
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simple design to achieve red-to-near-infrared emissive cationic Ir(III) emitters and their use in light emitting electrochemical cells†
Amlan K. Pala,
David B. Cordes b,
Alexandra M. Z. Slawinb,
Cristina Momblonac,
Antonio Pertegásc,
Enrique Ortíc,
Henk J. Bolink*c and
Eli Zysman-Colman*a
b,
Alexandra M. Z. Slawinb,
Cristina Momblonac,
Antonio Pertegásc,
Enrique Ortíc,
Henk J. Bolink*c and
Eli Zysman-Colman*a
aOrganic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, St Andrews, Fife KY16 9ST, UK. E-mail: eli.zysman-colman@st-andrews.ac.uk; Web: http://www.zysman-colman.com
bEaStCHEM School of Chemistry, University of St Andrews, St Andrews, Fife KY16 9ST, UK
cInstituto de Ciencia Molecular, Universidad de Valencia, C\Catedrático José Beltrán 2, 46980 Paterna, Spain
First published on 21st June 2017
Abstract
Two cationic Ir(III) complexes bearing 2-phenylpyridinato cyclometalating ligands and bithiazole-type ancillary ligands have been synthesized and optoelectronically characterised. These emitters exhibit unusually deep red-to-near-infrared emission at room temperature, thereby rendering them as attractive emitters in solution-processed light emitting electrochemical cell (LEEC) electroluminescent devices.
Near-infrared- (NIR-) emitting compounds are an important subclass of luminescent materials due to their potential applications in sensors, night-vision displays, optical cosmetology, telecommunication and photodynamic therapies.1 The most studied NIR-dyes in these contexts comprise semiconductor quantum dots,2 lanthanoid complexes,3 organic small-molecule dyes4 and transition metal complexes.1a,b,5 With rich photophysical behaviour, the NIR-emitting transition metal complexes are attractive candidates for solid-state lighting (SSL), as, with access to the triplet state, 100% of the electrically generated excitons can be harvested. Iridium complexes are well known phosphors for SSL due to their relatively short excited-state lifetimes, high photoluminescence quantum yields (ΦPL) and remarkable colour tunability, particularly across the visible spectrum.6 While stable green and yellow emissive iridium complexes are relatively well documented and have been successfully incorporated into electroluminescent (EL) devices, there is a dearth of examples of red-to-NIR emissive iridium complexes.1a,b,7 Cationic Ir complexes of the form [Ir(C^N)2(N^N)]+ (where C^N = cyclometalating ligand and N^N = ancillary ligand) are the most widely studied class of emitters for LEECs.1b In these charged complexes, the highest occupied molecular orbital (HOMO) typically resides on both the C^N ligand and metal, while the lowest unoccupied molecular orbital (LUMO) resides on the N^N ligand.8 The common strategies of achieving red-to-NIR emission (λem > 620 nm) include: (a) destabilisation of the HOMO through introduction of electron-donating substituents7a,9 onto the C^N ligands and/or stabilisation of the N^N ligand-based LUMO by incorporation of electron-withdrawing substituents,10 and (b) introduction of π-conjugated systems either on one or both of the C^N or N^N ligands11 thereby decreasing the HOMO–LUMO gap. Based on these principles, several charged red-to-NIR emitting Ir(III) complexes have been reported, a representative selection of which are shown in Chart 1 (sections a and b).7a,9
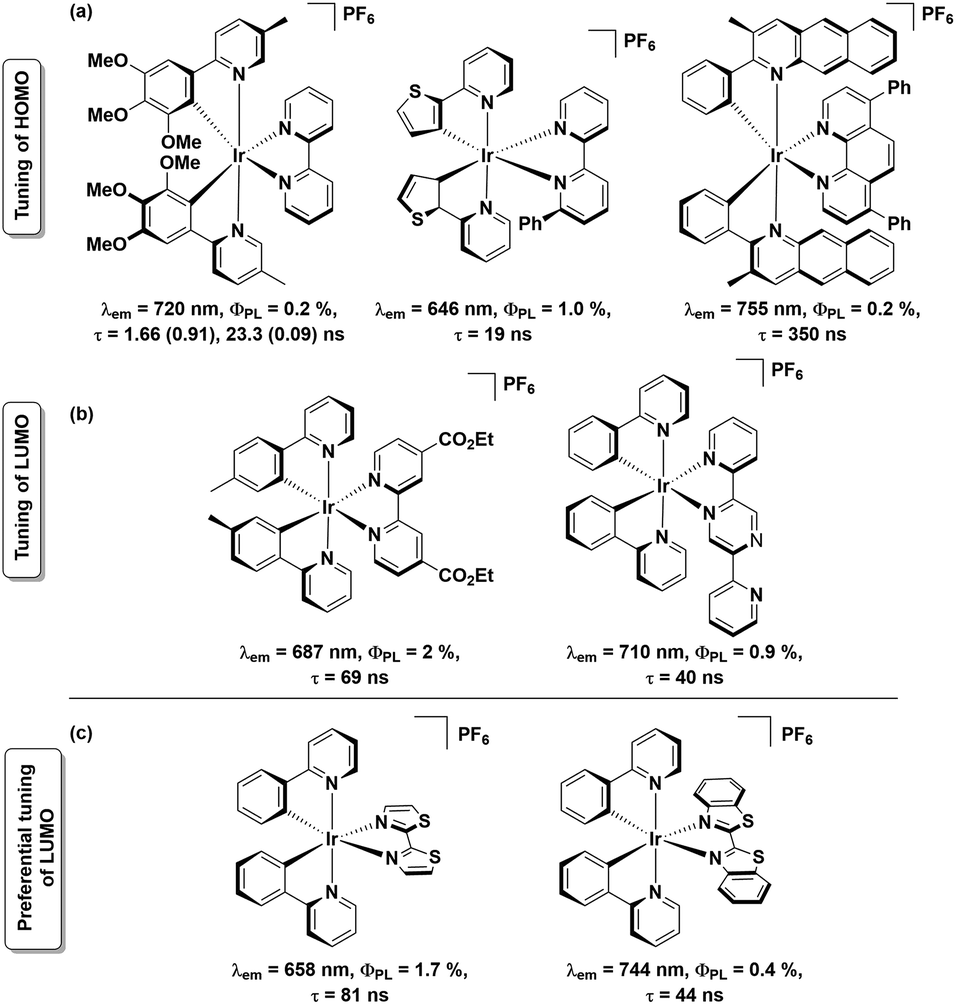 | ||
| Chart 1 Strategies of achieving red-to-NIR emissive cationic Ir(III) complexes by (a) destabilisation of HOMO (top row), (b) stabilisation of LUMO (middle row). Complexes in current study are shown in (c) bottom row. | ||
Of the strategies outlined above, that involving LUMO stabilization is the most underexplored, despite its potential to produce red-to-NIR emissive cationic Ir(III) complexes.10,11 In this context, herein, we report the syntheses of two red-to-NIR cationic Ir(III) complexes, [Ir(ppy)2(L1)][PF6], 1, and [Ir(ppy)2(L2)][PF6], 2, possessing remarkably simple ligand architectures. These complexes bear 2-phenylpyridinato-C2,N′ as the C^N ligand and strongly π-accepting ligands 2,2′-bithiazole (L1) and 2,2′-bibenzo[d]thiazole (L2) as the N^N ligands (Chart 1c). Coincident with the present study, complex 2 was recently reported by Ertl et al.,12 though no Light Emitting Electrochemical Cell (LEEC) device data was reported. Herein, the optoelectronic properties of complexes 1 and 2 are reported along with the LEEC device performance.
Ligands L1 and L2 were synthesized by Pd-catalysed homocoupling of 2-bromothiazole or 2-bromobenzothiazole, respectively, in moderate yields (41% for L1 and 51% for L2, ESI†).13 Complexes 1 and 2 were obtained by cleavage of the [Ir(ppy)2Cl]2 dimer in the presence of 2.3 equiv. of the corresponding ancillary ligands followed by an anion metathesis with aqueous NH4PF6 in 91% and 95%, respectively (ESI†). Ligands L1 and L2 and complexes 1 and 2 were characterised by 1H and 13C NMR spectroscopy, HRMS, melting point determination and elemental analyses. The 1H and 13C NMR spectra confirmed the inherent C2 symmetry present in both 1 and 2 (Fig. S1–S4, ESI†). Single crystal XRD studies of 1 and 2 corroborated the microanalysis and the geometry of the complexes (Fig. S5, Tables S1 and S2, ESI†).
In both crystal structures, the coordinatively saturated Ir(III) ions occupy a distorted octahedral geometry. The C^N carbon atoms are mutually cis disposed, similar to the solid-state structure of [Ir(ppy)2(bpy)]+, R1 (where bpy = 2,2′-bipyridine).14 The ancillary ligands coordinate in a bidentate N′,N′-mode through the hard N-donors instead of the soft S-donors. The Ir–CC^N bond distances [ranging from 2.003(7) to 2.007(6) Å in 1 and 2.009(4) to 2.009(5) Å in 2] are similar to those observed in R1 [ranging from 2.004(4) to 2.025(4) Å]. The Ir–NC^N bond distances are likewise similar across the three complexes [ranging from 2.046(5) to 2.048(5) Å in 1, 2.049(4) to 2.058(4) Å in 2 and 2.042(3) to 2.048(3) Å in R1]. The Ir–NN^N length [2.148(6) to 2.149(6) Å in 1 and 2.180(4) to 2.195(4) Å in 2] resemble closely those in R1 [2.129(3) to 2.137(3) Å in 2].
The electrochemical properties of 1 and 2 were monitored by cyclic- (CV) and differential pulse- (DPV) voltammetry and the CV and DPV traces in MeCN are shown in Fig. S6 (ESI†). The detailed CV data vs. SCE (Fc/Fc+ = 0.38 V in MeCN)15 are summarized in Table S3 (ESI†) while only first redox potentials are listed in Table 1. In the anodic scan, complexes 1 and 2 exhibit quasi-reversible monoelectronic oxidation processes at 1.24 V and 1.37 V, respectively. These oxidation processes are assigned to the Ir(III)/(IV) redox couple with contributions from the C^N ligands, assignments supported by DFT calculations (Fig. S7†). While the oxidation potential of complex 1 (EOx1/2 = 1.24 V) is similar to that of R1 (EOx1/2 = 1.27 V),16 that of complex 2 (EOx1/2 = 1.37 V) is anodically shifted by 100 mV compared to R1, which aligns with the stabilized nature of the calculated HOMO of 2 (EHOMO = −5.80 eV) compared to that found in 1 (EHOMO = −5.58 eV) and R1 (EHOMO = −5.56 eV). The more positive oxidation potential accounted for complex 2 is probably due to the modulation of the electron density of the Ir(III) ion by L2 as the latter is a better π–acceptor than L1 or 2,2′-bpy and thereby rendering the Ir(III) centre more electron deficient compared to that in complex 1.
| λem/nm | ΦPL/% | τe/μs | kr/105 s−1 | knr/105 s−1 | E1 Ox1/2/V (ΔEp, mV) | E1 Red1/2/V (ΔEp, mV) | |
|---|---|---|---|---|---|---|---|
| a ΦPL measured at 298 K using [Ru(bpy)3]Cl2 (ΦPL = 4% in H2O, aerated) as the reference.19 kr = ΦPL/τe and knr = (1 − ΦPL)/τe. CV values are vs. SCE.15 ΔEp = |Epa − Epc|, where Epa = anodic peak potential and Epc = cathodic peak potential; E1/2 = (Epa + Epc)/2. A silver wire was used as the pseudoreference electrode; a glassy-carbon electrode was used for the working electrode and a Pt electrode was used as the counter electrode.b ΦPL measured at 298 K using [Ru(bpy)3](PF6)2 (ΦPL = 9.5% in MeCN) as the reference.20 | |||||||
| 1 | 658 | 1.72 | 0.081 | 0.21 | 123.2 | 1.24 (110) | −1.15 (74) |
| 2 | 744 | 0.35 | 0.044 | 0.08 | 227.2 | 1.37 (169) | −0.82 (88) |
| R1b | 602 | 9.29 | 0.275 | 3.4 | 33 | 1.27 | −1.38 |
Upon scanning to negative potential, complex 1 exhibits two quasi-reversible one-electron reduction processes up to −2 V, while three other irreversible and quasi-reversible reduction waves could also be observed at higher negative potentials. Complex 2, on the other hand, displayed four mono-electronic quasi-reversible waves up to −3 V (Table S3 and Fig. S5, ESI†). DFT calculations show that the calculated LUMOs of 1 and 2 are localized on the N^N ligand and therefore the first reduction processes are ascribed to radical anion formation on the ancillary ligand. The first reduction potentials for 1 and 2 (ERed1/2 = −1.15 V for 1 and −0.82 V for 2) are anodically shifted by 230 mV and 560 mV, respectively, compared to that for R1, demonstrating the significantly stronger π-accepting nature of ligands L1 and L2 compared to bpy. The CV data mirrors the DFT results, which show significant LUMO stabilization from R1 (ELUMO = −2.31 eV) to 1 (ELUMO = −2.76 eV) to 2 (ELUMO = −3.07 eV). The trend in the calculated decreasing HOMO–LUMO gap from R1 (3.25 eV) to 1 (2.82 eV) to 2 (2.73 eV) is in good agreement to the decreasing redox gap observed for R1 (2.65 V) to 1 (2.38 V) to 2 (2.19 V). The second quasi-reversible and third irreversible reductions (Table S3, ESI†) of complex 1 could be ascribed to the reductions of the pyridine moiety of the C^N ligand based on the coarse approximation that both the LUMO+1 and LUMO+2 reside on the C^N ligand as predicted by DFT calculations (Fig. S7, ESI†) and similar assignments are also valid for the second and third quasi-reversible reductions of complex 2 (Table S3, ESI†).
The UV-vis absorption spectra of complexes 1 and 2 were obtained in dry MeCN and the values are tabulated in Table S4.† Overlays of experimentally observed and theoretically predicted absorption data by TD-DFT are shown in Fig. S8 (ESI†). The UV region is dominated by spin allowed 1π → π* transitions in both ligand moieties centered around 240–280 nm (Tables S4 and S5, ESI†). For complex 2, singlet ligand-centered π → π* transitions (1LC) extend up to 407 nm, as predicted by TD-DFT, due to increased conjugation in L2 (Table S5†). The set of absorption bands clustered around 315 nm in 1 are significantly red-shifted in 2 at ca. 363 nm. The character of these band in 1 is an admixture of singlet metal-to-ligand charge transfer (1MLCT) from Ir(dπ) → ppy(π*) and ppy-based 1LC transitions while in 2 the nature of these transitions is more CT in character and comprises 1MLCT from Ir(dπ) → L2(π*) and singlet ligand-to-ligand charge transfer (1LLCT) from ppy(π) → L2(π*), as suggested by TD-DFT. The greater CT character coupled with the lower lying LUMO is responsible for this large bathochromic shift. The lowest energy absorption maxima for 1 and 2 appear at 500 nm and 599 nm, respectively. These bands are assigned as a mixture of spin-allowed and spin-forbidden 1MLCT/3MLCT and ligand-to-ligand charge transfer (1LLCT/3LLCT) transitions. These hypochromic bands at lower energy are the result of poor spatial overlap between the HOMO and LUMO and are typical for cationic Ir complexes.6,17 Concomitant to the larger calculated HOMO–LUMO gap for 1 (ΔE = 2.82 eV) than 2 (ΔE = 2.73 eV) (Fig. S7†), the absorption onset of 2 is also red-shifted by 99 nm (3.31 × 103 cm−1) compared to that of 1. The UV-vis absorption spectrum of complex 2 in CH2Cl2 solution exhibits intense absorption bands extending into the visible region (around 450 nm), as observed by Ertl et al.12 The observed red-shift of 149 nm of the lowest energy transition of complex 2 in comparison to the study of Ertl et al. is presumably because of the solvent polarity where the MeCN used in this study stabilises both Frontier MOs, perhaps more pronounced for the LUMO. In comparison to R1 (lowest-energy λabsmax = 420 nm),16 both complexes show significantly red-shifted absorption spectra.
The steady-state emission spectra of 1 and 2 were recorded in degassed MeCN at 298 K and are shown in Fig. S9 (ESI†). The photophysical data are summarised in Table 1. The emission profiles are broad and featureless, indicative of mixed 3CT emission, as also observed by Ertl et al.12 The spin density distribution of the excited-state corroborates this assignment (Fig. S9 and S10†). The emission maximum (λemmax = 744 nm) of complex 2 is red-shifted by 86 nm (1.76 × 103 cm−1) compared to that of complex 1 (λemmax = 658 nm). The DFT predicted emission maxima for 1 (λemDFT = 712 nm) and 2 (λemDFT = 761 nm) also follow the observed trend in emission maxima of complexes 1 and 2, with relative errors of 7.6% and 2.2%, respectively. The emission maxima of 1 and 2 are significantly red-shifted compared to that of R1 (λemmax = 602 nm).16 In comparison to the observed emission maximum at 686 nm in CH2Cl2 solution of complex 2 by Ertl et al.,12 the same for complex 2 is red-shifted by 58 nm in our study, which is because of the effect of solvent polarity in emission.18 With the exception of the cationic Ir(III) complex bearing 3-methyl-2-phenylbenzo[g]quinolinato C^N ligands reported by Tao et al.7a (Chart 1a), complex 2 is, to the best of our knowledge, the most red-shifted emitting cationic Ir(III) complex.7a,9–11 The ΦPL in degassed MeCN of 1 and 2 are 1.7% and 0.4%, respectively (Table 1). The corresponding excited-state lifetimes, τe, are very short at 81 and 44 ns, for 1 and 2, respectively. The lower ΦPL and shorter τe values compared to R1 are a stark consequence of energy-gap law as evidence by the associated ∼4-fold (for 1) and 7-fold (for 2) increase in the non-radiative decay rate constant, knr, compared to that in R1. The observed ΦPL and τe values of complex 2 in CH2Cl2 solution, reported by Ertl et al., are 3.6% and 126 ns, respectively.12 The lower ΦPL and τe values in our study compared to the study of Ertl et al. is again due to a consequence of energy-gap law for a NIR emitter like 2.
The emission maxima of the thin films of 1 and 2 mixed with 1-butyl-3-methylimidazolium hexafluorophosphate, [Bmim][PF6] are 658 and 707 nm, respectively (λexc = 320 nm). The thin film ΦPL values of 14% and 5% for 1 and 2, respectively, are enhanced compared to solution values. The PL spectra and photophysical data of the films are displayed in Fig. S11 and Table S9 (ESI†).
Light emitting electrochemical cells (LEECs) were prepared and the electroluminescence properties of the complexes were evaluated. The preparation of LEECs were done by spin-coating on pre-cleaned ITO substrates, which were first coated with a 80 nm thick film of PEDOT:PSS. The active layer was then deposited from dichloromethane solution. Recently, a first attempt was done for the LEEC fabrication with complex 2.12 At that time, the bad film morphology led to poor LEEC performance. In this work, a different solvent was used to improve the film morphology of the emissive layer. Here, the complex was dissolved in dichloromethane, which led to good films completely covering the substrates after spin-coating. After the active layer deposition, the top electrode (70 nm thick film of aluminium) was thermally evaporated. The fabricated LEECs were measured using a pulsed current driving (1 kHz, 50% duty cycle and block wave). The current densities applied to the LEECs were 800 and 1600 A m−2, which corresponds to an average current density of 400 and 800 A m−2, respectively, as a function of the duty cycle. For simplicity, the LEECs fabricated with 1 and 2 are called LEEC-1 and LEEC-2.
The luminance for LEEC-1 or light output for LEEC-2 and average voltage versus time measured under average current density of 400 A m−2 is depicted in Fig. 1 while the measurements at 800 A m−2 are shown in Fig. S12 (ESI†). In the inset of Fig. 1, the electroluminescence (EL) spectrum of each LEEC is shown and all the device data are summarized in Table S11 for LEEC-1 and Table S12 for LEEC-2, respectively (ESI).† LEEC-1 exhibits a red electroluminescence centred at 661 nm while LEEC-2 shows a near-infrared emission at 705 nm, in both cases, in good agreement with the PL spectra registered in thin film (658 nm for 1 and 707 nm for 2). The applied voltage rapidly diminishes due to the ionic motion over operation, which decreases the electrical resistance. As this characteristic is directly related with the ionic mobility, generally, the time to reach the maximum light emission (tmax) in LEECs varies from seconds to hours. Despite the similarity structure of complexes 1 and 2 as well as the presence of the same counter-anion (PF6), tmax is significantly shorter for LEEC-1 (<5 seconds) than LEEC-2 (0.8 and 4.3 hours for 400 and 800 A m−2, respectively). Moreover, differences are observed between LEEC-1 and LEEC-2 when comparing the device lifetime, usually defined as the time to reach one-half of the maximum light emission (t1/2). While LEEC-1 shows a t1/2 of 0.3 hours, LEEC-2 was significantly more stable at above 80 hours. The differences observed seem to be related to the presence of the annulated benzenes in the ancillary ligand. A possible explanation is related to the larger cation in complex 2, which reduces the ionic movement in the active layer during operation. LEEC-2 exhibits an improved efficiency compared to that based on 1. While LEEC-1 achieved an external quantum efficiency (EQE) of 0.23% and 0.13%, LEEC-2 reached 0.37% and 0.30% at 400 and 800 A m−2, respectively. Hence, the efficiency of LEEC-1 is more strongly affected by increasing the current density than LEEC-2. Thus, the slower ionic movement in LEEC-2 leads to more balanced carrier. To the best of our knowledge, only a few examples of near-infrared emitting LEECs have been reported, generally with low EQE (EQE < 0.1%)6 due to the energy gap law. Recently, new examples have overcome the EQE of 1%,5d,21 but none of these is based on a cationic iridium complex as the emitter. Considering the low emission quantum yield of complex 2 (0.4%) and the high current density applied (up to 800 A m−2) the achieved efficiencies for LEEC-2 are very impressive and close to the theoretical achievable one assuming a typical light outcoupling of 20%.
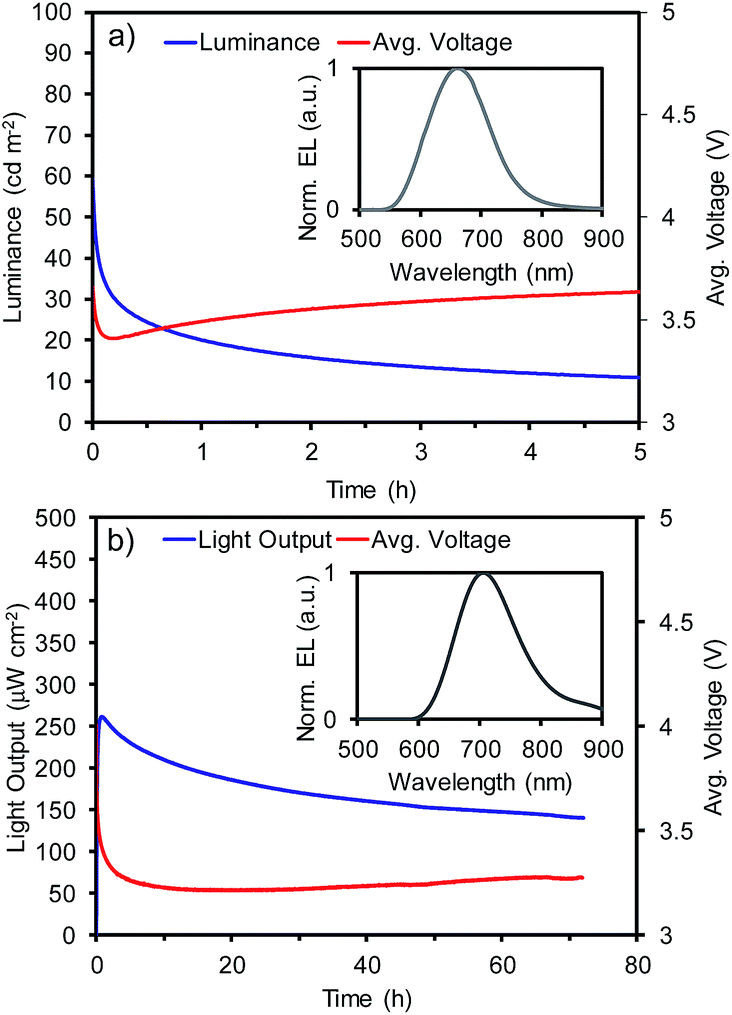 | ||
| Fig. 1 Device performance versus time and electroluminescence (inset) for (a) LEEC-1 and (b) LEEC-2 under a pulsed current of 400 A m−2 (average current density), 1 kHz and 50% of duty cycle. | ||
In summary, we demonstrate dramatic red-shifted emission for both 1 and 2 as a function of the use of strongly π-accepting bithiazole-type ancillary ligands compared to the archetype complex R1. Notably, to the best of our knowledge, complex 2 displays the most red-shifted emission maximum for a cationic iridium complex, especially considering the simplicity of the ligand structure and synthesis. LEEC devices show deep red and NIR emission using 1 and 2, respectively. LEEC-2 shows improved efficiency compared to LEEC-1 and represents the first example of a NIR LEEC employing a cationic iridium complex emitter. Current efforts are focussed on pushing the emission even further into the near-IR through addition of strongly conjugated C^N ligands.
Acknowledgements
We are grateful to the University of St Andrews and EPSRC for financial support from grant EP/M02105X/1. We thank the EPSRC UK National Mass Spectrometry Facility at Swansea University for analytical services. We thank Umicore AG for the gift of IrCl3. We also thank the Spanish Ministry of Economy and Competitiveness (MINECO) via the Unidad de Excelencia María de Maeztu MDM-2015-0538, MAT2014-55200, CTQ2015-71154-P and PCIN-2015-255, the Generalitat Valenciana (Prometeo/2016/135) and European FEDER funds (CTQ2015-71154-P). C. M. thanks MINECO for her predoctoral contract.References
- (a) A. Barbieri, E. Bandini, F. Monti, V. K. Praveen and N. Armaroli, Top. Curr. Chem., 2016, 374, 47 CrossRef PubMed; (b) A. F. Henwood and E. Zysman-Colman, Chem. Commun., 2017, 53, 807–826 RSC; (c) J. Yellol, S. A. Perez, G. Yellol, J. Zajac, A. Donaire, G. Vigueras, V. Novohradsky, C. Janiak, V. Brabec and J. Ruiz, Chem. Commun., 2016, 52, 14165–14168 RSC; (d) G. Qian and Z. Y. Wang, Chem.–Asian J., 2010, 5, 1006–1029 CrossRef CAS PubMed.
- C. Wang, X. Gao and X. Su, Anal. Bioanal. Chem., 2010, 397, 1397–1415 CrossRef CAS PubMed.
- S. V. Eliseeva and J. C. Bunzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- (a) E. Zysman-Colman, S. S. Ghosh, G. Xie, S. Varghese, M. Chowdhury, N. Sharma, D. B. Cordes, A. M. Z. Slawin and I. D. W. Samuel, ACS Appl. Mater. Interfaces, 2016, 8, 9247–9253 CrossRef CAS PubMed; (b) J. O. Escobedo, O. Rusin, S. Lim and R. M. Strongin, Curr. Opin. Chem. Biol., 2010, 14, 64–70 CrossRef CAS PubMed; (c) O. A. Melville, B. H. Lessard and T. P. Bender, ACS Appl. Mater. Interfaces, 2015, 7, 13105–13118 CrossRef CAS PubMed.
- (a) A. K. Pal, S. Serroni, N. Zaccheroni, S. Campagna and G. S. Hanan, Chem. Sci., 2014, 5, 4800–4811 RSC; (b) H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128–6185 RSC; (c) D. A. Ross, P. A. Scattergood, A. Babaei, A. Pertegas, H. J. Bolink and P. I. Elliott, Dalton Trans., 2016, 45, 7748–7757 RSC; (d) J. H. Hsu and H. C. Su, Phys. Chem. Chem. Phys., 2016, 18, 5034–5039 RSC; (e) B. N. Bideh, C. Roldan-Carmona, H. Shahroosvand and M. K. Nazeeruddin, J. Mater. Chem. C, 2016, 4, 9674–9679 RSC.
- A. F. Henwood and E. Zysman-Colman, Top. Curr. Chem., 2016, 374, 36 CrossRef PubMed.
- (a) R. Tao, J. Qiao, G. Zhang, L. Duan, L. Wang and Y. Qiu, J. Phys. Chem. C, 2012, 116, 11658–11664 CrossRef CAS; (b) D. Ma, T. Tsuboi, Y. Qiu and L. Duan, Adv. Mater., 2017, 29, 1603253 CrossRef PubMed.
- The reported complex is [(ppy-F2)2Ir(dtBubpy)]+, where ppy-F2 is 2,4-difluorophenylpyridinato. H. J. Bolink, E. Coronado, R. n. D. Costa, N. Lardiés and E. Orti, Inorg. Chem., 2008, 47, 9149–9151 CrossRef CAS PubMed.
- (a) K. Hasan, A. K. Bansal, I. D. W. Samuel, C. Roldán-Carmona, H. J. Bolink and E. Zysman-Colman, Sci. Rep., 2015, 5, 12325 CrossRef CAS PubMed; (b) A. M. Bunzli, H. J. Bolink, E. C. Constable, C. E. Housecroft, J. M. Junquera-Hernandez, M. Neuburger, E. Orti, A. Pertegas, J. J. Serrano-Perez, D. Tordera and J. A. Zampese, Dalton Trans., 2014, 43, 738–750 RSC.
- (a) L. Donato, C. E. McCusker, F. N. Castellano and E. Zysman-Colman, Inorg. Chem., 2013, 52, 8495–8504 CrossRef CAS PubMed; (b) J. L. Rodriguez-Redondo, R. D. Costa, E. Orti, A. Sastre-Santos, H. J. Bolink and F. Fernandez-Lazaro, Dalton Trans., 2009, 9787–9793 RSC; (c) D. Ma, L. Duan and Y. Qiu, J. Mater. Chem. C, 2016, 4, 5051–5058 RSC.
- (a) Y. Li, N. Dandu, R. Liu, Z. Li, S. Kilina and W. Sun, J. Phys. Chem. C, 2014, 118, 6372–6384 CrossRef CAS; (b) A. M. Bünzli, E. C. Constable, C. E. Housecroft, A. Prescimone, J. A. Zampese, G. Longo, L. Gil-Escrig, A. Pertegás, E. Ortí and H. J. Bolink, Chem. Sci., 2015, 6, 2843–2852 RSC; (c) X. Zhu, L. Lystrom, S. Kilina and W. Sun, Inorg. Chem., 2016, 55, 11908–11919 CrossRef CAS PubMed.
- C. D. Ertl, C. Momblona, A. Pertegás, J. M. Junquera-Hernández, M.-G. La-Placa, A. Prescimone, E. Ortí, C. E. Housecroft, E. C. Constable and H. J. Bolink, J. Am. Chem. Soc., 2017, 139, 3237–3248 CrossRef CAS PubMed.
- B. Fu, C.-Y. Wang, B. D. Rose, Y. Jiang, M. Chang, P.-H. Chu, Z. Yuan, C. Fuentes-Hernandez, B. Kippelen, J.-L. Brédas, D. M. Collard and E. Reichmanis, Chem. Mater., 2015, 27, 2928–2937 CrossRef CAS.
- R. D. Costa, E. Ortì, H. J. Bolink, S. Graber, S. Schaffner, M. Neuburger, C. E. Housecroft and E. C. Constable, Adv. Funct. Mater., 2009, 19, 3456–3463 CrossRef CAS.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- For examples see: S. Ladouceur, D. Fortin and E. Zysman-Colman, Inorg. Chem., 2011, 50, 11514–11526 CrossRef CAS PubMed.
- Y.-C. Chiu, J.-Y. Hung, Y. Chi, C.-C. Chen, C.-H. Chang, C.-C. Wu, Y.-M. Cheng, Y.-C. Yu, G.-H. Lee and P.-T. Chou, Adv. Mater., 2009, 21, 2221–2225 CrossRef CAS.
- M. A. Haidekker, T. P. Brady, D. Lichlyter and E. A. Theodorakis, Bioorg. Chem., 2005, 33, 415–425 CrossRef CAS PubMed.
- H. Ishida, S. Tobita, Y. Hasegawa, R. Katoh and K. Nozaki, Coord. Chem. Rev., 2010, 254, 2449–2458 CrossRef CAS.
- S. Ladouceur and E. Zysman-Colman, Eur. J. Inorg. Chem., 2013, 2013, 2985–3007 CrossRef CAS.
- (a) C.-C. Ho, H.-F. Chen, Y.-C. Ho, C.-T. Liao, H.-C. Su and K.-T. Wong, Phys. Chem. Chem. Phys., 2011, 13, 17729–17736 RSC; (b) C.-L. Lee, C.-Y. Cheng and H.-C. Su, Org. Electron., 2014, 15, 711–720 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic, crystallographic, optoelectronic and DFT studies. CCDC 1538393 and 1538394. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra06347d |
| This journal is © The Royal Society of Chemistry 2017 |