DOI:
10.1039/C7RA06439J
(Paper)
RSC Adv., 2017,
7, 33293-33304
Sn–H bond additions to asymmetric trigonal phosphinidene-bridged dimolybdenum complexes†
Received
8th June 2017
, Accepted 26th June 2017
First published on 30th June 2017
Abstract
The reactions between organotin hydrides HSnR3 (R = Bu, Ph) and the asymmetric trigonal phosphinidene-bridged complexes [Mo2Cp(μ-κ1:κ1,η5-PC5H4)(CO)2(η6-HMes*)] (1), [Mo2Cp2(μ-PH)(CO)2(η6-HMes*)] (2), and [Mo2Cp2(μ-PMe)(CO)2(η6-HMes*)] (3) were examined, and found to be strongly dependent on the parent complex (Mes* = 2,4,6-C6H2tBu3). Compound 1 reacted with HSnR3 upon moderate heating (353–363 K), to give the corresponding derivatives [Mo2Cp{μ-κ1:κ1,η5-P(H)C5H4}(SnR3)(CO)2(η6-HMes*)], following from a formal 1,2-addition of the Sn–H bond across the Mo–P double bond of the parent complex, with specific formation of Sn–Mo and H–P bonds. Complex 2 reacted analogously, but much faster, to yield the corresponding phosphanyl-bridged derivatives [Mo2Cp2(μ-PH2)(SnR3)(CO)2(η6-HMes*)] at room temperature. However, these products progressively degraded to the corresponding mononuclear phosphine complexes trans-[MoCp(SnR3)(CO)2(PH3)] in the presence of trace amounts of water. The methylphosphinidene-bridged complex 3 (Mo–P = 2.550(3) and 2.281(3) Å) was prepared upon reaction of MeLi with the phosphide-bridged complex [Mo2Cp2(μ-P)(CO)2(η6-HMes*)](BAr′4), (Ar′ = 3,5-C6H3(CF3)2). Its reaction with HSnR3 proceeded at room temperature with H–Sn bond addition now accompanied by spontaneous dehydrogenation, to yield the corresponding cyclopentadienylidene–phosphanyl derivatives [Mo2Cp{μ-κ1:κ1,η5-P(Me)C5H4}(SnR3)(CO)2(η6-HMes*)]. The structures of the new complexes were analyzed using spectroscopic, diffractometric and, in some cases, density functional theory methods. The Sn–H bond cleavages leading to the complexes eventually isolated in these reactions were proposed to be initiated by σ-bond coordination of the organotin reagent, via its Sn–H bond, to the metal atom of the MoCp(CO)2 fragment in the parent compounds, this being followed by a H-shift to the P atom of the bridging phosphinidene ligand.
Introduction
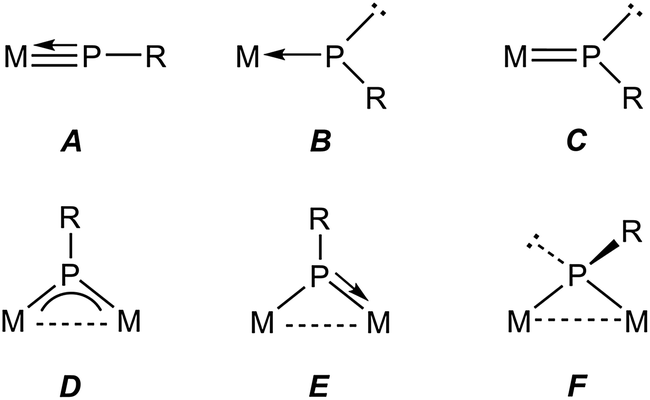
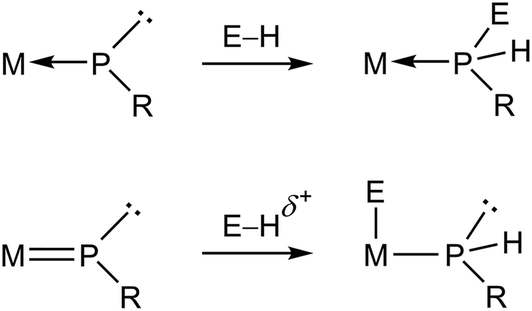
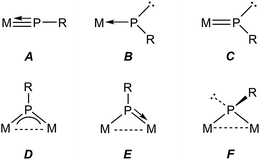
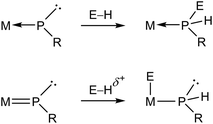
Metal complexes having phosphinidene ligands (PR) have been a subject of interest over the years. Particular attention has been paid to the mononuclear complexes displaying bent-terminal ligands (B and C in Chart 1), which are reactive toward many different organic reagents, whereby a large number of unusual organophosphorus molecules have been built on.1,2 Because of the presence at the P site of both a lone electron pair and a low-energy empty orbital, these molecules meet the criteria for P-based activation of even single, relatively unreactive H–E bonds (E = p-block element).3 Indeed, transient electrophilic complexes of type B have been found to undergo E–H bond addition at the P site to yield the corresponding phosphine derivatives (E = B, C, Si, N, O; Scheme 1).1a In contrast, the nucleophilic complex of type C [ZrCp2(PMes*)(PMe3)] (Mes* = 2,4,6-C6H2tBu3) underwent E–H bond addition to the double M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P bond, with specific binding of the P atom to the positively polarized H atom (E = O, S, N, P; Scheme 1).4 We note that no studies appear to have been carried out on the reactions of nucleophilic complexes with molecules bearing E–H bonds with negatively charged H atoms, as it is the case of silanes and stannanes.
P bond, with specific binding of the P atom to the positively polarized H atom (E = O, S, N, P; Scheme 1).4 We note that no studies appear to have been carried out on the reactions of nucleophilic complexes with molecules bearing E–H bonds with negatively charged H atoms, as it is the case of silanes and stannanes.
 |
| | Chart 1 | |
 |
| | Scheme 1 | |
In relative terms, the chemistry of binuclear phosphinidene-bridged complexes has been less explored, although this situation has been changing in the last years.5 As concerning the activation of E–H single bonds, we might consider the pyramidal phosphinidene complexes of type F as poorly reactive, if we exclude protonation processes, because of the lack of a suitable low-energy empty orbital. Yet, the diiron complexes [Fe2Cp2(μ-PR)(μ-CO)(CO)2] (Cy, Ph, Mes*) were able to induce intermolecular O–H and H–H bond cleavages,6 as well as an intramolecular C–H bond cleavage,7 to give phosphine derivatives of type [Fe2Cp2(μ-CO)2(CO)(PRHE)], although the H–H and C–H bond activations were attributed to the presence in solution of small amounts of an isomer having a bent terminal phosphinidene ligand. The symmetric trigonal phosphinidene complexes of type D might be more reactive towards molecules with E–H single bonds, because of the presence in that case of a low-lying empty orbital having π*(M–P) character,8,9 but only a limited number of studies in this direction have been carried out so far. The reactions of the ditungsten complex [W2(μ-PCp*)(CO)10] with primary and secondary phosphines indeed involved the activation of P–H bonds and formation of new P–P bonds, but also the active participation of the Cp* ring,10 while the reaction of [Mo2Cp2(μ-PMes*)(μ-CO)2] (Mo![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Mo) with PH2Cy involved the regioselective addition of a P–H bond to the Mo–P multiple bond of the parent complex, to yield the phosphanyl derivative [Mo2Cp2(μ-PHMes*)(μ-PHCy)(CO)2].9 In contrast, reactions of the discandium complex [Sc2(μ-PXyl)2L2] (L = diiminate ligand) with several disubstituted boranes led to the cleavage of B–O or B–H bonds depending on the reagent.11 We also note that the photochemical isomerisation of [Mo2Cp2(μ-PMes*)(CO)4] involves the intramolecular activation of a C–H bond in one of the tBu groups of the Mes* substituent.12
Mo) with PH2Cy involved the regioselective addition of a P–H bond to the Mo–P multiple bond of the parent complex, to yield the phosphanyl derivative [Mo2Cp2(μ-PHMes*)(μ-PHCy)(CO)2].9 In contrast, reactions of the discandium complex [Sc2(μ-PXyl)2L2] (L = diiminate ligand) with several disubstituted boranes led to the cleavage of B–O or B–H bonds depending on the reagent.11 We also note that the photochemical isomerisation of [Mo2Cp2(μ-PMes*)(CO)4] involves the intramolecular activation of a C–H bond in one of the tBu groups of the Mes* substituent.12
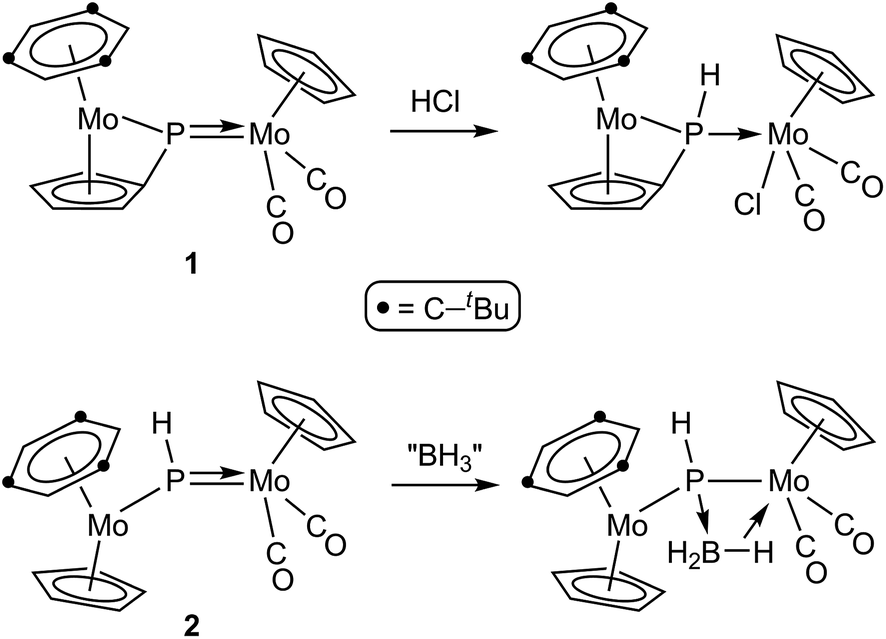
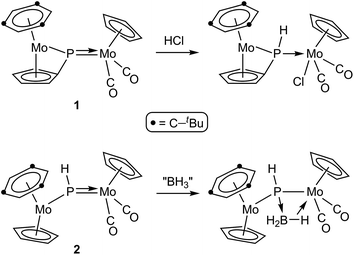
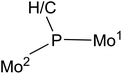
As for asymmetric trigonal phosphinidene complexes of type E, only two reactions involving the activation of E–H bonds have been actually reported (Scheme 2). Firstly, the reaction of the cyclopentadienylidene–phosphinidene complex [Mo2Cp(μ-κ1:κ1,η5-PC5H4)(CO)2(η6-HMes*)] (1) with HCl resulted in formal 1,2-addition of the reagent to the Mo–P multiple bond of the complex, with specific formation of a P–H bond.13 On the other hand, the reaction of the phosphinidene complex [Mo2Cp2(μ-PH)(CO)2(η6-HMes*)] (2) with BH3·THF gave a phosphinidene–borane derivative, also following from specific attachment of the P atom to the positively polarized part of the reagent (here, the B atom), although a B–H bond is not actually cleaved in the process, but just weakened as a result of an agostic-like interaction with the metal centre.14 These results are in agreement with the general view that the Mo–P double bond in these complexes behaves as expected for polar bonds with nucleophilic P sites and electrophilic M sites,5 also substantiated through the reactions of complexes 1 and 2 with metal-based electrophiles such as [AuCl(THT)], [Au(PR3)(THT)]+ and HgI2.15 In this paper we report the preparation of the Me-substituted analogue of 2, the methylphosphinidene complex [Mo2Cp2(μ-PMe)(CO)2(η6-HMes*)] (3), and the results of the reactions of compounds 1 to 3 with the organotin hydrides HSnR3 (R = Bu, Ph), which turned to be strongly dependent on the parent phosphinidene complex. We note that no previous studies appear to have been carried out on the reactions of organotin hydrides with phosphinidene complexes of any kind, while only one study addressed the reactivity of silanes (in that case, by using a mononuclear complex of type B).16 As it will be shown below, the reactions of compounds 1 to 3 with HSnR3 all seem to result in formal 1,2-addition of the Sn–H bond across the Mo–P double bond of the parent complex, with specific formation of Sn–Mo and H–P bonds, although a rare dehydrogenation involving a cyclopentadienyl ligand then takes place for the more congested PMe-bridged complex.
 |
| | Scheme 2 | |
Results and discussion
Synthesis, structural characterization and electronic structure of the methylphosphinidene complex 3
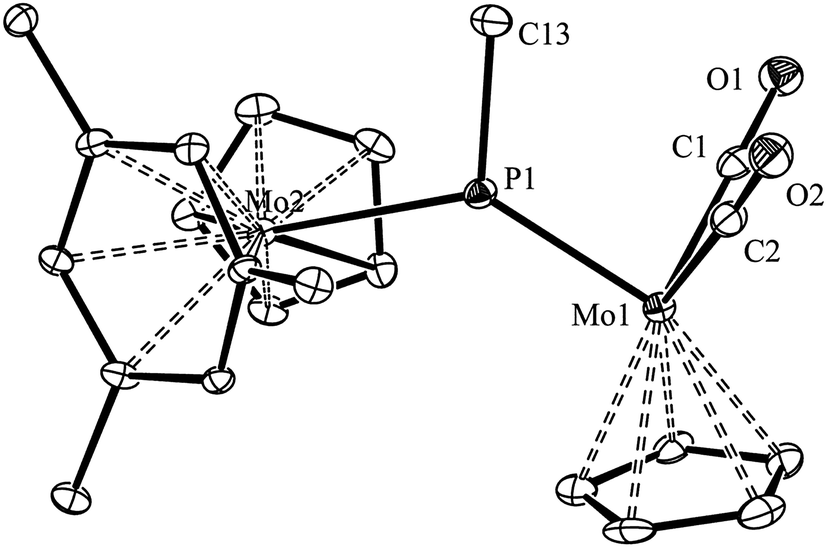
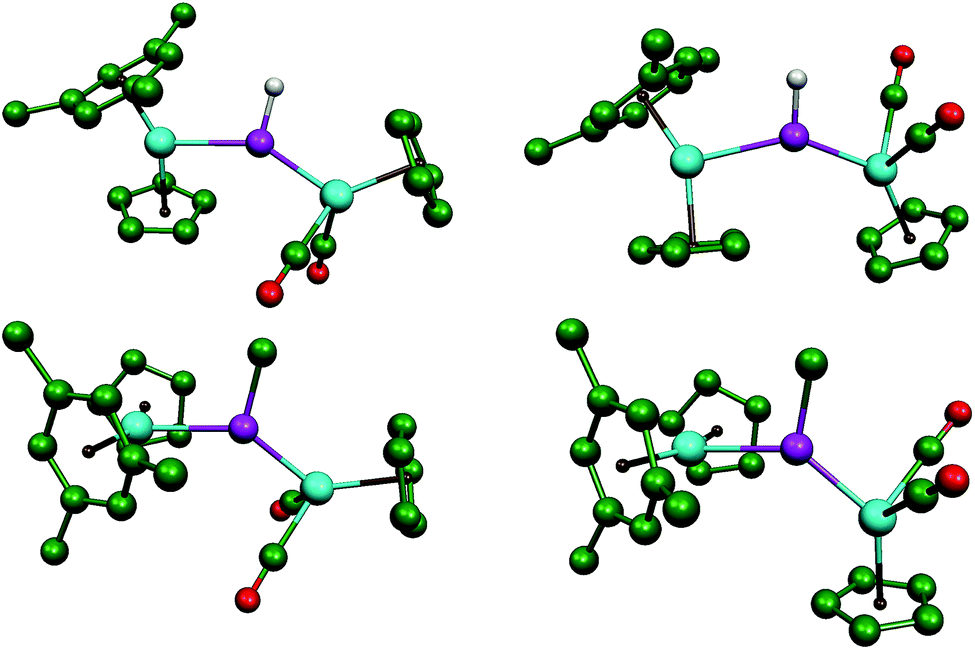
We have previously shown that the phosphinidene complex 2 is obtained through hydride addition on the phosphide-bridged cation [Mo2Cp2(μ-P)(CO)2(η6-HMes*)](BAr′4), by using Li[HBEt3] as the hydride transfer reagent (Ar′ = 3,5-C6H3(CF3)2).14,17 In an analogous way, we have now found that the above cationic complex reacts readily with MeLi in tetrahydrofuran solution at 193 K to give the related methylphosphinidene complex [Mo2Cp2(μ-PMe)(CO)2(η6-HMes*)] (3) in good yield. Although quite air-sensitive, we managed to grow single crystals of 3 in order to obtain details of its molecular structure (Fig. 1 and Table 1).
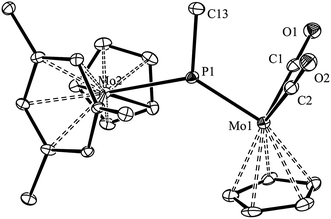 |
| | Fig. 1 ORTEP diagram (30% probability) of compound 3, with H atoms and tBu groups (except their C1 atoms) omitted for clarity. | |
Table 1 Selected bond lengths (Å) and angles (°) for compound 3
| Mo1–P1 |
2.264(1) |
Mo1–P1–Mo2 |
134.07(6) |
| Mo2–P1 |
2.538(1) |
Mo1–P1–C13 |
118.4(2) |
| Mo1–C1 |
1.936(5) |
Mo2–P1–C13 |
106.5(2) |
| Mo1–C2 |
1.944(5) |
C1–Mo1–P1 |
87.3(2) |
| P1–C13 |
1.857(5) |
C2–Mo1–P1 |
89.8(2) |
| |
|
C1–Mo1–C3 |
102.6(2) |
The structure of 3 in the crystal is built up from MoCp(η6-HMes*) and MoCp(CO)2 fragments connected via a trigonal planar PMe ligand (ΣXPY = 359.0°), which is asymmetrically bound to the Mo atoms according to their distinct electronic needs (1 and 3 electrons respectively). Thus, the P–Mo2 length of 2.538(1) Å is quite long, as expected for a single bond, while the P–Mo1 length of 2.264(1) Å is much shorter, and falls within the range of 2.20–2.30 Å displayed by terminal 3-electron donor phosphanyl ligands in Mo/W cyclopentadienyl complexes (cf. 2.284(4) Å in [WCp(PtBu2)(CO)2],18 but 2.204(1) Å in [MoCp(PFMes*)(CO)2]).19 This supports a description of the short interaction as a double bond, also in agreement with Density Functional Theory (DFT) calculations to be discussed below. For comparison, the corresponding distances in the related phosphinidene complex [W2Cp2(μ-PMes)(CO)4(PH2Mes)] were 2.550(3) and 2.281(3) Å, respectively.20 It should be noticed that the Mo2P plane in 3 bisects the Mo2Cp(CO)2 fragment, as found in the mentioned phosphanyl complexes, thus allowing for the required π-bonding Mo–P overlap,21 whereas the orientation of the Cp and arene rings of the metallocene fragment, on opposite sides of that plane, precludes any delocalization of such a bonding interaction over the Mo–P–Mo skeleton, as actually found in the PC5H4 complex 1.17 We finally note that the Me substituent at phosphorus in 3 occupies a transoid (or anti) positioning with respect to the Cp ring of the MCp(CO)2 fragment, relative to the MoP double bond, a matter to be addressed below.
Spectroscopic data in solution for 3 are similar to those of the PH-bridged analogue 2 (Table 2 and Experimental section). It expectedly displays an even more deshielded 31P NMR resonance (δP 569.0 ppm), which is a characteristic signature of trigonal phosphinidene-bridged complexes,5 and its IR spectrum displays two bands with relative intensities (very strong and strong, in order of decreasing frequencies) denoting the presence of M(CO)2 oscillators defining C–M–C angles below 90°,22 in agreement with its solid-state structure (C–Mo–C = 77.5°). An unexpected finding, however, was that the C–O stretching frequencies observed for 3 were some 12 cm−1 higher than those of the PH-bridged analogue 2. To uncover the origin of this anomaly we carried out DFT calculations (see the Experimental section for details) that revealed that two isomers of similar energy were possible for both 2 and 3, they mainly differing in the arrangement of the H or Me groups with respect to the Cp ring of the M(CO)2 fragment, relative to the double MoP bond (Fig. 2 and Table 3). This circumstance had passed unnoticed for us in an earlier DFT calculation of the structure of compound 2.17 We have now found that the transoid (or anti) arrangement was slightly favoured (by 5 kJ mol−1) for the PMe-bridged complex 3, and corresponds to the isomer actually found in the crystal, discussed above. In contrast, the cisoid (or syn) isomer was the one preferred for the PH-bridged complex 2. Moreover, the conformation of the metallocene fragments with respect to the Mo2P plane may not be identical in these two compounds. For the more encumbered PMe complex 3, the Cp and arene rings of the metallocene fragments occupy opposite sides relative to the Mo–P–Mo plane, presumably to minimize steric repulsions. For the less crowded PH-bridged complex 2, however, isomers with the above conformation (syn-2 and anti-2) and the corresponding rotamers in which that plane almost bisects the mentioned rings (denoted as syn-2R and anti-2R) are true energy minima, with relative Gibbs free energies being respectively 8, 16, 0 and 13 kJ mol−1 in the gas phase (see the ESI†). This rotational change, only possible when the P atom bears just the small hydrogen atom, allows for some delocalization of the π(Mo–P) bonding interaction over the Mo–P–Mo chain (see below), and seems to provide a modest increase in stability of some 10 kJ mol−1, but little geometrical changes. Moreover, interconversion between these rotamers and even between syn and anti isomers seems to be feasible in solution at room temperature (a maximum barrier of some 55 kJ mol−1 was estimated for compound 2, see the ESI†), so that such an isomerism likely has no chemical consequences. We finally note that the geometrical parameters computed for anti-3 are in good agreement with the X-ray data of 3 discussed above, if we allow for the usual overestimation of distances involving the metal atoms.23 Interestingly, in both compounds the C–O stretching frequencies computed for the syn isomers are some 20 cm−1 lower than the corresponding values for the anti isomers. On the other hand, replacement of H with Me at the P atom expectedly causes only a modest reduction of 3–8 cm−1 in the C–O stretching frequencies of the Mo(CO)2 fragment, while rotation of the metallocene fragment in 2 has little effect on the C–O stretches (see the ESI†). Our experimental IR data, therefore, allow us to unambiguously conclude that the anti isomer is the species actually present also in solution for the PMe-bridged complex 3, while a syn isomer would be the species present in solution for the PH-bridged complex 2. This is also in agreement with the relative energies computed for these isomers in the gas phase, which identify anti-3 and syn-2R as the most stable isomers in each case.
Table 2 Selected IR,a 31P{1H} and 1H NMR datab for new compounds
| Compound |
ν(CO) |
δ(P) [JPSn] |
δ(HP) [JHP] |
| Recorded in petroleum ether solution, with C–O stretching bands [ν(CO)] in cm−1. Recorded in C6D6 solution at 162.12 (31P) or 400.13 MHz (1H), and 298 K unless otherwise stated, with chemical shifts (δ) in ppm, and P–Sn [J(P–119Sn) ≈ J(P–117Sn) = JPSn] and H–P couplings (JHP) in Hz. Data taken from ref. 12a, recorded in dichloromethane. Data taken from ref. 17. In CD2Cl2 solution. In dichloromethane solution. |
| [Mo2Cp(μ-κ1:κ1,η5-PC5H4)(CO)2(η6-HMes*)] (1) |
1908 (vs), 1827 (s)c |
519.0c |
|
| [Mo2Cp2(μ-PH)(CO)2(η6-HMes*)] (2)d |
1908 (vs), 1833 (s) |
503.3 |
12.47 [183] |
| [Mo2Cp2(μ-PMe)(CO)2(η6-HMes*)] (3) |
1918 (vs), 1845 (s) |
569.0 |
|
| [Mo2Cp{μ-κ1:κ1,η5-P(H)C5H4}(SnBu3)(CO)2(η6-HMes*)] (4a) |
1899 (m), 1836 (vs) |
10.0 [102] |
4.12 [280]e |
| [Mo2Cp{μ-κ1:κ1,η5-P(H)C5H4}(SnPh3)(CO)2(η6-HMes*)] (4b) |
1905 (m), 1835 (vs)f |
0.9 [120]e |
4.08 [290]e |
| [Mo2Cp2(μ-PH2)(SnBu3)(CO)2(η6-HMes*)] (5a) |
1884 (m), 1818 (vs) |
−111.7 [100] |
3.63 [263] |
| [Mo2Cp2(μ-PH2)(SnPh3)(CO)2(η6-HMes*)] (5b) |
1901 (m), 1830 (vs) |
−113.8 [120] |
3.61 [266] |
| [MoCp(SnBu3)(CO)2(PH3)] (6a) |
1916 (m), 1857 (vs) |
−104.6 [103] |
3.71 [340] |
| [MoCp(SnBu3)(CO)2(PH3)] (6b) |
1931 (m), 1870 (vs) |
−105.9 [127] |
3.54 [347] |
| [Mo2Cp{μ-κ1:κ1,η5-P(Me)C5H4}(SnBu3)(CO)2(η6-HMes*)] (7a) |
1894 (m), 1828 (vs) |
49.6 [115] |
|
| [Mo2Cp{μ-κ1:κ1,η5-P(Me)C5H4}(SnPh3)(CO)2(η6-HMes*)] (7b) |
1906 (m), 1839 (vs) |
43.9 [145] |
|
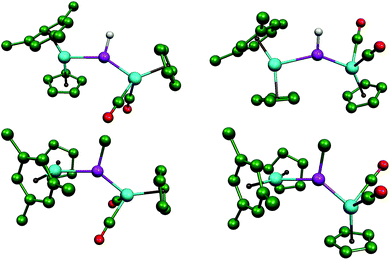 |
| | Fig. 2 DFT-optimized structures of the most stable isomers syn (left) and anti (right) of compounds 2 (above) and 3 (below), with most H atoms and tBu groups (except their C1 atoms) omitted for clarity. | |
Table 3 Selected data for DFT-optimized geometries of 2 and 3a
| Parameter |
syn-2R |
anti-2R |
syn-3 |
anti-3 |
| Bond lengths (Å) and angles (deg) according to the labelling shown in the figure; see the ESI† for other isomers of compound 2. |
| Mo1–P |
2.301 |
2.314 |
2.300 |
2.285 |
| Mo2–P |
2.590 |
2.600 |
2.603 |
2.609 |
| P–H/C |
1.426 |
1.419 |
1.886 |
1.875 |
| Mo1–P–Mo2 |
146.4 |
144.4 |
138.4 |
134.1 |
| Mo1–P–H/C |
108.6 |
111.7 |
116.2 |
119.6 |
| Mo2–P–H/C |
104.8 |
103.9 |
105.3 |
105.5 |
 |
Since the conformation displayed by the PH-bridged complex 2 in solution (syn) is different from the one displayed by 3 (anti), we then checked for possible differences in the corresponding Frontier orbitals that could be of significance to the reactivity of these molecules (see Fig. 3 and the ESI†). As expected, replacement of H with Me has no significant influence on the Frontier orbitals for a given isomer, and we also found that, for a given compound, syn and anti isomers have similar Frontier orbitals and geometrical parameters. The most significant difference is found in the π(Mo–P) bonding interaction, which is fully localized on the short Mo–P bond of isomers syn or anti (Mo–P ca. 2.30 Å), but partially delocalized over the longer Mo–P bond for isomers syn-2R or anti-2R due to a more favourable orientation of the metallocene fragment (cf. MO140 for anti-3 and MO136 for syn-2R in Fig. 3). This difference, however, has little influence on the relative strengths of the corresponding bonds, which display comparable lengths for the short (ca. 2.30 Å) and long (ca. 2.60 Å) Mo–P connections. In contrast, such a delocalization is more pronounced for the PC5H4-bridged complex 1, a molecule where the Mo2P plane exactly bisects the metal fragments, and displays less dissimilar Mo–P lengths of ca. 2.27 and 2.51 Å (exp. 2.252(2) and 2.403(1) Å).17 Perhaps this difference is at the origin of the lower reactivity of 1 (compared to 2 and 3) with HSnR3, discussed below. Irrespective of these differences, however, the LUMOs in all these molecules are similar to each other and correspond to a π*(Mo–P) orbital involving the P atom and the MoCp(CO)2 fragment.
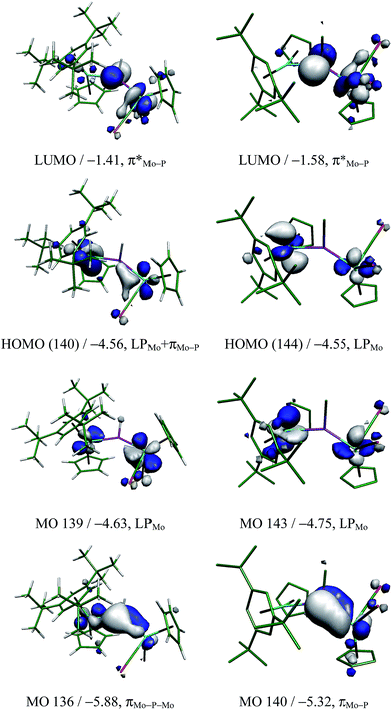 |
| | Fig. 3 Selected molecular orbitals of isomers syn-2R (left) and anti-3 (right, H atoms omitted), with their energies (in eV) and main bonding character indicated below. See the ESI† for the corresponding orbitals in other isomers of these compounds. | |
Reactions of the phosphinidene complexes 1–3 with organotin hydrides
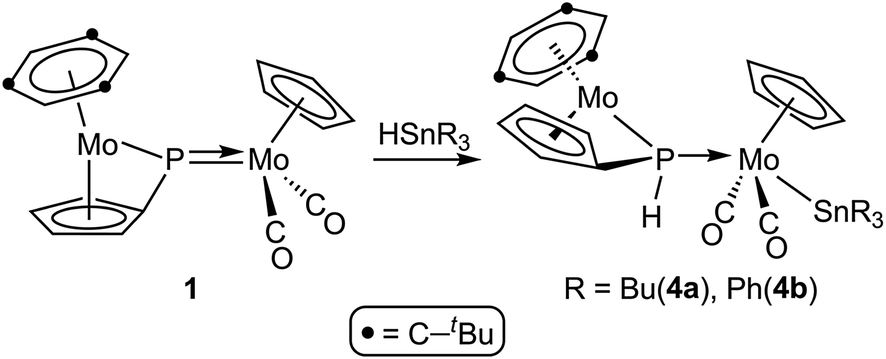
The results of these reactions were strongly dependent on the parent phosphinidene complex. The PC5H4 complex 1 reacted with HSnR3 only upon moderate heating (353–363 K), to give the corresponding phosphanyl-bridged derivatives [Mo2Cp{μ-κ1:κ1,η5-P(H)C5H4}(SnR3)(CO)2(η6-HMes*)] (R = Bu (4a), Ph (4b)), following from a formal 1,2-addition of the Sn–H bond across the Mo–P double bond of the parent complex, with specific formation of Sn–Mo and H–P bonds (Scheme 3).
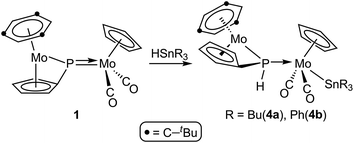 |
| | Scheme 3 | |
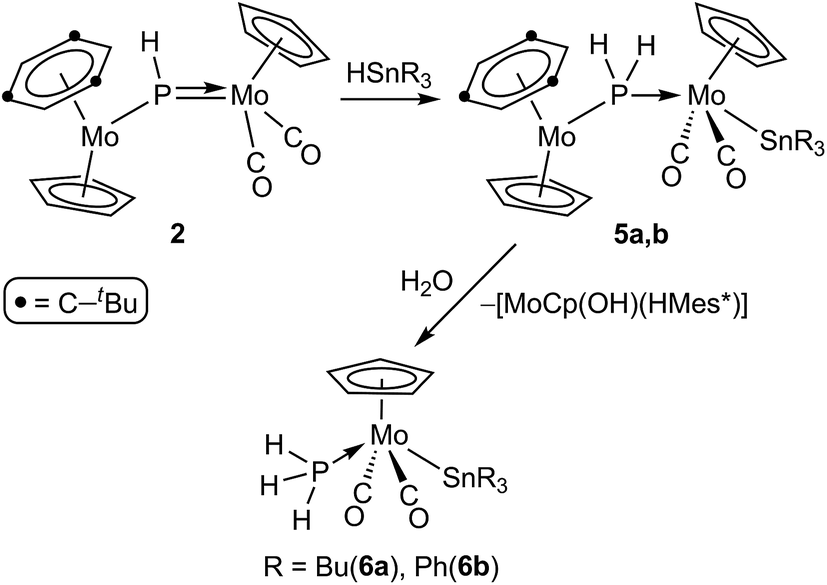
The phosphinidene complex 2 and the methylphosphinidene complex 3 were significantly more reactive than 1, and their reactions with the same organotin hydrides were completed at room temperature in ca. 1.5 h, although with different results. In the case of 2, the reactions ran parallel to those of 1, since they formally result in a regiospecific 1,2-addition of the Sn–H bond across the Mo–P double bond of the phosphinidene complex, to yield the corresponding phosphanyl-bridged derivatives [Mo2Cp2(μ-PH2)(SnR3)(CO)2(η6-HMes*)] (R = Bu (5a), Ph (5b)). These complexes, however, were quite susceptible to hydrolysis, and progressively degraded to the corresponding mononuclear phosphine complexes trans-[MoCp(SnR3)(CO)2(PH3)] (R = Bu (6a), Ph (6b)) in the presence of trace amounts of water (Scheme 4). The process could be reproduced on a synthetic scale by stirring tetrahydrofuran solutions of 5 with a ten-fold excess of water at room temperature, and also involved the formation of a less soluble solid, previously found by us in the hydrolysis of different heterometallic derivatives of complex 2, and thought to be the hydroxo complex [MoCp(OH)(η6-HMes*)].17 In all, the formation of complexes 6 completes an overall hydrogenation of the phosphinidene ligand under mild conditions, by using a H−(HSnR3)/H+(H2O) stepwise sequence.
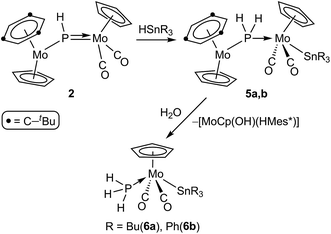 |
| | Scheme 4 | |
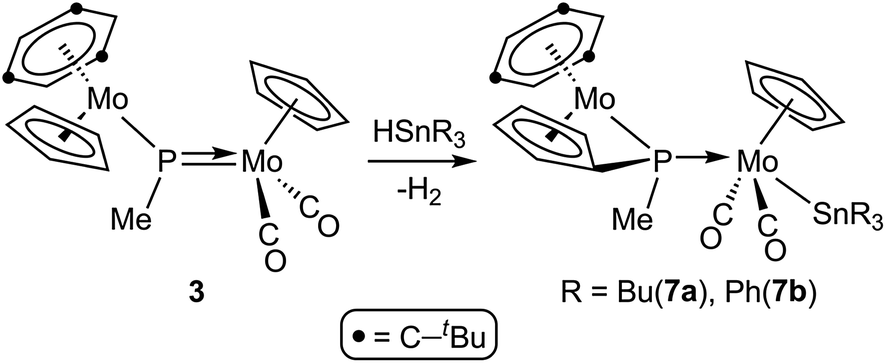
In contrast, reactions of the PMe-bridged complex 3 with HSnR3 proceeded with H–Sn bond addition accompanied by spontaneous dehydrogenation, to yield the corresponding cyclopentadienylidene–phosphanyl derivatives [Mo2Cp{μ-κ1:κ1,η5-P(Me)C5H4}(SnR3)(CO)2(η6-HMes*)] (R = Bu (7a), Ph (7b)), which are analogous to complexes 4 (Scheme 5). No intermediates were detected by IR or 31P NMR monitoring of these reactions, which obviously must be of a multistep nature.
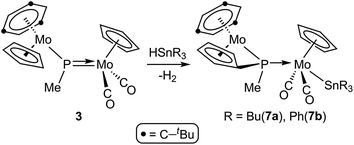 |
| | Scheme 5 | |
Structural characterization of compounds 4 and 7
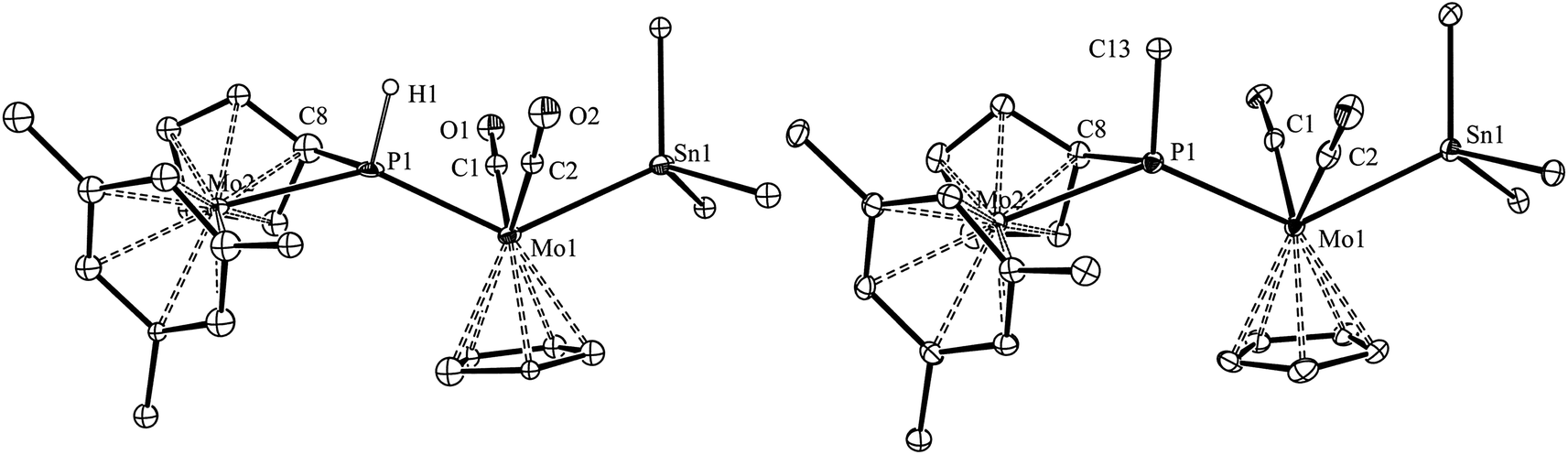
The molecular structure of the phosphanyl-bridged complex 4b and that of its methylphosphanyl analogue 7b were determined by single-crystal X-ray diffraction methods, which revealed that both complexes display very similar structures (Fig. 4 and Table 4). However, the low quality of the diffraction data for 4b and the presence of two independent, although similar, molecules in the corresponding crystal lattice, precluded the production of a precise structural model in this case; therefore, only the geometrical parameters for 7b will be discussed in detail here.
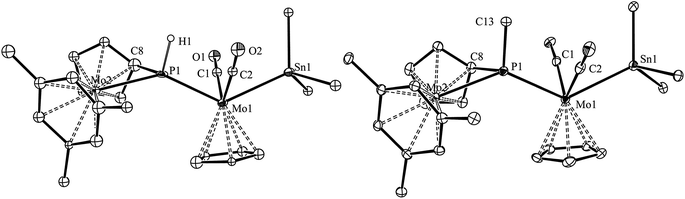 |
| | Fig. 4 ORTEP diagram (30% probability) of compounds 4b (left) and 7b (right), with Ph and tBu groups (except their C1 atoms), and most H atoms omitted for clarity. For compound 4b, only one of the independent molecules present in the unit cell is shown. | |
Table 4 Selected bond lengths (Å) and angles (°) for compounds 4b and 7b
| Parameter |
4b |
7b |
| Mo1–Sn1 |
2.745(2) |
2.7631(4) |
| Mo1–P1 |
2.480(5) |
2.487(1) |
| Mo2–P1 |
2.519(5) |
2.571(1) |
| Mo1–C1 |
1.91(2) |
1.956(4) |
| Mo1–C2 |
1.90(2) |
1.964(4) |
| P1–C8 |
1.79(2) |
1.785(4) |
| P1−Η1/C13 |
1.41(2) |
1.842(4) |
| P1–Mo1–Sn1 |
130.6(1) |
130.70(2) |
| P1–Mo1–C1 |
77.6(6) |
76.1(1) |
| P1–Mo1–C2 |
76.9(5) |
78.3(1) |
| C1–Mo1–C2 |
106.4(8) |
101.8(2) |
| Mo1–P1–Mo2 |
140.4(2) |
134.15(4) |
| Mo1–P1−Η1/C13 |
104(2) |
113.8(1) |
| Mo2–P1−Η1/C13 |
115(2) |
111.5(1) |
| C8–P1−Η1/C13 |
102(3) |
103.6(2) |
The molecule of 7b can be viewed as derived from that of the precursor 3 upon addition of a SnPh3 group at the MoCp(CO)2 fragment trans to the P atom, while one H atom of the Cp group in the metallocene fragment is removed, and the corresponding C atom is attached to phosphorus (P–C8 = 1.785(4) Å), thus yielding a bifunctional P(C5H4)Me phosphanyl ligand, analogous to the P(C5H4)H ligand present in compounds 4. The P atom of this bridging phosphanyl ligand formally contributes with 2 and 1 electrons respectively to the Mo1 and Mo2 atoms and, accordingly, the Mo–P separation to the metallocene fragment is the longest one (2.571(1) vs. 2.487(1) Å); in any case, both distances are much longer than the double-bond length of 2.264(1) Å measured in the parent compound 3. The local environment around the Mo1 atom is of the transoid four-legged piano stool type (P–Mo–Sn = 130.70(2)°, C1–Mo1–C2 = 101.8(2)°), and the only remarkable feature is the relatively short Mo1–Sn distance of 2.7631(4) Å, significantly shorter than the reference figure for a single bond between these atoms (2.93 Å),24 or the value of 2.8719(3) Å measured in the mononuclear complex [W(κ1,η5-C5H4SntBu2SntBu2)(CO)2(PMe3)],25 which also displays a trans arrangement of P and Sn atoms around a very similar metal centre. A second remarkable feature in the structure of 7b (also present in 4b) is the environment around the P atom, of a distorted trigonal pyramidal type, with the Mo, P and C(Me) atoms almost in the same plane (ΣXPY = 359.5°), and the cyclopentadienylidene C8 atom occupying the apex of the pyramid. This distortion, also found in different derivatives of compound 1, seems to optimize the different orbital overlaps within the geometrical restrictions imposed by the bifunctional PC5H4 group.26
Spectroscopic data in solution for compounds 4 and 7 (Table 2 and Experimental section) are consistent with the structures discussed above. Their IR spectra display in all cases two C–O stretches with relative intensities (medium and very strong, in order of decreasing frequencies) denoting the presence of transoid M(CO)2 oscillators defining C–M–C angles above 90°,22 in agreement with their solid-state structures (C–Mo–C = 102–106°). On the other hand, their 31P NMR resonances are dramatically shielded (by some 500 ppm) relative to their parent phosphinidene precursors, and fall in the range 0–50 ppm, which is a reasonable shift for phosphanyl ligands bridging Mo atoms not connected by intermetallic bonds.27 The PH(C5H4) complexes 4 expectedly display one-bond coupling to a single H atom, but the corresponding values of 280–290 Hz are lower than the usual figures found for P atoms in tetrahedral environments (cf. 340–350 Hz in the phosphine complexes 6). This suggests that the distorted pyramidal environment around the P atom found in the solid state for these compounds is also retained in solution, because this implies Mo–P–H angles around 110° at the “base” of the pyramid, therefore reduced s orbital character in the corresponding P–H bond and hence lower coupling. This is a spectroscopic effect particularly strong for compound 2 (1JPH = 183 Hz; Mo–P–H angles ca. 107°) and persistent in many of its derivatives.17
Structural characterization of compounds 5 and their phosphine derivatives 6. Spectroscopic data for the PH2-bridged complexes 5a,b are comparable to those of compounds 4 and 7 (Table 2 and Experimental section), suggesting a close structural relationship. They expectedly give rise to a much more shielded 31P NMR resonance (δP ca. −110 ppm) which displays one-bond coupling to two equivalent H atoms (δH ca. 3.6 ppm, 1JPH ca. 265 Hz); the P–H coupling again is somewhat low and indicative of a distorted environment around the P atom, as discussed for compounds 4. The fact that both P-bound H atoms appear as equivalent might be due to the adoption in solution of a conformation with the metallocene fragment and MoCp(CO)2 fragments being bisected by the Mo2P plane, but less symmetric conformations related by fast rotation around the Mo–P single bonds of compounds 5 would also account for the observed chemical equivalence (i.e. the gas phase DFT-optimized geometry for its SnMe3 analogue 5-mod, see the ESI†).Spectroscopic data for complexes 6 (Table 2 and Experimental section) were not very different from those of their parent compounds 5, except for the disappearance of the resonances characteristic of the metallocene rings and the replacement of the PH2 resonances by similar ones, but now corresponding to Mo-bound PH3 ligands (δP ca. −105 ppm, δH ca. 3.6 ppm, JPH ca. 350 Hz). Thus, there is no doubt that the hydrolysis of compounds 5 proceeds with retention of the transoid geometry at the MoCp(CO)2 fragment, as otherwise expected.
Mechanism of addition of Sn–H bonds across the Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) P bond of phosphinidene complexes 1–3. As discussed above, in spite of their geometric differences, the electronic structures of the asymmetric phosphinidene-bridged complexes 1 to 3 are rather similar to each other. In particular, the LUMO in all these substrates has π*(Mo–P) character, it actually being the antibonding partner of the π bonding interaction connecting the P atom with the MoCp(CO)2 fragment. This would render either the P or Mo atoms as potential sites for initial nucleophilic attack of the organotin reagent, perhaps via its negatively polarized hydride atom. Attempts to computationally model an approach at the P site of these complexes by using HSnMe3, however, failed to find a suitable intermediate or transition state of energy low enough to justify the experimental reactions with HSnBu3 and HSnPh3. This is likely due to steric factors, since the P site is a relatively crowded position in these molecules, as indicated by the fact that the simple replacement of H with Me at the P atom (i.e. from 2 to 3) imposes rotational restrictions to the corresponding metallocene fragments.
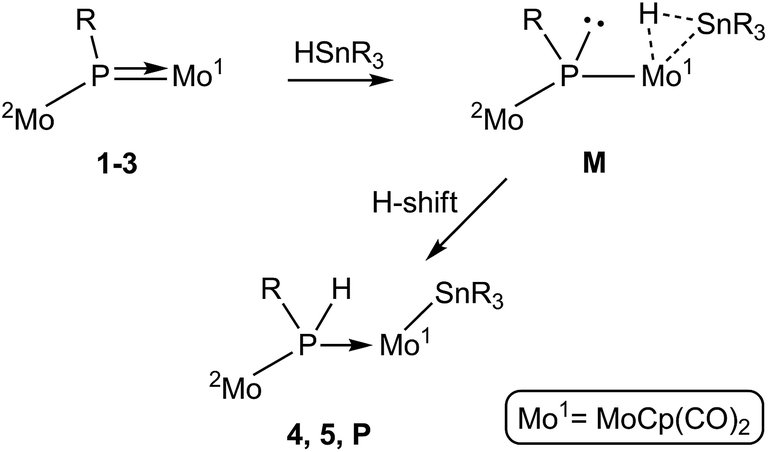
P bond of phosphinidene complexes 1–3. As discussed above, in spite of their geometric differences, the electronic structures of the asymmetric phosphinidene-bridged complexes 1 to 3 are rather similar to each other. In particular, the LUMO in all these substrates has π*(Mo–P) character, it actually being the antibonding partner of the π bonding interaction connecting the P atom with the MoCp(CO)2 fragment. This would render either the P or Mo atoms as potential sites for initial nucleophilic attack of the organotin reagent, perhaps via its negatively polarized hydride atom. Attempts to computationally model an approach at the P site of these complexes by using HSnMe3, however, failed to find a suitable intermediate or transition state of energy low enough to justify the experimental reactions with HSnBu3 and HSnPh3. This is likely due to steric factors, since the P site is a relatively crowded position in these molecules, as indicated by the fact that the simple replacement of H with Me at the P atom (i.e. from 2 to 3) imposes rotational restrictions to the corresponding metallocene fragments.We then examined an initial interaction of the tin hydride with the phosphinidene complexes at the Mo site. Indeed, inspection of the Frontier orbitals of these complexes reveals a high participation of the corresponding Mo atom not only in the LUMO but also in occupied Frontier orbitals (Fig. 3). This would enable an initial approach of the Sn–H bond to this site and its eventual cleavage. In fact, we have shown previously that compound 1 reacts at the MoCp(CO)2 fragment with different donors L to give derivatives [Mo2Cp(μ-κ1:κ1,η5-PC5H4)(CO)2(η6-HMes*)L] bearing a pyramidal phosphinidene ligand (L = PR3,26 CO,28 CNR, alkyne).29 In a related way, the tin reagent would be able to bind the MoCp(CO)2 fragment in complexes 1 to 3 via a three-centre Mo–H–Sn interaction to give intermediate species M bearing a pyramidal phosphinidene ligand, with the right geometry to facilitate the H transfer to the P atom (via the Mo atom) that yields complexes 4 and 5 (Scheme 6). In the case of 3, however, this process would not yield a stable compound, but an intermediate species P (not detected) to be further discussed below. A precedent for a related Sn–H bond cleavage being eventually followed by a P–H bond formation step is found in the reaction of the unsaturated phosphanyl-bridged complex [Mo2Cp2(μ-H)(μ-PCy2)(CO)2] with HSnPh3, which at low temperature yields the phosphine derivative [Mo2Cp2(μ-H)(SnPh3)(CO)2(PHCy2)].30
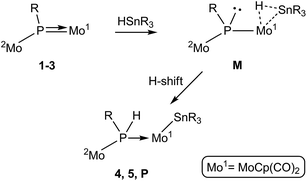 |
| | Scheme 6 | |
The observed addition of the H–Sn bond to the MoP double bond in the phosphinidene complexes 1–3, on the other hand, might be considered analogous to the one observed in the reaction of the terminal phosphanyl complex [Ru(PCy2)(η5-indenyl)(PPh3)] with the silane HSiEt3, which results in the selective formation of P–H and Ru–Si bonds.31 Interestingly, this phosphanyl complex reacted similarly with H2, and a subsequent DFT study revealed that the latter reaction proceeds via initial coordination of the H2 molecule at the metal atom to give a dihydrogen intermediate [Ru(PCy2)(η5-indenyl)(η2-H2)(PPh3)] bearing a pyramidal phosphanyl ligand, a species obviously related to the σ-bond intermediates M proposed by us. The H2 complex then evolves via H transfer to P, to give the phosphine-hydride derivative [RuH(η5-indenyl)(PHCy2)(PPh3)] eventually isolated in that case.32 Unfortunately, we have not been able to reproduce computationally the whole pathway resulting in eventual H transfer to P as proposed in Scheme 6, due to the large number of geometrical variables in our systems. Yet we have found that coordination of a HSnMe3 molecule at the MoCp(CO)2 fragment of compound 1 would yield a σ-bond intermediate of type M (M1-mod) with a Gibbs free energy 118 kJ mol−1 above the corresponding reactants in the gas phase (see the ESI†), a species therefore accessible in a reaction taking place in solution at 363 K.
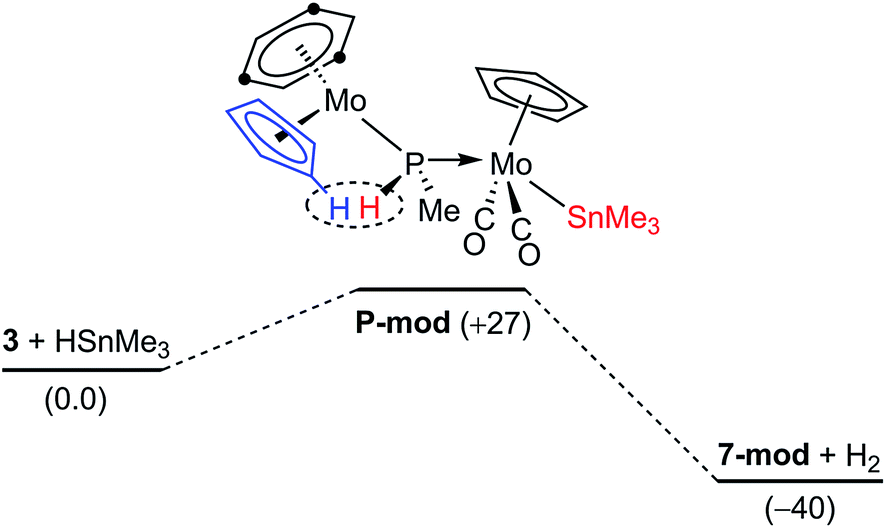
The dehydrogenation pathway in the reactions of the methylphosphinidene complex 3. As noted in the preceding sections, the reactions of 3 with HSnR3 take place with spontaneous dehydrogenation involving a cyclopentadienyl ligand to eventually yield compounds 7, which are analogous to the PH(C5H4)-bridged complexes 4. This process is likely to occur after the initial Sn–H bond cleavage has taken place (Fig. 5). We have computed that the phosphanyl intermediate of type P (P-mod) resulting from initial Sn–H bond addition of HSnMe3 to 3·would have a structure analogous to those of the phosphanyl complexes 5, but its energy would be higher than the corresponding reactants by some 27 kJ mol−1 (in contrast, the computed energy for the SnMe3 analogue of compounds 5 (5-mod) was 8 kJ mol−1 below the reagents). However, upon dehydrogenation to give the SnMe3 analogue of compounds 7 (7-mod) the Gibbs free energy balance falls to −40 kJ mol−1, in agreement with the experimental results (see the ESI†). An inspection of the structures of P-mod and 5-mod reveals more steric congestion in the former (PMe⋯tBu contacts), which forces the P–H atom to approach the Cp ring eventually being dehydrogenated (computed H⋯H separation 2.11 Å, to be compared with 2.71 Å in 5-mod). This might be at the origin of the unfavourable thermodynamics of P-mod relative to the corresponding reactants, while it would also facilitate its dehydrogenation, also resulting in the formation of the P–C bond that renders the PC5H4 group eventually present in compounds 7.
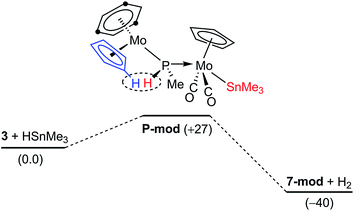 |
| | Fig. 5 DFT-computed thermodynamic profile (in kJ mol−1) for the reaction of 3 (anti) with HSnMe3. | |
Concluding remarks
The reactions of the asymmetric trigonal phosphinidene-bridged complexes 1 to 3 with organotin hydrides HSnR3 result in formal 1,2-addition of the Sn–H bond across the Mo–P double bond of the parent dimolybdenum complexes, with specific formation of Sn–Mo and H–P bonds. Apparently, this is in contrast with previous knowledge on the reactivity of this sort of complexes, which seem to behave as having polar Mo–P double bonds with nucleophilic P sites and electrophilic M sites, therefore leading to the prediction that Sn–P and H–Mo bonds should have been formed in reactions with HSnR3 reagents. However, our data suggest that there is no actual deviation from the general behaviour of these phosphinidene complexes, because their reactions with HSnR3 would be initiated in each case by the coordination of the organotin reagent to the Mo atom of the MoCp(CO)2 fragment (then effectively acting as the electrophilic site) via a three-centre Mo–H–Sn interaction. This would give intermediate species bearing a pyramidal phosphinidene ligand with the right geometry to facilitate the H transfer to the P atom that completes the formal addition of the Sn–H bond across the Mo–P double bond. Our data suggest that direct interaction of the organotin reagents with the P site of the parent phosphinidene complexes is likely prevented by the severe steric crowding of this site. Moreover, steric factors also seem to be responsible for the de-stabilization of the addition products of the PMe-bridged complex 3, and for the geometrical distortion that enables a dehydrogenation process involving the newly formed P–H bond with a C–H bond in a close cyclopentadienyl ligand at the addition product.
Experimental section
General procedures and starting materials
All manipulations and reactions were carried out under an argon (99.995%) atmosphere using standard Schlenk techniques. All experiments were carried out using Schlenk tubes equipped with Young's valves. Solvents were purified according to literature procedures and distilled prior to use.33 Petroleum ether refers to that fraction distilling in the range 338–343 K. Compounds [Mo2Cp(μ-κ1:κ1,η5-PC5H4)(CO)2(η6-HMes*)] (1) (Mes* = 2,4,6-C6H2tBu3),12a,26 [Mo2Cp2(μ-PH)(CO)2(η6-HMes*)] (2),17 and [Mo2Cp2(μ-P)(CO)2(η6-HMes*)](BAr′4), (Ar' = 3,5-C6H3(CF3)2),12a,34 were prepared as described previously, while all other reagents were obtained from the usual commercial suppliers and used as received, unless otherwise stated. Chromatographic separations were carried out using jacketed columns cooled by tap water (ca. 288 K) or by a closed 2-propanol circuit, kept at the desired temperature with a cryostat. Commercial aluminium oxide (activity I, 70-290 mesh) was degassed under vacuum prior to use. The latter was mixed under argon with the appropriate amount of water to reach activity IV. IR stretching frequencies were measured in solution and are referred to as ν(solvent) and given in wave numbers (cm−1). Nuclear magnetic resonance (NMR) spectra were routinely recorded at 400.13 (1H), 162.12 (31P{1H}), or 100.62 MHz (13C{1H}) at 290 K unless otherwise stated. Chemical shifts (δ) are given in ppm, relative to internal tetramethylsilane (1H, 13C), or external 85% aqueous H3PO4 (31P). Coupling constants (J) are given in Hz.
Preparation of [Mo2Cp2(μ-PMe)(CO)2(η6-HMes*)] (3). A solution of MeLi (54 μL, 1.5 M in Et2O, 0.080 mmol) was added to a tetrahydrofuran solution (10 mL) of [Mo2Cp2(μ-P)(CO)2(η6-HMes*)](BAr′4)(0.120 g, 0.080 mmol) at 195 K, and the mixture was stirred for 2 min to give a dark brown solution. The solvent was then removed under vacuum, the residue washed with petroleum ether (2 × 5 mL) and then extracted with 15 mL of a toluene/petroleum ether mixture (3/1). The extract was then filtered using a cannula, and the solvents removed from the filtrate to give compound 3 as a dark-brown microcrystalline solid (0.050 g, 92%). The crystals used in the X-ray diffraction study were grown through the slow diffusion of a layer of petroleum ether into a concentrated tetrahydrofuran solution of the complex at 253 K. Anal. calcd for C31H43Mo2O2P: C, 55.53; H, 6.46. Found: C, 55.48; H, 6.42. 1H NMR (C6D6): δ 5.48, 4.34 (2s, 2 × 5H, Cp), 4.19 (s, 3H, C6H3), 2.93 (d, JHP = 12, 3H, Me), 1.05 (s, 27H, tBu). 31P{1H} NMR (C6D6): δ 569.0 (s). 31P{1H} NMR (CD2Cl2): δ 596.4 (s). 13C{1H} NMR (C6D6): δ 244.2 (s, br, MoCO), 112.9 [s, C(C6H3)], 95.1, 85.6 (2s, Cp), 67.3 [s, CH(C6H3)], 47.7 (d, JCP = 25, PMe), 35.1 [s, C1(tBu)], 30.9 [s, C2(tBu)].
Preparation of [Mo2Cp{μ-κ1:κ1,η5-P(H)C5H4}(SnBu3)(CO)2(η6-HMes*)] (4a). Neat HSnBu3 (25 μL, 0.093 mmol) was added to a toluene solution (3 mL) of compound 1 (0.023 g, 0.035 mmol). The valve of the Schlenk tube was then closed and the mixture was stirred at 353 K for 20 min to give a brown-greenish solution. The solvent was then removed under vacuum and the residue extracted with dichloromethane/petroleum ether (1/9) and chromatographed on alumina at 288 K. A green band was eluted with the same solvent mixture which gave, after removal of solvents, compound 4a as a brown microcrystalline solid (0.025 g, 76%). Anal. calcd for C42H67Mo2O2PSn: C, 53.35; H, 7.14. Found: C, 53.10; H, 7.05. 1H NMR (CD2Cl2): δ 5.15, 5.05 (2m, 2 × 1H, C5H4), 5.00 (s, 5H, Cp), 4.88 (d, JHP = 4, 3H, C6H3), 4.64, 4.51 (2m, 2 × 1H, C5H4), 4.12 (dd, JHP = 280, JHH = 1, 1H, PH), 1.53, 1.33, 1.02 [3m, 3 × 6H, CH2], 1.20 (s, 27H, tBu), 0.90 (t, JHP = 7, 9H, CH3). 13C{1H} NMR (CD2Cl2): δ 233.7 [d, JCP = 22, MoCO], 233.6 [d, JCP = 22, MoCO], 108.5 [s, C(C6H3)], 96.3 [d, JCP = 14, C1(C5H4)], 89.6 [s, CH(C5H4)], 88.3 (s, Cp), 87.7 [d, JCP = 8, CH(C5H4)], 87.3 [s, CH(C5H4)], 84.2 [d, JCP = 14, CH(C5H4)], 73.0 [s, CH(C6H3)], 34.6 [s, C1(tBu)], 31.7 [s, C2(tBu)], 30.7 [s, J119SnC ≈ J117SnC = 19, C3(Bu)], 28.2 [s, J119SnC ≈ J117SnC = 56, C2(Bu)], 14.0 [s, C4(Bu)], 13.9 [s, J119SnC = 236, J117SnC = 225, C1(Bu)].
Preparation of [Mo2Cp{μ-κ1:κ1,η5-P(H)C5H4}(SnPh3)(CO)2(η6-HMes*)] (4b). Solid HSnPh3 (0.024, 0.068 mmol) was added to a toluene solution (3 mL) of compound 1 (0.023 g, 0.035 mmol). The valve of the Schlenk tube was then closed and the mixture was stirred at 363 K for 45 min to give a brown-greenish solution. The solvent was then removed under vacuum and the residue extracted with dichloromethane/petroleum ether (1/4) and chromatographed on alumina at 288 K. A green band was eluted with the same solvent mixture which gave, after removal of solvents, compound 4b as a brown microcrystalline solid (0.027 g, 77%). The crystals used in the X-ray diffraction study were grown through the slow diffusion of a layer of petroleum ether into a concentrated diethyl ether solution of the complex at 253 K. Anal. calcd for C48H55Mo2O2PSn: C, 57.36; H, 5.51. Found: C, 57.60; H, 5.65. 1H NMR (CD2Cl2): δ 7.56, 7.29 (2m, 15H, Ph), 5.19, 5.08 (2m, 2 × 1H, C5H4), 5.01 (d, JHP = 1, 5H, Cp), 4.90 (d, JHP = 4, 3H, C6H3), 4.62 (m, 2H, C5H4), 4.08 (dd, JHP = 290, JHH = 1, 1H, PH), 1.18 (s, 27H, tBu).
Preparation of [Mo2Cp2(μ-PH2)(SnBu3)(CO)2(η6-HMes*)] (5a). Neat HSnBu3 (25 μL, 0.093 mmol) was added to a tetrahydrofuran solution (4 mL) of compound 2 (0.026 g, 0.040 mmol), and the mixture was stirred at room temperature for 1 h to give a brown-yellowish solution. The solvent was then removed under vacuum and the residue extracted with petroleum ether and chromatographed on alumina at 253 K. A green band was eluted with the same solvent mixture which gave, after removal of the solvent, compound 5a as a brown microcrystalline solid (0.031 g, 82%). Anal. calcd for C42H69Mo2O2PSn: C, 53.24; H, 7.34. Found: C, 53.18; H, 7.19. 1H NMR (C6D6): δ 4.91 (s, 5H, Cp), 4.59 (d, JHP = 6, 3H, C6H3), 4.53 (d, JHP = 5, 5H, Cp), 3.63 (d, JHP = 263, J119SnH ≈ J117SnH = 9, 2H, μ-PH2), 1.99 (m, 6H, SnCH2), 1.67–1.49 (m, 12H, CH2), 1.10 (s, 27H, tBu), 1.08 (t, JHH = 7, 9H, Me).
Preparation of [Mo2Cp2(μ-PH2)(SnPh3)(CO)2(η6-HMes*)] (5b). The procedure is analogous to the one described for compound 5a, but using HSnPh3 instead (0.024 g, 0.068 mmol). After similar workup, compound 5b was isolated as a brown microcrystalline solid (0.034 g, 84%). Anal. calcd for C48H57Mo2O2PSn: C, 57.22; H, 5.70. Found: C, 57.11; H, 5.61. 1H NMR (C6D6): δ 8.13, 7.67 (2m, 15H, Ph), 4.86 (s, 5H, Cp), 4.53 (d, JHP = 6, 3H, C6H3), 4.39 (d, JHP = 5, 5H, Cp), 3.61 (d, JHP = 266, J119SnH ≈ 3J117SnH = 9, 2H, μ-PH2), 1.03 (s, 27H, tBu).
Preparation of [MoCp(SnBu3)(CO)2(PH3)] (6a). Degassed water (6 μL, 0.33 mmol) was added to a tetrahydrofuran solution (3 mL) of compound 5a (0.030 g, 0.032 mmol), and the mixture was stirred at room temperature for 8 h to give a brown solution. The solvent was then removed under vacuum and the residue extracted with petroleum ether and chromatographed on alumina at 253 K. A yellow band was eluted with the same solvent mixture which gave, after removal of the solvent, compound 5a as a yellow solid (0.012 g, 69%). The solid residue left after extraction yielded NMR resonances in C6D6 solution at 4.72 (s, 5H, Cp), 4.69 (s, 3H, C6H3) and 1.21 (s, 27H, tBu), and was tentatively identified as the hydroxo complex [MoCp(OH)(η6-HMes*)] (no OH resonance detected, see discussion). Data for 6a: anal. calcd for C19H35MoO2PSn: C, 42.17; H, 6.52. Found: C, 42.37; H, 6.70. 1H NMR (C6D6): δ 4.54 (d, JHP = 2, 5H, Cp), 3.71 (d, JHP = 340, J119SnH ≈ 3J117SnH = 6, 3H, PH3), 1.85, 1.56, 1.42 (3m, 3 × 6H, CH2), 1.04 (t, JHH = 7, 9H, Me).
Preparation of [MoCp(SnPh3)(CO)2(PH3)] (6b). The procedure is analogous to the one described for compound 6a, but using compound 5b instead (0.030 g, 0.030 mmol). After similar workup, compound 6b was isolated as a yellow solid (0.014 g, 78%). Anal. calcd for C25H23MoO2PSn: C, 49.96; H, 3.86. Found: C, 49.82; H, 3.79. 1H NMR (C6D6): δ 7.92, 7.60 (2m, 15H, Ph), 4.45 (d, JHP = 2, 5H, Cp), 3.54 (d, JHP = 347, J119SnH ≈ 3J117SnH = 8, 3H, PH3).
Preparation of [Mo2Cp{μ-κ1:κ1,η5-P(Me)C5H4}(SnBu3)(CO)2(η6-HMes*)] (7a). Neat HSnBu3 (25 μL, 0.093 mmol) was added to a tetrahydrofuran solution (4 mL) of compound 3 (0.027 g, 0.040 mmol), and the mixture was stirred at room temperature for 90 min to give a brown solution. The solvent was then removed under vacuum and the residue extracted with dichloromethane/petroleum ether (1/3) and chromatographed on alumina at 253 K. A green band was eluted with the same solvent mixture which gave, after removal of solvents, compound 7a as a brown microcrystalline solid (0.034 g, 88%). Anal. calcd for C43H69Mo2O2PSn: C, 53.82; H, 7.25. Found: C, 53.69; H, 7.14. 1H NMR (C6D6): δ 4.99 (d, JHP = 1, 5H, Cp), 4.77 (m, 1H, C5H4), 4.66 (m, 2H, C5H4), 4.56 (d, JHP = 4, 3H, C6H3), 4.42 (m, 1H, C5H4), 1.96 (m, 6H, SnCH2), 1.81 (d, JHP = 8, 3H, PMe), 1.67–1.50 (m, 12H, CH2), 1.10 (s, 27H, tBu), 1.08 (t, JHH = 7, 9H, Me).
Preparation of [Mo2Cp{μ-κ1:κ1,η5-P(Me)C5H4}(SnPh3)(CO)2(η6-HMes*)] (7b). The procedure is analogous to the one described for compound 7a, but using HSnPh3 instead (0.024 g, 0.068 mmol). After similar workup, compound 7b was isolated as a brown solid (0.031 g, 76%). The crystals used in the X-ray diffraction study were grown through the slow diffusion of a layer of petroleum ether into a concentrated diethyl ether solution of the complex at 253 K. Anal. calcd for C49H57Mo2O2PSn: C, 57.73; H, 5.64. Found: C, 57.90; H, 5.09. 1H NMR (C6D6): δ 8.08 [m, 6H, o-H(Ph)], 7.35–7.18 (m, 9H, Ph), 4.93 (s, 5H, Cp), 4.75 (m, 1H, C5H4), 4.61 (m, 2H, C5H4), 4.52 (d, JHP = 4, 3H, C6H3), 4.36 (m, 1H, C5H4), 1.77 (d, JHP = 8, 3H, PMe), 0.98 (s, 27H, tBu).
X-ray structure determination of compound 3
Data collection was performed at 100 K on a Nonius KappaCCD single crystal diffractometer, using graphite-monochromated Mo Kα radiation. Images were collected at a 29 mm fixed crystal-detector distance, using the oscillation method, with 2° oscillation and 20 s exposure time per image. Data collection strategy was calculated with the program Collect.35 Data reduction and cell refinement were performed with the programs HKL Denzo and Scalepack.36 A semi-empirical absorption correction was applied using the program SORTAV.37 Using the program suite WINGX,38 the structure was solved by Patterson interpretation and phase expansion, and refined with full-matrix least squares on F2 using SHELXL2016.39 All non-hydrogen atoms were refined anisotropically, and all hydrogen atoms were geometrically placed and refined using a riding model.
X-ray structure determination of compounds 4b and 7b
Data collection for these compounds was performed at 100 K on an Oxford Diffraction Xcalibur Nova single crystal diffractometer, using Cu Kα radiation. Images were collected at a 65 mm fixed crystal-detector distance, using the oscillation method, with 1° oscillation and variable exposure time per image. Data collection strategy was calculated with the program CrysAlis Pro CCD40 and data reduction and cell refinement was performed with the program CrysAlis Pro RED.40 An empirical absorption correction was applied using the SCALE3 ABSPACK algorithm as implemented in the latter program. Using the program suite WINGX,38 the structures were solved with direct methods using SIR92,41 and refined with full-matrix least squares on F2 using SHELXL2016,39 to give the residuals collected in Table 5. For compound 7b all non-hydrogen atoms were refined anisotropically, and all hydrogen atoms were geometrically placed and refined using a riding model. For compound 4b, two independent molecules of the complex were present in the unit cell. Due to the poor quality of the diffraction data, only the heavy atoms of both independent molecules were refined anisotropically, while all other non-hydrogen atoms were refined isotropically to prevent their temperature factors from becoming non-positive definite. Hydrogen atoms were geometrically placed and refined using a riding model, except for the P-bound H(1) and H(1B) atoms, which were located in the Fourier map and refined analogously. In that case, a restraint on the corresponding P–H bond length (1.40 ± 0.02 Å) was necessary to obtain a satisfactory refinement of their positions.
Table 5 Crystal data for new compounds
| |
3 |
4b |
7b |
| R = ∑||Fo| − |Fc||/∑|Fo|. wR = [∑w(|Fo|2 − |Fc|2)2/∑w|Fo|2]1/2. w = 1/[σ2(Fo2) + (aP)2 + bP] where P = (Fo2 + 2Fc2)/3. a = 0.1079, b = 9.3484. a = 0.1328, b = 0.0000. a = 0.0677, b = 0.0000. |
| Mol. formula |
C31H43Mo2O2P |
C48H55Mo2O2PSn |
C49H57Mo2O2PSn |
| Mol. wt |
670.50 |
1005.46 |
1019.48 |
| Cryst. syst. |
Monoclinic |
Triclinic |
Monoclinic |
| Space group |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
| Radiation (λ, Å) |
0.71073 |
1.54184 |
1.54184 |
| a, Å |
12.3155(3) |
10.8296(8) |
13.2651(1) |
| b, Å |
16.7360(4) |
18.5024(12) |
22.5628(2) |
| c, Å |
15.9369(6) |
24.3014(16) |
14.5925(1) |
| α, deg |
90 |
78.476(6) |
90 |
| β, deg |
116.248(1) |
80.063(6) |
94.564(1) |
| γ, deg |
90 |
76.777(6) |
90 |
| V, Å3 |
2946.09(15) |
4603.4(6) |
4353.65(6) |
| Z |
4 |
4 |
4 |
| Calcd density, g cm−3 |
1.512 |
1.451 |
1.555 |
| Absorp coeff, mm−1 |
0.931 |
9.266 |
9.806 |
| Temperature, K |
100(2) |
100(2) |
100(2) |
| θ range (deg) |
1.84–25.24 |
2.84–69.99 |
3.03–73.71 |
| Index ranges (h, k, l) |
−17, 13; −20, 0; −19, 13 |
−12, 13; −22, 22; −29, 27 |
−16, 14; −27, 21; −11, 17 |
| No. of reflns collected |
22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 174 174 |
41823 |
22458 |
| No. of indep reflns (Rint) |
5292 (0.0365) |
16732 (0.1646) |
8547 (0.0317) |
| No. of reflns with I > 2σ(I) |
3952 |
6112 |
6907 |
| R Indexes [data with I > 2σ(I)]a |
R1 = 0.0450 |
R1 = 0.0958 |
R1 = 0.0282 |
| wR2 = 0.1186b |
wR2 = 0.2538c |
wR2 = 0.0717d |
| R Indexes (all data)a |
R1 = 0.0656 |
R1 = 0.2011 |
R1 = 0.0394 |
| wR2 = 0.1571b |
wR2 = 0.3114c |
wR2 = 0.0913d |
| GOF |
0.971 |
0.955 |
0.915 |
| No. of restraints/parameters |
0/335 |
4/473 |
0/506 |
| Δρ (max., min.), eÅ−3 |
2.130/−1.754 |
6.008/−1.888 |
1.181/−0.979 |
| CCDC deposition no. |
1536451 |
1536452 |
1536453 |
Computational details
All DFT computations were carried out using the GAUSSIAN03 package,42 in which the hybrid method B3LYP was used with the Becke three-parameter exchange functional43 and the Lee–Yang–Parr correlation functional.44 A pruned numerical integration grid (99, 590) was used for all the calculations via the keyword Int = Ultrafine. Effective core potentials and their associated double-ζ LANL2DZ basis set were used for the metal atoms (Mo and Sn).45 The light elements (P, O, C, N and H) were described with the 6-31G* basis.46 Geometry optimizations were performed under no symmetry restrictions, and frequency analyses were performed for all the stationary points to ensure that minimum structures with no imaginary frequencies were achieved. Molecular orbitals and vibrational modes were visualized using the Molekel program.47
Acknowledgements
We thank the Gobierno del Principado de Asturias for a grant (to J. S.) and financial support (Project GRUPIN14-011), the MINECO of Spain and FEDER for financial support (Project CTQ2015-63726-P), and the CMC of the Universidad de Oviedo for access to computing facilities.
Notes and references
- Recent reviews:
(a) F. Mathey and Z. Duan, Dalton Trans., 2016, 45, 1804 RSC;
(b) H. Aktas, J. C. Slootweg and K. Lammertsma, Angew. Chem., Int. Ed., 2010, 49, 2102 CrossRef CAS PubMed;
(c) R. Waterman, Dalton Trans., 2009, 18 Search PubMed;
(d) F. Mathey, Dalton Trans., 2007, 1861 RSC;
(e) K. Lammertsma, Top. Curr. Chem., 2003, 229, 95 CrossRef CAS;
(f) K. Lammertsma and M. J. M. Vlaar, Eur. J. Org. Chem., 2002, 1127 CrossRef CAS;
(g) R. Streubel, Coord. Chem. Rev., 2002, 227, 175 CrossRef CAS;
(h) F. Mathey, N. H. T. Huy and A. Marinetti, Helv. Chim. Acta, 2001, 84, 2938 CrossRef CAS;
(i) D. W. Stephan, Angew. Chem., Int. Ed., 2000, 39, 314 CrossRef;
(j) S. Shah and J. D. Protasiewicz, Coord. Chem. Rev., 2000, 210, 181 CrossRef CAS.
- K. B. Dillon, F. Mathey and J. F. Nixon, Phosphorus: The Carbon-Copy, Wiley, Chichester, 1998, p. 19 Search PubMed.
-
(a) P. P. Power, Acc. Chem. Res., 2011, 44, 627 CrossRef CAS PubMed;
(b) P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed.
- T. L. Breen and D. W. Stephan, Organometallics, 1996, 15, 4223 CrossRef CAS.
- M. E. García, D. García-Vivó, A. Ramos and M. A. Ruiz, Coord. Chem. Rev., 2017, 330, 1 CrossRef , and references therein..
-
(a) M. A. Alvarez, M. E. García, R. González, A. Ramos and M. A. Ruiz, Organometallics, 2010, 29, 1875 CrossRef CAS;
(b) M. A. Alvarez, M. E. García, D. García-Vivó, A. Ramos and M. A. Ruiz, Inorg. Chem., 2012, 51, 3698 CrossRef CAS PubMed.
- M. A. Alvarez, M. E. García, R. González, A. Ramos and M. A. Ruiz, Organometallics, 2011, 30, 1102 CrossRef CAS.
- G. Huttner and K. Evertz, Acc. Chem. Res., 1986, 19, 406 CrossRef CAS.
- M. A. Alvarez, I. Amor, M. E. García, D. García-Vivó, A. Ramos, M. A. Ruiz, D. Sáez, H. Hamidov and J. C. Jeffery, Inorg. Chim. Acta, 2015, 424, 103 CrossRef CAS.
-
(a) M. Stubenhofer, C. Kuntz, G. Balazs, M. Zabel and M. Scheer, Chem. Commun., 2009, 1745 RSC;
(b) M. Scheer, C. Kuntz, M. Stubenhofer, M. Zabel and A. Y. Timoshkin, Angew. Chem., Int. Ed., 2010, 49, 188 CrossRef CAS PubMed;
(c) M. Stubenhofer, G. Lassandro, G. Balazs, A. Y. Timoshkin and M. Scheer, Chem. Commun., 2012, 48, 7262 RSC;
(d) M. Stubenhofer, C. Kuntz, M. Bodensteiner, A. Y. Timoshkin and M. Scheer, Organometallics, 2013, 32, 3521 CrossRef CAS.
- W. Wang, Y. Lv, X. Gou, X. Leng and Y. Chen, Chin. J. Chem., 2014, 32, 752 CrossRef CAS.
-
(a) I. Amor, M. E. García, M. A. Ruiz, D. Sáez, H. Hamidov and J. C. Jeffery, Organometallics, 2006, 25, 4857 CrossRef CAS;
(b) M. E. García, V. Riera, M. A. Ruiz, D. Sáez, J. Vaissermann and J. C. Jeffery, J. Am. Chem. Soc., 2002, 124, 14304 CrossRef.
- M. A. Alvarez, I. Amor, M. E. García, D. García-Vivó and M. A. Ruiz, Inorg. Chem., 2007, 46, 6230 CrossRef CAS PubMed.
- I. Amor, D. García-Vivó, M. E. García, M. A. Ruiz, D. Sáez, H. Hamidov and J. C. Jeffery, Organometallics, 2007, 26, 466 CrossRef CAS.
- B. Alvarez, M. A. Alvarez, I. Amor, M. E. García, M. A. Ruiz and J. Suárez, Inorg. Chem., 2014, 53, 10325 CrossRef CAS PubMed.
- K. Vaheesar, T. M. Bolton, A. L. L. East and B. T. Sterenberg, Organometallics, 2010, 29, 484 CrossRef CAS.
- M. A. Alvarez, I. Amor, M. E. García, D. García-Vivó, M. A. Ruiz and J. Suárez, Organometallics, 2010, 29, 4384 CrossRef CAS.
- K. Jörg, W. Malisch, W. Reich, A. Meyer and U. Schubert, Angew. Chem., Int. Ed., 1986, 25, 92 CrossRef.
- M. Alonso, M. E. García, M. A. Ruiz, H. Hamidov and J. C. Jeffery, J. Am. Chem. Soc., 2004, 126, 13610 CrossRef CAS PubMed.
- W. Malisch, U. A. Hirth, T. A. Bright, H. Käb, T. S. Ertel, S. Hückmann and H. Bertagnolli, Angew. Chem., Int. Ed. Engl., 1992, 31, 1525 CrossRef.
- L. Rosenberg, Coord. Chem. Rev., 2012, 256, 606 CrossRef CAS.
- P. S. Braterman, Metal Carbonyl Spectra, Academic Press, London, U. K., 1975 Search PubMed.
-
(a) W. Koch and M. C. Holthausen, A Chemist's Guide to Density Functional Theory, Wiley-VCH, Weinheim, Germany, 2nd edn, 2002 Search PubMed;
(b) C. J. Cramer, Essentials of Computational Chemistry, Wiley, Chichester, U. K., 2nd edn, 2004 Search PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echevarría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- H. Braunschweig, R. Dörfler, K. Gruss, J. Köhler and K. Radacki, Organometallics, 2011, 30, 305 CrossRef CAS.
- I. G. Albuerne, M. A. Alvarez, M. E. García, D. García-Vivó and M. A. Ruiz, Inorg. Chem., 2015, 54, 9810 CrossRef CAS PubMed.
- A. J. Carty, S. A. MacLaughlin and D. Nucciarone, in Phosphorus-31 NMR Spectroscopy in Stereochemical Analysis, ed. J. G. Verkade, L. D. Quin, VCH, Deerfield Beach, FL, 1987, ch. 16 Search PubMed.
- M. A. Alvarez, M. E. García, D. García-Vivó, A. Ramos, M. A. Ruiz and J. Suárez, Inorg. Chem., 2012, 51, 34 CrossRef CAS PubMed.
-
(a) M. A. Alvarez, M. E. García, M. A. Ruiz and J. Suárez, Angew. Chem., Int. Ed., 2011, 50, 6383 CrossRef CAS PubMed;
(b) M. A. Alvarez, I. Amor, M. E. García, D. García-Vivó, M. A. Ruiz and J. Suárez, Organometallics, 2012, 31, 2749 CrossRef CAS.
- M. A. Alvarez, M. E. García, A. Ramos and M. A. Ruiz, Organometallics, 2006, 25, 5374 CrossRef CAS.
- E. J. Derrah, D. A. Pantazis, R. McDonald and L. Rosenberg, Organometallics, 2007, 26, 1473 CrossRef CAS.
- M.-A. M. Hoyle, D. A. Pantazis, H. M. Burton, R. McDonald and L. Rosenberg, Organometallics, 2011, 30, 6458 CrossRef CAS.
- W. L. F. Armarego and C. Chai, Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, U. K, 7th edn, 2012 Search PubMed.
- M. E. García, V. Riera, M. A. Ruiz, D. Sáez, H. Hamidov, J. C. Jeffery and T. Riis-Johannessen, J. Am. Chem. Soc., 2003, 125, 13044 CrossRef PubMed.
- B. V. Nonius, Collect, Delft, The Netherlands, 1997–2004 Search PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307 CAS.
- R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33 CrossRef.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
-
(a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 5 Search PubMed.
- CrysAlis Pro, Oxford Diffraction Limited, Ltd., Oxford, U. K., 2006 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Gualardi, J. Appl. Cryst., 1993, 26, 343 CrossRef.
- M. J. Frisch, et al., Gaussian 03, Revision B.02, Gaussian, Inc., Wallingford, CT, 2004, see the ESI† for the complete reference.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
-
(a) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS;
(b) G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081 CrossRef CAS;
(c) G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193 CrossRef CAS.
- S. Portmann and H. P. Luthi, Chimia, 2000, 54, 766 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: A CIF file containing full crystallographic data for compounds 3, 4b, and 7b (CCDC 1536451–1536453), a PDF file containing results of DFT calculations (drawings, molecular orbitals and energies, IR data) and the complete ref. 42, and an XYZ file including the Cartesian coordinates for all computed species. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra06439j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Inmaculada Amor,
M. Esther García
,
Inmaculada Amor,
M. Esther García