
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A mechanistic study of ethanol transformation into ethene and acetaldehyde on an oxygenated Au-exchanged ZSM-5 zeolite†
Yuwanda Injongkolab,
Thana Maihom *ab,
Saowapak Choomwattanac,
Bundet Boekfaab and
Jumras Limtrakuld
*ab,
Saowapak Choomwattanac,
Bundet Boekfaab and
Jumras Limtrakuld
aDepartment of Chemistry, Faculty of Liberal Arts and Science, Kasetsart University, Kamphaeng Saen Campus, Nakhon Pathom 73140, Thailand. E-mail: faastnm@ku.ac.th
bCenter for Advanced Studies in Nanotechnology for Chemical, Food and Agricultural Industries, KU Institute for Advanced Studies, Kasetsart University, Bangkok 10900, Thailand
cCenter of Data Mining and Biomedical Informatics, Faculty of Medical Technology, Mahidol University, Salaya Campus, Nakhon Pathom 73170, Thailand
dDepartment of Materials Science and Engineering, Vidyasirimedhi Institute of Science and Technology, Rayong 21210, Thailand
First published on 2nd August 2017
Abstract
Ethanol transformation to ethene and acetaldehyde over low- and high-spin state oxygenated Au-exchanged ZSM-5 zeolite has been investigated using a well-validated density functional method, M06-L. The reaction initiates from the ethanol O–H bond dissociation leading to the formation the ethoxide–hydroxide intermediate with the activation energy of 9.5 kcal mol−1. This intermediate can be then decomposed to either ethene or acetaldehyde products. In the ethene production pathway, the decomposition of the ethoxide–hydroxide intermediate proceeds via the β-H–C scission with the activation energy of 40.5 kcal mol−1. For the acetaldehyde production pathway, the ethoxide–hydroxide intermediate transforms to acetaldehyde via α-H–C scission with the activation barrier of 10.6 kcal mol−1 which is significantly lower than the ethene pathway. The reaction rate for acetaldehyde formation is also found to be higher than the ethene one. The results suggest that the acetaldehyde product is thermodynamically and kinetically favored over ethene for the transformation of the ethanol on oxygenated Au-exchanged ZSM-5 zeolite.
1. Introduction
Bioethanol, derived from energy crops, has recently received remarkable attention from the chemical industry for the development of clean technology.1–4 It is ideal for converting ethanol to more valuable chemicals, especially ethene and acetaldehyde. They are important feedstocks for many processes in the industry.1,4 Ethene can be produced via ethanol dehydration, which is preferably performed over solid acids such as acidic zeolites.5,6 Acetaldehyde is obtained from the dehydrogenation reaction catalyzed by metals and metal-supported or basic oxides such as magnesium oxide.7–12 Both reactions are found to be competitive on transition metals oxide catalysts such as Fe2O3, Mn2O3 and CeO2.11,13Supported gold (Au) catalysts have been additionally found to be favorable for these processes. With various supported materials including silica, high activity and good selectivity can be enhanced for various reactions of alcohol transformation.14–19 Zeolites are considered to be useful silica supports for metals. Au-exchanged zeolites, including NaY, Na–ZSM-5 and ZSM-5, were previously reported to display high activity for several reactions such as N2O and NO decomposition, the water gas shift (WGS) reaction and oxidation of CO, ethanol, cyclohexane and benzyl alcohol.20–28 Moreover, we also previously theoretically demonstrated the utilization of Au-exchanged zeolites for catalyzing the reaction including methane C–H bond activation, the direct conversion of carbon dioxide and methane to acetic acid, nitrous oxide decomposition, and the conversion of carbon dioxide and ethane to propanoic acid.29–32 The surface oxygen on Au-exchanged ZSM-5 zeolite promoting the ethanol O–H bond dissociation and the conversion of ethoxide to acetaldehyde is an example of how supported zeolite facilitate the Au activity in catalyzing reactions.33 To the best our knowledge, ethanol dehydration and dehydrogenation on the catalyst have not been studied in detail.
In the article, we report the comparison of ethanol dehydration and dehydrogenation on Au exchanged zeolites with the presence of surface oxygen. The reaction mechanism of both reactions together with their structures and energetics of reaction intermediates were investigated using Density Functional Theory (DFT) calculations with M06-L functional as well as the transition states were discussed to predict the preferred one. The possibilities of spin crossing were also additionally examined throughout the competitive reactions.
2. Model and methods
The ZSM-5 zeolite was represented by the 34T H-ZSM-5 cluster model (T refers to Si or Al atoms) generated from their lattice structures.34 Due to the Si/Al of 33, this model therefore are expected to represent the modulate Si/Al ZSM-5 zeolite. It covers the intersection between straight and zigzag channels, illustrated in Fig. 1a. In this model, a silicon atom at the T12 site was substituted by an aluminium atom to generate the Brønsted acid site. This site is the most stable Al substitution site and has been generally used to model the active site of H-ZSM-5 zeolite.35 The Brønsted site was then replaced with the Au cation to generate the Au–zeolite (Au/ZSM-5). Our previous studies on adsorption and hydrogen exchange of unsaturated aliphatic, aromatic and heterocyclic compounds employed the model and reported reasonable adsorption and activation energies, which are compared to experimental values.36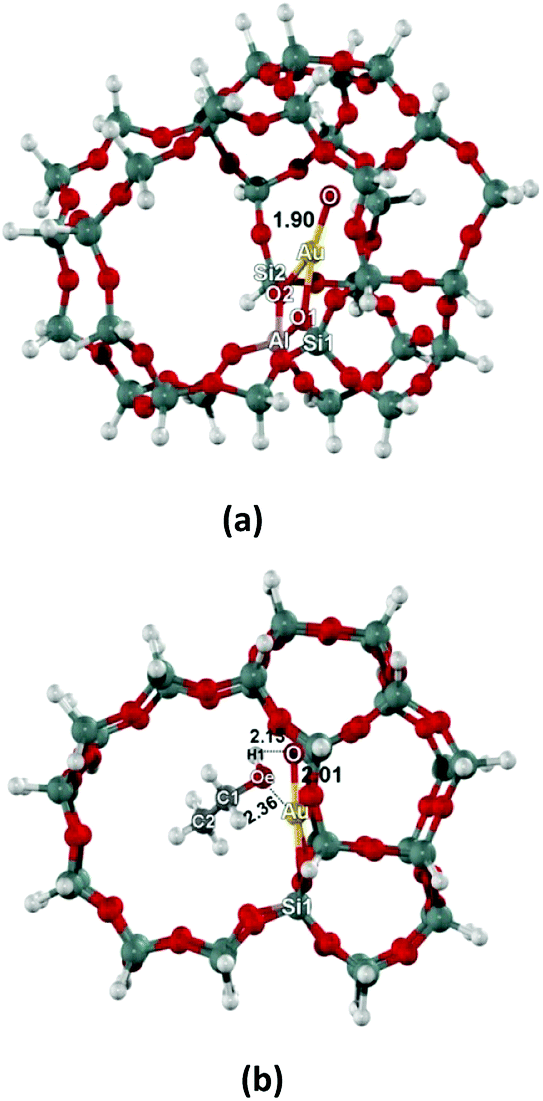 | ||
| Fig. 1 Optimized structures of AuO/ZSM-5 model (a) and ethanol adsorbed on AuO/ZSM-5 (b). | ||
All structure optimizations were performed with the M06-L density functional. This method was developed by Zhao and Truhlar37,38 and included van der Waals interactions in the parameterization. It has been successfully used for investigating the adsorption and reaction mechanisms over zeolites23,29–33,39–45 and also over other materials such as metals-organic frameworks.46–51 The 6-31G(d,p) basis set was used to treat Al, Si, C, O and H atoms, while the Au atom was described by the Stuttgart Effective Core Potential basis (ECP).52 During the geometry optimizations, only the 5T cluster of the active site region [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)3(AuO)Al(OH)Si] and the adsorbate molecule were allowed to relax, while the remaining atoms were fixed at the crystallographic coordinates. Transition states were confirmed by performing the frequency calculations at the same level of theory to guarantee that the transition state structure has only one imaginary frequency. The calculated relative adsorption energies was defined as follows:
SiO)3(AuO)Al(OH)Si] and the adsorbate molecule were allowed to relax, while the remaining atoms were fixed at the crystallographic coordinates. Transition states were confirmed by performing the frequency calculations at the same level of theory to guarantee that the transition state structure has only one imaginary frequency. The calculated relative adsorption energies was defined as follows:
| ΔE = E(complex) − E(zeolite) − E(adsorbate) |
The rate constants are also calculated using classical transition-state theory (TST) consistent with the following equation:
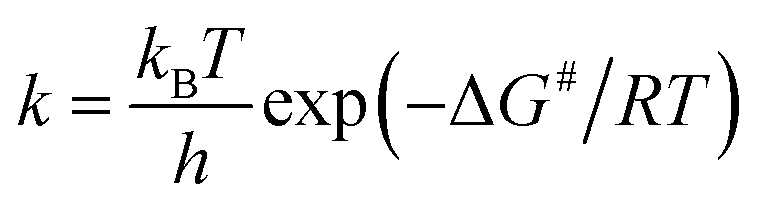
3. Results and discussion
3.1 Oxygenated Au-exchanged ZSM-5 zeolite model and ethanol adsorption
The singlet and triplet spin states of the oxygenated Au-exchanged ZSM-5 zeolite (AuO/ZSM-5) are first considered. We found that the triplet state is 19.0 kcal mol−1 more stable than the singlet state. We therefore investigate the structure of AuO/ZSM-5 and its interaction with ethanol molecule in the triplet state. The optimized structures of AuO/ZSM-5 are shown in Fig. 1. The Au–O active site can be produced by decomposition of oxidizing agents such as nitrous oxide (N2O) on Au/ZSM-5 leading to AuO/ZSM-5.55,56 The Au–O active site is coordinated to two oxygen bridging atoms of zeolite with the Au⋯O1 and Au⋯O2 distances of 2.23 and 2.30 Å, respectively (Fig. 1a). The calculated distance of Al⋯Au is 3.14 Å. The Au–O bond distance in the surface oxygen species is 1.90 Å. The value is compared well with the [AuO]+ bond length calculated with the highly accurate method of CCSD(T)57 and also approximately equivalent the Au–O bond distance of 1.89 Å in AuO–ZSM-5 calculated from B3LYP method with the 6-31G(d,p) basis set for light atoms and LANL2DZ for Au.56 The NBO analysis shows the overlap orbital interaction between Au s-type orbital (s = 86%, d = 14%) and O sp-type orbital (s = 14%, p = 86%). The partial charges of Au and O are +0.88e and −0.29e, respectively. We also found the spin density located on the O atom of the Au–O active site (O = 1.38 and Au = 0.44) which shows the free radical character of this surface O on Au. This might be responsible for the hydrogen abstraction in the further steps.The optimized structure of ethanol adsorption on the Au–O site of the Au/ZSM-5 zeolite are shown in Fig. 1b. The ethanol interacts with the active site via the hydroxyl group (OH) of ethanol and the Au–O active site. The same location was previously reported in methanol and ethanol adsorption on Fe–O/ZSM-5 and also for GaO/ZSM-5.43,58 The Au–O bond distance of the Au–O active site is elongated upon the adsorption of ethanol from 1.90 to 2.01 Å and the length of the alcoholic O–H bond increases by 0.02 Å. The intermolecular distance of Oe⋯Au is 2.36 Å. Because of the electron transfers from Oe to the Au site, the negative partial charge of Oe decreases from −0.64e to −0.51e. The adsorption energy is calculated to be −17.3 kcal mol−1.
To ensure the convergence of 34T cluster model used in this work, the single point calculations at the same level of theory for ethanol adsorption based on the 34T cluster optimized structures were performed on different cluster sizes i.e. 5T, 12T, 46T and the extended 120T models. The cluster models and their ethanol adsorption energies are showed in the Fig. S1.† The calculated adsorption energy is proportional to the cluster size due to the increasing contribution of the zeolite frameworks. However, the adsorption energies are almost equal from the calculations in the original 34T to the extended 46T and 120T models, indicating that the model size increasing from 34T does not affect the energetics of the system. The 34T cluster model should be therefore practical and large enough to represent interactions between the adsorbate and the zeolite frameworks. The 34T model is then employed to investigate the reaction mechanisms of ethanol dehydration and dehydrogenation in the subsequent sections.
3.2 Reaction mechanisms of ethanol dehydration and dehydrogenation
The proposed reaction mechanism of ethanol transformation to ethene and acetaldehyde initiates with the ethanol O–H bond dissociation to form the ethoxide–hydroxide intermediate on the Au site. The intermediate can be then transformed either to ethene or acetaldehyde products. It is well known that the spin state crossing especially for the reactions containing transition metals is often observed and may play a role in the catalytic reactions. We therefore analyze the reaction pathways along with two possible spin states of singlet and triplet. From the results of two spin states, the intersystem spin-crossing occurrence is found in the first step of the ethoxide–hydroxide intermediate formation after the ethanol O–H bond dissociation, leading to the lowering endothermicity of intermediate formation (Fig. S2†). Moreover, the spin crossing is also found before the transition state for the intermediate transformation to ethene product, while the spin state remains singlet throughout the acetaldehyde route (Fig. S3 and S4†). In the following discussion, only the most stable spin state for each complex is considered in reaction coordinates. The superscript numbers 1 and 3 in the energy profiles and tables refer to the singlet and triplet spin states. The full reaction mechanisms with two-dimensional chemical structure are also provided in Fig S5.†Optimized structures for the first step of the ethanol O–H bond dissociation are displayed in Fig. 2 and selected geometrical parameters are shown in Table 1. The reaction starts with the ethanol interacting to the Au–O site of Au–O/ZSM-5 zeolite with the adsorption energy of −17.3 kcal mol−1 as reported above. Then, the adsorbed ethanol is converted to the ethoxide intermediate through the transition state (TS_1). Its alcoholic O–H bond of the ethanol molecule is dissociated and H1 simultaneously transfers to the O of the Au–O active site of zeolite. The Oe–H1 bond distance is elongated from 0.98 to 1.29 Å, whereas the interatomic distance of O⋯H1 is decreased from 2.13 to 1.16 Å. The transition state is confirmed by normal mode analysis with one imaginary frequency at −1532.5 cm−1, which corresponds to the simultaneous Oe–H1 bond spitting and H1–O1 bond forming. The activation energy of this step is 9.5 kcal mol−1. It is considered to be lower than the ethanol O–H bond dissociation on FeO/ZSM-5 zeolite in a previous theoretical study.43 This suggests that the [Au–O] site is more active than that [Fe–O] site on ZSM-5 zeolite due mainly to the unpaired electrons localized on the active O of the [Au–O] site that helps to abstract the hydrogen from the ethanol hydroxyl group. These results agree with the theoretical study of the methanol O–H bond dissociation on Au(111) surface and the experimental observation which reported that the surface O on Au was essential for the activation of the O–H bond in alcohols on the Au surface.59–62
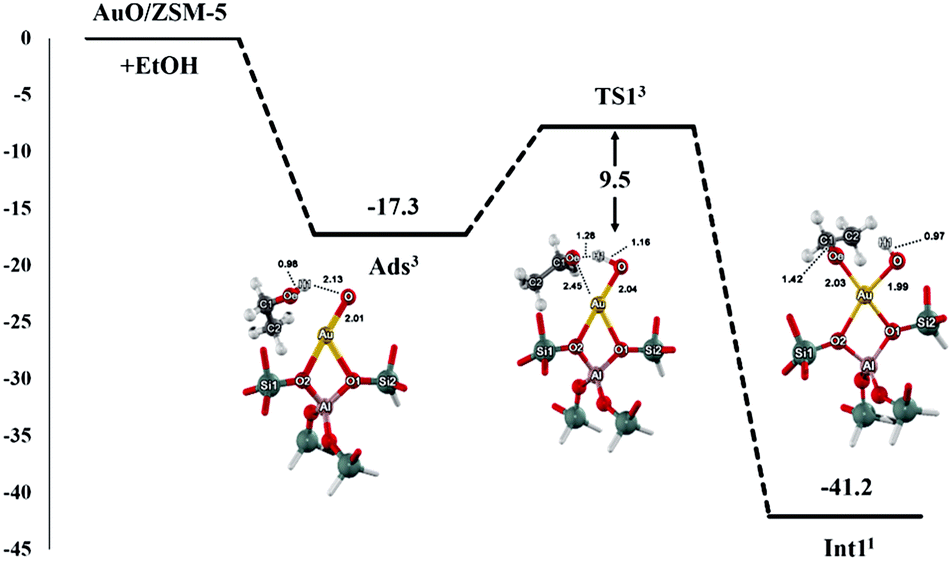 | ||
| Fig. 2 Energy profile and optimized structures of adsorption, transition state and intermediate involved the ethoxide–hydroxide intermediate formation (energies are in kcal mol−1). The superscript numbers refer to the most stable spin state of each reaction complex. | ||
| Parameters | Triplet | Singlet | ||||
|---|---|---|---|---|---|---|
| Ads | TS1 | Int1 | Ads | TS1 | Int1 | |
| Distance (Å) | ||||||
| Au–Al | 3.08 | 3.13 | 3.13 | 3.09 | 3.04 | 3.03 |
| Au–O | 2.01 | 2.04 | 2.14 | 1.88 | 1.92 | 1.99 |
| Au–Oe | 2.37 | 2.45 | 2.27 | 2.91 | 2.38 | 2.03 |
| O–H1 | 2.13 | 1.16 | 0.97 | 2.23 | 1.75 | 0.97 |
| Oe–H1 | 0.98 | 1.29 | 2.41 | 0.97 | 1.00 | 2.19 |
| Oe–C1 | 1.45 | 1.41 | 1.40 | 1.42 | 1.44 | 1.42 |
| C1–C2 | 1.51 | 1.51 | 1.51 | 1.50 | 1.50 | 1.52 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| Angle (°) | ||||||
| Si1–O1–Al | 128.3 | 126.6 | 128.0 | 127.9 | 128.6 | 127.4 |
| Si2–O2–Al | 136.8 | 138.1 | 137.3 | 135.7 | 136.0 | 139.1 |
| Oe–Au–O | 74.7 | 61.8 | 60.6 | 64.9 | 71.2 | 81.9 |
| Oe–H1–O | 112.7 | 145.9 | 67.5 | 109.1 | 131.6 | 105.9 |
The ethoxide–hydroxide intermediate (Int_2) is then generated in the zeolite pore which is comparable to the reaction of alcohols O–H bond activation on the FeO/ZSM-5 and GaO/ZSM-5 systems.43,58,61 In this complex, the spin state is changed from triplet to singlet (cf. Fig. S2†). Furthermore, the result of the spin-crossing phenomenon leads to the lowering of the exothermicity of the reaction to −41.2 kcal mol−1. The Au⋯Oe and Au⋯O distances are 2.03 and 1.99 Å, respectively. Since the complexation energy of this intermediate over AuO/ZSM-5 zeolite is less stable than that of the GaO site of GaO/ZSM-5 zeolite (−68 kcal mol−1),58 the intermediate is more readily converted over AuO/ZSM-5 zeolite compared to GaO/ZSM-5 zeolite.
The reaction is followed by the decomposition of ethoxide–hydroxide intermediate to either ethene or acetaldehyde products as shown in Fig. 3. For the ethene pathway, the spin crossing is found between the singlet-state intermediate and the second transition state (TS2_E) in triplet state (see Fig. S3†). The H2 proton of TS2_E is transferred to the oxygen (O) of the hydroxyl group. The C2–H2 bond distance is elongated from 1.09 to 1.30 Å, while the O⋯H2 distance decreases to 1.28 Å for the bond forming (see Table 2). This transition state of this step is very similar to the carbenium-ion complex i.e. the hybridization of C1 changes from tetrahedral (sp3) to planar (sp2). The transition state is confirmed by the frequency calculation with one imaginary frequency at 1592.2i cm−1, which relates to the breaking of the C2–H2 bond and the movement of the H2 to O. The predicted activation energy of this step is 40.5 kcal mol−1. After the migration, the ethene is formed on the Au–ZSM-5 zeolite (Prod_E) with the calculated complexation energy of −4.6 kcal mol−1.
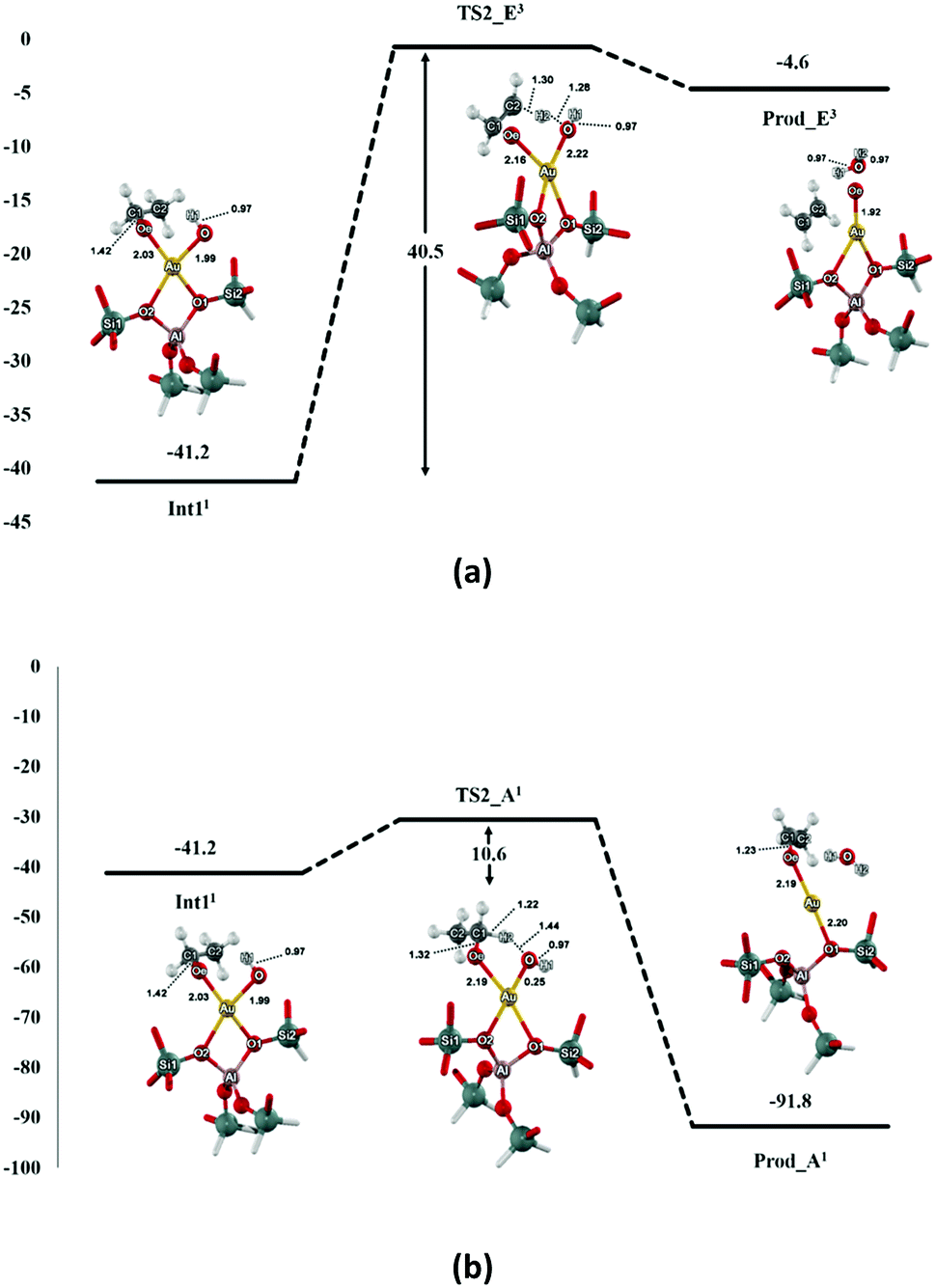 | ||
| Fig. 3 Energy profile and optimized structures of the intermediate, transition state and product involved the formation of ethene (a) and acetaldehyde (b) (energies are in kcal mol−1). The superscript numbers refer to the most stable spin state of each reaction complex. | ||
| Parameters | Ethene pathway | Acetaldehyde pathway | ||||||
|---|---|---|---|---|---|---|---|---|
| TS2_E3 | Prod_E3 | TS2_E1 | Prod_E1 | TS2_A3 | Prod_A3 | TS2_A1 | Prod_A1 | |
| Distances (Å) | ||||||||
| Au–Al | 3.09 | 3.12 | 3.00 | 3.11 | 3.11 | 3.09 | 2.96 | 3.18 |
| Au–O1 | 2.36 | 2.17 | 2.25 | 2.32 | 2.34 | 2.25 | 2.31 | 2.20 |
| Au–O2 | 2.27 | 2.30 | 2.15 | 2.21 | 2.26 | 2.33 | 2.24 | 2.74 |
| Au–O | 2.22 | 2.28 | 2.05 | 3.21 | 2.24 | 2.46 | 2.05 | 2.97 |
| Au–Oe | 2.16 | 1.92 | 1.97 | 1.90 | 2.20 | 2.11 | 2.19 | 2.19 |
| Oe–H1 | 2.97 | 2.65 | 2.00 | 2.14 | 3.02 | 2.31 | 3.55 | 2.54 |
| Oe–C1 | 1.41 | 4.04 | 2.25 | 3.19 | 1.34 | 1.34 | 1.32 | 1.23 |
| O–H2 | 1.28 | 2.26 | 1.22 | 0.97 | 1.32 | 0.97 | 1.44 | 0.96 |
| C2–H2 | 1.30 | 3.90 | 1.40 | 2.38 | 2.15 | 4.06 | 2.20 | 4.98 |
| C1–C2 | 1.50 | 1.33 | 1.38 | 1.33 | 1.52 | 1.50 | 1.51 | 1.47 |
|
||||||||
| Angles (°) | ||||||||
| O–Au–Oe | 84.2 | 53.8 | 77.0 | 36.8 | 76.6 | 76.5 | 76.2 | 63.1 |
| O–H2–C2 | 166.4 | 28.4 | 166.7 | 48.7 | 119.4 | 25.3 | 139.2 | 24.2 |
In the acetaldehyde production pathway, there is no spin crossing for this reaction path and the reaction will follow the singlet state (see Fig. S4†). Acetaldehyde can be generated via the second carbenium ion transition state (TS2_A2) in which the C1–H3 bond is broken and the α-proton (H3) is transferred to the oxygen atom O of the intermediate OH group. In the transition state, the C1–H3 intramolecular distance lengthens from 1.10 to 1.22 Å, whereas the O⋯H3 distance is decreased to 1.44 Å (see Table 2). One imaginary frequency at −809.0 cm−1 is revealed from this transition state. It relates the movement of the H3 to O and also the C1–H3 bond breaking. With respect to the intermediate complex, 10.6 kcal mol−1 is the estimated activation energy of this step, which is lower than that of the ethene production pathway reported above. The acetaldehyde product is then formed (Prod_A) and adsorbed on the Au active site with the neighboring water molecule. The distances of the Au–Oe and the Au⋯O are 2.19 and 2.97 Å, respectively. Energy of −91.8 kcal mol−1 is gained from the formation of this product complex.
From the energy profile for the preferred spin state in all reaction complexes shown in Fig. 4, it can be seen that the ethoxide decomposition to the acetaldehyde product is more facile than to the ethene one. The reaction barrier for the acetaldehyde pathway is found to be 29.9 kcal mol−1 lower than the rate-determining step of the ethene pathway. The reason is that the secondary carbocation transition state obtained from the β-H–C scission in the acetaldehyde pathway is practically more stable than the primary carbocation transition state occurred via the α-H–C scission for the ethene pathway (Fig. 4). In kinetic aspect, the reverse reaction is possible. From the outperformance of the forward reaction rate (kr+) over the reverse reaction rate (kr−) in the acetaldehyde pathway (cf. Table 3), the forward reaction to the acetaldehyde product is favored over the reverse one. In contrast, the ethene pathway prefers the reverse reaction (kr− > kr+). Therefore, the formation of acetaldehyde via ethanol transformation on the oxygenated Au exchanged ZSM-5 zeolite should be more kinetically and thermodynamically over the ethene one. This is in agreement with the experimental findings for ethanol dehydrogenation and dehydration catalyzed by Au and also metals oxide surfaces catalysts.63–65
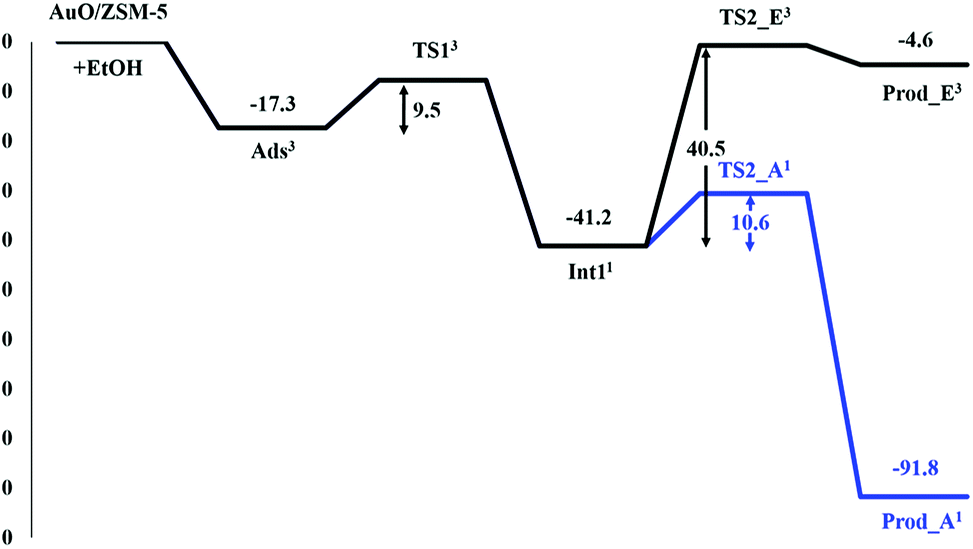 | ||
| Fig. 4 Energy profile for ethanol transformation to either ethene (black line) or acetaldehyde (blue line) over AuO/ZSM-5 zeolite (energies are in kcal mol−1). The superscript numbers in reaction profiles refer to the most stable spin state of each reaction complex. | ||
| Reaction path | Rate constants (s−1) | |
|---|---|---|
| Forward (kr+) | Reverse (kr−) | |
| Ethene (α C–H scission) | 2.33 × 10−12 | 2.16 × 1010 |
| Acetaldehyde (β C–H scission) | 1.53 × 109 | 4.65 × 10−31 |
We also investigate the zeolite frameworks effect by calculating the studied reaction with the preferred spin state on the small zeolite 12T cluster model (cf. Fig. S6†). The relative energies involved in the reaction on both cluster models are summarized in Table 4. For example, the adsorption energy of ethanol on 12T model of AuO/ZSM-5 is −14.4 kcal mol−1 (−17.3 kcal mol−1 in 34T cluster model). The framework contributes only around 17% because of the strong interaction between ethanol and the AuO active site of Au–ZSM-5 zeolite. The same trend is also found in all species along the reaction coordinates in the range of 3–7 kcal mol−1. The activation energies of both ethene and acetaldehyde production pathways remain virtually unchanged by the size of ZSM-5 zeolite framework.
| Reaction coordinate | Relative energy (kcal mol−1) | |
|---|---|---|
| 12T | 34T | |
| Ads3 | −14.4 | −17.3 |
| TS13 | −5.4 (Ea1 = 9.0) | −7.8 (Ea1 = 9.5) |
| Int1 | −36.0 | −41.2 |
|
||
| Ethene path | ||
| TS2_E3 | 2.0 (Ea2 = 38.0) | −0.7 (Ea2 = 40.5) |
| Prod_E3 | 0.5 | −4.6 |
|
||
| Acetaldehyde path | ||
| TS2_A1 | −28.6 (Ea2 = 7.4) | −30.6 (Ea2 = 10.6) |
| Prod_A1 | −84.4 | −91.8 |
4. Conclusions
The reaction mechanism of ethanol transformation to ethene and acetaldehyde over AuO/ZSM-5 was investigated by utilizing the 34T cluster model and the density functional M06-L method. The spin crossing along the reaction coordinate was also considered. First, the ethanol O–H bond breaks to form the ethoxide intermediate with the activation barrier of 9.5 kcal mol−1. Consequently, the intermediate is decomposed into either ethene or acetaldehyde products. The ethene production pathway requires 40.5 kcal mol−1 for the decomposition via α-H–C scission to produce water and ethene. For the acetaldehyde production pathway, the ethoxide intermediate is decomposed via β-H–C scission into an acetaldehyde with the activation energy of 10.6 kcal mol−1. It is significantly lower than that of the ethene production one. Due to the more stable secondary transition state, the ethoxide species conversion to acetaldehyde is more favorable than that of the ethene product. We also found higher reaction rate for acetaldehyde production compared to ethene one. Overall, it implies that ethanol transformation on the oxygenated Au-exchange zeolites prefers to produce acetaldehyde over ethene. The effect of the zeolite framework in all reacting species was also highlighted.Acknowledgements
This research was supported in part by grants from the National Science and Technology Development Agency (NANOTEC Center for Nanoscale Materials Design for Green Nanotechnology funded by the National Nanotechnology Center), the PTT group (PTT Public Company Limited, PTT Exploration & Production, PTT Global Chemical, IRPC Thai oil), the Commission on Higher Education, Ministry of Education (the “National Research University Project of Thailand (NRU)” to Y. I.), Kasetsart University Research and Development Institute (KURDI) and as well as the Thailand Research Fund (TRF) to TM (TRG5880248). The authors also acknowledge National e-Science Infrastructure Consortium for providing computing resources that have contributed to the research results reported within this paper. S. C. would also like to thank Mahidol University and the Talent Management program.Notes and references
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS PubMed.
- A. Haryanto, S. Fernando, N. Murali and S. Adhikari, Energy Fuels, 2005, 19, 2098–2106 CrossRef CAS.
- R. K. Niven, Renewable Sustainable Energy Rev., 2005, 9, 535–555 CrossRef CAS.
- Y. Gucbilmez, T. Dogu and S. Balci, Ind. Eng. Chem. Res., 2006, 45, 3496–3502 CrossRef CAS.
- C. B. Phillips and R. Datta, Ind. Eng. Chem. Res., 1997, 36, 4466–4475 CrossRef CAS.
- J. N. Kondo, K. Ito, E. Yoda, F. Wakabayashi and K. Domen, J. Phys. Chem. B, 2005, 109, 10969–10972 CrossRef CAS PubMed.
- N. Takezawa, C. Hanamaki and H. Kobayashi, J. Catal., 1975, 38, 101–109 CrossRef.
- J. M. Vohs and M. A. Barteau, Surf. Sci., 1989, 221, 590–608 CrossRef CAS.
- F. W. Chang, W. Y. Kuo and K. C. Lee, Appl. Catal., A, 2003, 246, 253–264 CrossRef CAS.
- R. M. Rioux and M. A. Vannice, J. Catal., 2003, 216, 362–376 CrossRef CAS.
- T. Zaki, J. Colloid Interface Sci., 2005, 284, 606–613 CrossRef CAS PubMed.
- R. Issaadi, F. Garin and C. E. Chitour, Catal. Today, 2006, 113, 166–173 CrossRef CAS.
- M. Li, Z. Wu and S. H. Overbury, J. Catal., 2013, 306, 164–176 CrossRef CAS.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552–560 CrossRef CAS.
- S. Biella and M. Rossi, Chem. Commun., 2003, 3, 378–379 RSC.
- A. Abad, P. Concepción, A. Corma and H. García, Angew. Chem., Int. Ed., 2005, 44, 4066–4069 CrossRef CAS PubMed.
- Y. Guan and E. J. M. Hensen, Appl. Catal., A, 2009, 361, 49–56 CrossRef CAS.
- Z. Martinez-Ramirez, J. A. Gonzalez-Calderon, A. Almendarez-Camarillo and J. C. Fierro-Gonzalez, Surf. Sci., 2012, 606, 1167–1172 CrossRef CAS.
- O. A. Simakova, E. Smolentseva, M. Estrada, E. V. Murzina, S. Beloshapkin, S. M. Willför, A. V. Simakov and D. Y. Murzin, J. Catal., 2012, 291, 95–103 CrossRef CAS.
- S. Qiu, R. Ohnishi and M. Ichikawa, J. Chem. Soc., Chem. Commun., 1992, 1425–1427 RSC.
- S. Qiu, R. Ohnishi and M. Ichikawa, J. Phys. Chem., 1994, 98, 2719–2721 CrossRef CAS.
- T. M. Salama, T. Shido, R. Ohnishi and M. Ichikawa, J. Chem. Soc., Chem. Commun., 1994, 2749–2750 RSC.
- M. M. Mohamed, T. M. Salama and M. Ichikawa, J. Colloid Interface Sci., 2000, 224, 366–371 CrossRef CAS PubMed.
- Z. X. Gao, Q. Sun, H. Y. Chen, X. Wang and W. M. H. Sachtler, Catal. Lett., 2001, 72, 1–5 CrossRef CAS.
- J. C. Fierro-Gonzalez and B. C. Gates, J. Phys. Chem. B, 2004, 108, 16999–17002 CrossRef CAS.
- G. Li, D. I. Enache, J. Edwards, A. F. Carley, D. W. Knight and G. J. Hutchings, Catal. Lett., 2006, 110, 7–13 CrossRef CAS.
- R. Zhao, D. Ji, G. Lv, G. Qian, L. Yan, X. Wanga and J. Suo, Chem. Commun., 2004, 904–905 RSC.
- H. Chen, X. Jia, Y. Li, C. Liu and Y. Yang, Catal. Today, 2015, 256, 153–160 CrossRef CAS.
- W. Panjan, J. Sirijaraensre, C. Warakulwit, P. Pantu and J. Limtrakul, Phys. Chem. Chem. Phys., 2012, 14, 16588–16594 RSC.
- S. Wannakao, C. Warakulwit, K. Kongpatpanich, M. Probst and J. Limtrakul, ACS Catal., 2012, 2, 986–992 CrossRef CAS.
- T. Maihom, S. Wannakao, B. Boekfa and J. Limtrakul, Chem. Phys. Lett., 2013, 556, 217–224 CrossRef CAS.
- W. Sangthong, M. Probst and J. Limtrakul, ChemPhysChem, 2014, 15, 514–520 CrossRef CAS PubMed.
- T. Maihom, M. Probst and J. Limtrakul, J. Phys. Chem. C, 2014, 118, 18564–18572 CAS.
- H. V. Koningsvel, H. V. Bekkkum and J. C. Jansen, Acta Crystallogr., Sect. A: Found. Crystallogr., 1987, 48, 127–132 Search PubMed.
- S. Lonsinger, A. Chakraborty, D. Theodorou and A. Bell, Catal. Lett., 1991, 11, 209 CrossRef CAS.
- B. Boekfa, S. Choomwattana, P. Khongpracha and J. Limtrakul, Langmuir, 2009, 25, 12990–12999 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. C, 2008, 112, 6860–6868 CAS.
- C. Kumsapaya, K. Bobuatong, P. Khongpracha, Y. Tantirungrotechai and J. Limtrakul, J. Phys. Chem. C, 2009, 113, 16128–16137 CAS.
- T. Maihom, B. Boekfa, J. Sirijaraensre, T. Nanok, M. Probst and J. Limtrakul, J. Phys. Chem. C, 2009, 113, 6654–6662 CAS.
- T. Maihom, P. Pantu, T. Chaiwat, M. Probst and J. Limtrakul, J. Phys. Chem. C, 2010, 114, 7850–7856 CAS.
- S. Wannakao, P. Khongpracha and J. Limtrakul, J. Phys. Chem. A, 2011, 115, 12486–12492 CrossRef CAS PubMed.
- T. Maihom, P. Khongpracha, J. Sirijaraensre and J. Limtrakul, ChemPhysChem, 2013, 14, 101–107 CrossRef CAS PubMed.
- T. Maihom, M. Probst and J. Limtrakul, ChemPhysChem, 2015, 16, 3334–3339 CrossRef CAS PubMed.
- P. Charoenwiangnuea, T. Maihom, P. Kongpracha, J. Sirijaraensre and J. Limtrakul, RSC Adv., 2016, 6, 105888–105894 RSC.
- S. J. Kolmann, B. Chan and M. J. T. Jordan, Chem. Phys. Lett., 2008, 467, 126–130 CrossRef CAS.
- R. B. Getman, J. H. Miller, K. Wang and R. Q. Snurr, J. Phys. Chem. C, 2011, 115, 2066–2075 CAS.
- T. Maihom, S. Choomwattana, P. Khongpracha, M. Probst and J. Limtrakul, ChemPhysChem, 2012, 13, 245–249 CrossRef CAS PubMed.
- T. Maihom, S. Wannakao, B. Boekfa and J. Limtrakul, J. Phys. Chem. C, 2013, 117, 17650–17658 CAS.
- C. Raksakoon, T. Maihom, M. Probst and J. Limtrakul, J. Phys. Chem. C, 2015, 119, 3564–3571 CAS.
- T. Maihom, S. Choomwattana, S. Wannakao, M. Probst and J. Limtrakul, ChemPhysChem, 2016, 17, 3416–3422 CrossRef CAS PubMed.
- M. Dolg, H. Stoll and H. Preuss, J. Phys. Chem., 1993, 97, 5852–5859 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision C01, Gaussian Inc., Wallingford CT, 2010 Search PubMed.
- A. Sierraalta, R. Hernandez-Andara and E. Ehrmann, J. Phys. Chem. B, 2006, 110, 17912–17917 CrossRef CAS PubMed.
- M. F. Fellah and I. Onal, Catal. Today, 2011, 171, 52–59 CrossRef CAS.
- F. X. Li, K. Gorham and P. B. Armentrout, J. Phys. Chem. A, 2010, 114, 11043–11052 CrossRef CAS PubMed.
- E. A. Pidko, E. J. M. Hensen and R. A. Van Santen, J. Phys. Chem. C, 2007, 111, 13068–13075 CAS.
- J. Gong, D. W. Flaherty, R. A. Ojifinni, J. M. White and C. B. Mullins, J. Phys. Chem. C, 2008, 112, 5501–5509 CAS.
- B. Xu, X. Liu, J. Haubrich, R. J. Madix and C. M. Friend, Angew. Chem., Int. Ed., 2009, 48, 4206–4209 CrossRef CAS PubMed.
- M. F. Fellah, J. Catal., 2011, 282, 191–200 CrossRef CAS.
- B. Xu, J. Haubrich, T. A. Baker, E. Kaxiras and C. M. Friend, J. Phys. Chem. C, 2011, 115, 3703–3708 CAS.
- A. Beste and S. H. Overbury, J. Phys. Chem. C, 2015, 119, 2447–2455 CAS.
- J. Gong and C. B. Mullins, J. Am. Chem. Soc., 2008, 130, 16458–16459 CrossRef CAS PubMed.
- T. Takei, N. Iguchi and M. Haruta, Catal. Surv. Asia, 2011, 15, 80–88 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 the ethanol adsorption energies of the AuO/ZSM-5 as a function of the cluster model sizes. Fig. S2–S4. Energy profile for the ethoxide–hydroxide conversion on AuO/ZSM-5 for singlet state (dot line) and triplet state (solid line). Fig. S5. The 12T cluster model of AuO/ZSM-5 zeolite. See DOI: 10.1039/c7ra06313j |
| This journal is © The Royal Society of Chemistry 2017 |