7 days, non-adherent and loosely adherent cells were harvested and used for in vitro experiments.
2.6. Cell viability assay
To evaluate the cytotoxicity of OVA, PCL/OVA and PCL/CpG/OVA NPs on BMDCs, the cell viability was measured by propidium iodide (PI) staining using flow cytometry. BMDCs were seeded in 12-well plate at a density of 5 × 105 cells per well in 1 mL RPMI 1640 medium and cultured at 37 °C in a 5% CO2 atmosphere overnight. The OVA, PCL/OVA and PCL/CpG/OVA NPs were freshly prepared before the experiment. Then the BMDCs were incubated with PBS, OVA, PCL/OVA and PCL/CpG/OVA NPs (the final OVA concentration in the well was 5 μg mL−1) at 37 °C in 5% CO2 atmosphere for 24 h. Then the cells were collected and stained with APC-anti-CD11c and PI in 4 °C for 30 min. The cells were tested with a Guava EasyCyte flow cytometer (Guava Technologies).
2.7. Analysis of cell uptake
Cell uptake experiment was carried out by flow cytometry. Briefly, BMDCs were seeded in 12-well plates at a density of 5 × 105 cells per well in 1 mL RPMI 1640 medium and cultured at 37 °C in a 5% CO2 atmosphere overnight. The OVA, PCL/OVA and PCL/CpG/OVA NPs were freshly prepared before the experiment (FITC-OVA was used for NPs preparation and tracking). Then the BMDCs were incubated with PBS, OVA, PCL/OVA and PCL/CpG/OVA NPs (the final FITC-OVA concentration in the well was 5 μg mL−1) at 37 °C in 5% CO2 atmosphere for 4 h. Then the cells were collected and tested with Guava EasyCyte flow cytometer (Guava Technologies).
2.8. Confocal laser scanning microscopy
BMDCs were seeded on coverslips in 6-well plates at a density of 5 × 105 cells per well and cultured overnight. OVA, PCL/OVA and PCL/CpG/OVA NPs were freshly prepared (FITC-OVA was used for NPs preparation and intracellular tracking) and added into each well. After 4 h of incubation, the cells were washed with PBS and fixed with 3.7% paraformaldehyde for 15 min at room temperature. The cell nuclei were stained by 4′-6-diamidino-2-phenylindole (DAPI, 1 mg mL−1, 1 μL per well) for 15 min. At last, the coverslips were carefully taken out and placed on the slides, enclosed with glycerol. The samples were observed by confocal laser scanning microscopy (CLSM, ZEISS LSM780, Germany).
2.9. Cytokine and maturation assay
BMDCs were cultured in 12-well plates at a density of 1 × 106 cells per well and incubated overnight. OVA, PCL/OVA and PCL/CpG/OVA NPs were freshly prepared and added into the wells and incubated for 24 h (the final OVA concentration in the well was 5 μg mL−1). For testing the cytokine secretion, the culture supernatants were harvested and analyzed for TNF-α and IL-6 by using cytokine-specific ELISA kits according to the manufacturer's instructions. To test the effect of OVA, PCL/OVA and PCL/CpG/OVA NPs on the expression of BMDCs maturation markers, the cells were collected and stained with APC-anti-CD11c, PE-anti-CD80 and PE-anti-CD86 in 4 °C for 30 min. Cells treated with PBS and LPS (lipopolysaccharide) were respectively used as negative and positive controls. The cells were tested with a Guava EasyCyte flow cytometer (Guava Technologies).
2.10. Measurement of OVA-specific-IgG and IFN-γ producing T-cells
All animal studies were conducted in accordance with the principles and procedures outlined in “Regulations for the Administration of Affairs Concerning Laboratory Animals” approved by the National Council of China, and “The National Regulation of China for Care and Use of Laboratory Animals” promulgated by the National Science and Technology Commission of China. The protocols were approved under the guidance of the Animal Care and Use Committee of Northeast Normal University (China). C57BL/6 mice (n = 5 per group) were immunized with OVA, PCL/OVA and PCL/CpG/OVA NPs (in a total volume of 100 μL, containing 25 μg of OVA) by subcutaneous injection at day 0, 7 and 14. Serum samples and spleens were collected from the immunized mice at day 21.
The OVA-specific-IgG antibody was measured by ELISA. 96-well ELISA plate was coated with OVA (5 μg mL−1) at 4 °C for 24 h. Then the wells were blocked with BSA and washed with PBST, serial dilutions (1:100, 1:1000, 1:5000, 1:10000, 1:20000, 1:40000, 1:80000, 1:160000, 1:320000) of sera samples were added (sera samples of control group treated with PBS were diluted to 1:100). The plate was incubated at 37 °C for 2 h and washed three times, the antibody dilutions of anti-mouse IgG (1:2000) were added and incubated at 37 °C for 1 h. After washing, the substrate was added into the well and incubated for 30 min at room temperature. The reaction was further stopped by 0.5 N sulfuric acid.
The IFN-γ secreting T-cells were measured by using a mouse IFN-γ enzyme-linked immunosorbent spot (ELISPOT) assay. First, an ELISPOT plate was coated with an anti-mouse IFN-γ capture antibody (5 μg mL−1) in PBS at 4 °C overnight. The harvested splenocytes (5 × 106 per mL) were blocked and cultured in the 96-well ELISPOT plate with 10 μg mL−1 OVA. The plate was incubated for 60 h at 37 °C and washed with PBST. After that the plate was incubated with a biotin-conjugated detection antibody for 2 h at room temperature, and further incubated with a streptavidin-conjugated HRP for 1 h. Substrate was added into the well and incubated for 15 min at room temperature. The colorimetric reaction was terminated by washing with water. After the plate was dried, the spots were automatically counted.
2.11. Statistical analysis
The data were expressed as mean ± standard deviation (SD). Student's t-test was performed to compare the statistical significance. *p < 0.05 was considered statistically significant. **p < 0.01 and ***p < 0.001 were considered extremely significant.
3. Results and discussions
3.1. Preparation and characterization of the NPs
PCL/CpG/OVA NPs were easily prepared by the electrostatic binding of PCL polycation with antigen OVA and adjuvant CpG (Fig. 1a). The zeta potential and particle size of the OVA, PCL/OVA and PCL/CpG/OVA NPs were measured, and the results were respectively shown in Fig. 1b and c. The results showed that the zeta potential of PCL/OVA and PCL/CpG/OVA NPs was +22.17 mV and +19.35 mV. The data displayed that by adding negatively charged CpG ODNs the zeta potential of the NPs was slightly decreased. In addition, the particle size of free OVA, PCL/OVA and PCL/CpG/OVA NPs was 15563 nm, 401 nm and 203 nm, and the polydispersity index (PDI) of them was 0.350 ± 0.051, 0.268 ± 0.031 and 0.236 ± 0.034, respectively. And the particle size of PCL/CpG/OVA NPs was much smaller than PCL/OVA NPs. This was because of the increased ionic interactions between the positively charged PCL and the negatively charged CpG ODNs, leading the PCL/CpG/OVA NPs to be more compacted than the PCL/OVA NPs. The morphology of the PCL/OVA and PCL/CpG/OVA NPs were observed by TEM (Fig. 1d). Compared with PCL/OVA NPs, the PCL/CpG/OVA showed much smaller and more homogeneous particle morphology. The in vitro stability result of PCL/OVA and PCL/CpG/OVA NPs was shown in Fig. 1e. Both the sizes of the PCL/OVA NPs and the PCL/CpG/OVA NPs grew slightly with the incubation time during the first 24 h, which meant that the PEG-contained NPs had good stability. Meanwhile, the growth rate of PCL/OVA NPs was higher than that of the PCL/CpG/OVA NPs. This might be due to the CpG could increase the electrostatic binding force inside the NPs, which contributed to keep the PCL/CpG/OVA NPs more stable. Although both of the two kinds of NPs became a little bigger at 48 h, their sizes were still at the nanoscale which was acceptable for in vivo use, because the DCs could phagocytize the particles with the size distribution ranged from nanoscale to microscale.24–26
 |
| | Fig. 1 Schematic illustration and characterization of PCL/CpG/OVA NPs. (a) Schematic illustration for the preparation of PCL/CpG/OVA NPs and the use of this vaccine delivery system to induce humoral and cellular immunity response. (b) The zeta potential of PCL/CpG/OVA NPs. (c) The particle size of PCL/CpG/OVA NPs. (d) The TEM images of the PCL/OVA and PCL/CpG/OVA NPs. (e) The in vitro stability of the PCL/OVA and PCL/CpG/OVA NPs. | |
3.2. In vitro release of OVA and CpG
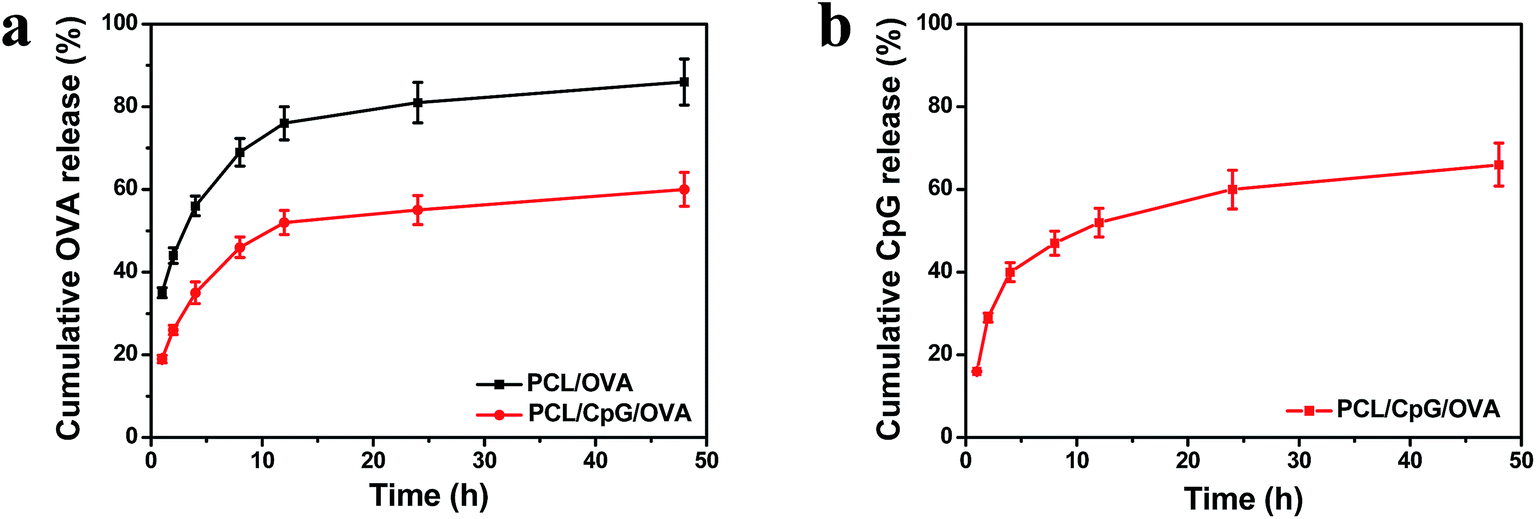
The in vitro release of OVA and CpG was tested, and the results were shown in Fig. 2. The OVA gradually released from the PCL/OVA or PCL/CpG/OVA NPs, and the cumulative OVA release of PCL/OVA was as high as 86% at 48 h, which was higher than that of PCL/CpG/OVA NPs (Fig. 2a). The increased ionic interactions between the positively charged PCL and the negatively charged CpG made the PCL/CpG/OVA NPs more compact and more stable than the PCL/OVA NPs, so the OVA release of PCL/CpG/OVA NPs was lower than that of the PCL/OVA NPs. The cumulative release profile of CpG was shown in Fig. 2b, and up to 66% of CpG was released from the PCL/CpG/OVA NPs during the 48 h. The results clearly verified that the OVA and CpG could be released from the PCL/CpG/OVA NPs in an appropriate manner.
 |
| | Fig. 2 In vitro release of OVA (a) from PCL/OVA and PCL/CpG/OVA NPs, and CpG (b) from PCL/CpG/OVA NPs. | |
3.3. Cell viability assay
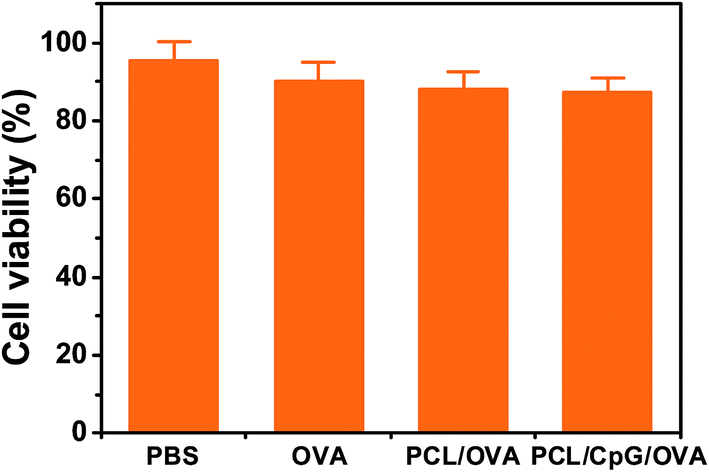
The cytotoxicity of OVA, PCL/OVA and PCL/CpG/OVA NPs were investigated in BMDCs. The results (Fig. 3) showed that the cell viability of free OVA was around 90%. When further loaded the OVA in the PCL, the cell viability of the PCL/OVA NPs only decreased slightly, which meant that the polycationic PCL was biocompatible. The cell viability of PCL/CpG/OVA NPs was similar with the PCL/OVA NPs, and both of them did not remarkably reduce the cell viability of BMDCs.
 |
| | Fig. 3 Cytotoxicity of the OVA, PCL/OVA and PCL/CpG/OVA NPs in BMDCs. | |
3.4. Analysis of cellular uptake
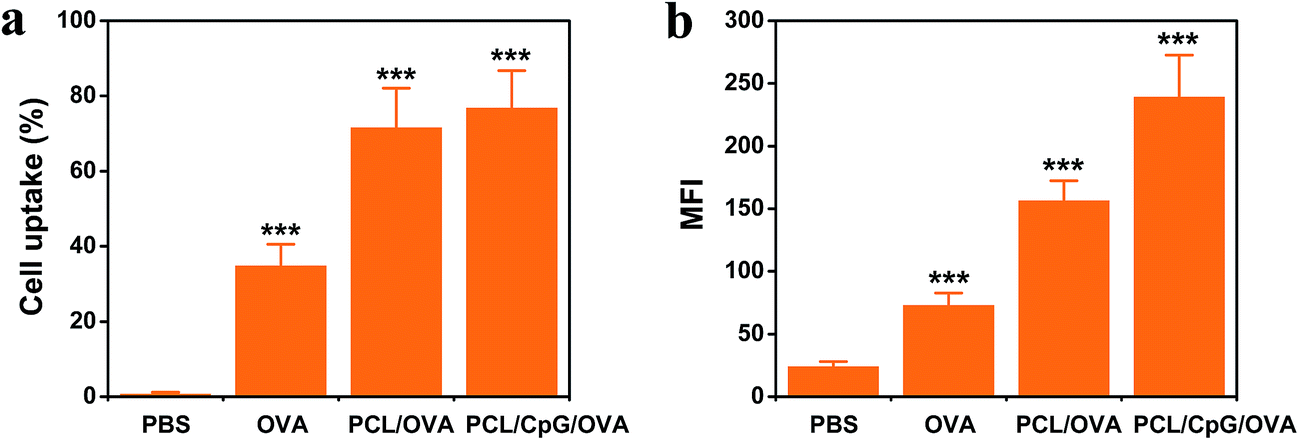
Cell uptake experiment was carried out by flow cytometry and the results were shown in Fig. 4. The cell uptakes of PCL/OVA NPs and PCL/CpG/OVA NPs were much higher than that of free OVA. After the negatively charged OVA was loaded in the positively charged PCL, the PCL/OVA NPs and PCL/CpG/OVA NPs were positively charged, which were taken up by the BMDCs more easily via the electrostatic attractive forces between NPs and negatively charged cell membranes. Moreover, the PCL/CpG/OVA NPs had much higher cell uptake and mean florescence intensity (MFI) than PCL/OVA NPs, this might because of the PCL/CpG/OVA NPs had much smaller particle size, which would be better for BMDCs uptake.
 |
| | Fig. 4 Cell uptake (a) and MFI (b) of BMDCs incubated with OVA, PCL/OVA and PCL/CpG/OVA NPs for 4 h. | |
3.5. Confocal laser scanning microscopy
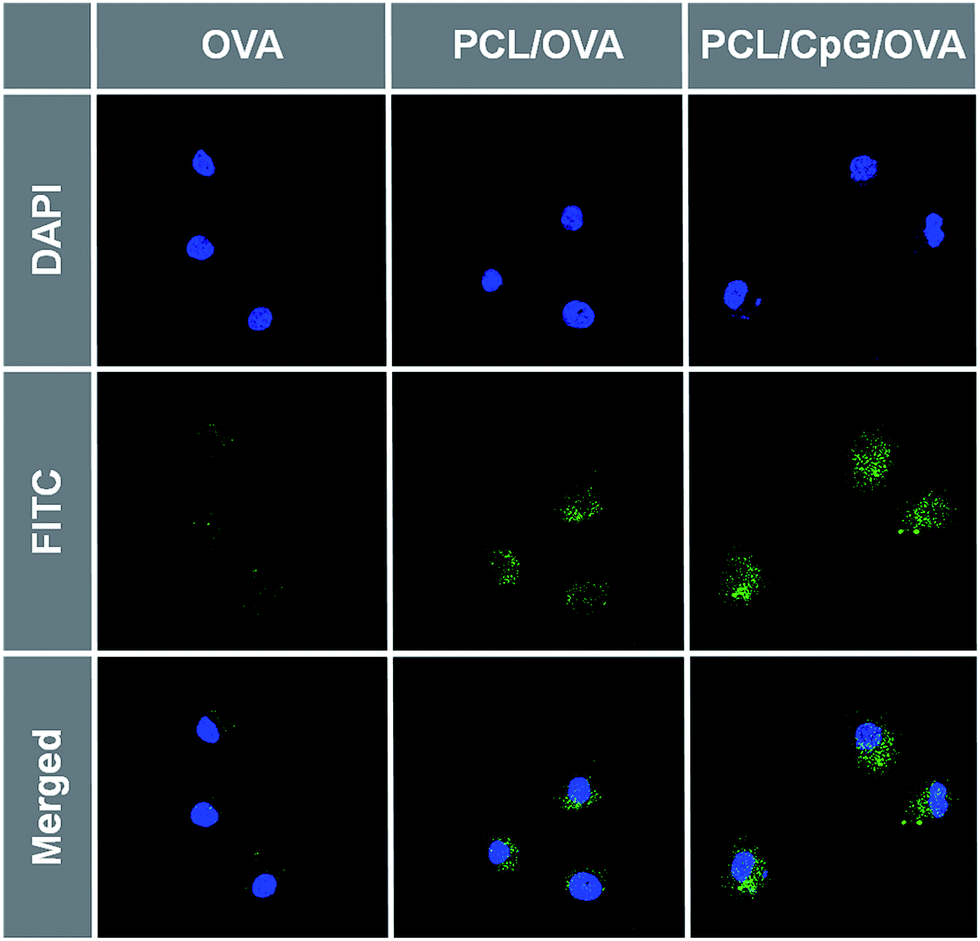
The intracellular delivery of OVA, PCL/OVA and PCL/CpG/OVA NPs in BMDCs was assessed by confocal laser scanning microscopy (CLSM). The fluorescent dye FITC conjugated OVA was used to track the internalization of the NPs in BMDCs. As shown in Fig. 5, green signals were both observed in BMDCs incubated with OVA, PCL/OVA and PCL/CpG/OVA NPs. However, the fluorescence intensities were really different. Only very weak green signals were found in BMDCs incubated with free OVA, the negatively charged OVA alone was not easy to be internalized in BMDCs. Strong fluorescence signals were observed in BMDCs that incubated with PCL/OVA and PCL/CpG/OVA NPs. Moreover, the PCL/CpG/OVA NPs also showed much higher fluorescence signals than PCL/OVA NPs, which was consistent with the cell uptake result, the PCL/CpG/OVA NPs had higher internalization in BMDCs.
 |
| | Fig. 5 CLSM images of BMDCs incubated with OVA, PCL/OVA and PCL/CpG/OVA NPs for 4 h. The cell nuclei were stained with DAPI (blue), and FTIC-labeled OVA generated green fluorescence. | |
3.6. Cytokine and maturation assay
To assess the BMDCs activation and maturation in vitro, the surface maturation markers and proinflammatory cytokine secretions were characterized in BMDCs. The CpG loaded in the PCL/CpG/OVA NPs was a potent immune adjuvant which was a TLR9 ligand. And the PCL/CpG/OVA NPs could protect the CpG and OVA from directly contacting with the nucleases and proteases during delivery process. When the NPs were internalized in the endosomes, the CpG could interact with the TLR9 on the endosome membrane and help to stimulate the BMDC maturation. As is known, CD86 and CD80 are costimulatory molecules of BMDCs for mediating the interaction with T cells. To confirm the maturation effect, the expressions of surface maturation markers were analyzed using flow cytometry (Fig. 6a–f). The free OVA and PCL/OVA NPs only showed similar CD80 and CD86 expression levels compared with PBS. However, the results showed that the maturation marker expressions of BMDCs that treated with PCL/CpG/OVA NPs were much higher than the PBS, OVA and PCL/OVA NPs treated groups. The CD80 and CD86 co-expression results also showed high levels for PCL/CpG/OVA NPs treated BMDCs (Fig. 6e and f). The secretion of proinflammatory cytokines from BMDCs were also measured (Fig. 6g and h). TNF-α and IL-6 play important roles in effective immune responses. The cytokine secretion of the PCL/CpG/OVA NPs was significantly higher than that of the free OVA and PCL/OVA NPs. These results clearly demonstrated that PCL/CpG/OVA NPs containing immunostimulating CpG could enhance the BMDCs activation and maturation, which would influence the downstream immune responses.
 |
| | Fig. 6 Cytokine and maturation assay of the NPs on BMDCs. (a–f) Expressions of the maturation markers of CD86 and CD80. (g and h) Proinflammatory cytokine secretion of TNF-α and IL-6. | |
3.7. Measurement of OVA-specific IgG and IFN-γ producing T-cells
Inspired by the in vitro results that the PCL/CpG/OVA NPs had high cell uptake and ability to stimulate BMDCs maturation, we further investigated the potentials of the PCL/CpG/OVA NPs to induce OVA-specific humoral and cellular immune responses in vivo by measuring the OVA-specific IgG (humoral immunity) and IFN-γ producing T-cells (cellular immunity). The C57BL/6 mice were immunized with OVA, PCL/OVA and PCL/CpG/OVA NPs by subcutaneous injection at day 0, 7 and 14. The serum samples and spleens were collected from the immunized mice at day 21 to determine the OVA-specific antibody level and IFN-γ producing T-cells (Fig. 7). The level of OVA-specific IgG was very low in mice immunized with OVA alone. And PCL/OVA NPs showed increased OVA-specific IgG production than free OVA. It was exciting to find that the mice immunized with PCL/CpG/OVA NPs had much higher level of OVA-specific IgG than the other groups (Fig. 7a). To further test OVA-specific cellular immune responses, IFN-γ ELISPOT assay was applied to evaluate the IFN-γ secreting T-cells from mouse spleens when restimulated with the OVA (Fig. 7b). PCL/CpG/OVA NPs showed much higher numbers of IFN-γ secreting T-cells, were about 5-fold higher compared with OVA alone. Taken the in vivo experiment results together, PCL/CpG/OVA NPs could strongly induce OVA-specific antibody production and IFN-γ producing T-cells, which might be largely due to the superior stimulating capacity of PCL/CpG/OVA for DC maturation. It was well-known that the mature DCs could activate and induce the proliferation and differentiation of the T cells which were mainly responsible for the cellular immune responses.27 Meanwhile, the generated T helper cells could secrete lymphokines for stimulating the B cells to produce antibodies for humoral immune responses.28,29 Both the in vitro and the in vivo results have clearly suggested that our PCL/CpG/OVA system could effectively stimulate the maturation and proinflammatory cytokine secretion of DCs and further induce powerful humoral and cellular immune responses potently.
 |
| | Fig. 7 Measurement of OVA-specific IgG and IFN-γ producing T-cells. (a) The OVA-specific-IgG secretion in mouse serum measured by ELISA analysis. (b) The percentage of IFN-γ producing T-cells from spleen samples. | |
4. Conclusions
In conclusion, we have developed a simple and powerful vaccine delivery system by electrostatic binding of chitosan-based polycation PCL with antigen OVA and immune adjuvant CpG. The vaccine delivery system could increase the cell uptake of OVA into BMDCs. Moreover, the PCL/CpG/OVA NPs showed much higher efficiency to stimulate BMDCs maturation and proinflammatory cytokine secretion. Finally, the PCL/CpG/OVA NPs induced enhanced OVA-specific antibody production and increased IFN-γ producing T-cells, suggesting that this simple vaccine delivery system had the ability to powerfully raise both humoral and cellular immune responses. We believe that the PCL/CpG/OVA NPs will have great promise to be applied in vaccine delivery for the treatment of infectious diseases and cancers in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are thankful to the National Natural Science Foundation of China (21153003), Jilin Science & Technology Department (20140204017GX, 20150520025JH), China Postdoctoral Science Foundation (no. 2014M561268), Science & Technology Bureau of Changchun City project (2013060), the Education Department of Jilin Province science project (JJKH20170606KJ) for financial support to this work.
References
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- A. Bolhassani, S. Safaiyan and S. Rafati, Mol. Cancer, 2011, 10, 3 CrossRef CAS PubMed.
- N. D. Sonawane, F. C. Szoka and A. S. Verkman, J. Biol. Chem., 2003, 278, 44826–44831 CrossRef CAS PubMed.
- A. des Rieux, V. Fievez, M. Garinot, Y. J. Schneider and V. Préat, J. Controlled Release, 2006, 116, 1–27 CrossRef CAS PubMed.
- S. Y. Kim, M. B. Heo, G. S. Hwang, Y. Jung, D. Y. Choi, Y. M. Park and Y. T. Lim, Angew. Chem., Int. Ed., 2015, 54, 8139–8143 CrossRef CAS.
- S. Y. Kim, H. Phuengkham, Y. W. Noh, H. G. Lee, S. H. Um and Y. T. Lim, Adv. Funct. Mater., 2016, 26, 8072–8082 CrossRef CAS.
- Q. Gan and T. Wang, Colloids Surf., B, 2007, 59, 24–34 CrossRef CAS.
- K. L. Douglas, C. A. Piccirillo and M. Tabrizian, J. Controlled Release, 2006, 115, 354–361 CrossRef CAS PubMed.
- M. George and T. E. Abraham, J. Controlled Release, 2006, 114, 1–14 CrossRef CAS PubMed.
- H. D. Han, L. S. Mangala, J. W. Lee, M. M. Shahzad, H. S. Kim, D. Shen and S. J. Lee, Clin. Cancer Res., 2010, 16, 3910–3922 CrossRef CAS PubMed.
- K. H. Noh, Y. M. Park, H. S. Kim, T. H. Kang, K. H. Song and Y. H. Lee, BMC Immunol., 2014, 15, 1–7 CrossRef PubMed.
- L. Qi and Z. Xu, Bioorg. Med. Chem. Lett., 2006, 16, 4243–4245 CrossRef CAS PubMed.
- R. Hejazi and M. Amiji, J. Controlled Release, 2003, 89, 151–165 CrossRef CAS PubMed.
- M. Amidi, S. G. Romeijn, G. Borchard, H. E. Junginger, W. E. Hennink and W. Jiskoot, J. Controlled Release, 2006, 111, 107–116 CrossRef CAS PubMed.
- Y. T. Lim, S. M. Shim, Y. W. Noh, K. S. Lee, D. Y. Choi, H. Uyama and H. Poo, Small, 2011, 7, 3281–3286 CrossRef CAS PubMed.
- H. D. Han, Y. Byeon, J. H. Jang, H. N. Jeon, G. H. Kim, M. G. Kim and Y. J. Lee, Sci. Rep., 2016, 6, 38348 CrossRef CAS PubMed.
- M. N. V. R. Kumar, React. Funct. Polym., 2000, 46, 1–27 CrossRef CAS.
- P. K. Dutta, J. Dutta and V. S. Tripathi, J. Sci. Ind. Res., 2004, 63, 20–31 CAS.
- Y. Han, Q. Duan, Y. Li, Y. Li and J. Tian, Adv. Polym. Technol., 2016 DOI:10.1002/adv.21782.
- A. M. Krieg, Annu. Rev. Immunol., 2002, 20, 709–760 CrossRef CAS PubMed.
- D. M. Klinman, Nat. Rev. Immunol., 2004, 4, 249–259 CrossRef CAS.
- A. M. Krieg, Nat. Rev. Drug Discovery, 2006, 5, 471–484 CrossRef CAS PubMed.
- G. K. Mutwiri, A. K. Nichani, S. Babiuk and L. A. Babiuk, J. Controlled Release, 2004, 97, 1–17 CrossRef CAS PubMed.
- A. Mant, F. Chinnery, T. Elliott and A. P. Williams, Immunology, 2012, 136, 163–175 CrossRef CAS PubMed.
- B. San Román, J. M. Irache, S. Gómez, N. Tsapis, C. Gamazo and M. S. Espuelas, Eur. J. Pharm. Biopharm., 2008, 70, 98–108 CrossRef PubMed.
- X. Q. Zhang, C. E. Dahle, G. J. Weiner and A. K. Salem, J. Pharm. Sci., 2007, 96, 3283–3292 CrossRef CAS PubMed.
- M. C. Raff, Nature, 1973, 242, 19–23 CrossRef CAS PubMed.
- S. Crotty, Nat. Rev. Immunol., 2015, 15, 185–189 CrossRef CAS PubMed.
- D. H. Katz and B. Benacerraf, Adv. Immunol., 1972, 15, 1–94 CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Jian Tian*ab
*a and
Jian Tian*ab