
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChicoric acid supplementation ameliorates cognitive impairment induced by oxidative stress via promotion of antioxidant defense system
Yutang Wang†
,
Zhijun Diao†,
Jing Li,
Bo Ren,
Di Zhu,
Qian Liu,
Zhigang Liu and
Xuebo Liu *
*
Laboratory of Functional Chemistry and Nutrition of Food, College of Food Science and Engineering, Northwest A&F University, 28. Xi-nong Road, Yangling 712100, China. E-mail: xueboliu@aliyun.com; Fax: +86-29-87092325; Tel: +86-29-87092325
First published on 21st July 2017
Abstract
Chicoric acid, a nutritional component found in the Mediterranean vegetable chicory, possesses multiple biological properties including antioxidant activities, anti-obesity effects, anti-HIV, and others. However, there have been very few studies of its effects to counter oxidative stress-induced cognitive loss. To address this, the goal of this study was to investigate the effects of chicoric acid on oxidative stress induced both in the brain of C57BL/6J mice and in SH-SY5Y cells. The C57BL/6J mice induced by D-galactose (D-gal; 300 mg kg−1 d−1 body weight, intraperitoneal injection for 2 months) and SH-SY5Y cells induced by H2O2 were used to evaluate protective effects of chicoric acid (100 mg kg−1 d−1 body weight in drinking water for 8 weeks). The learning and memory abilities were assessed using the Y-maze and Morris maze tests. Chicoric acid attenuated neuron damage in D-gal-treated mice as revealed through histological examination in the hippocampus region of the mouse brain. The levels of inflammatory mediators, such as TNF-α and IL-1β, as well as malondialdehyde levels, were markedly reduced after chicoric acid treatment. In contrast, the activity of CAT and the level of GSH were significantly elevated in serum by chicoric acid. The results also revealed that chicoric acid treatment noticeably activated the Nrf2 antioxidative defense system by up-regulation of downstream antioxidant enzyme expression both in the animal and the cell models. The chicoric acid regulation mechanism may be associated with balancing cellular redox status, reversing mitochondrial dysfunction, decreasing inflammation, and neuron apoptosis caused by oxidative stress. These results suggested that chicoric acid supplementation ameliorated cognitive impairment induced by D-gal and SH-SY5Y cell apoptosis induced by H2O2 via promotion of the Keap1/Nrf2 signal pathway and its downstream antioxidant enzymes, indicating that chicoric acid has great potential for use as part of a nutritional preventive strategy to counter oxidative stress-related cognitive impairment.
1. Introduction
Over the past two decades, oxidative stress has been identified as a pivotal mediator in neurodegenerative disease and in the normal aging process.1–3 Studies have described the reactive oxygen species (ROS)-induced oxidative modification of biomolecules, such as cellular lipids, proteins, or DNA damage, which impairs normal cellular activity. These modifications deregulate the homeostatic system of neurons, ultimately leading to neuronal cell death in several neurodegenerative disorders,4,5 including Alzheimer's disease (AD), Parkinson's disease (PD) and amyotrophic lateral sclerosis (Lou Gehrig's disease). Thus, the pathogenesis of many neurodegenerative diseases is thought to be associated with oxidative stress owing to the accumulation of ROS. These ROS effects on neuronal deregulation factors also contribute to the pathology of stroke,6 characterized by hippocampal neurogenesis impairments, deficits in cognitive function, and difficulties remembering newly acquired information.7,8A major mechanism in the cellular defense against oxidative stress is activation of the Nrf2-antioxidant response element signaling pathway, which controls the expression of genes whose protein products are involved in the detoxification and elimination of reactive oxidants by enhancing cellular antioxidant capacity.9 The redox-sensitive transcription factor nuclear factor erythroid 2 p45 (NF-E2)-related factor (Nrf2) upregulates peroxiredoxins and phase II detoxification enzymes,10 including hemeoxygenase-1 (HO-1), NAD(P)H:quinone oxidoreductase-1 (NQO1), and thioredoxins (Trxs), as well as antioxidant enzymes, including superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase (CAT). Thus Nrf2 exerts a variety of cytoprotection effects, including anti-inflammatory response and anti-carcinogenicity.11
Preclinical and clinical studies using in vitro and in vivo models suggest that dietary supplementation with natural antioxidants, such as polyphenols, exerts potent neuroprotective effects, relieving brain aging and neurodegenerative diseases.12–15 Polyphenols contain one or more phenol groups. For instance, phenolic acids hold one carboxylic acid function and two different carbon frameworks, giving rise to the hydroxycinnamic (e.g., p-coumaric, caffeic, ferulic, and sinapic acids) and hydroxybenzoic structures.16 Their structure greatly enhances the antioxidant activity. Flavonoids are, therefore, particularly potent antioxidants.17 Chlorogenic acid and caffeic acid have been reported to scavenge superoxide anion free radicals in vitro.18 Caffeic acid significantly reduced glutathione depletion and inhibited lipid peroxidation during tert-butyl hydroperoxide treatment in U937 human monocytic cells.19 Studies have also found that rutin increased the GSH/GSSG ratio and SOD activity and reduced GSSG and MDA levels in AD transgenic mice.20 Chicoric acid is a natural dicaffeyltartaric acid and is found in a large number of edible plants and vegetables, including the Mediterranean vegetable chicory, Cichorium intybus L., dandelion, basil, iceberg lettuce, Echinacea purpurea, and Orthosiphon stamineus Benth.21–26 Chicoric acid has strong free radical scavenging and antioxidant activities.27–29 Our previous study reported that chicoric acid and, to a lesser extent, its metabolites, caffeic acid and caftaric acid, inhibited DPPH˙, ˙OH, and ABTS˙+ free radicals.30 A recent study found that chicoric acid could significantly inhibit obesity induced by a high fat diet in C57BL/6J mice by enhancing the body's antioxidant defense system.31 Liu et al. illustrated that chicoric acid attenuated inflammatory responses via redox-sensitive signaling, including Nrf2 pathways in LPS-stimulated neuroinflammation in BV2 microglial cells.32 They also illustrated that chicoric acid could alleviate systemic inflammation-induced amyloidogenesis and cognitive deficits by preventing neuron damage, suppressing glia activation, and down-regulating inflammatory responses in CNS.33 In addition, chicoric acid was shown to distribute rapidly in various tissues after gavage administration and was able to cross the blood–brain barrier.34 Based on these significant conclusions, we hypothesized that chicoric acid has great potential as part of a nutritional preventive strategy to inhibit oxidative stress-induced aging-/AD-related cognitive impairment in the brain.
Therefore, the aim of this study was to evaluate the effects of chicoric acid on oxidative stress in vivo and in vitro by: (a) investigating the effects of chicoric acid on cognitive deficits and neuron damage; (b) determining effects of chicoric acid on D-gal-elicited redox status imbalance including inflammatory cytokine generation and on effects on the levels of antioxidant enzymes in mice brain; and (c) examining effects of chicoric acid on H2O2-induced SH-SY5Y cell viability and mitochondria function through regulation of the Keap1/Nrf2 antioxidative defense signaling pathway. This study should improve our understanding of the inhibitory effects of chicoric acid on cognitive impairment induced by D-galactose, clarify its antioxidant mechanisms, and provide the scientific basis for the design and development of promising functional foods and formulations based on chicoric acid to prevent oxidative stress-induced memory impairment. Fig. 8 illustrates the effects of chicoric acid on neuroprotection against D-gal-induced memory impairment via inflammation and oxidative stress.
2. Materials and methods
2.1. Animals and treatments
Two-month-old male C57BL/6J mice were purchased from Beijing Vital River Laboratory Animal Technology Company (Beijing, China) and maintained in the animal facility under standard conditions (temperature 22 ± 2 °C, 12/12 h light–dark cycle, and humidity 50 ± 15%) with food provided ad libitum. After one week of adaptation, mice were randomly divided into three groups (n = 10/group): (1) control group (vehicle control): physiological saline (intraperitoneally injected, i.p.) + purified water without chicoric acid; (2) D-gal group: D-gal (i.p.) + purified water without chicoric acid; or (3) D-gal + chicoric acid group: D-gal (i.p.) + chicoric acid (100 mg per kg per day in drinking water). The concentration of chicoric acid (purity ≥ 98%) (Fig. 1A), purchased from Weikeqi Biological Technology Co., Ltd (Sichuan, China), was 100 mg kg−1 for the in vivo experiments based on previous studies.35 Mice in the D-gal group and the D-gal + chicoric acid group were intraperitoneally injected (i.p.) with D-gal (in physiological saline, 150 mg mL−1) at a volume of 2 mL kg−1 body weight for a daily dose of 300 mg kg−1 body weight; mice in the control group were treated with the same volume of physiological saline (2 mL kg−1 body weight) daily. After injection, mice in the chicoric acid treatment group drank freely and received chicoric acid AT 100 mg kg−1 body weight once daily via oral administration. At the end of the treatment, the mice were sacrificed after being anaesthetised with 10% chloralichydras (in physiological saline, 4 mL kg−1 body weight). The brains of the mice were carefully and quickly removed and put into cold physiological saline. The hippocampi were immediately separated from the cerebra on a cold plate and stored in 4% phosphate-buffered paraformaldehyde (in 0.1 M phosphate buffer, pH 7.4). The rest of the brain was stored at −80 °C for subsequent experiments (Fig. 1B). Blood samples were clotted for 1 hour and obtained by removal of the mouse eyeball, then subjected to centrifugation at 3000 rpm for 10 min at 4 °C. The animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals: Eighth Edition, ISBN 310: 0330931539634, and approved by the Northwest A&F University Animal Care and Use Committee (Yangling, Shaanxi, China). Every effort was made to minimize animal suffering.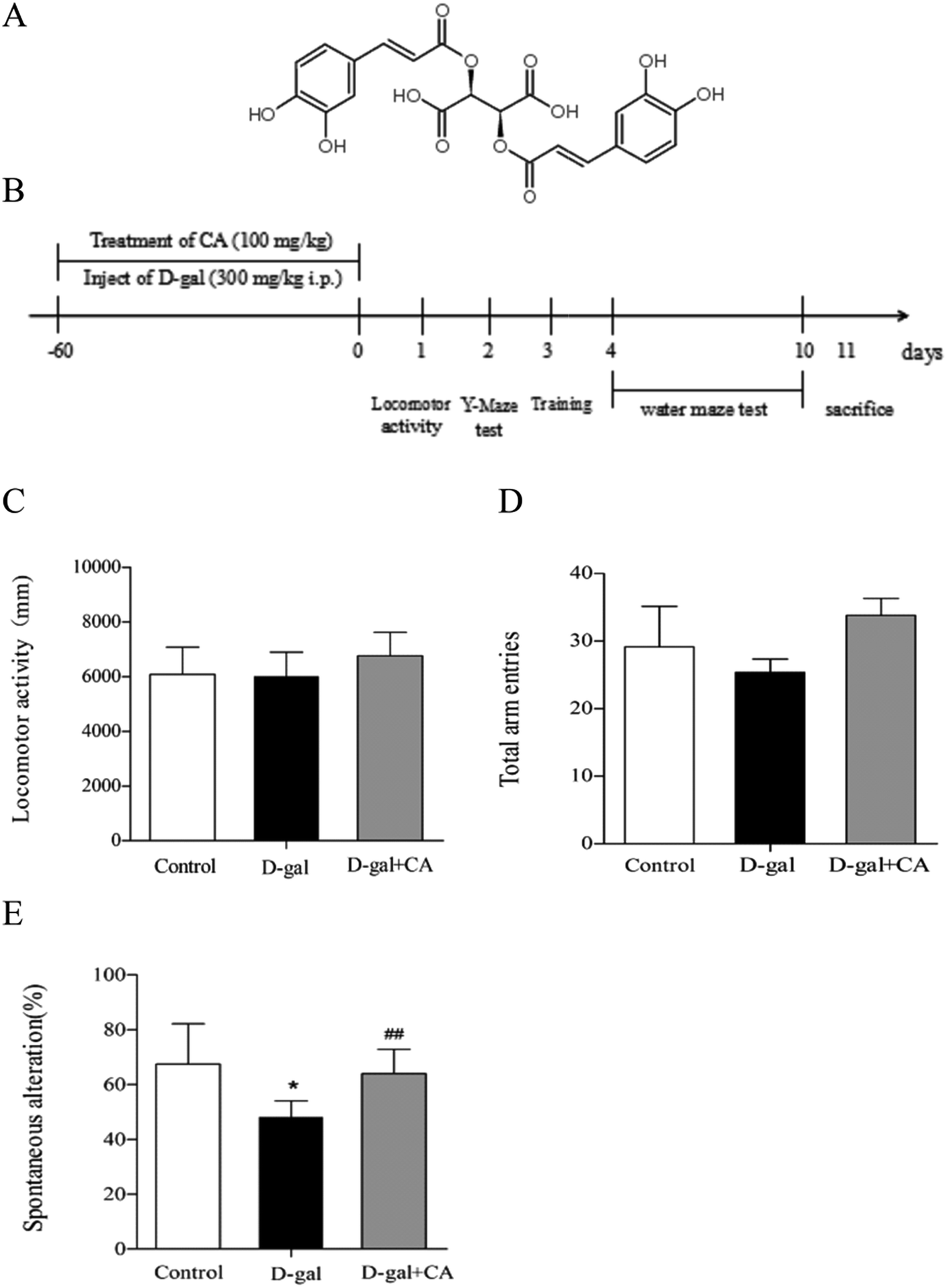 | ||
| Fig. 1 Timeline illustrating chicoric acid (CA) treatment and assessments of cognitive functions of mice. (A) The chemical structure of chicoric acid (CA). (B) Experimental scheme for effect of chicoric acid against D-gal-induced memory impairment. Male C57BL/6J mice were treated with CA (100 mg per kg per day) in drinking water for 8 weeks. D-Gal (300 mg kg−1) was administered intraperitoneally once daily for 2 months. The animals performed assessments of cognitive functions. (C) Effect of CA on the locomotor activity of mice. Effect of CA on the (D) number of total arm entries and (E) spontaneous alternation in the Y-maze task of mice. Data presented as mean ± SEM, n = 10. *p < 0.05, **p < 0.01, versus control group, #p < 0.05, ##p < 0.01 versus D-gal group. | ||
2.2. Open-field test
An open field (36 cm × 36 cm) was constructed by enclosure of an area of tiled floor with 40 cm high plywood planks, with the floor divided into 16 equal squares by red lines. The animals were carefully placed in the center of the open field and their behavior was recorded for 5 min. To investigate the effects of any treatment on spontaneous locomotor activity, the distances of horizontal (crossings) performed by each mouse during the observation period were counted using a video tracking system (Super Maze software; Shanghai Xinruan Information Technology, Shanghai, China).2.3. Y-maze task
Each arm of the maze was 35 cm long, 15 cm high, and 5 cm wide at the bottom and 10 cm wide at the top. Each mouse was placed at the center of the apparatus and allowed to move freely through the maze for 8 min. The series of arm entries was visually observed. Spontaneous alteration was defined as the successive entry of the mouse into the three arms of overlapping triplet sets. The percentage spontaneous alteration (%) was calculated as [successive triplet sets (entries into three different arms consecutively)/total number of arm entries − 2] × 100%. All data were recorded using video tracking software (XR-XM101, Shanghai Xinruan Information Technology Co. Ltd, Shanghai, China).2.4. Morris water maze (MWM) test
The MWM test was used to evaluate memory functions and was performed as described previously with some modifications.34 The experimental apparatus consisted of a circular water tank (150 cm in diameter, 35 cm in height), containing water (23 ± 1 °C) to a depth of 15.5 cm, which was rendered opaque by addition of white, non-toxic, food-grade titanium dioxide. The pool was divided into four quadrants of equal area. A transparent escape platform (4.5 cm in diameter, 14.5 cm in height) was hidden 1 cm under the water surface and placed at the midpoint of one quadrant. Four prominent visual cues were located on each side of four quadrants of the pool. The swimming route of the mice, from the start position to the platform, was monitored and analyzed by a video tracking system (Super Maze software, Shanghai Information Technology, Co. Ltd, China). Each mouse received training per day for six consecutive days using a single hidden platform in one quadrant with three quadrants of rotational starting. The ability to escape from the water maze (finding the submerged escape platform) was calculated for each trial. If animals did not locate the platform within 60 s, the experimenter guided the mouse by hand to the platform, then the mouse was kept on the escape platform for 30 s and an escape latency of 60 s was recorded. On the seventh day, a probe test was performed to evaluate memory consolidation. In the probe test, the platform was removed and each mouse was allowed to swim freely for 60 s. The amount of time spent in the target quadrant (where the platform was located during hidden platform training) was measured. All data were recorded using video tracking software (XR-XM101, Shanghai Xinruan Information Technology Co. Ltd, Shanghai, China).2.5. Hematoxylin-eosin and immunohistochemical staining
The brain tissue samples were fixed in 4% (v/v) paraformaldehyde/PBS, embedded in paraffin, cut into 5 μm sections using a microtome, and dried overnight. The brain sections were dehydrated in decreasing concentrations of ethanol (100, 90, 80, and 70% ethanol) for 5 min in each grade, followed by three washes in PBS (pH 7.4) for each 5 min. For hematoxylin-eosin (HE) staining, the brain slides were stained with HE. For immunohistochemistry (IHC) staining, the Tris–EDTA Buffer Epitope Retrieval Method was used to retrieve the antigen.36 The slice was then treated with 3% H2O2 for 10 min to eliminate endogenous peroxidase. Sections were then rinsed with PBS. The brain sections were blocked for 20 min in normal goat serum blocking solution to block the non-specific binding of immunoglobulin. Sections were incubated overnight at 4 °C with appropriate primary antibodies: BDNF (SC-2546, Thermo Fisher Scientific, Waltham, MA, USA) or β-amyloid (1-42 specific) (Aβ1-42) (1:1600; Cell Signaling Technology, Danvers, MA, USA) and for 30 min at room temperature with secondary antibodies. Samples were then rinsed in PBS for 3 × 5 min. Brain sections were washed and visualized by chromogen DAB reaction (DAB kit, Zhongshan Golden Bridge biotechnology Co. Ltd, Beijing, China) for up to 10 min. Finally, these sections were counterstained with hematoxylin. Brain sections were dehydrated in ethanol, cleared in xylene, mounted with Permount, and evaluated by light microscopy (Olympus IX71, Tokyo, Japan; × 200).2.6. Measurement of activities of antioxidant enzymes and proinflammatory cytokines
The levels of GSH and MDA and the enzyme activities of CAT in mice serum were assayed using kits purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). MPO, TNF-α, and lL-1β levels were measured using ELISA kits (Mouse MPO kit, Mouse TNF-α kit, Mouse IL-1β kit; Xinle Biology Technology).2.7. RNA preparation and RT-qPCR
Total RNA was extracted from brain tissue using the RNA Extraction Kit (TaKaRa MiniBEST Universal RNA Extraction Kit, Dalian, China) as previously described.37 For quality control, the RNA purity and integrity were evaluated using a Quawell 5000 UV-vis Spectrophotometer (Quawell Technology, San Jose, CA, USA). RNA was stored at −80 °C prior to further analysis by microarray and real-time quantitative PCR (RT-qPCR).Total RNA was reverse transcribed into cDNA using the PrimeScript™ RT Master Mix reverse transcription kit (TaKaRa PrimeScript RT Master Mix, Dalian, China), and the mRNA expression was quantified by RT-qPCR using the SYBR green PCR kit (TaKaRa SYBR® Premix Ex Taq™ II, Dalian, China) and the CFX96™ real time system (Bio-Rad, Hercules, CA). Gene-specific mouse primers were used as listed in Table 1. Ct values were normalized to GAPDH, and the relative gene expression was calculated with the 2−ΔΔCt method.
| Forward primer | Reverse primer | |
|---|---|---|
| Nqo1 | GGATTGGACCGAGCTGGAA | AATTGCAGTGAAGATGAAGGCAAC |
| Ho-1 | ATGTGGCCCTGGAGGAGGAGA | CGCTGCATGGCTGGTGTGTAG |
| Gapdh | TGGAGAAACCTGCCAAGTATGA | TGGAAGAATGGGAGTTGCTGT |
2.8. Cell culture
The human neuroblastoma SH-SY5Y cell line was provided by the cell bank of the Chinese Academy of Science (http://www.cellbank.org.cn) and cultured in High-DMEM (Hyclone Co., USA) with 10% fetal bovine serum (FBS), 100 IU mL−1 penicillin, and 100 μg mL−1 streptomycin at 37 °C in a humidified atmosphere with 5% CO2. The cultured cells were treated with 100 μmol L−1 H2O2 and various concentrations of chicoric acid (0, 10, 25, and 50 μM) for 24 h. The cells were then collected for further analysis.2.9. Measurement of cell viability
Cell viability was determined by the MTT assay. Cells were seeded at a density of 1 × 106 cells per mL in 96-well polystyrene culture plates at 37 °C with 5% (v/v) CO2. After various treatments, the medium was removed and the cells were incubated with 0.5% (w/v) MTT for 4 h at 37 °C. The formazan crystals formed by live cells were solubilized by addition of 100 μL of dimethyl sulfoxide and the absorbance at 490 nm was measured with a microplate reader (Bio-Rad Laboratories, China). Cell viability was expressed as a percentage of the control group (untreated cells).2.10. Detection of intracellular redox status
The redox status of the SH-SY5Y cells was detected by fluorescence dye H2DCFDA.35 Cells were incubated with 100 μM H2O2 for 24 h, followed by treatment with chicoric acid for 24 h, then 10 μM H2DCFDA dye, diluted into serum free medium, was added. After incubation for 30 min at 37 °C, cells were washed with PBS three times, and an inverted fluorescence microscope was used to observe the cellular oxidation status (Olympus IX71, Tokyo, Japan; × 200). ROS levels were measured according to the method of Xiao et al.38 Fluorescence levels were observed by using an inverted fluorescence microscope and quantified using a multimode microplate reader at 485 nm excitation and 538 nm emission wavelengths.2.11. Analysis of mitochondrial membrane potential (Δψ)
Mitochondrial membrane potential (Δψ) was determined using the mitochondrion-specific lipophilic cationic fluorescence dye JC-1. After the specific treatment described above, cells were treated with 5 μg mL−1 JC-1 at 37 °C for 30 min and were washed twice with PBS and observed by fluorescence microscope (Olympus IX71, Tokyo, Japan). Fluorescence intensity was separately measured using a multimode microplate reader (Molecular Devices Co., Sunnyvale, CA, USA) at 485 nm excitation and 585 or 538 nm emission wavelengths. The values are expressed as the OD585/OD538 ratio.2.12. SDS-PAGE and western blot analysis
The brains of the mice were homogenized with ice-cold normal sodium to form the brain homogenate. The homogenate was then centrifuged at 4000 × g for 10 min and the supernatant was used for subsequent tests. The treated SH-SY5Y cells were harvested and lysed with cell lysis buffer (Beyotime Institute of Biotechnology, Jiangsu, China) and nuclear extraction reagent (Beyotime Institute of Biotechnology, Jiangsu, China) to yield a cytosolic extract (cytosol) and nuclear extract (nucleus). Brain tissue homogenates or cell lysates were solubilized in SDS sample buffer and then heated at 95 °C for 10 min. The protein concentration in the samples was measured using a commercially available BCA protein assay kit. Samples were separated by SDS-PAGE and transferred onto PVDF membranes. Using appropriate antibodies, the immune reactive bands were visualized with an enhanced chemiluminescence reagent. Antibodies against lamin B (SC-6217), HO-1 (SC-1796), NQO-1 (SC-16464), Nrf2 (SC-722), and GAPDH (SC-225778) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).2.13. Statistical analysis
All experiments were performed at least three times and the data are presented as the means ± SEM. Significant differences between mean values were determined by one-way ANOVA followed by student t-test by GraphPad Prism 16.0 software. All p-values less than 0.05 were considered statistically significant.3. Results
3.1. The locomotor activity and Y-maze test
The chemical structure of chicoric acid is shown in Fig. 1A. The behavior tests were performed as scheduled (Fig. 1B). As shown in Fig. 1, the locomotor activity (Fig. 1C) and the behavioral performance of mice in the Y-maze task did not significantly differ among the groups for the number of total arm entries (Fig. 1D), suggesting that treatment with D-gal or chicoric acid did not affect mice motor activities. However, the behavioral performance of mice in the Y-maze task differed. The D-gal-treated mice exhibited decreased spontaneous alternation compared with the control group, showing less working memory. Chicoric acid treatment significantly increased the spontaneous alternation in the D-gal-treated mouse group compared to the mice that did not receive chicoric acid treatment (D-gal + chicoric acid group vs. the D-gal-treated group, Fig. 1E).3.2. The Morris water maze test
The effects of chicoric acid on spatial learning and memory in D-gal mice were analyzed using the Morris water maze test (MWM). Fig. 2A shows the observed decreased swimming speed in D-gal-treated mice compared with that of the saline-treated group on day 5. Interestingly, chicoric acid significantly increased swim speeds in the D-gal + chicoric acid group after day 3, restoring the swimming speed to that of the control group. Similarly, there was a significant difference in the escape distance for the different groups (Fig. 2B). Mice that received 100 mg kg−1 of chicoric acid used a shorter route to find the platform on testing day 5 than mice in the D-gal group, similar to the ability of mice in the control group for escape distance. As shown in Fig. 2C, compared to the control group, mice in the D-gal group took a longer time to find the platform on testing days 3–5. Meanwhile, supplementation with chicoric acid significantly decreased the escape latency time in D-gal-treated mice.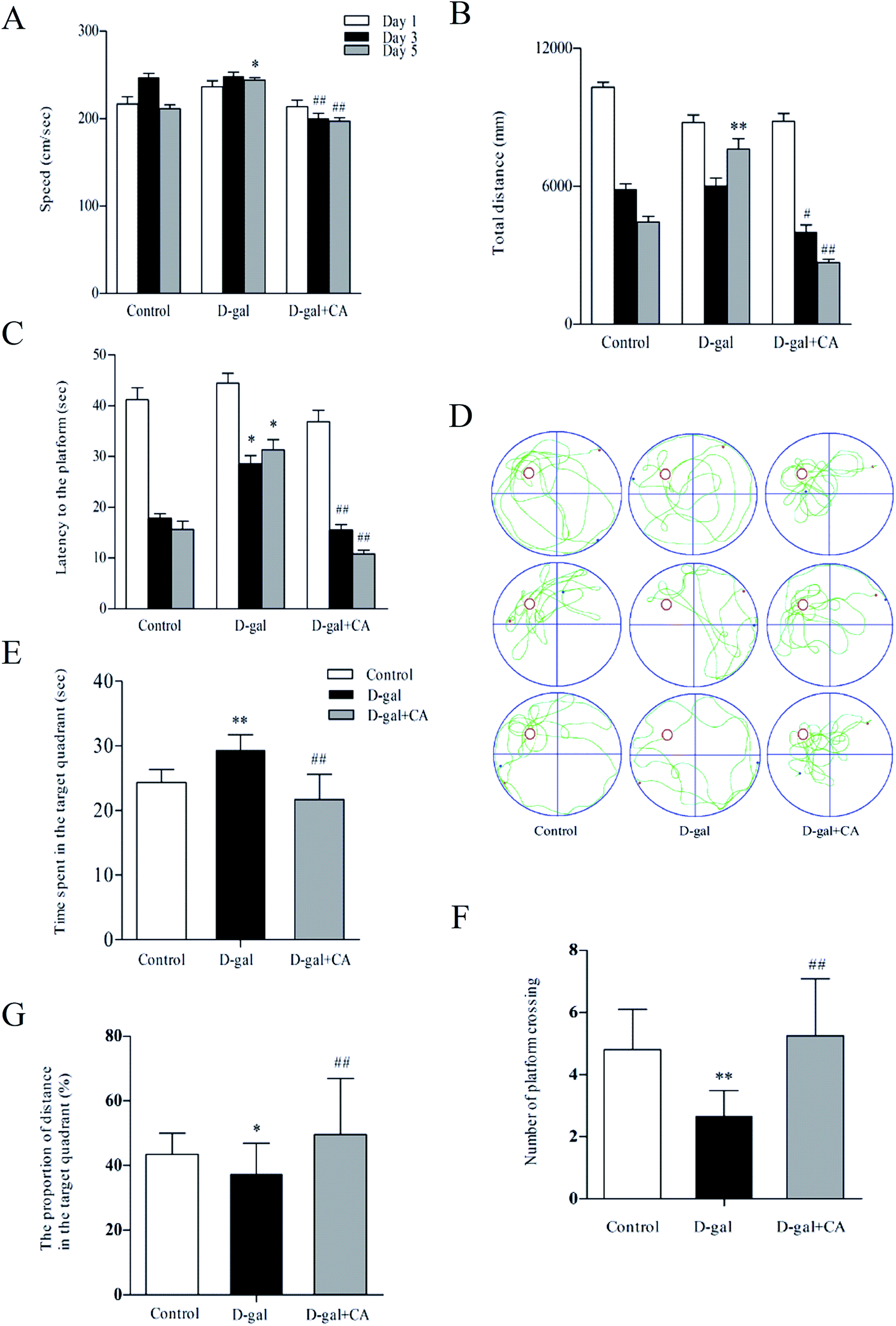 | ||
| Fig. 2 Chicoric acid (CA) ameliorated D-gal-induced learning and spatial memory loss in the Morris water maze test. The Morris water maze tests were performed as described in the methods section. (A) Changes in the swimming speed during place navigation test. (B) Total distance as well as (C) escape latency time to reach the hidden platform were recorded during the 5 test days. (D) Representative swimming paths of three mice of each treatment group at days 3–5 of the spatial probe trial test. (E) The time spent in target quadrant, (F) the number of platform crossings, and (G) the proportion of distance in the target quadrant during the spatial probe trial. Data presented as mean ± SEM, n = 10. *p < 0.05, **p < 0.01, versus control group, #p < 0.05, ##p < 0.01 versus D-gal group. | ||
A probe trial was performed 24 h after the MWM test for all experimental groups. The hidden platform was removed during this trial, and the average time spent in the target quadrant was measured. As shown in Fig. 2D, the escape route of the control mice was short and direct; however, in D-gal-treated mice, the swimming pattern was confined more to side walls rather than to the center of the pool favored by the control group mice. The swim pattern of chicoric acid-treated mice was better than that in the D-gal-treated group, indicating better learning and memory. These results indicate that D-gal-induced learning and memory deficits were improved in mice that were treated with chicoric acid. As shown in Fig. 2E–G, D-gal-injected mice swam across the entire pool and spent less time in the target quadrant with fewer platform crossings than mice in the control group. Mice that received the chicoric acid plus D-gal treatment reversed the D-gal-induced memory impairment because these mice exhibited a significant increase in the average time spent in the target quadrant with more platform crossings.
3.3. Effects of chicoric acid on preventing neuron damage
To determine whether chicoric acid supplementation protected neurons from D-gal-induced damage, HE staining was used to examine any histopathological changes in the hippocampus. Histological features of the mouse hippocampus are depicted in Fig. 3A. In the D-gal treated group, there were damaged neurons in mice DG and hippocampal CA1 and CA3 regions. The hippocampus neurons showed pronounced shrinkage of the neuronal bodies and the loss of the nuclei regular outlines compared with the control group. Chicoric acid supplementation significantly reduced the number of degenerated neurons in the mice hippocampus.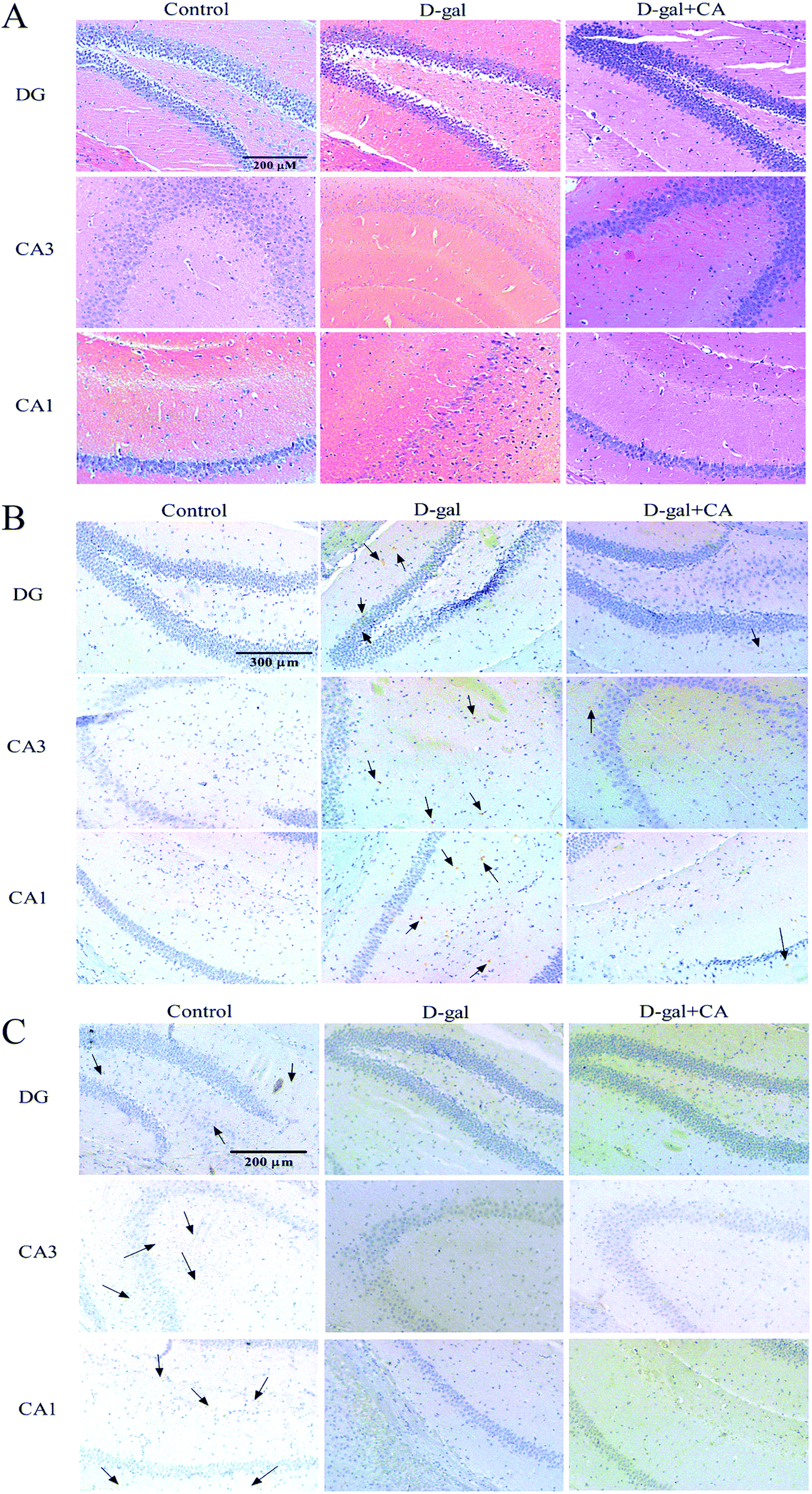 | ||
| Fig. 3 Chicoric acid (CA) prevented D-gal-induced neuron damage and amyloidogenesis. HE staining of mice hippocampus, including DG, CA3 and CA1 region (A). Accumulation of Aβ was detected by Aβ1-42 immunostaining in hippocampus (marker of Aβ) (B), as well as representative images of immunohistochemical images for neurotrophic factors BDNF (marker of BDNF) (C). | ||
3.4. Effect of chicoric acid on the expressions of β-amyloid (1-42 specific) (Aβ1-42) and brain-derived neurotrophic factor (BDNF) in the hippocampus
Aβ accumulation is known to be a contributor to AD. Therefore, we next examined whether chicoric acid reduced D-gal-induced Aβ accumulation and thus recovered memory function. To do this, Aβ1-42 IHC staining was used to detect Aβ accumulation. As shown in Fig. 3B, the higher accumulation of Aβ was detected in the brains of D-gal treatment group mice, and lower accumulation of Aβ was observed in the chicoric acid treatment group. As expected, the immunohistochemistry results for Aβ1-42 demonstrated that chicoric acid inhibited D-gal-induced Aβ1-42 accumulation in hippocampus of mice brain.BDNF plays a key role in regulating the cognition, formation, and storage of memories.39,40 The expression of BDNF in the hippocampus was also measured by an IHC method. The changes of BDNF expression were obvious in the hippocampus and the results are shown in Fig. 3C. In the control group, significant positive staining was dispersed in the hippocampus, indicating that BDNF was expressed to high levels in the hippocampus of the control group. However, positive staining in the hippocampus of the D-gal group was obviously decreased compared to the control group, which suggested that the expression of BDNF was decreased in the hippocampus of aging mice. Interestingly, the hippocampi of the chicoric acid treatment group mice were similar to those observed in the D-gal group and lower than those of the control group, suggesting chicoric acid treatment did not restore the expression of BDNF in the hippocampus of aging mice.
3.5. Effects of chicoric acid on D-gal-induced oxidative stress in mice serum
Many studies have shown that oxidative stress affects the biological aging process. To determine the effects of chicoric acid on preventing D-gal-induced oxidative stress, several key antioxidants including CAT, GSH, and MPO were detected in mice serum. As shown in Fig. 4A–C, chicoric acid significantly enhanced the activities of CAT (Fig. 4A) (p < 0.01) and serum level of GSH (Fig. 4B) (p < 0.01), and suppressed the level of MPO (Fig. 4C) (p < 0.05), compared with the D-gal group. Moreover, as shown in Fig. 4D–F, D-gal treatment obviously increased the serum levels of TNF-α (Fig. 4D) and IL-1β (Fig. 4E) compared with the control group. Additionally, the D-gal group mice showed increased MDA levels compared to the control group, consistent with D-gal-induced production of lipid peroxidation products (Fig. 4F) (p < 0.05). The expression levels of these inflammatory mediators were significantly attenuated by administration of chicoric acid (p < 0.05). The results suggested that chicoric acid treatment significantly improved CAT activity and GSH content and decreased the levels of MPO, TNF-α, IL-1β, and MDA in D-gal-treated mice.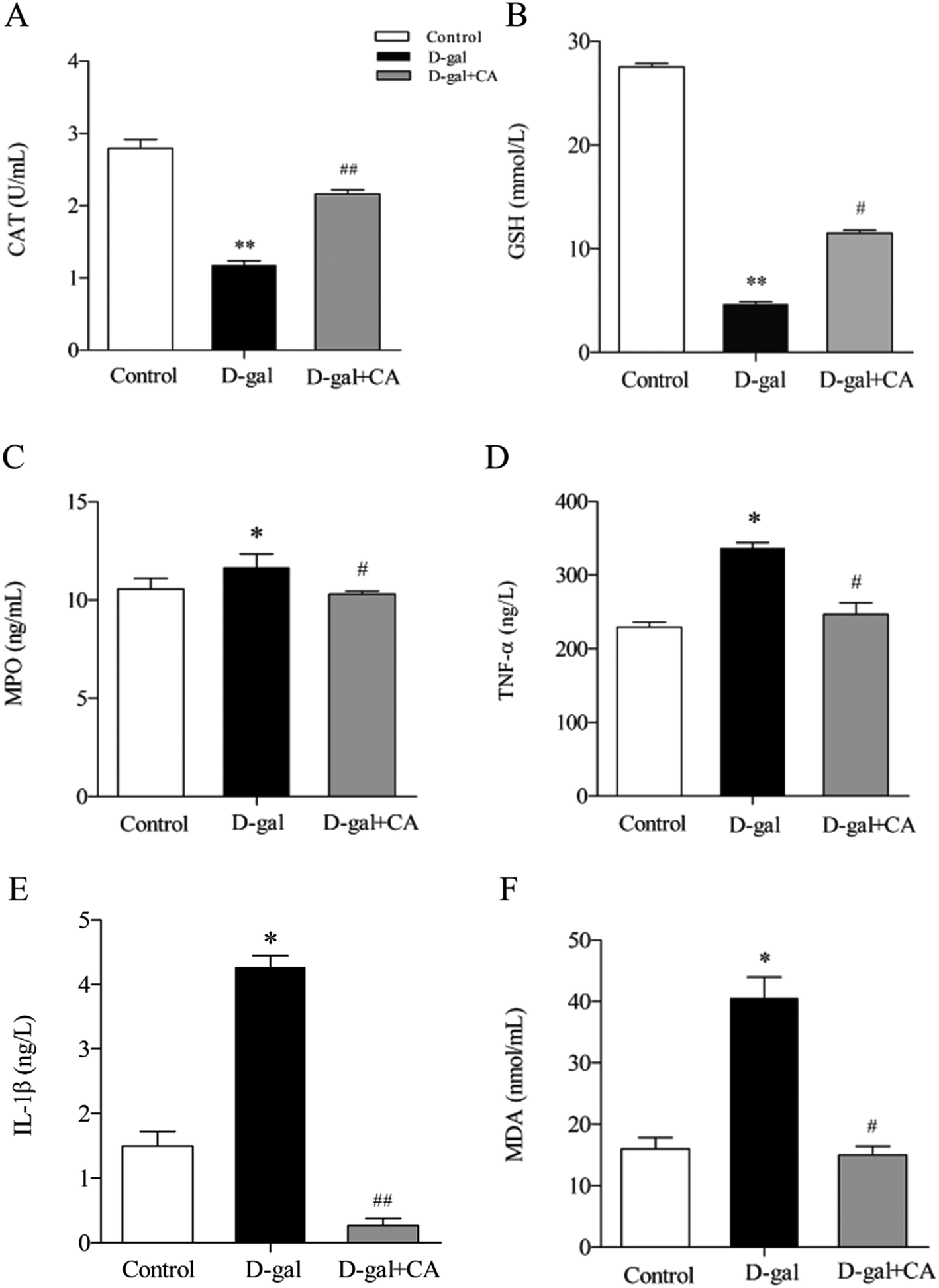 | ||
| Fig. 4 Effects of chicoric acid (CA) on the expression of antioxidant enzymes and the release of inflammatory mediators in mice serum. Activity of (A) CAT and relative levels of (B) GSH and (F) MDA in mice serum determined using assay kits. Levels of (D) TNF-α and (E) IL-1β, and activity of (C) MPO in serum of mice were measured by ELISA. Data presented as the mean ± SEM, n ≥ 5. *p < 0.05 and **p < 0.01, versus control group; #p < 0.05 and ##p < 0.01, versus D-gal group. | ||
3.6. Effects of chicoric acid on the Keap1/Nrf2 antioxidative signaling pathway in mice brains
To determine whether chicoric acid supplementation activated the Keap1/Nrf2 signaling pathway in chicoric acid-treated mice brains, expression of antioxidant defense enzymes in downstream pathways, including HO-1 and NQO1, was detected using western blot and RT-qPCR. The results (Fig. 5A & B) showed that chicoric acid resulted in noticeably increased expression levels of HO-1, NQO-1 (p < 0.05), and Keap1 (p < 0.01) compared with the D-gal-treated group. Chicoric acid enhanced Nrf2 expression in the nucleus (p < 0.05) compared to the D-gal group. Consistently, the mRNA expression levels of NQO1 and HO-1 in the chicoric acid supplementation group were also up-regulated compared with those in D-gal-treated mice (p < 0.05).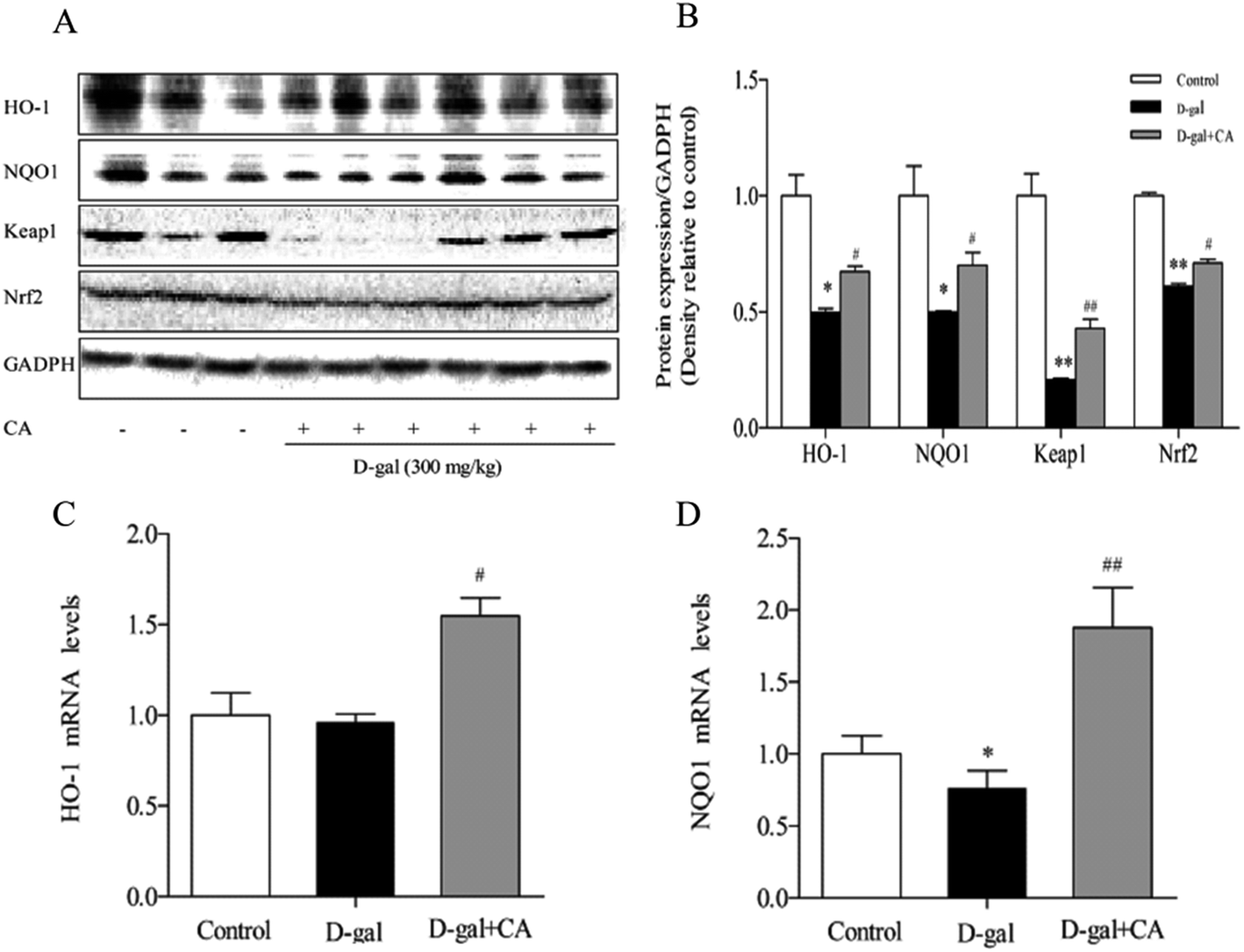 | ||
| Fig. 5 Effects of chicoric acid (CA) on the antioxidative signaling pathway Keap1/Nrf2 in the D-gal-treated mice brains. (A) Representative western blots of cytoprotective Keap1/Nrf2 signaling pathway in the mice brains, and GAPDH was used as loading control. Densitometry analysis of (B) mRNA levels of (C) HO-1 and (D) NQO1 in mice brains was carried out by RT-qPCR. Data presented as mean ± SEM, n ≥ 5. *p < 0.05, **p < 0.01, versus control group, #p < 0.05, ##p < 0.01 versus the D-gal group. | ||
3.7. Effects of chicoric acid on cell viability and mitochondria function in H2O2-induced SH-SY5Y cells
As demonstrated in Fig. 6A, SH-SY5Y cells were exposed to different concentrations of H2O2 (0–1000 μM) for 0–48 h, causing a dose- and time-dependent decrease in cell viability. There was no notable change in the viability of SH-SY5Y cells treated with 10–50 μM of H2O2 for 0–12 h, but high concentrations of H2O2 (100 μM to 1000 μM) for 24–48 h significantly decreased cell viability. More significantly, there was 62.5% viability of SH-SY5Y cells treated with 100 μM at 24 h. Based on the above results, we used a concentration of H2O2 of 100 μM for 24 h for the following experiments. To evaluate the protective efficacy of chicoric acid against H2O2-induced apoptotic cell death of SH-SY5Y, cells were treated with 100 μM H2O2 for 24 h, followed by various doses of chicoric acid (0, 10, 25, or 50 μM) for 12 h and 24 h. The viabilities of SH-SY5Y cells treated with 10, 25, and 50 μM of chicoric acid for 24 h were 66.28%, 90.12%, and 91.53%, respectively, compared to the 37.51% of the H2O2 treatment group as shown in Fig. 6B. The results showed that chicoric acid significantly inhibited the H2O2-induced cell viability decrease, and this suppression was correlated with chicoric acid concentration.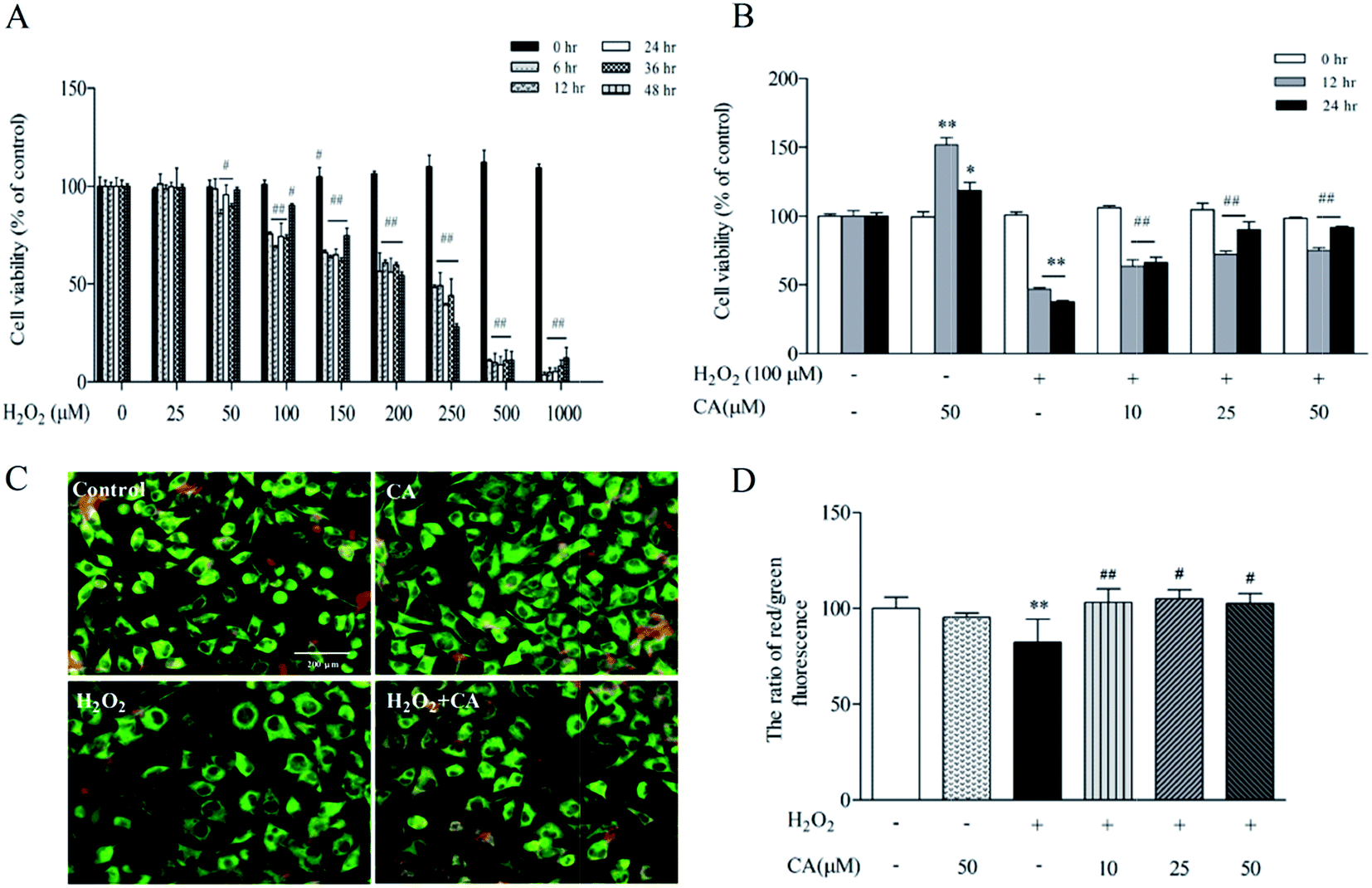 | ||
| Fig. 6 Effects of chicoric acid (CA) on cell viability and mitochondria function in H2O2-induced SH-SY5Y cells. (A) Effect of H2O2-induced SH-SY5Y cell viability. SH-SY5Y cells were treated with H2O2 (0–1000 μM) for 0–48 h. (B) Effect of CA on H2O2-induced SH-SY5Y cell viability. SH-SY5Y cells were treated with H2O2 (100 μM) for 24 h with or without treatment of CA (10, 25, 50 μM) for 24 h. Cell viability was measured by MTT assay. Mitochondrial dysfunction was detected by dyeing with 10 μM JC-1 for 30 min at 37 °C in the dark. The fluorescence was observed by using an inverted fluorescence microscope (C) and quantified using a multimode microplate reader at 485 nm excitation, 585 nm (red) and 538 nm (green) emission for JC-1 staining (D). Data presented as mean ± SEM, n ≥ 5. *p < 0.05, **p < 0.01, versus control group, #p < 0.05, ##p < 0.01 versus H2O2-treated group. | ||
Mitochondria serve as cellular “energy factories” and also play a central role in regulating cellular redox status by releasing free radicals, a second messenger that mediates different cell signaling pathways.32 As illustrated in Fig. 6C & D, JC-1 staining results also suggested that chicoric acid improved H2O2 (100 μM)-elicited mitochondrial membrane potential loss, which is a hallmark of mitochondrial function and reflects the normal cellular redox status.
3.8. Regulation effects of chicoric acid on cellular redox status and Keap1/Nrf2 antioxidant defense pathway in H2O2-treated neuroblastoma SH-SY5Y cells
ROS play a key role in cell life processes as signaling molecules. However, excessive ROS can induce cell apoptosis and autophagy. As shown in Fig. 7A, H2DCFDA staining results indicated that chicoric acid significantly decreased intracellular ROS levels and alleviated H2O2 (100 μM)-induced cellular oxidation status. According to the results in Fig. 7B, H2O2 stimulated the generation of ROS by 39.22% compared with the control group, but chicoric acid significantly quenched intracellular ROS to the normal level (p < 0.01).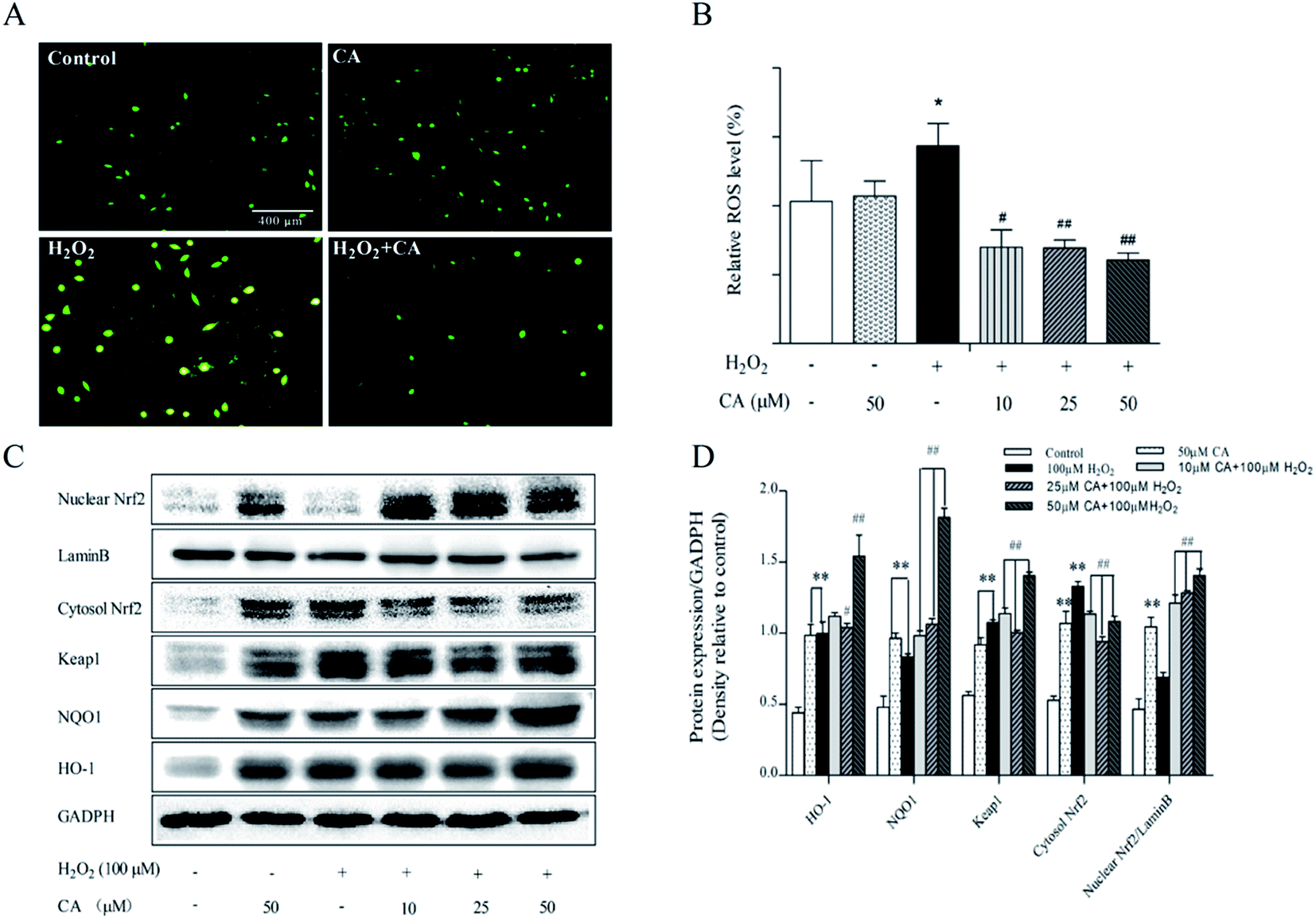 | ||
| Fig. 7 Regulation effects of chicoric acid (CA) on cellular redox status and Keap1/Nrf2 antioxidant defense pathway in H2O2-treated neuroblastoma SH-SY5Y cells. Intracellular redox status was detected by dyeing with 10 μM H2DCFDA for 30 min at 37 °C in the dark. (A) The fluorescence was observed by using an inverted fluorescence microscope and (B) quantified using a multimode microplate reader at 485 nm excitation and 538 nm emission for H2DCFDA dyeing. The expression of (C) Nrf2 in the cytosol and nucleus, keap1, HO-1 and NQO1 were measured by western blotting, and GAPDH and lamin B were used as loading controls in the cytosol and nucleus, respectively, and densitometry analysis is presented in (D). Data presented as mean ± SEM, n ≥ 5. *p < 0.05, **p < 0.01, versus control group, #p < 0.05, ##p < 0.01 versus H2O2-treated group. | ||
 | ||
| Fig. 8 Graphic for table of contents. Illustration of effects of chicoric acid on neuroprotection against D-gal-induced memory impairment via inflammation and oxidative stress. | ||
Previous studies have demonstrated that Nrf2 regulates expression of many antioxidant enzymes, such as HO-1 and NQO-1, which contributes to maintenance of redox homeostasis in the mouse brain. Similar results were also found for in vitro studies. The results showed that chicoric acid treatment significantly promoted the translocation of nuclear Nrf2 in H2O2-induced SH-SY5Y cells, enhanced Nrf2 expression in the nucleus and decreased its expression in the cytosol, and at the same time increased expression of antioxidant enzymes NQO1 and HO-1 in SH-SY5Y cells in a dose-dependent manner (Fig. 7C & D).
4. Discussion
In this study, the regulation effects of chicoric acid on oxidative stress and underlying molecular mechanisms were investigated. The results demonstrated that chicoric acid attenuated D-gal-induced learning and memory loss and SH-SY5Y cell apoptosis induced by H2O2 via promotion of the Keap1/Nrf2 signal pathway. Significantly, chicoric acid alleviated D-gal-stimulated memory deficits, restrained neuronal damage, suppressed the excessive release of inflammatory mediators, and enhanced antioxidant enzyme activities, all activities that may be related to cognitive function enhancement. Furthermore, the results demonstrated that chicoric acid significantly reversed H2O2-elicited SH-SY5Y cell viability decrease and mitochondrial dysfunction, serving to balance the cellular redox status.The activities of chicoric acid, a major antioxidant of caffeic acid derived from chicory, have been well documented. Chicoric acid has been used as a nutraceutical food ingredient due to its excellent antioxidant bioactivities. Our previous study demonstrated that chicoric acid was partly metabolized to caffeic acid and caftaric acid on cytochrome P450s in rat liver microsomes, and the antioxidant activities of chicoric acid were significantly higher than those of its metabolites.30 Metabolic and distribution studies showed that chicoric acid could penetrate the blood–brain barrier34 and showed strong capacities for scavenging free radicals, indicating that chicoric acid is a potential phytochemical to increase antioxidant activity in the brain.
Various models of aging and neurodegenerative disease are associated with oxidative stress. The brain is the most susceptible organ to oxidative damage.41 Neurodegenerative diseases and aging-related dementia are characterized by cognitive dysfunction, including memory impairment and difficulties learning new information. Many studies have indicated that D-gal administration over long periods of time can lead to an enzymatic overload, which impairs the body's natural ability to catalyze galactose into glucose, causing an increase of galactitol and an activation of aldose reductase. D-Gal creates biochemical abnormalities in experimental animals, including the accumulation of reactive oxygen species, reduction of antioxidant enzymes, increased mitochondrial deficits, and neuroinflammation. The intraperitoneal or subcutaneous injections of D-gal led to spatial learning impairments, a gradual loss of cognitive performance, memory, and spatial ability.42 These changes in rodents are similar to those that occur in the aging human brain.43–45 Thus, D-gal treated animals are widely studied as an ideal animal model of memory impairment to mimic many characteristics of the natural aging process, such as cognitive deficits and neurologic impairments,42,46,47 allowing the testing of new therapeutics.48 The present study focused on the role of chicoric acid in minimizing D-gal-induced accelerated aging in the mouse model with impaired memory. In this study, chronic injection of D-gal impaired working memory and reference memory in the Y-maze and Morris water maze tests, respectively, without changing locomotive activity, with prolonged crossing times and escape latency, and shortened target quadrant occupancy in mouse behavioral studies. However, chicoric acid reversed the D-gal-elicited deterioration of spatial learning abilities at a daily dose of 100 mg kg−1 body weight for 8 weeks.
Many factors contribute to the development of cognitive impairment. Tissue-accumulated D-gal can react with the amino group of peptides and proteins to form advanced glycation end-products (AGEs). AGEs bound to their receptors (RAGE) play roles in aging, neuroinflammation, and neurodegeneration, such as in AD.49–51 RAGE acts as the receptor for Aβ transport across the blood–brain barrier, which results in cerebral Aβ accumulation and plaque formation.52 Various studies have demonstrated that D-gal-induced increased expression of RAGE in the brain is accompanied by astrocytes and microglial cells activation, which induces neuroinflammation and neurodegeneration.53,54 Significantly, Liu et al.33 found that chicoric acid treatment markedly decreased the expression of APP and BACE1 in the whole brain, which partly explains the inhibition by chicoric acid of Aβ1-42 accumulation in the cortex and hippocampus. Our study found that chicoric acid treatment inhibited the increased expression of Aβ proteins in the hippocampus of the D-gal-treated mice by minimizing oxidative stress, suggesting that chicoric acid treatment effectively ameliorates oxidative damage and improves indexes related to aging and AD in D-gal-treated mice. Recent studies showed that neurotrophic factors such as BDNF and NGF may reduce amyloidogenesis in AD.51 Unexpectedly, we found that chicoric acid treatment was unable to restore levels of BDNF as evidenced by the expression of neurotrophic factors in the hippocampus of oxidative stress mice as measured by an IHC method. These results were also seen in our previous studies, in which there were no changes in BDNF, NGF, NT3, and NT4 mRNA expression of neurotrophic factors after chicoric acid treatment. Additionally, several studies reported that cytokines including TNF-α and IL-1β exert their effects directly on synaptic plasticity and neurogenesis, aspects of cognitive processes.55,56 Chicoric acid significantly inhibited D-gal induced excretion of inflammation signallers, including IL-1β as well as TNF-α, and mediated lipid peroxidation products, such as MDA and the level of MPO, but increased CAT activity and GSH content in mouse serum, suggesting another underlying mechanism for chicoric acid to promote learning and spatial cognitive abilities.
Nrf2 plays a crucial role in the modulation of redox homeostasis and regulation of inflammatory conditions through the induction of many stress-responsive and cytoprotective enzymes and related proteins, including HO-1 and NQO-1. Nrf2 regulation maintains redox homeostasis in healthy cells. However, this regulation is perturbed during pathological conditions in which inflammation and oxidative stress are main partners, such as age-related diseases, cancer, diabetes, and neurodegenerative disorders.57 Recent studies showed that caffeic acid, the metabolite of chicoric acid, prevented acetaminophen-induced liver injury by activating the Nrf2 antioxidative defense system. It was suggested that caffeic acid may decrease the expression of Keap1 and may interact with the Nrf2 binding site in the Keap1 protein. Li et al.58 demonstrated that D-gal-induced oxidative stress decreased Nrf2 translocation and the expression of its targeted antioxidants. One possible explanation is the different responses of Nrf2 to acute or chronic oxidative stress. In the present study, the efficacy of the up-regulated expression of antioxidant defense enzymes with chicoric acid was investigated. We found that chicoric acid could improve expression of phase II antioxidant enzymes including HO-1 and NQO-1 in D-gal-induced mice brain both in terms of protein levels and mRNA levels. Additionally, various studies have demonstrated that H2O2 induces apoptosis in a variety of cells and acts as a precursor of ROS to induce neurotoxicity.59 Moreover, our study found that chicoric acid significantly enhanced CAT activity and GSH content in mouse serum but inhibited the excretion of inflammation signallers, such as IL-1β and TNF-α. A large number of studies have indicated that the balance between the ROS system and the autoxidation system determines the degree of oxidative stress.60 These antioxidant enzymes catalyse the decomposition of H2O2 into H2O and O2,61 which is regarded as the first line of defense against the ROS generated during oxidative stress.60 To make the results more convincing, H2O2 was used to stimulate D-gal-induced oxidative damage. The results demonstrated that chicoric acid significantly reversed H2O2-elicited SH-SY5Y cell viability decrease and mitochondrial dysfunction, and eventually disrupted cellular function.62–66 Haskew-Layton et al.67 reported that subtoxic doses of H2O2 fail to activate nuclear factor erythroid 2-related factor (Nrf2) in astrocytes and trigger Nrf2-independent responses that protect cocultured neurons. Meanwhile, many studies also showed that mild oxidative insults, including subtoxic H2O2, activate Nrf2/antioxidant response element (ARE)-dependent gene expression, which contributes to neuroprotection.68 Similarly, the in vitro study indicated that the mild concentration of H2O2 (100 μM) failed to markedly invoke nuclear translocation of Nrf2 in SH-SY5Y cells. However, it slightly activated the Nrf2 antioxidative defense system and released Nrf2 from Keap1, upregulating the expressions of HO-1 and NQO1 via stimulating the cellular self-protection system to resist the external stimulation. More significantly, based on the self-protection mechanism of the biological system, our studies also indicated that chicoric acid strongly promoted nuclear translocation of Nrf2 from the cytoplasm to the nucleus in SH-SY5Y cells and enhanced the expression of antioxidant enzymes, including HO-1 and NQO-1, activating the Nrf2 pathway via balancing cellular redox status in H2O2-induced SH-SY5Y cells.
5. Conclusion
In summary, chicoric acid prevented D-gal-induced neural loss and deficits in cognitive function, regulating the oxidative stress-signaling pathway by promotion of Keap1/Nrf2 activation and up-regulated expression of antioxidant defense enzymes, both in vivo and in vitro. Chicoric acid shows promise for future application in preventive and therapeutic strategies aimed to slow the growth and progression of oxidative stress. Further research is needed to evaluate the mechanism of chicoric acid action in various neurodegenerative disorders.Author contributions
Conception and design of research: YW, ZD and XL. Performed experiments: YW, ZD, JL, BR, DZ. Analyzed data: YW, ZD. Interpretation of results of experiments: YW, ZD. Prepared figures: YW, ZD. Drafted manuscript: YW, ZD, QL. All authors read and approved the final manuscript.Abbreviations
| CA | Chicoric acid |
| D-gal | D-Galactose |
| AD | Alzheimer disease |
| BDNF | Brain-derived neurotrophic factor |
| DG | Dentate gyrus |
| CA1 | Cornu ammonis region 1 |
| CA3 | Cornu ammonis region 3 |
| HE | Hematoxylin and eosin stained |
| IHC | Immunohistochemistry |
| qPCR | Quantitative PCR |
| IL-1β | Interleukin-1 beta |
| TNF-α | Tumor necrosis factor-α |
| Nrf2 | Nuclear factor E2-related factor 2 |
| NQO1 | NADPH:quinone oxidoreductase 1 |
| HO-1 | Hemeoxygenase-1 |
| CAT | Catalase |
| GSH | Glutathione |
| MDA | Malonic dialdehyde |
| MPO | Myeloperoxidase |
| MWM | Morris water maze |
| ROS | Reactive oxygen species |
| Aβ1-42 | β-Amyloid (1-42 specific) |
| MTT | 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide |
| CNS | Central nervous system |
| JC-1 | 5,5,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide |
| H2DCFDA | 2′,7′-Dichlorodihydro fluorescein diacetate |
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 31671859) and China Postdoctoral Science Foundation (k3080215804).References
- D. A. Linseman, Antioxid. Redox Signaling, 2008, 11, 421–424 CrossRef PubMed.
- A. Reynolds, C. Laurie, R. L. Mosley and H. E. Gendelman, Int. Rev. Neurobiol., 2007, 82, 297 CrossRef CAS PubMed.
- W. R. Markesbery, Free Radical Biol. Med., 1997, 23, 134–147 CrossRef CAS PubMed.
- T. Finkel and N. J. Holbrook, Nature, 2000, 408, 239–247 CrossRef CAS PubMed.
- K. J. Barnham, C. L. Masters and A. I. Bush, Biomed. Pharmacother., 2004, 58, 39 CrossRef.
- W. Dröge, Bangladesh J. Med. Sci., 2010, 23, 29–36 Search PubMed.
- J. P. Lister and C. A. Barnes, Arch. Neurol., 2009, 66, 829–833 CrossRef PubMed.
- M. P. Mattson, W. A. Pedersen, W. Duan, C. Culmsee and S. Camandola, Ann. N. Y. Acad. Sci., 1999, 893, 154–175 CrossRef CAS PubMed.
- H. Tanaka, T. Nishi and Y. Wada, J. Biol. Chem., 2009, 284, 13291 CrossRef PubMed.
- K. Itoh, T. Chiba, S. Takahashi, T. Ishii, K. Igarashi, Y. Katoh, T. Oyake, N. Hayashi, K. Satoh and I. Hatayama, Biochem. Biophys. Res. Commun., 1997, 236, 313 CrossRef CAS PubMed.
- A. Giudice and M. Montella, BioEssays, 2006, 28, 169–181 CrossRef CAS PubMed.
- J. A. Joseph, B. Shukitt-Hale and G. Casadesus, Am. J. Clin. Nutr., 2005, 81, 313S CAS.
- M. E. Obrenovich, N. G. Nair, A. Beyaz, G. Aliev and V. P. Reddy, Rejuvenation Res., 2010, 13, 631–643 CrossRef CAS PubMed.
- S. Z. Zhong, Q. H. Ge, R. Qu, Q. Li and S. P. Ma, J. Neurol. Sci., 2009, 277, 58 CrossRef CAS PubMed.
- Y. Pan, L. D. Kong, Y. C. Li, X. Xia, H. F. Kung and F. X. Jiang, Pharmacol., Biochem. Behav., 2007, 87, 130–140 CrossRef CAS PubMed.
- A. Galano, G. Mazzone, R. A. Diduk, T. Marino, J. R. A. Idaboy and N. Russo, Annu. Rev. Food Sci. Technol., 2016, 7, 335–352 CrossRef CAS PubMed.
- S. Oliver, O. Vittorio, G. Cirillo and C. Boyer, Polym. Chem., 2016, 7, 1529–1544 RSC.
- Y. Sato, S. Itagaki, T. Kurokawa, J. Ogura, M. Kobayashi, T. Hirano, M. Sugawara and K. Iseki, Int. J. Pharm., 2011, 403, 136–138 CrossRef CAS PubMed.
- M. Nardini, P. Pisu, V. Gentili, F. Natella, F. M. Di, E. Piccolella and C. Scaccini, Free Radical Biol. Med., 1998, 25, 1098 CrossRef CAS PubMed.
- P. X. Xu, S. W. Wang, X. L. Yu, Y. J. Su, T. Wang, W. W. Zhou, H. Zhang, Y. J. Wang and R. T. Liu, Behav. Brain Res., 2014, 264, 173–180 CrossRef CAS PubMed.
- C. Liu, B. Abbasi, M. Gao, S. Murch and P. Saxena, J. Agric. Food Chem., 2006, 54, 8456 CrossRef CAS PubMed.
- K. Schütz, D. R. Kammerer, R. Carle and A. Schieber, Rapid Commun. Mass Spectrom., 2005, 19, 179–186 CrossRef PubMed.
- J. M. Lee and C. F. Scagel, Food Chem., 2009, 115, 650–656 CrossRef CAS.
- S. Baur, R. G. Klaiber, A. Koblo and R. Carle, J. Agric. Food Chem., 2004, 52, 7017–7025 CrossRef CAS PubMed.
- M. Innocenti, S. Gallori, C. Giaccherini, F. Ieri, F. F. Vincieri and N. Mulinacci, J. Agric. Food Chem., 2005, 53, 6497 CrossRef CAS PubMed.
- N. K. Olah, L. Radu, C. Mogoşan, D. Hanganu and S. Gocan, J. Pharm. Biomed. Anal., 2003, 33, 117–123 CrossRef CAS PubMed.
- Z. Q. Ou, D. M. Schmierer, T. Rades, L. Larsen and A. Mcdowell, J. Pharm. Pharmacol., 2013, 65, 271–279 CrossRef CAS PubMed.
- A. Schlernitzauer, C. Oiry, R. Hamad, S. Galas, F. Cortade, B. Chabi, F. Casas, L. Pessemesse, G. Fouret and C. Feilletcoudray, PLoS One, 2013, 8, e78788 CAS.
- A. Schlernitzauer, C. Oiry, F. Casas, B. Chabi, G. Cros, R. Magous, G. Cabello and C. Cabello, Diabetes Metab., 2013, 39, A41 Search PubMed.
- Q. Liu, Y. Wang, C. Xiao, W. Wu and X. Liu, Food Funct., 2015, 6, 1928 CAS.
- H. Xiao, G. Xie, J. Wang, X. Hou, X. Wang, W. Wu and X. Liu, Food Res. Int., 2013, 54, 345–353 CrossRef CAS.
- Q. Liu, Y. Hu, Y. Cao, G. Song, Z. Liu and X. Liu, J. Agric. Food Chem., 2017, 65(2), 338–347 CrossRef CAS PubMed.
- Q. Liu, Y. Chen, C. Shen, Y. Xiao, Y. Wang, Z. Liu and X. Liu, FASEB J., 2017, 31, 1494–1507 CrossRef CAS PubMed.
- Y. Wang, X. Guo, L. Qian, D. Xiang, Z. Liu and X. Liu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1031, 139–145 CrossRef CAS PubMed.
- H. Wang and J. A. Joseph, Free Radical Biol. Med., 1999, 27, 612–616 CrossRef CAS PubMed.
- Z. Liu, Y. Chen, Q. Qiao, Y. Sun, Q. Liu, B. Ren and X. Liu, Mol. Nutr. Food Res., 2017, 61, 5–16 Search PubMed.
- Z. Liu, Q. Qiao, Y. Sun, Y. Chen, B. Ren and X. Liu, Mol. Nutr. Food Res., 2016 DOI:10.1002/mnfr.201600717.
- H. Xiao, J. Wang, L. Yuan, C. Xiao, Y. Wang and X. Liu, J. Agric. Food Chem., 2013, 61, 1509–1520 CrossRef CAS PubMed.
- J. M. Conner, J. C. Lauterborn, Q. Yan, C. M. Gall and S. Varon, J. Neurosci., 1997, 17, 2295 CAS.
- T. Numakawa, S. Suzuki, E. Kumamaru, N. Adachi, M. Richards and H. Kunugi, Histol. Histopathol., 2010, 25, 237 CAS.
- R. J. Reiter, FASEB J., 1995, 9, 526–533 CAS.
- X. Cui, P. Zuo, Q. Zhang, X. Li, Y. Hu, J. Long, L. Packer and J. Liu, J. Neurosci. Res., 2006, 84, 647–654 CrossRef CAS PubMed.
- H. Hueimin, W. M. Wu and M. L. Hu, Food Chem. Toxicol., 2009, 47, 625–632 CrossRef PubMed.
- A. Kumar, A. Prakash and S. Dogra, J. Neurosci. Res., 2011, 347569 Search PubMed.
- F. Ullah, T. Ali, N. Ullah and M. O. Kim, Neurochem. Int., 2015, 90, 114–124 CrossRef CAS PubMed.
- Q. Zhang, X. Li, X. Cui and P. Zuo, Neurol. Res., 2005, 27, 552–556 CrossRef CAS PubMed.
- J. Lu, Y. L. Zheng, D. M. Wu, L. Luo, D. X. Sun and Q. Shan, Biochem. Pharmacol., 2007, 74, 1078–1090 CrossRef CAS PubMed.
- H. F. Wei, L. Li, Q. J. Song, H. X. Ai, J. Chu and W. Li, Behav. Brain Res., 2005, 157, 245–251 CrossRef CAS PubMed.
- J. W. Baynes, Exp. Gerontol., 2001, 36, 1527 CrossRef CAS PubMed.
- V. Srikanth, A. Maczurek, T. Phan, M. Steele, B. Westcott, D. Juskiw and G. Münch, Neurobiol. Aging, 2011, 32, 763 CrossRef CAS PubMed.
- T. Viviana, S. Valentina, B. Giulia, C. M. Teresa, P. Piergiuseppe, A. C. Bruni, C. Chiara, M. Raffaele, F. Marco and N. Robert, Aging Cell, 2016, 15, 661–672 CrossRef PubMed.
- S. J. Kim, J. W. Ahn, H. Kim, H. J. Ha, S. W. Lee, H. K. Kim, S. Lee, H. S. Hong, Y. H. Kim and C. Y. Choi, Biochem. Biophys. Res. Commun., 2013, 439, 252–257 CrossRef CAS PubMed.
- J. Tian, K. Ishibashi, K. Reiser, R. Grebe, S. Biswal, P. Gehlbach and J. T. Handa, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11846 CrossRef CAS PubMed.
- M. Lei, X. Hua, M. Xiao, J. Ding, Q. Han and G. Hu, Biochem. Biophys. Res. Commun., 2008, 369, 1082–1087 CrossRef CAS PubMed.
- J. Mcafoose and B. T. Baune, Neurosci. Biobehav. Rev., 2008, 33, 355–366 CrossRef PubMed.
- N. Terrando, C. Monaco, D. Ma, B. M. Foxwell, M. Feldmann and M. Maze, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20518–20522 CrossRef CAS PubMed.
- H. Lassmann and H. J. Van, Biochim. Biophys. Acta, 2015, 1862, 506–510 CrossRef PubMed.
- Y. N. Li, Y. Guo, M. M. Xi, P. Yang, X. Y. Zhou, S. Yin, C. X. Hai, J. G. Li and X. J. Qin, Oxid. Med. Cell. Longevity, 2014, 320513 Search PubMed.
- T. B. Sherer, R. Betarbet, A. K. Stout, S. Lund, M. Baptista, A. V. Panov, M. R. Cookson and J. T. Greenamyre, J. Neurosci., 2002, 22, 7006 CAS.
- B. Poljsak, D. Suput and I. Milisav, Oxid. Med. Cell. Longevity, 2013, 2013, 11 Search PubMed.
- H. Chen, M. Yu and M. Li, et al., Mol. Cell. Biochem., 2012, 363, 85–91 CrossRef CAS PubMed.
- S. J. Tsai and M. C. Yin, Eur. J. Pharmacol., 2012, 689, 81–88 CrossRef CAS PubMed.
- A. M. Gardner, F. H. Xu, C. Fady, F. J. Jacoby, D. C. Duffey, Y. Tu and A. Lichtenstein, Free Radical Biol. Med., 1997, 22, 73–83 CrossRef CAS PubMed.
- A. M. Gorman, A. Mcgowan, C. O. Neill and T. Cotter, J. Neurol. Sci., 1996,(suppl. 139), 45 CrossRef CAS.
- E. D. Hall and J. M. Braughler, Free Radical Biol. Med., 1989, 6, 303–313 CrossRef CAS PubMed.
- J. M. Mccord, N. Engl. J. Med., 1985, 312, 159–163 CrossRef CAS PubMed.
- R. E. Haskew-Layton, et al., Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17385–17390 CrossRef CAS PubMed.
- K. F. Bell, B. Almubarak, J. H. Fowler, P. S. Baxter, K. Gupta, T. Tsujita, S. Chowdhry, R. Patani, S. Chandran and K. Horsburgh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3–4 CrossRef PubMed.
Footnote |
| † These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2017 |