
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold nanoparticle bioconjugates labelled with 211At for targeted alpha therapy
L. Dziawera,
P. Koźmińskia,
S. Męczyńska-Wielgosza,
M. Pruszyńskia,
M. Łyczkoa,
B. Wąsb,
G. Celichowskic,
J. Grobelnyc,
J. Jastrzębskid and
A. Bilewicz *a
*a
aInstitute of Nuclear Chemistry and Technology, Dorodna 16, 03-195 Warsaw, Poland. E-mail: a.bilewicz@ichtj.waw.pl
bInstitute of Nuclear Physics, Polish Academy of Sciences, Radzikowskiego 152, 31-342 Cracow, Poland
cFaculty of Chemistry, University of Lodz, Pomorska 163, 90-236 Lodz, Poland
dHeavy Ion Laboratory, University of Warsaw, Pasteura 5A, 02-093 Warszawa, Poland
First published on 22nd August 2017
Abstract
Alpha particle emitting isotopes are of considerable interest for radionuclide therapy because of their high cytotoxicity and short path length. Due to the relatively high availability, 211At is actually the most prospective α emitter for targeted radiotherapy. The factor limiting the use of 211At in targeted therapy is the low in vivo stability of the obtained astatinated bioconjugates. The goal of this study was to elaborate a new approach that can be applied for labelling biomolecules with 211At. The new method consists in the use of gold nanoparticles as a carrier for 211At. It's well known that iodide (lighter homolog of astatine) is strongly adsorbed on noble metal surfaces such as Au, Pd and Pt forming strong surface covalent bonds. In recent study we verified our hypothesis that due to the similarity between iodine and astatine, adsorption of 211At proceeds according to the same reaction. Gold nanoparticles (AuNPs) with 5 and 15 nm diameter were modified with Substance P(5-11), a peptide fragment which targets the NK1 receptors on the glioma cells, through the HS–PEG–NHS linker. Bioconjugates were synthesized with high yield in a two-step procedure, and the products were characterized by transmission electron microscopy (TEM), dynamic light scattering (DLS) and chromatography (HPLC). It was determined that 47 and 3300 molecules of Substance P(5-11) were attached to one AuNP of 5 nm and 15 nm size, respectively. The obtained AuNP–S–PEG–SP(5-11) conjugates were labelled with 211At by chemisorption on the gold surface. The labelled bioconjugates almost quantitatively retain 211At in human serum and cerebrospinal fluid at 37 °C for 24 h. The synthesized 211At–AuNP–S–PEG–SP(5-11) radiobioconjugate exhibited a high cytotoxic effect in vitro on glioma cells.
Introduction
Alpha-particle-emitting radionuclides have several physical characteristics that make them attractive for radionuclide therapy. The emitted α particles are much heavier than other subatomic particles emitted from decaying radionuclides and thus they have a much shorter range in tissues. The high linear energy transfer of α particles induces significantly more DNA double strand breaks than do β-particles.1 Furthermore, the relative biological effectiveness of α particles does not depend on hypoxia or cell cycle considerations.2 There are only a few α particle emitting radionuclides that have properties suitable for developing therapeutic radiopharmaceuticals: generator-obtained 212Bi (t1/2 = 60 min), 213Bi (t1/2 = 46 min.), 226Th (t1/2 = 30 min), 225Ac (t1/2 = 10 d), 227Th (t1/2 = 18.7 d), 223Ra (t1/2 = 11. d), and cyclotron-produced 211At (t1/2 = 7.2 h). However, these radionuclides have serious shortcomings: in the case of 212Bi, 213Bi and 226Th short half-lives often limit the application of these radionuclides to situations when the tumour cells are rapidly accessible to the targeting agent. Crucial for radiopharmaceuticals stability are bifunctional ligands which can be attached to the targeting biomolecules and can bind a radioactive metal cation rapidly and selectively. The targeted α-radionuclide therapy (TAT), in particular, requires a very stable attachment of the radionuclide to the carrier molecule, because unbound α-emitters may target healthy tissues, thus leading also to the irradiation of healthy organs. In the case of 225Ac, 223Ra and 227Th the designed ligand must form chemically stable complexes both with the parent and also with the decay radionuclides. 225Ac decays directly to 221Fr (alkali metal), which has a half-life of 4.9 minutes, and escapes from 225Ac-radiobioconjugates. Similar situation appears in the case of 227Th and 223Ra where the decay product, the gaseous 219Rn is easily liberated from 227Th and 223Ra radiobioconjugates. Till now only 223Ra in the simple form of RaCl2, with its natural affinity to build into bones, as does calcium, finds application in the treatment of bone metastases from breast and prostate cancers.3 Additionally, current supplies of medically useful α-emitters remain limited by isolated by-products from nuclear weapons processing and power plants development within the USA and Soviet Union, where the parental stock is preserved for disposal. Actually, only three sources are available for obtaining “clinical grade” 225Ac (213Bi): Oak Ridge National Laboratory (USA), Institute of Physics and Power Engineering (Obninsk, Russia), and Institute for Transuranium Elements (Karlsruhe, Germany). The actual level of 225Ac production (∼1.7 Ci per year) is sufficient only for preclinical studies and for limited number of clinical trials.4 Therefore, a cyclotron-produced 211At seems to be the most promising candidate for TAT, because its 7.2 h half-life assures sufficient time for its transportation, synthetic chemistry, multistep labelling, quality control, and clinical application without problems caused by the relatively long-living daughters emitting α-particle. Another merit of this radionuclide is simplicity of its production in cyclotrons in the 209Bi(α, 2n) 211At nuclear reaction, followed by simple dry distillation and isolation from irradiated bismuth target.Attaching 211At to biomolecules targeting cancer cells is crucial for its application in the radionuclide therapy. Although astatine is generally treated as a halogen, it also shows a significant metallic character in certain conditions.5 The strength of aryl carbon–halogen bond for astatine is significantly lower than that for iodine6 which precludes the use of standard direct radio-iodination methods for labelling monoclonal antibodies (mAbs) with 211At. Such methods lead to unstable products resulting in rapid in vivo loss of 211At. More stable astatinated proteins have been prepared by acylation of astatobenzoic acid derivatives prepared from trialkylstannyl precursors.7,8 Unfortunately, biomolecules labelled by this method have been found to be not always stable with respect to in vivo deastatination.9 Because of these difficulties, nontraditional methods of labelling based on carboranes,10 Rh[16aneS4-diol] precursor,11,12 calixarenes13 and hypervalent compounds14 with 211At have been reported. One from the recently developed approaches is based on application of small nanoparticles as vehicles for 211At. Liposomes, spherical vesicles of lipid bilayers which can range from 100 to 800 nm in diameter, are the most widely used nanostructures for cancer therapy.15 211At-loaded liposomes were capable to deliver high doses of radiation to xenografted tumours in mice.16 Hartman et al.17 have developed single walled carbon nanotubes labelled with 211AtCl, which was bonded by noncovalent van der Waals interactions within the interior side walls of the nanotubes. In another approach Kućka et al.18 and Cędrowska et al.19 used remarkable affinity of astatine towards metallic silver and proposed application of astatinated silver-containing particles coated with poly(ethylene oxide) for cancer therapy. Bochvarova et al.20 and Wunderlich et al.21 have found that elemental tellurium exhibits high affinity for astatine. In acidic and neutral solutions adsorption of 211At on tellurium was complete. The 211At adsorbed on Te grains was used to study therapeutic effect of α particles by injection of 211At–Te grains directly into a tumour in the mouse model.22
It is well known that iodine has high affinity for the noble metal surface. The degree of specific adsorption of halides on the gold surface increases in the order F− < Cl− < Br− < I−, indicating the decreasing energy of a solvation of these species. In particular, F− has the lowest affinity and only nonspecifically or weakly specifically adsorbs on the metal surface. The Cl−, Br−, and I− are able to chemisorb on the gold surface to form an Au–X bond with increasingly covalent character.23 This phenomenon was applied to elaboration of new method for iodination of biomolecules using the gold nanoparticle as a metal bridge between 125I and a biomolecule.24 Taking into account the increasing order F− < Cl− < Br− < I− in formation of strong bonds with Au surface we can assume that astatide, as the heaviest halogen anion, should bind stronger to the Au surface.
In this study we propose a new type of 211At radiopharmaceuticals, based on gold nanoparticles modified by attached peptides. As a model peptide we have selected Substance P(5-11), which is a fragment of regulatory neuropeptide that belongs to the tachykinin family and shows affinity towards neurokinin type 1 receptors (NK1). This receptor has been shown to be consistently overexpressed in all primary malignant gliomas of astrocytic, oligodendrocytic, and mixed histotype of all WHO gradings. NK-1 receptors have also been detected on tumour cells infiltrating the intratumoral and peritumoral vasculature. Besides physiological expression on distinct interneuron populations NK-1 receptors have been detected only at restricted sites within the central nervous system.25
Malignant gliomas are among the most radioresistant tumours; even 90 Gy externally applied radiation dose is not sufficient to sterilize glioblastoma multiforme in vivo.26 Recently, successful pilot studies on the therapy of gliomas critically located in brain by using Substance P (SP) radiolabelled with the α emitter 213Bi were performed.27,28 Treatment of GBM is done through intratumoral or intracavitary injection of radiopharmaceutical, which than can readily penetrate brain parenchyma and target widely disseminated GBM cells. Replacement of the short-lived 213Bi (t1/2 = 46 min.) by the longer-lived 211At gives the possibility of deeper and more effective irradiation of the tumour and of distant metastases. Fragment SP(5-11) is a metabolite of native SP and is still biologically active because the binding site with NK1 receptors is localized in the region containing 7-11 amino acids.29 To obtain astatinated radiobioconjugate we decided to choose Substance P(5-11) fragment because it contains L-glutamine in position 5 what allows coupling with gold nanoparticles through the NHS–PEG-thiol linker and next labelling with 211At. Herein, we report results obtained for the synthesis of the 211At–AuNP–S–PEG–SP(5-11) radiobioconjugate, its stability studies in human serum (HS) and cerebrospinal fluid (CSF) and toxicity to human glioma cells T98G.
Results and discussion
Although a large number of peptides, monoclonal antibodies and other molecules suitable for targeted radiotherapy have been developed, applying them in a cancer therapy is possible only when the radionuclide forms a stable bond with a biomolecule. This is particularly important in the case of α emitters, because highly toxic radionuclide released from radiobioconjugate may accumulate in critical organs. Due to the relatively high availability, 211At is actually the most perspective α emitter for targeted therapy. However, many astatine compounds that have been synthesized are unstable in vivo, providing motivation for seeking other 211At labelling strategies. Therefore, without a good chemical method of attaching 211At to potential cancer targeting agents which will provide high in vivo stability of the formed conjugate, the application of this radionuclide in targeted radiotherapy will be limited. Herein, we have proposed an alternative method of labelling molecules with 211At in order to solve stability problems of formed bioconjugates. The new method consists in the use of gold nanoparticles as a carrier for 211At. Before labelling with 211At, the AuNPs are modified with biomolecules in a simple way by using thiol-PEG linker.It is known that iodide is strongly adsorbed on noble metal surfaces such as Au, Pd and Pt forming strong surface covalent bond according to the reaction:30
| Iaq− + H2O + Au → AuI + 1/2H2 + OH− |
The determined by X-ray Photoelectron Spectroscopy (XPS) zero valency of both surface metal and adsorbed iodine atoms can only mean that iodine is covalently bonded to the metal surface.31 We believe, that due to the similarity between iodine and astatine adsorption of 211At undergoes according to the same reaction. Because of the increasing tendency of halides from F− to I− to form covalent bond with gold surface we expect that chemisorption of At will be the strongest in the halogen group. Therefore, in this paper we propose to use gold nanoparticles as prosthetic group for astatination.
AuNPs, 5 and 15 nm median diameter, were synthesized by using sodium citrate and tannic acid as stabilizing agents. Synthesized AuNPs are stable in solution because the citrate stopping layer of the AuNPs provides an electrostatic repulsion force resulting from the electric double layer.32
The biologically active fragment of Substance P(5-11), named shortly as SP(5-11), was conjugated to the gold nanoparticles to obtain bioconjugates targeting NK1 receptors on glioma cells. Previously it was found that Substance P in cerebrospinal fluid exhibits a stability of 84% after 24 hours, while the serum residence time is only 1 to 2 minutes due to the rapid degradation.33 We have also shown that SP(5-11) has similar affinity to NK1 receptors as the whole peptide (IC50 = 38 nM).34 The PEG linker (2000 kDa) comprising the disulfide bridge and the N-hydroxysuccinimide esters (NHS) at the ends was used for synthesis of the SP(5-11)–PEG–SS–PEG–SP(5-11) conjugate. The process of nanoparticles biofunctionalization is summarized in Fig. 1.
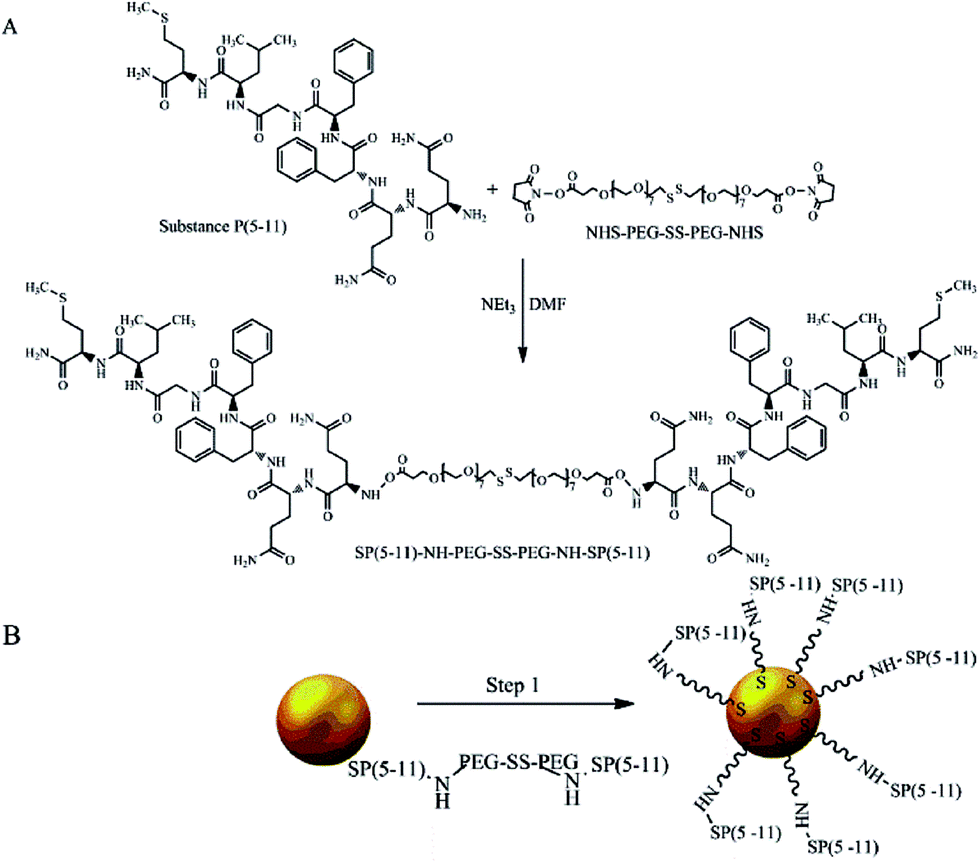 | ||
| Fig. 1 Synthesis of AuNP–S–PEG–SP(5-11): conjugation of NHS–PEG–SS–PEG–NHS with SP(5-11) (A) and attaching SP(5-11)–PEG–SS–PEG–SP(5-11) to AuNPs surface (B). | ||
The synthesized SP(5-11)–PEG–SS–PEG–SP(5-11) conjugates were spontaneously attached to the AuNPs surface. When the disulfide bridge is disrupted sulphur atoms can be easily attached to the surface of gold nanoparticles to form stable Au–S bond. To calculate the number of SP(5-11) molecules attached to the AuNPs surface, we applied the gel filtration chromatography technique. As shown in Fig. 2 this method allows observation of well separated peaks associated with AuNPs, SP(5-11)–PEG–SS–PEG–SP(5-11) and AuNPs–PEG–SP(5-11) species. Peak area comparison allows to estimate the degree of SP(5-11) conjugation to the AuNPs surface, which exceeds 93% yield. On the basis of the obtained results the number of S–PEG–SP(5-11) coating molecules per one Au nanoparticle can be estimated. The calculation was performed based on the mass of taken S–PEG–SP(5-11), known reaction yield and under assumption that the nanoparticles are spherical with medium diameter of 5 and 15 nm, as measured by TEM and that the density of gold is 19.28 g cm−3. The obtained results show that one 5 nm AuNP contains an average 47 SP(5-11) molecules on the surface (0.6 molecules per nm2) and that 15 nm AuNP contains about 3300 molecules of SP(5-11) (4.7 molecules per nm2).
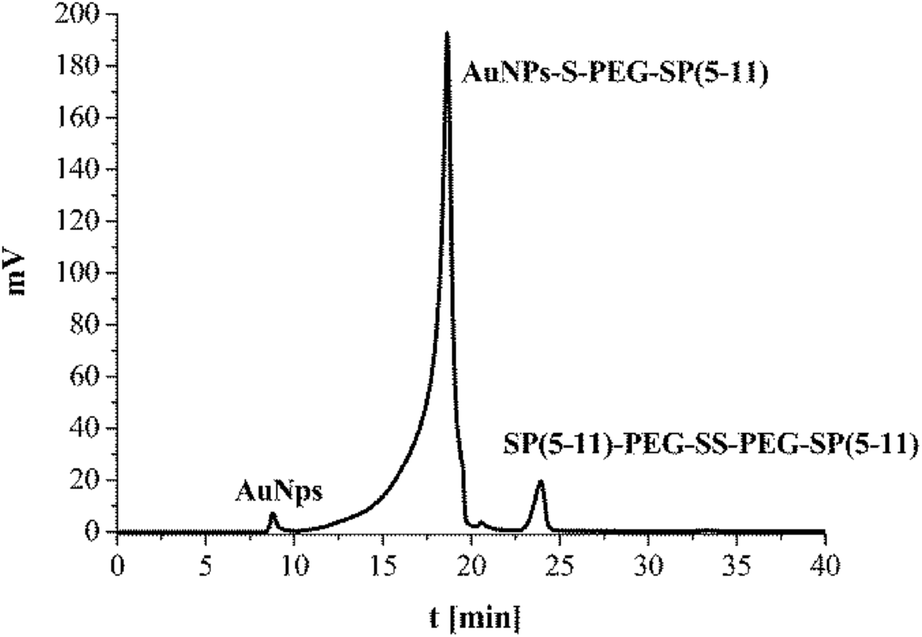 | ||
| Fig. 2 HPLC chromatogram of the reaction mixture of AuNPs (15 nm) with SP(5-11)–PEG–SS–PEG–SP(5-11); aqueous gel filtration column Polysep-GFC-P-Linear (7.8 mm × 300 mm); elution system 2. | ||
Taking into account that total surface area of 5 nm AuNP is 78.5 nm2 and that the area occupied on Au by S atoms is 1.6 nm2 we found that approximately 98% of the AuNP area remains for 211At adsorption. In the case of 15 nm AuNP sulphur atoms occupy about 112 nm,2 whereas 85% of AuNP area remains free for 211At adsorption.
The obtained AuNP–S–PEG–SP(5-11) bioconjugates were characterized by TEM and DLS methods. The results are presented in Table 1 and in Fig. 3. The transmission electron micrograph of AuNPs–S–PEG–Substance P(5-11) shows that the particles are uniformly dispersed with significantly narrow size equal to 5 nm and to 15 nm (Fig. 3). It was not possible to observe the “corona” around the gold nanoparticle due to poor interaction of the electron beam with the peptide molecules (low electron density), in contrast to the strong scattering of the electron beam when it interacted with metallic nanoparticles.
| 5 nm AuNPs | 15 nm AuNPs | |
|---|---|---|
| Hydrodynamic diameter (nm) | ||
| AuNP | 12.9 | 19.3 |
| AuNP–S–PEG–CH3 | 13.0 | 21.6 |
| AuNP–S–PEG–SP(5-11) | 20.2 | 24.6 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Zeta potential (mV) | ||
| AuNP | −20.2 | −41.1 |
| AuNP–S–PEG–CH3 | −18.1 | −37.3 |
| AuNP–S–PEG–SP(5-11) | −42.1 | −38.8 |
 | ||
| Fig. 3 TEM image of: (left) Au(5 nm) NPs–S–PEG–SP(5-11) (right) Au(15 nm) NPs–S–PEG–SP(5-11). | ||
In order to additionally confirm the presence of SP(5-11) on the nanoparticles surface hydrodynamic diameters and zeta potentials of AuNPs, AuNP–S–PEG–CH3 and AuNP–S–PEG–SP(5-11) were measured at pH 7 (Table 1). The particles size, determined by DLS, was significantly larger than that observed by TEM. This is caused by fact that the DLS technique measures the mean hydrodynamic diameter of the AuNPs core bounded by the organic and solvation layers, and this hydrodynamic diameter is affected by the viscosity and concentration of the medium. TEM, however, gives the diameter of the core alone. The increase of the hydrodynamic diameter after addition of polyethylene glycol (PEG) or a peptide is commonly observed after coating nanoparticles with biomolecules, like it was in the case of AuNPs modified by folic acid.35
The difference between the zeta potential of citrate AuNPs and AuNP–S–PEG–SP(5-11) additionally confirms the surface modification. The negative zeta potential value of around −40 mV for AuNP–S–PEG–SP(5-11) conjugate indicates that the particles repel each other and that there is no tendency for the particles to aggregate as confirmed by monitoring changes in hydrodynamic diameter during 7 days.
The obtained AuNP–S–PEG–SP(5-11) conjugates were labelled with 131I and 211At by chemisorption on the gold surface. The radioactivity of 211At was in the range of 100 to 150 MBq upon arrival to the laboratory. Isolation of 211At was carried out using dry-distillation of the target material, so that the recovery yield equal to 50% of the initial 211At activity was routinely obtained within about 40 min preparation time. Aliquots of the recovered astatine were evaporated, dissolved in water and immediately used for labelling AuNP–PEG–SP(5-11) bioconjugates. The presence of the anionic form of astatine was confirmed by paper electrophoresis through comparison with the behaviour of iodide anion.
The percent of labelling yield was determined using TLC analysis with MeOH as a developing solvent. As shown in Fig. 4 under these conditions the labelled 211At–AuNP–S–PEG–SP(5-11) radiobioconjugates remain at the bottom of the ITLC strip (Rf = 0), while free 211At, released from the radiobioconjugates, moves with the solvent front (Rf = 0.9).
 | ||
| Fig. 4 TLC radiochromatograms of free 211At (A) and radiolabelled 211At–AuNP–S–PEG–SP(5-11) radiobioconjugate (B). | ||
The labelling yields of AuNPs and AuNP–S–PEG–SP(5-11) with 131I and 211At radionuclides in water at pH = 6 are shown in Table 2. Also labelling efficiency in the solutions at pH 2 and 10 were performed. In all cases the labelling yield exceeded 99%, but in solutions of pH 2 and 10 the agglomeration of the nanoconjugates was observed. Therefore, further labelling of bioconjugates were conducted at pH 6.
| NPs | % of labelling | |
|---|---|---|
| 131I | 211At | |
| AuNPs 5 nm | 93.2 | 99.5 |
| AuNPs 15 nm | 98.1 | 99.9 |
| AuNP–S–PEG–SP(5-11) 5 nm | 92.1 | 99.3 |
| AuNP–S–PEG–SP(5-11) 15 nm | 89.3 | 99.7 |
The labelling yield of AuNPs with 211At is much higher (>99%) than in the case of another commonly used prosthetic group N-succinimidyl-3-(tri-n-butylstannyl) benzoate where the labelling yield is between 60–70%.36 For the boron precursor and for the Rh[16aneS4-diol] group the yields are equal to 80–89% (ref. 36 and 37) and 80%,11 respectively. As presented in Table 2, the 211At and 131I absorption on AuNP–S–PEG–SP(5-11) conjugates was nearly the same as in the case of naked AuNPs, indicating that the attachment of PEG–SP(5-11) molecules to the AuNPs surface only little changes adsorption properties of AuNPs. This is expected, as previous calculations revealed that attached PEG–SP(5-11) molecules occupy only a small percentage of the AuNPs surface. The results obtained for 131I and 211At adsorption on AuNPs agree well with the prior reports of high affinity and strong binding of heavy halide ions to the noble metals surface.23,24 In the case of iodine radionuclides this specific adsorption is well documented in the field of surface chemistry. In our work we have demonstrated for the first time that astatine also forms strong bond with the gold surface.
Stability of 211At–AuNP–S–PEG–SP(5-11) in human serum and in cerebrospinal fluid was studied at different time points (Table 3 and 4). The percent of liberated 211At was determined using TLC analysis with MeOH as a developing solvent. Under these conditions the intact 211At–AuNP–S–PEG–SP(5-11) radiobioconjugates remain at the bottom of the ITLC strip (Rf = 0), while free 211At, released from the radiobioconjugates, moves with the solvent front (Rf = 0.9). The blank tests with human serum and cerebrospinal fluid with free 211At has shown that free 211At does not bind to proteins present in both media.
| % of leakage | |||
|---|---|---|---|
| 2 h | 4 h | 24 h | |
| 5 nm 211At–AuNP–S–PEG–SP(5-11) | |||
| Human serum | 0.5 | 1.1 | 3.9 |
| Cerebrospinal fluid | 0.3 | 0.6 | 2.3 |
|
|||
| 15 nm 211At–AuNP–S–PEG–SP(5-11) | |||
| Human serum | 0.3 | 0.4 | 0.7 |
| Cerebrospinal fluid | 0.2 | 0.7 | 0.5 |
| % of leakage | |||
|---|---|---|---|
| 2 h | 4 h | 24 h | |
| 5 nm 131I–AuNP–S–PEG–SP(5-11) | |||
| Human serum | 3.4 | 3.5 | 3.2 |
| Cerebrospinal fluid | 4.0 | 3.7 | 5.2 |
|
|||
| 15 nm 131I–AuNP–S–PEG–SP(5-11) | |||
| Human serum | 4.8 | 6.9 | 7.2 |
| Cerebrospinal fluid | 18.6 | 16.0 | 17.9 |
The radiobioconjugates were found to be very stable towards dissociation – almost no release of 211At was observed during 24 h, corresponding to more than 3 half-lives. As shown in Tables 3 and 4 the astatinated radiobioconjugates exhibit better stability than the iodinated analogues. This confirms our assumption that astatine, as the biggest halogen in the group, forms strongest bond with Au surface.
The in vitro studies were carried out on human gliomas tumour T98G cells overexpressing NK1 receptors on the cell membrane. At first, the internalization of AuNP–S–PEG–SP(5-11) conjugates was evaluated. In these experiments AuNP(5 nm)–S–PEG–SP(5-11) and AuNP(15 nm)–S–PEG–SP(5-11) were labelled with 131I, which was used as the radioisotopic marker. The results are presented in Fig. 5. The internalization of 131I–AuNP(5 nm)-SP(5-11) radiobioconjugate and alone 131I–Au(5 NPs(5 nm)) was equal to 4.4% and 2.0%, respectively. In the case of conjugates based on 15 nm AuNP internalization was significantly greater due to much higher contents of SP(5-11) molecules on nanoparticle surface. This statistically significant (student t-test, n = 6, p < 0.05) difference indicates that over 6% of totally internalized 131I–AuNP–S–PEG–SP(5-11) is due to specific (NK1 receptor) binding to T98G cells.
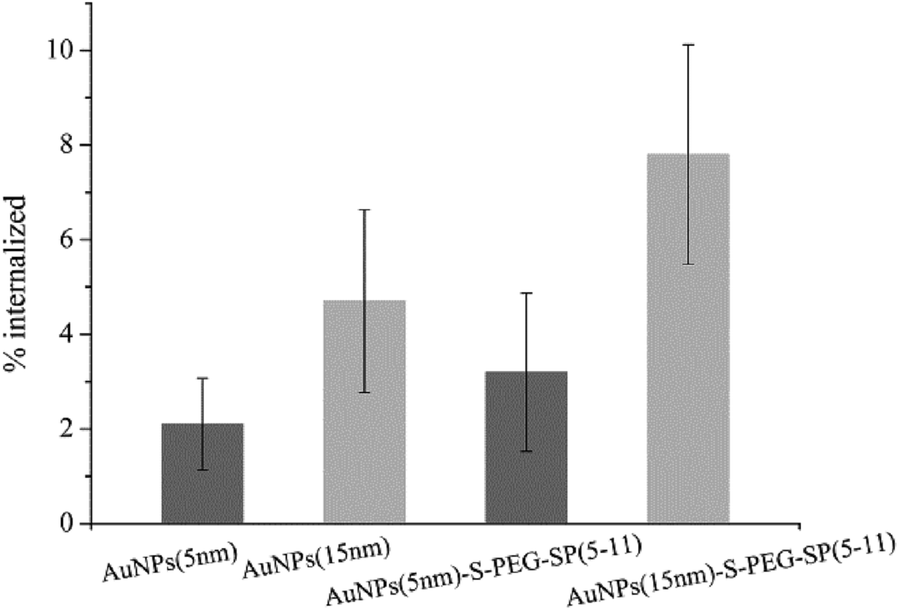 | ||
| Fig. 5 Internalization of 5 and 15 nm 131I–AuNPs and 131I–AuNP–S–PEG–SP(5-11) by T98G cells. | ||
Subsequently, the influence of 5 and 15 nm 211At–AuNP–S–PEG–SP(5-11) radiobioconjugates on cell viability was investigated. Cells were exposed for 24 h to different activities of 211At–AuNPs, 211At–AuNP–S–PEG–SP(5-11) and to free 211At-astatide. We also have measured cytotoxicity of non-radioactive AuNPs and AuNP–S–PEG–SP(5-11) bioconjugates in the function of concentration. Cells were treated with radioactivity for over 24 h and the metabolic activity was assessed by colorimetric MTT assay in comparison to non-treated cells, as presented in Fig. 6 and 7.
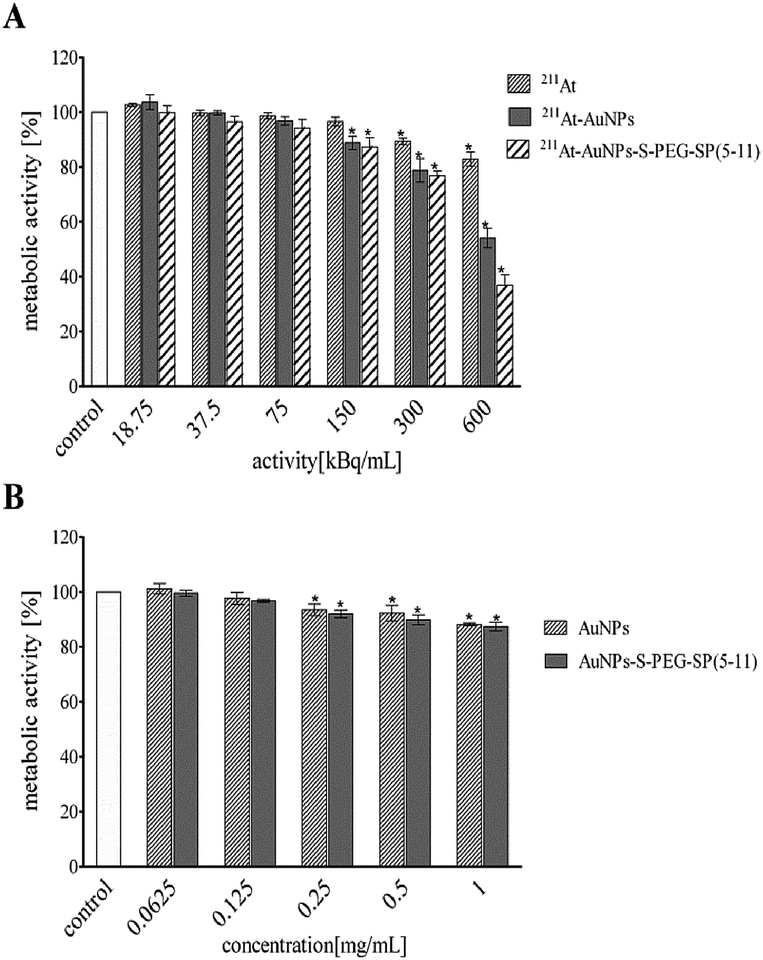 | ||
| Fig. 6 Metabolic activity of T98G cells assessed by colorimetric MTT assay after 24 h incubation: (A) cells treated with different activities of 211At–AuNP(15 nm), 211At–AuNP(15 nm)–S–PEG–SP(5-11) and free 211At-astatide anions alone; (B) cells treated with different concentrations of non-radioactive AuNPs and AuNP–S–PEG–SP(5-11) bioconjugates. | ||
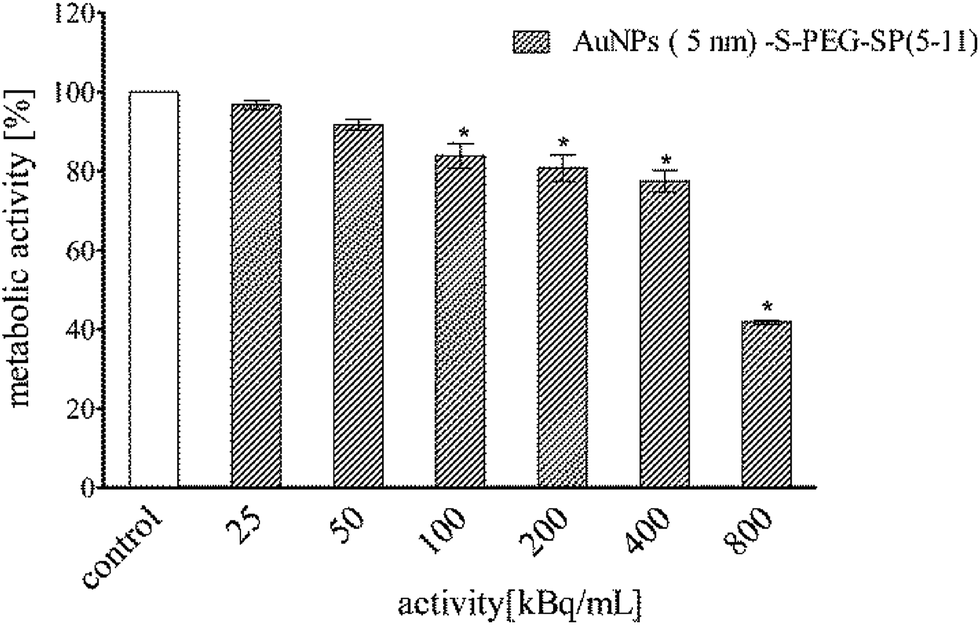 | ||
| Fig. 7 Metabolic activity of T98G cells assessed by colorimetric MTT assay after 24 h incubation with different activities of 211At–AuNP(5 nm)–S–PEG–SP(5-11). | ||
In Fig. 6 and 7 data are presented as the average percent ± SD from three independent experiments (*denotes statistically significant differences from unexposed control, P < 0.05). The obtained results demonstrate that 211At–AuNPs(15 nm) and 211At–AuNP(15 nm)–S–PEG SP(5-11) significantly reduce metabolic activity of T98G in a dose dependent manner reaching adequately 52% (211At–AuNPs) and 36% (radiobioconjugate) of survived fraction at the highest radioactivity. At the same time alone 211At-astatide was also reducing the cell viability (∼80%), although the level was not so spectacular as with the targeting vector (Fig. 6A).
In the case of 211At–AuNP(5 nm)–S–PEG–SP(5-11) (Fig. 7) cell viability (ca. 40%) was reached at dose around 800 kBq mL−1, a dose higher than for radiolabelled AuNP(15 nm)–SP(5-11). This result is probably due to the much lower number of attached HS–PEG–SP(5-11) molecules on the surface of 5 nm compare to 15 nm nanoparticles.
Performed cytotoxicity studies with the control probes of non-radioactive AuNPs and AuNP–S–PEG–SP(5-11) bioconjugate exhibited their almost non-toxic effect. The survived fraction of cells was high, equal to almost 88%, even after exposure to concentration as high as 1 mg mL−1 (Fig. 5B). It should be mentioned that amount of 211At–AuNP–S–PEG–SP(5-11) carrier used in the cells studies was equal to 1 mg mL−1 concentration of the unlabeled AuNP–S–PEG–SP(5-11) bioconjugate; therefore, possible additional toxic effect from non-radioactive conjugate was negligible. As shown in Fig. 6 and 7 the cytotoxicity of 5 nm 211At–AuNP–S–PEG–SP(5-11) was nearly the same as that of 15 nm 211At–AuNP–S–PEG–SP(5-11) radiobioconjugate, indicating that the size of the radiolabelled nanoparticles does not affect the cytotoxicity of radiobioconjugate.
Conclusions
In this study we described new concept of the preparation of astatinated bioconjugates using the covalent adsorption of astatine atoms on the surface of gold nanoparticles. The conjugated probes were stable for 24 h in human serum and cerebrospinal fluid. Radiobioconjugate 211AuNP–S–PEG–SP(5-11) has been shown high toxicity towards glioma cells expressing NK-1 receptor and exhibits properties suitable for treatment of glioma cancer cells by intertumoral or post-resection injection. The intravenous injection of the 211AuNP–S–PEG–SP(5-11) radiobioconjugate for glioma treatment is excluded, because of its relatively large size and high hydrophilicity, which prevent it from crossing the blood–brain barrier (BBB). The BBB is especially limited to larger molecules following systemic application. As mentioned by Cordier et al. cumulative organ toxicity using α emitter labelled targeting agents severely limit the systemic approach of gliomas.25 These limitations have stimulated the development of strategies for local drug application.Experimental
Radionuclides
Astatine-211 was produced through the 209Bi(α, 2n) 211At reaction in the U-200 cyclotron at the Heavy Ion Laboratory of the Warsaw University. The separation of 211At was carried out by dry distillation under nitrogen, at the gas flow rate of 120 cm3 min−1. After flushing the apparatus with nitrogen for 15 min, the active bismuth target was placed in the quartz tube inside the resistance furnace, and heating was switched on. The temperature of 650 °C, at which the 211At releasing was carried out, was achieved within 30 min, and kept constant for the next 15 min. The evolved 211At was collected in a cold polyether ether ketone (PEEK) tube immersed in ethanol and cooled down with liquid nitrogen to the temperature between −55 °C and −50 °C. The collected 211At was eluted from PEEK with 200–400 μL of methanol or in Na2SO3/methanol solution.Because 131I is much more easily available than 211At and moreover because iodine and astatine have similar properties, we used in several experiments the 131I radionuclide instead of 211At. No-carrier-added sodium [131I]iodide in 0.01 M NaOH with the specific activity of about 550 GBq mg−1 was purchased from POLATOM Radioisotope Centre, Świerk, Poland. All radioactive materials were handled according to protocols approved at the Institute of Nuclear Chemistry and Technology.
Reagents
The following chemical reagents were used: gold(III) chloride trihydrate (AuCl3 3H2O), trisodium citrate dihydrate (Na3C6H5O7), polyethylene glycol N-succinimidyl ester disulfide (NHS–PEG–SS–PEG–NHS), polyethylene glycol methyl ether thiol (CH3–PEG–SH) and triethylamine (TEA) of 99% purity were purchased from Sigma-Aldrich. Methanol (99.9%), nitric acid, hydrochloric acid were purchased from POCH, Poland. Anhydrous dimethylformamide (DMF) and dichloromethane (DCM) of ACS grade were purchased from BDH Prolabo VWR. Substance P(5-11) was obtained from Bachem.Serum aliquots were prepared from blood samples taken from healthy volunteers and stored at −20 °C. Volunteers were informed about the research conducted and conscious volunteers consent was obtained. The studies were conducted in accordance with institutional regulations. Cerebrospinal fluid (CSF) was a gift from the Central Clinical Hospital belongings to the Ministry of the Interior and Administration in Warsaw.
In cell studies the following materials were used: DMEM medium (Gibco, Poland), phosphate-buffered saline (PBS), dimethylsulfoxide (DMSO), 3-(4,5-dimetyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) from Sigma Aldrich, (Poland). Fetal calf serum was the product of Biological Industries (Israel). T98G cells were purchased from the American Type Tissue Culture Collection (ATCC, Rockville, MD) and maintained according to the ATCC protocol.
Instrumentations
Shape, diameter and morphology of the nanoparticles and nanoparticle bioconjugates were determined by transmission electron microscopy (TEM, LEO 912B). The apparent hydrodynamic diameter and zeta potential (ζ) were measured by dynamic light scattering (DLS, Malvern, UK). The radioactivity of collected samples was measured with the gamma spectrometer containing germanium detector (Canberra, Meriden, CT, USA). The detector had a resolution of 0.8 at 5.9 keV, 1.0 at 123 keV and 1.9 at 1332 keV. Radioactivity of 211At was quantified by its 89.69 keV γ-ray.The oxidation state of astatine ions was determined by paper electrophoresis (Sigma-Aldrich horizontal electrophoresis) on Whatman GF83 Glass Paper at the potential gradient 12 V cm−1 for 20 min with 0.01 M NaNO3 as the electrolyte. Deionized water was prepared by the Hydrolab water purification system (Hydrolab, Poland).
TLC analyses were performed on Alufolien sheets RP-18 Merck 7.5 cm using MeOH as a developing solvent. The distribution of radioactivity on the paper electrophoresis or TLC sheets was measured by Storage Phosphor System Cyclone Plus (Perkin-Elmer Life and Analytical Sciences) and analyzed using Optiquant software (version 5.0) provided by the manufacturer.
High-performance liquid chromatography (HPLC) was performed using the VWR-Hitachi LaChrom Elite HPLC system which consisted of a pump L2130, column thermostate L-2350, UV diode array detector (DAD) L-2455 and the EZChrom Elite data system. The radioactivity was monitored using a 3 × 3′′ NaI(Tl) scintillation detector Raytest Gabi Star (Straubenhardt, Germany). The separation of SP(5-11)–PEG–SS–PEG–SP(5-11) from reaction mixture was accomplished on the LiChrospher® 100 RP-18 analytical column (5 μm particle size, 4.6 mm × 250 mm) from Merck (Germany). The solvent and gradient conditions were as follows: solvent A 0.1% (v/v) trifluoroacetic acid (TFA) in water, solvent B 0.1% (v/v) trifluoroacetic acid (TFA) in acetonitrile. System 1: 0% to 70% B in 0–15 min, 70% to 95% B in 15–20 min, 95% B in 20–30 min, 1 mL min−1; UV detection (220–400 nm). The identification and separation of Au nanoparticles decorated with (5-11)SP–PEG–SS–PEG–SP(5-11) were performed using aqueous gel filtration column Polysep-GFC-P-Linear (7.8 mm × 300 mm) from Phenomenex (USA). Isocratic elution was performed at a flow rate of 0.5 mL min−1 using water (system 2).
Synthesis of 15 nm gold nanoparticles
Gold nanoparticles of 15 nm diameter were synthesized by the modified Turkevich method.38 For this purpose 39.38 mg of gold(III) chloride trihydrate was dissolved in 100 mL of distilled water and heated under reflux in round bottom flask. Next 111.79 mg of trisodium citrate dihydrate was dissolved in 10 mL of distilled water. During boiling the solution of gold(III) chloride trihydrate the whole amount of trisodium citrate dihydrate was added rapidly and the mixture was heated for additional 15 minutes. The cooled down flask with gold nanoparticles was wrapped with aluminium foil and stored at 4–8 °C. Correctly prepared solution of gold nanoparticles with size 15 nm has the colour of red wine. The size of nanoparticles was checked by TEM and DLS techniques.Synthesis of 5 nm gold nanoparticles
AuNPs with 5 nm diameter were synthesized in water by chemical reduction as described before.39 Aqueous solution of chloroauric acid (93.80 g of 1.84 10−2 wt%) was boiled, and next the mixture of sodium citrate (4.48 g, 0.877 wt%) and tannic acid (1.73 g, 1 wt%) was added. Following the addition of the mixture the solution color changed from yellow to dark red. The suspension was stirred for additional 15 min and cooled down to room temperature.AuNP PEG functionalization
The synthesized 5 nm and 15 nm gold nanoparticles were coated with CH3–PEG–SH with average molar mass 800 at different molar ratios of PEG to gold nanoparticles in order to determine its effect on the labelling efficiency. The ratios were equal to 2500:1, 1500:1, 500:1 for AuNPs 15 nm and 50:1, 100:1, 200:1 for AuNPs 5 nm. The CH3–PEG–SH was mixed with nanoparticles and stirred for 24 h. The size and zeta potential of Au–PEG NPs were checked by DLS technique.
Synthesis of AuNP–S–PEG–SP(5-11) bioconjugate
The synthesis of AuNP–S–PEG–SP(5-11) bioconjugates was performed in two steps. In the first step NHS–PEG–SS–PEG–NHS was conjugated with Substance P(5-11). Briefly, to the solution of NHS–PEG–SS–PEG–NHS (1.6 μmol dissolved in 0.1 mL of DMF) equal molar amounts of Substance P(5-11) and triethylamine (6.4 μmol) were added. The reaction mixture was stirred for 24 h at room temperature under argon atmosphere. After completion of the reaction (checked by HPLC, system 1) the solvent was removed under vacuum. SP(5-11)–PEG–SS–PEG–SP(5-11) peptide was separated from unreacted substrates by HPLC, alkalized to pH 7 and lyophilized. In the second step, the SP(5-11)–PEG–SS–PEG–SP(5-11) peptide was attached to the surface of gold nanoparticles. For example, 1 mg of bioconjugate was added to 1 ml of deionized water, mixed and next suitable amount of this solution was added to gold nanoparticles solution and stirred for 24 h at room temperature. The suitable amount of bioconjugate solution was calculated for desirable molar ratio of PEG–SP(5-11) in analogy discussed in the previous point.Labeling of AuNP–S–PEG–SP(5-11) bioconjugate with 211At and 131I
211At–AuNP–S–PEG–SP(5-11) and 131I–AuNP–S–PEG–SP(5-11) radiobioconjugates were prepared by adsorption of 211At and 131I on gold nanoparticle conjugates. Aliquots of the eluted from PEEK astatine (200–400 μL) in Na2SO3/methanol solution were evaporated, dissolved in water. Next 20 μL of 211At− activity (5–10 MBq) was added to each 200 μL sample of AuNP–S–PEG–SP(5-11) bioconjugate solution in the Eppendorf tube. After adjusting the pH to 6 by dropwise addition of 0.001–0.1 M HNO3 or NaOH the solution was stirred for 1 h at room temperature. The same procedure was performed for labelling with 131I, where 3 MBq Na 131I in 0.01 M NaOH was used. The efficiency of the labelling was measured by the TLC technique with MeOH as a developing solvent. In this condition free 211At− or 131I− moves with the solvent front and 211At and 131I attached to AuNPs remain at the bottom of the TLC strip. The distribution of radioactivity on the TLC sheets was measured by cutting the strips into 1 cm pieces and counting them in a NaI(Tl) well counter or by Storage Phosphor System Cyclone Plus (Perkin-Elmer Life and Analytical Sciences) and analyzed using Optiquant software (version 5.0) provided by the manufacturer.Stability studies of radiolabelled AuNP–S–PEG–SP(5-11) bioconjugates
The 20 μL of 211At–AuNP–S–PEG–SP(5-11) or 131I–AuNP–S–PEG–SP(5-11) solution was mixed with 200 μL of human serum (HS) or cerebrospinal fluid (CSF) and incubated at room temperature. Stability studies were carried out after 2 h, 4 h and 24 h on three analogous samples using the TLC technique. The blank tests with the HS or CSF and radionuclide solutions were carried out to estimate the percentage of possible binding of free 211At to proteins present in both.AuNPs and AuNPs–SP(5-11) internalization
The internalization studies were performed using AuNPs and AuNPs–SP(5-11) conjugate labelled with 131I chemisorbed on AuNP surface. The cells were seeded into 6-well plates (TPP, Israel) at 5 × 105 cells per well and cultured for 24 h. Then, cells were incubated at 37 °C for 2 h with 50 μg mL−1 of 5 and 15 nm 131I–AuNPs or 131I–AuNPs–SP(5-11). After treatment, the growth medium of each well was collected, the cells were washed twice with 750 μL of PBS and incubated with 500 μL of pre-warmed trypsin solution at 37 °C for 10 min. Subsequently, the cells were collected and activity of each fraction was measured. The results are expressed as a percentage of the total radioactivity added.Cytotoxicity evaluation
The impact of AuNPs, AuNP–S–PEG–SP(5-11), free 211At, 211At–AuNP and 211At–AuNP–S–PEG–SP(5-11) on metabolic activity and proliferation of T98G cells was measured by colorimetric MTT assay. The assay was performed as described.40 In brief, T98G cells were seeded a day before an experiment in 96-well microplates (TPP) at a density of 4 × 103 cells per well in 100 μL of culture medium. Next, cells were treated for 24 h with increasing concentration of the cold AuNPs and AuNP–S–PEG–SP(5-11) or with radiobioconjugate labelled with 211At (1–800 kBq mL−1) or only with 211At-astatide (1–800 kBq mL−1). After 24 h incubation, the cell culture medium was removed and 100 μL of 3 mg mL−1 MTT solution was added to each well. After 3 h incubation at 37 °C the MTT solution was removed. Remaining insoluble formazan crystals were dissolved in 100 μL DMSO and absorbance of the solution was measured at 570 nm in the plate reader spectrophotometer Infinite M200 (Tecan). At least three independent experiments in six replicate wells were conducted.Statistical analysis
At least three independent experiments in six replicate wells were conducted for each toxicity point. Difference between samples and control were evaluated using GraphPad Prism 5.0 software (GraphPad Software Inc., USA). Toxicological data were evaluated by Kruskal-Wallis One Way Analysis of Variance on Ranks (ANOVA) followed by post hoc Dunnet's method. Differences were considered statistically significant when the p-value was less than <0.05.Conflicts of interest
The are no conflicts to declare.Acknowledgements
This work was supported by grant 2013/11/B/ST4/00516 from the National Science Centre of Poland.References
- M. R. Zalutsky, Radionuclide therapy, in Radiochemistry and Radiopharmaceutical Chemistry in Life Sciences, ed. F. Rösch, Kluwer Academic Publishers, Dordrecht, 2003, pp. 315–348 Search PubMed.
- M. R. Zalusky, D. A. Reardon, E. Pozi, G. Vaidyanathan and D. D. Bigner, Nucl. Med. Biol., 2007, 34, 779 CrossRef PubMed.
- NRC licensing decision, J. Nucl. Med., 2013, 54, 19N CrossRef PubMed.
- A. Morgenstern, F. Bruchertseifer and C. Apostolidis, Curr. Radiopharm., 2012, 5, 221 CrossRef CAS PubMed.
- J. Champion, M. Seydou, A. Sabatie-Gogova, E. Renault, G. Montavon and N. Galland, Phys. Chem. Chem. Phys., 2011, 13, 14984 RSC.
- K. Berei and L. Vasaros, Organic Chemistry of Astatine, Hungarian Academy of Sciences Report KFKI- 1981-10.
- F. Guerard, J. F. Gestin and M. W. Brechbiel, Cancer Biother. Radiopharm., 2013, 28, 1 CrossRef CAS PubMed.
- M. R. Zalutsky, D. A. Reardon, G. Akabani, R. E. Coleman, A. H. Friedman, H. S. Friedman, R. E. McLendon, T. Z. Wong and D. D. Bigner, J. Nucl. Med., 2008, 49, 30 CrossRef CAS PubMed.
- S. Hadley, D. S. Wilbur, M. A. Gray and R. W. Atcher, Bioconjugate Chem., 1991, 2, 171 CrossRef CAS PubMed.
- D. S. Wilbur, M. K. Chyan, D. K. Hamlin, B. B. Kegley, R. Risler, P. M. Pathare, J. Quinn, R. L. Vessella, C. Foulon, M. R. Zalutsky, T. J. Wedge and M. F. Hawthorne, Bioconjugate Chem., 2004, 15, 203 CrossRef CAS PubMed.
- M. Pruszyński, A. Bilewicz and M. R. Zalutsky, Bioconjugate Chem., 2008, 19, 958 CrossRef PubMed.
- M. Pruszyński, A. Bilewicz, M. Łyczko and M. R. Zalutsky, Nucl. Med. Biol., 2015, 42, 439 CrossRef PubMed.
- A. Y. Yordanov, K. Deal, K. Garmestani, H. Kobayashi, B. Herring, T. A. Waldmann and M. W. Brechiel, J. Labelled Compd. Radiopharm., 2000, 4, 1219 CrossRef.
- F. Guérard, H. Rajerison, A. F. Chauvet, J. Barbet, G. Meyer, I. D. Silva and F. Gestin, J. Nucl. Med., 2011, 52(supplement 1), 1486 Search PubMed.
- K. Kostarelos and D. Emfietzoglou, J. Liposome Res., 1999, 9, 429 CrossRef CAS.
- K. Kostarelos and D. Emfietzoglou, Anticancer Res., 2000, 20, 3339 CAS.
- K. B. Hartman, D. K. Hamlin, D. S. Wilbur and L. J. Wilson, Small, 2007, 3, 1496 CrossRef CAS PubMed.
- J. Kućka, M. Hrubý, C. Konák, J. Kozempel and O. Lebeda, Appl. Radiat. Isot., 2006, 64, 201 CrossRef PubMed.
- E. Cędrowska, M. Łyczko, A. Piotrowska, A. Bilewicz, A. Stolarz, A. Trzcińska, K. Szkliniarz and B. Wąs, Radiochim. Acta, 2016, 104, 267 CrossRef.
- M. Bochvarova, D. K. Tyung, I. Dudova, Y. V. Norseev and V. A. Khalkin, Soviet Radiochemistry, 1972, 14, 889 Search PubMed.
- G. Wunderlich, S. Fischer, R. Dreyer, W. Dobrenz and I. Dobrenz, Radiochim. Acta, 1989, 47, 153 CrossRef CAS.
- W. D. Bloomer, W. H. McLaughlin, R. D. Neirinckx, S. J. Adelstein, P. R. Gordon, T. J. Ruth and A. P. Wolf, Science, 1981, 212, 340 CAS.
- Z. Zhang, H. Li, F. Zhang, Y. Wu, Z. Guo, L. Zhou and J. Li, Langmuir, 2014, 30, 2648 CrossRef CAS PubMed.
- Y. H. Kim, J. Jeon, S. H. Hong, W. K. Rhim, Y. S. Lee, H. Youn, J. K. Chung, M. C. Lee, D. S. Lee, K. W. Kang and J. M. Nam, Small, 2011, 7, 2052 CrossRef CAS PubMed.
- D. Cordier, L. Krolicki, A. Morgenstern and A. Merlo, Semin. Nucl. Med., 2016, 46, 243 CrossRef PubMed.
- J. L. Chan, S. W. Lee, B. A. Fraass, D. P. Normolle, H. S. Greenberg, L. R. Junck, S. S. Gebarski and H. M. Sandler, J. Clin. Oncol., 2002, 20, 1635 CrossRef PubMed.
- D. Cordier, F. Forrer, F. Bruchertseifer, A. Morgenstern, C. Apostolidis, S. Good, J. Müller-Brand, H. Mäcke, J. C. Reubi and A. Merlo, Eur. J. Nucl. Med. Mol. Imaging, 2010, 37, 1335 CrossRef CAS PubMed.
- A. Morgenstern, L. Krolicki, J. Kunikowska, H. Koziara, B. Krolicki, M. Jakucinski, C. Apostolidis and F. Bruchertseifer, J. Nucl. Med., 2014, 55(supplement 1), 390 Search PubMed.
- S. K. Ozker, R. S. Hellman and A. Z. Krasnow, Appl. Radiat. Isot., 2002, 57, 729 CrossRef CAS PubMed.
- M. P. Soriaga, Chem. Rev., 1990, 90, 771 CrossRef CAS.
- G. M. Berry, M. E. Bothwell, B. G. Bravo, G. J. Cali, J. E. Harris, T. Mebrahtu, S. L. Michelhaugh, J. F. Rodriguez and M. P. Soriaga, Langmuir, 1989, 5, 707 CrossRef CAS.
- T. Kim, K. Lee, M. S. Gong and S.-W. Joo, Langmuir, 2005, 21, 9524 CrossRef CAS PubMed.
- S. Kneifel, D. Cordier, S. Good, M. S. Ionescu, A. Ghaffari, S. Hofer, M. Kretzschmar, M. Tolnay, C. Apostolidis, B. Waser, M. Arnold, J. Mueller-Brand, H. R. Maecke, J. C. Reubi and A. Merlo, Clin. Cancer Res., 2006, 12, 3843 CrossRef CAS PubMed.
- A. Piotrowska, S. Męczyńska-Wielgosz, A. Majkowska-Pilip, P. Koźmiński, G. Wójciuk, E. Cędrowska, F. Bruchertseifer, A. Morgenstern, M. Kruszewski and A. Bilewicz, Nucl. Med. Biol., 2017, 47, 10 CrossRef CAS PubMed.
- Z. Zhang, J. Jia, Y. Lai, Y. Ma, J. Weng and L. Sun, Bioorg. Med. Chem., 2010, 18, 5528 CrossRef CAS PubMed.
- D. S. Wilbur, M. S. Thakar, D. K. Hamlin, E. B. Santos, M.-K. Chyan, H. Nakamae, J. M. Pagel, O. W. Press and B. M. Sandmaier, Bioconjugate Chem., 2009, 20, 1983 CrossRef CAS PubMed.
- Y. Yang, R. Lin, N. Liu, J. Liao, M. Wei and J. Jin, J. Radioanal. Nucl. Chem., 2011, 288, 71 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- K. Soliwoda, E. Tomaszewska, B. Tkacz-Szczesna, E. Mackiewicz, M. Rosowski, A. Bald, C. Blancks, M. Schmutz, J. Novák, F. Schreiber, G. Celichowski and J. Grobelny, Langmuir, 2014, 30, 6684 CrossRef CAS PubMed.
- A. Lankoff, W. J. Sandberg, A. Wegierek-Ciuk, H. Lisowska, M. Refsnes, B. Sartowska, P. E. Schwarze, S. Meczynska-Wielgosz, M. Wojewodzka and M. Kruszewski, Toxicol. Lett., 2012, 208, 197 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |