
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phillygenin attenuates inflammatory responses and influences glucose metabolic parameters by inhibiting Akt activity
Wenjuan Liu a,
Guangcui Chua,
Nianwei Changa,
Xiaoyao Mab,
Min Jiang*b and
Gang Bai*b
a,
Guangcui Chua,
Nianwei Changa,
Xiaoyao Mab,
Min Jiang*b and
Gang Bai*b
aTianjin University of Traditional Chinese Medicine, Tianjin, 300193, People's Republic of China
bState Key Laboratory of Medicinal Chemical Biology, College of Pharmacy, Tianjin Key Laboratory of Molecular Drug Research, Nankai University, Haihe Education Park, 38 Tongyan Road, Tianjin 300353, People's Republic of China. E-mail: minjiang@nankai.edu.cn; gangbai@nankai.edu.cn
First published on 18th August 2017
Abstract
Phillygenin (Phi) is one of the main chemical constituents of the fruit of Forsythia suspensa (Thunb.) Vahl. It has various bioactivities, including anti-inflammatory, anti-obesity and antipyretic activities. However, its exact targets and molecular mechanism are still poorly understood. Bioinformatics tools were used to explore the potential targets of Phi, and 8 predicted targets, 4 primary pathways (MAPK, PI3K-Akt, T-cell receptor and m-TOR signaling pathway) related to the inflammatory response, and Akt as an important node was mentioned. Moreover, a Phi alkylated molecular probe was synthesized and used to capture the target proteins Akt. Then Akt and its downstream signaling pathway were verified by molecular docking, intracellular enzyme activity evaluation, and accurate pathway analysis. The results indicated that Phi targets an allosteric inhibit pocket on Akt; reduces Akt phosphorylation; alleviates multiple inflammatory-associated downstream signal transduction pathways, including IKKα/β and NF-κB; and influences glucose metabolic parameters associated with the downstream GSK3β protein and glucose uptake. The results suggest that Phi could reduce inflammatory responses and influence glucose metabolic parameters by inhibiting Akt phosphorylation. Moreover, these findings suggest a potential application for Phi in respiratory and metabolic diseases therapy.
1. Introduction
Forsythia Fructus (“Lianqiao” in Chinese) is the dried fruit of Forsythia suspensa (Thunb.) Vahl, which is widely distributed in China, Korea, Japan and many European countries.1 It is commonly used for pneumonia, boils and other infectious diseases.2 Phillygenin (Phi) is an aglycone of phillyrin, which is a major lignan compound of Forsythia Fructus that plays an important role in the anti-inflammatory, antiviral, antioxidant, anti-obesity activity and antipyretic activities.3–5 Pharmacokinetic studies have shown that phillyrin is rapidly metabolized to Phi in the body, which then has a pharmacological effect.6,7 Although there have been many reports on the anti-inflammatory and antipyretic effects of Phi, its exact targets and molecular mechanism remain unclear. Natural products are important sources of novel structures in new drug development, due to the remarkable structural diversity and impressive pharmacological activities.8,9 The present study highlights drug-target-pathway mechanism research on natural products and will be helpful in guiding the design of selective and effective drugs for disease therapy.10–12It is a challenging and important to identify the targets and mechanisms of active natural products. In recent years, progress in chemical biology research has accelerated the identification of target proteins of biologically active small molecules.13 The addition of azide and alkynyl moieties is a promising chemical biology method because these groups are highly reactive and totally biocompatible.14 The alkyne tag can serve as a benign handle for target identification by click chemistry to azides. The click chemistry refers to a group of chemical reactions that are fast, simple to carry out, easy to purify, regiospecific, and have high yields in joining diverse structures.15,16 Since its introduction, the concept of the click reaction has enabled the study of biomolecules, such as glycans and proteins, in real time in living systems without cellular toxicity.17,18 In addition, a fishing-rod strategy is also one of promising methods applicable for target identification of natural products.19 This prompted us to explore natural products using the effective approaches of molecular imaging and fishing-rod strategy.
In this study, a probe of alkylated phillygenin (aPhi) was prepared first, and then molecular imaging and target capture were carried out in living HEK 293T cells. Subsequently, molecular docking, cell co-localization and downstream signalling pathway evaluation were performed to evaluate the potential targets and the effects of Phi.
2. Materials and method
2.1. Materials and chemicals
Phillygenin (Phi) was purchased from National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Alkynylation of phillygenin (aPhi) was synthesized by WuXi AppTec (Beijing, China). Akt activator SC79 (SC) and Akt Inhibitor VIII (VIII) were obtained from Med Chem Express (Rocky Hill, NJ, USA). The NF-κB Dual-Luciferase ReporterR Assay System was purchased from Promega (Madison, WI, USA). The Lipofectamine 2000 transfection reagent was obtained from Invitrogen (Carlsbad, CA, USA). Human tumour necrosis factor alpha (TNF-α) was obtained from Pepro Tech (Rocky Hill, NJ, USA). The NF-κB luciferase reporter plasmid pGL4.32 and Renilla luciferase reporter vector plasmid pRL-TK were purchased from Promega (Madison, WI, USA). Dexamethasone (Dex), lipopolysaccharide (LPS) and dimethyl sulfoxide (DMSO) were purchased from Sigma Corporation (St. Louis, MO, USA). The polyclonal primary antibody against Akt and monoclonal antibodies against p-Akt (Ser473), NF-κB, p-NF-κB, p-GSK3β and β-actin were purchased from Cell Signaling Technology (Danvers, MA, USA). Alexa Fluor® 594-conjugated anti-rabbit antibody and monoclonal antibodies GSK3β, p-Akt (Thr308) and IKKα/β were purchased from Abcam (Cambridge, MA, UK), and polyclonal primary antibody p-IKKα was purchased from Assay Biotechnology Company, Inc. (San Francisco, CA, USA). The DAPI Fluoromount-G® was purchased from Southern Biotech Company, Inc. (Birmingham, AL, USA). A bicinchoninic acid (BCA) protein assay kit was purchased from Pierce Co. (Rockford, IL, USA). All other materials are commercially available.2.2. Target proteins-prediction
In silico drug target identification, Pharm Mapper server is a freely accessed web server designed to identify potential target candidates for the given small molecules via large-scale reverse pharmacophore mapping strategy.20 So the three-dimensional structure of Phi was saved as sdf format and uploaded to the Pharm Mapper database (http://59.78.96.61/pharmmapper/) for target prediction of Phi. In the process, we selected only human protein targets and set 100 reserved matched targets. Then, top 100 target proteins ranked by fit score in descending order were output from prediction of Pharm Mapper server. Next, we studied the top 30 target proteins of Phi one by one by uploading them to the String 9.1 website (http://string-db.org/), which is a curated knowledge database of known and predicted protein–protein interactions. In addition, the PATHWAY database of KEGG website (http://www.ge-nome.jp/kegg/) was used to further analyze the proteins and pathways associated with inflammation. The crystal structures of potential target proteins were obtained from the Protein Data Bank (http://www.rcsb.org/pdb). The structure of Phi and potential targets were constructed and minimized using the Molecular Operating Environment software package (Chemical Computing Group, Inc.). Then, Auto Dock version 4.2 (Olson Laboratory, La Jolla, CA) was used to simulate ligand-receptor binding using a hybrid lamarckian genetic algorithm (LGA). The step size parameters of quaternion and torsion were set to 30. For the compound, 30 independent runs were performed. Default values were used for the other parameters. The free energy of binding was calculated from the contributions of the hydrophobic, ionic, hydrogen bond, and van der Waals interactions between the protein and the ligand, as well as intramolecular hydrogen bonds and strains of the ligand. We observed that the docking poses were ranked by the binding free energy calculation in the S field.2.3. Cell culture
The HEK 293T cells and BEAS-2B cells were purchased from American Type Culture Collection (Rockville, MD) and were cultured in DMEM and RPMI 1640, respectively, supplemented with 10% FBS, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C and 5% CO2 in a humidified incubator.2.4. Dual-luciferase assay system for NF-κB inhibition
The HEK 293T cells were grown in 96-well culture plates for 24 h and then co-transfected with the NF-κB luciferase reporter plasmid PGL4.32 (100 ng per well) and the Renilla luciferase reporter vector plasmid pRL-TK (9.6 ng per well). Transfection was performed for 20 h using Lipofectamine 2000 according to the manufacturer's instructions. The cells were pretreated with TNF-α (5 ng mL−1) for 6 h. And the inhibition of NF-κB activity was detected according to a previously described method.212.5. Fishing for target proteins
A total of 2 mL of Fe3O4 amino magnetic microspheres (Tianjin baseline chromtech research centre, Tianjin, China; 5 mg mL−1) were added to a stirred solution of Sulfo-SADP (Bioworld, MN, USA; 1 mg, 22 μmol) in 10 mL borate buffer (pH 8.5). The reaction mixture was allowed to stand for 12 h at room temperature. Then, a magnetic separation was conducted using a magnet, and the beads were washed 3 times with distilled water. Then, aPhi (0.58 mg, 1.5 μmol) was added to the microspheres and incubated with catalyst solution (103 μmol L−1 CuSO4 and 103 μmol L−1 sodium ascorbate in pre-cooled PBS) at 25 °C for 12 h. After fully interacting, the microspheres were collected and rinsed with PBST three times. Next, cell lysates of HEK 293T cells were added to the microspheres and incubated at 4 °C for 10 h. After shocking, the microspheres were collected, rinsed three times with PBST for 10 min each, and then the captured proteins were released with 103 μmol L−1 DTT (100 μL) at 4 °C for 1 h, and the supernatants after enrichment were collected for SDS-PAGE or Western blot analysis.2.6. Co-localization of target protein and aPhi
After treatment with aPhi (0.1–1 μmol L−1) or Phi (1 μmol L−1) for 6 h, BEAS-2B cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature and were incubated with 1 μmol L−1 azide and catalyst solution (10 μmol L−1 CuSO4 and 10 μmol L−1 sodium ascorbate in pre-cooled PBS) at room temperature for 1 h. After blocking with 5% serum albumin for 1 h, the cells were incubated overnight at 4 °C with anti-Akt antibody (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :400) in 5% serum albumin, and then incubated with Alexa Fluor® 594-conjugated goat anti-rabbit IgG (1:500) and the DAPI Fluoromount-G®, successively according to the instructions. Images were captured with a spectral-type LSM 700 confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany). The excitation and emission wavelengths of cy3 employed were 590 nm and 617 nm, respectively. The excitation and emission wavelengths of the click reaction were 365 nm and 470 nm, respectively.
:400) in 5% serum albumin, and then incubated with Alexa Fluor® 594-conjugated goat anti-rabbit IgG (1:500) and the DAPI Fluoromount-G®, successively according to the instructions. Images were captured with a spectral-type LSM 700 confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany). The excitation and emission wavelengths of cy3 employed were 590 nm and 617 nm, respectively. The excitation and emission wavelengths of the click reaction were 365 nm and 470 nm, respectively.
2.7. Western blot analysis and Akt kinase assay
BEAS-2B cells were plated in 75 cm2 culture flasks at a density of 5 × 106 cells per flask and grown as described in the cell culture section. The cells were cultured in serum-free medium for 24 h for synchronization. Then, the cells were treated for 4 h with different drug groups. Phi, Akt inhibitor VIII (VIII) and Akt activator SC79 (SC) were dissolved in DMSO, and working concentrations of Phi (0.1–100 μmol L−1), VIII (5 μmol L−1), and SC (10 μmol L−1) were added directly to serum-free culture medium. The final concentration of DMSO did not exceed 0.1% (v/v) and did not affect cell viability. Then, the activity of Akt1/PKBα kinase in cells lysates was quantified as described in the manufacturer's manuals (Arlington, Ma, USA). The effects of Phi on Akt expression were determined using western blot analysis procedures described in the literature.22 Optical density analysis was performed in Image J (NIH, USA).2.8. Inflammatory cytokines assay, protein phosphorylation and NF-κB nucleus translocation
BEAS-2B cells were grown on glass coverslips in 96-well culture plates for 24 h, pre-treated or not pre-treated with Phi (10 μmol L−1) for 4 h, and then treated with 10 μg mL−1 LPS for different times for the observation of protein phosphorylation, NF-κB translocation analysis and the level of inflammatory cytokines.The effects of Phi on phosphorylation of the proteins Akt, IKKα/β, NF-κB and GSK3β were detected using western blot analysis procedures described in the literature.22 NF-κB translocation analysis was performed using cell immunofluorescence analysis procedures described in the literature.23 The concentrations of human IL-6 and IL-8 in the culture supernatants were detected with ELISA kits according to the manufacturer's instructions (Wes Tang Bio-Tech, Shanghai, China).
2.9. Determination of glucose uptake in HEK 293T cells
HEK 293T cells were plated in 96-well plates at a density of 1 × 105 cells per well, and after incubation for 24 h, the serum-free medium was replaced with Phi (0.1–100 μmol L−1) for 6 h. At the end of the incubation periods, cellular glucose uptake was assayed in 10 μL medium by enzymatic methods using a glucose assay kit. The glucose concentration of the wells with cells was subtracted from the glucose of the blank wells to obtain the glucose uptake amount.2.10. Statistical analysis
All experiments were performed in at least triplicate with similar results, and values are expressed as the mean ± SD. Significant differences between multiple groups of data were analysed by multiple t-tests with Bonferroni correction (Graph Pad Prism 5.0). P < 0.05 was considered statistically significant.3. Results and discussion
3.1. Predicting and fishing the target protein of Phi
To explore the protein targets of Phi, the first 30 candidate targets of Phi were filtered using bioinformatics tools. Next, we studied the candidate targets one by one by uploading them to the String 9.1 website. Eventually, 8 predicted targets (MAPK14, FGFR1, LCK, BRAF, PDPK1, AKT, HSP90AA1, and KDR) and 4 primary pathways (MAPK, PI3K-Akt, T-cell receptor and m-TOR signalling pathway) related to the inflammatory response were found through the compound-target-pathway network, as shown in Fig. 1A. Furthermore, we studied the interaction relationships between the forecasted 8 targets and Phi using the Auto Dock 4.2 software. The binding energy between Phi and Akt had the highest score and the 4 signalling pathways all contain the Akt protein, as shown in Fig. 1A. Therefore, we preliminarily inferred that Akt is the node target for Phi involved in anti-inflammatory responses.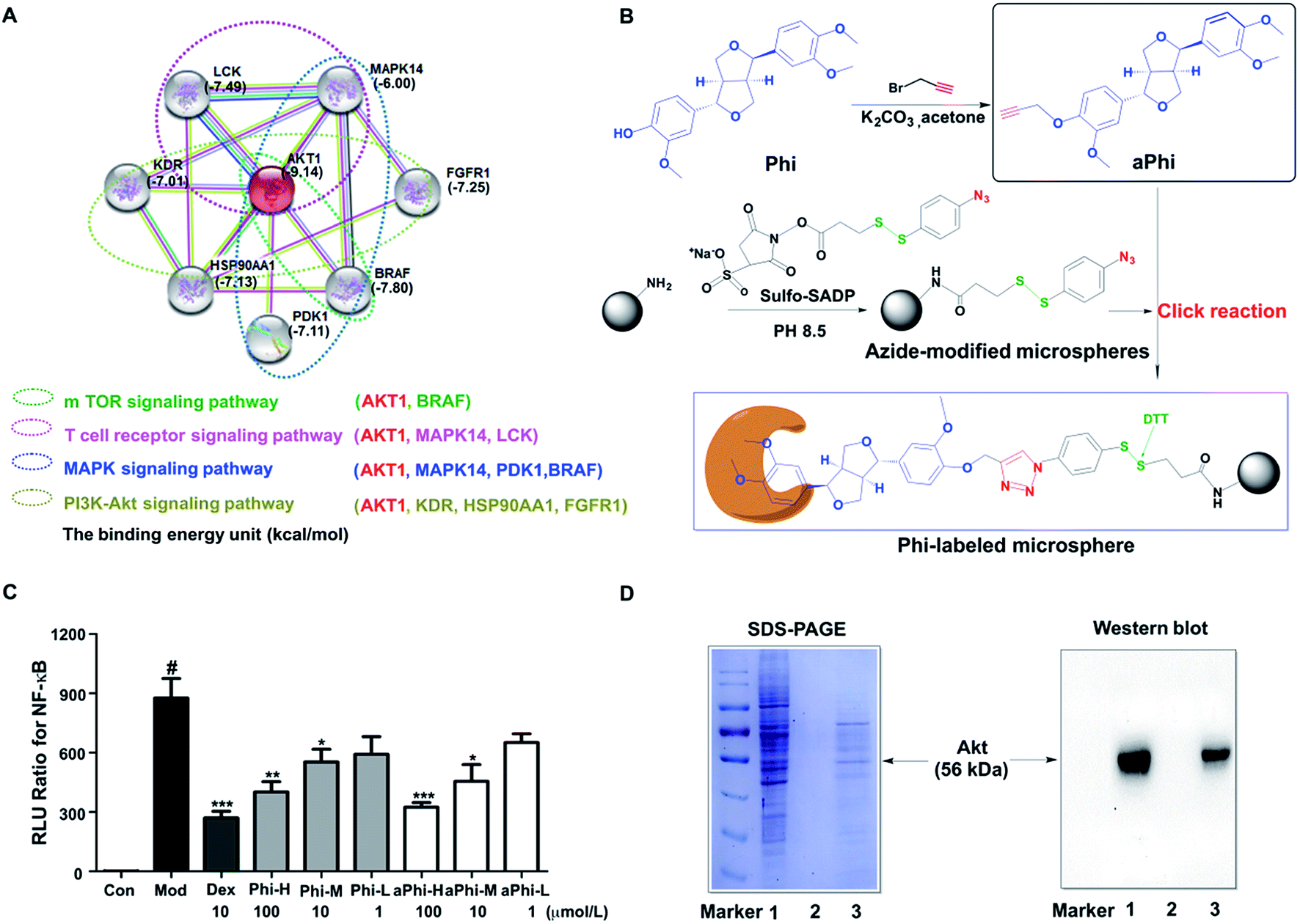 | ||
| Fig. 1 The predicting and fishing for the protein target of Phi. (A) Pharm Mapper and String 9.1 were used to predict and analyse the target protein interactions and function of Phi. (B) The process of labelling, the synthetic method for aPhi and azide-modified microspheres, and the linkage of these two compounds is shown. (C) The effects of Phi and aPhi on the NF-κB expression (each bar represents the mean ± SD; *P < 0.05, **P < 0.01, ***P < 0.001 compared to the model group. #P < 0.01 compared to the control group). (D) Fishing for and identifying the potential target protein. SDS-PAGE (left panel) and western blot analysis (right panel) were used to detect the proteins enriched by Phi microspheres. Lane 1 is HEK293T lysates, and lane 2 is lysates enriched by Sulfo-SADP-treated microspheres as a negative control, and lane 3 is lysate enriched by the Phi-labelled microspheres. Marker shows the molecular weight. The predicted protein target was analysed by western blot analysis for Akt. | ||
To further verify the above prediction, a chemical proteomics strategy was carried out to determine the relationship between Akt and the Phi. Initially, we modified Phi with alkynylation and verified drug the efficacy with Phi on NF-κB inhibition. Compared to Phi, aPhi showed no obvious changes in the inhibitory activity of NF-κB, as shown in the Fig. 1C. Therefore, we can conclude that aPhi does not change NF-κB related anti-inflammatory targets. After confirming aPhi has the same potency on the anti-inflammatory activity, we then used the aPhi probe to fish the potential targets of Phi according to the program outlined that we have designed in Fig. 1B. The magnetic-captured proteins were released by DTT reduction, and SDS-PAGE and western blotting were performed to evaluate capture efficiency. As shown in the Fig. 1D (left panel), some proteins were enriched from the lysate of HEK 293T cells by Phi-modified magnetic microspheres (lane 3), but these bands almost could not be detected by only azide-functionalized magnetic microspheres (lane 2, negative control group). This result indicates that Phi-related proteins were selectively enriched and released by this fishing strategy. To further verify the above prediction, western blots were carried out to determine the most likely target protein. As shown in Fig. 1D (right panel), an Akt protein band at approximately 56 kDa was obviously enriched compared to the negative control group (lane 2). Of course, the Akt protein band was also enriched at the same position compared to the positive control group (lane 1), which did not include magnetic microspheres. To explore the mechanism, a combined chemical biology approach was conducted to capture and evaluate potential targets of Phi, resulting in the identification of Akt.
AKT, also known as protein kinase B (PKB), is a serine–threonine kinase that functions as a key signalling node in the PI3K/Akt pathway and plays a central role in cell proliferation, migration, survival, and metabolism.24 Once activated, AKT can phosphorylate downstream substrates, such as Bad, IKKα/β and GSK3β. AKT-mediated modifications of above target genes function through a variety of mechanisms, and the signalling cascade eventually leads to cell proliferation and migration.25 AKT overexpression is an important driver of malignant progression and chemo-resistance in cancers.26
3.2. Phi down-regulated the level of Akt phosphorylation
To further confirm the relationship between Phi and Akt, we performed co-localization of the target protein and aPhi in BEAS-2B cells. As shown in Fig. 2A, chemical imaging of aPhi (green) preferentially merged with Akt proteins (red) and partially occurred in the cellular context (yellow). Moreover, this phenomenon could be competitively inhibited by Phi. This result showed that aPhi can bind Akt.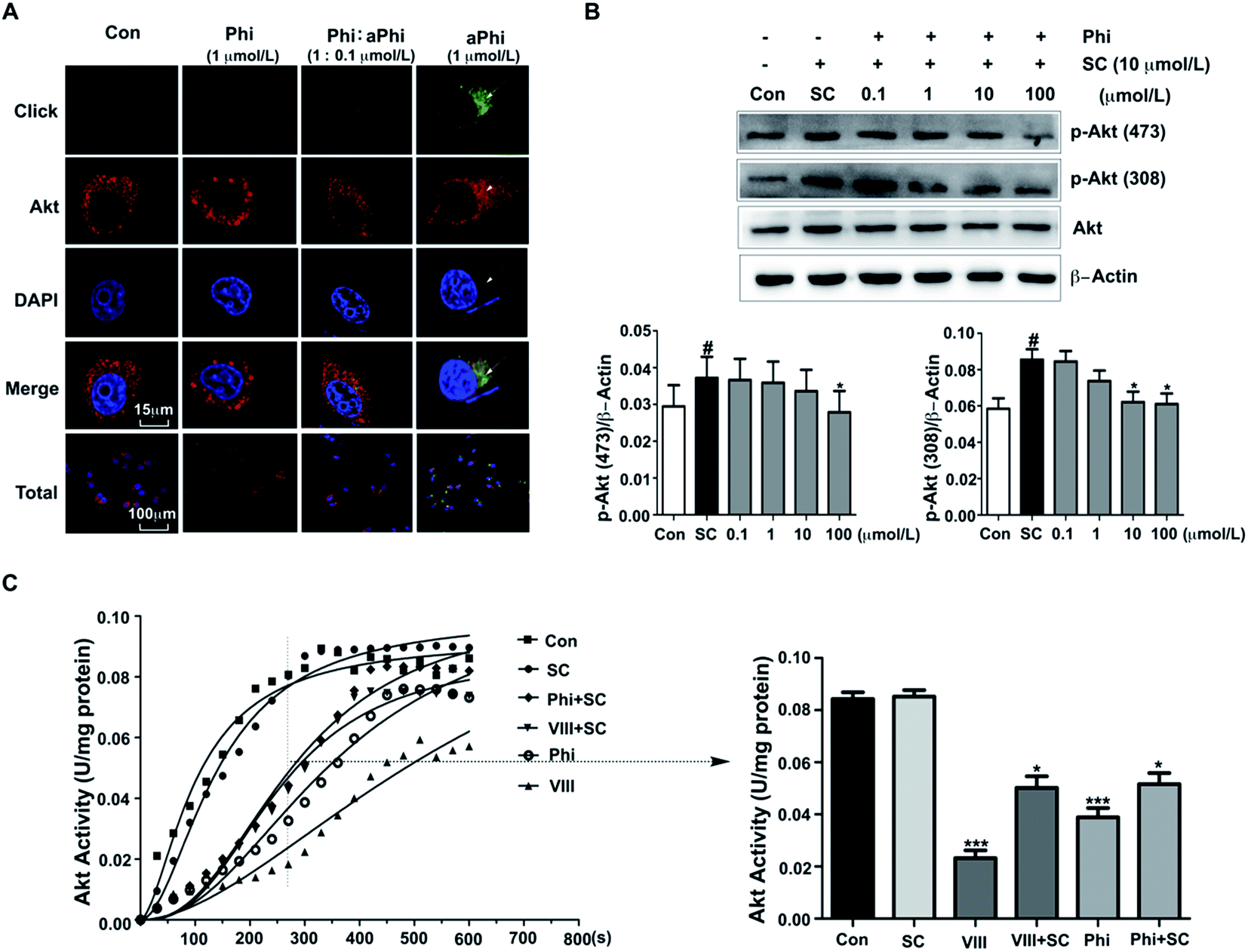 | ||
| Fig. 2 Phi binds to Akt and inhibits its activity by down-regulating the phosphorylation of Akt. (A) Co-localization of Akt and aPhi in BEAS-2B cells. After treatment of BEAS-2B cells with various conditions described below, cells were fixed and stained with the click reaction reagent aPhi (green) and rabbit anti-Akt (red) followed by Alexa Fluor® 594-conjugated anti-rabbit antibody. Cells were stained with the DAPI Fluoromount-G® to visualize nuclei (blue), and examined by fluorescence microscopy. Cells were treated with Con (DMSO; left column), 1 μmol L−1 Phi (second column on the left), 1 μmol L−1 Phi and 0.1 μmol L−1 aPhi (second column on the right) and 1 μmol L−1 aPhi (right column) for 4 h. Scale bars are 15 and 100 μm. (B) Phi modulates phosphorylation of Akt in BEAS-2B cells. BEAS-2B cells were serum-starved overnight and then treated with SC79 and different doses of Phi for 4 h. Thr 308 and Ser 473 phosphorylation of Akt and total Akt were detected by western blot. β-Actin was used as an internal control. The histograms present the dose-response effect of Phi on Ser 473 (left histogram) and Thr 308 (right histogram) phosphorylation of Akt. The band intensity was quantified by scanning densitometry and standardized with respect to β-actin from three independent experiments. Different superscript letters indicate statistically significant differences among groups (*P < 0.05 compared to the SC79 group. #P < 0.01 compared to the control group). (C) The pyruvate kinase and lactate dehydrogenase continuous loop response system was used to measure the activity of Akt. The histogram presents the activities of Akt at 5 min from three independent experiments. Drugs were added at concentrations of 10 μmol L−1. Different superscript letters indicate statistically significant differences among groups (*P < 0.05; ***P < 0.001 compared to the control group). | ||
Under physiological conditions, phosphorylation at Thr308 and Ser473 is essential for Akt activation.27 Therefore, we performed a western blot assay to investigate both Akt and its two activated forms with Thr308 and Ser473 phosphorylated Akt (p-Akt (308) and p-Akt (473), respectively). As shown in Fig. 2B, Akt expression exhibited no apparent changes, but compared to SC79-induced Akt phosphorylation, both p-Akt (308) and p-Akt (473) levels exhibited apparent decreases following Phi treatment with dosages ranging from 0.1 μmol L−1 to 100 μmol L−1. To further confirm the inhibitory effect, the activity of Akt was tested using an Akt kinase activity kit. Total Akt kinase activity was eliminated by 10 μmol L−1 Phi, which was the same dose for which inhibitor VIII (VIII) inhibited the activity of Akt (Fig. 2C). Therefore, we speculated that Phi may be a potential inhibitor of Akt.
3.3. Molecular docking of Phi with Akt
To identify the interaction relationships between Phi and Akt, we docked Phi and AKT inhibitor VIII (VIII) to the binding site of Akt (PDB: 3o96) using Auto Dock 4.2 software. VIII, an allosteric inhibitor of Akt, was used as a positive control. First, we calculated the binding energy of the Phi and VIII to AKT, and found that Phi could directly bind to Akt with an ideal binding energy (−9.14 kcal mol−1) that was comparable to VIII (−13.63 kcal mol−1). Then, we analysed the top-scoring poses of Phi and VIII in all cases, which are shown as 3D maps of the interaction of VIII and Phi with Akt (Fig. 3A). Akt consists of three conserved domains, a pleckstrin homology (PH) domain, a central kinase domain and a C-terminal hydrophobic motif (HM).28 As shown in Fig. 3A, Phi and VIII both established hydrogen bonds between the PH domain and the kinase domain. Docking of VIII showed one hydrogen bond formed with Thr291 located in the kinase domain and two hydrogen bonds formed with Asn54 and Trp80, which were located in the PH domain (left panel). Furthermore, the association of the PH domain residue Trp80 to VIII through the PH domain-induced cavity would block Akt in the inactive PH-in conformer, preventing Thr308 accessibility, and therefore its phosphorylation.29–31 When compared to VIII, Phi docking results showed that two hydrogen bonds formed with Trp80 and Thr211, which were located in the PH domain and the kinase domain, respectively (right panel). Phi was deeply and stably embedded in the cavity of Akt. A distance of 2.8 Å measured from the head of Phi to Trp80 suggested a possible interaction by hydrogen bonding. These results showed that Phi can directly bind to Akt and may inhibit Akt kinase activity by inhibiting its phosphorylation.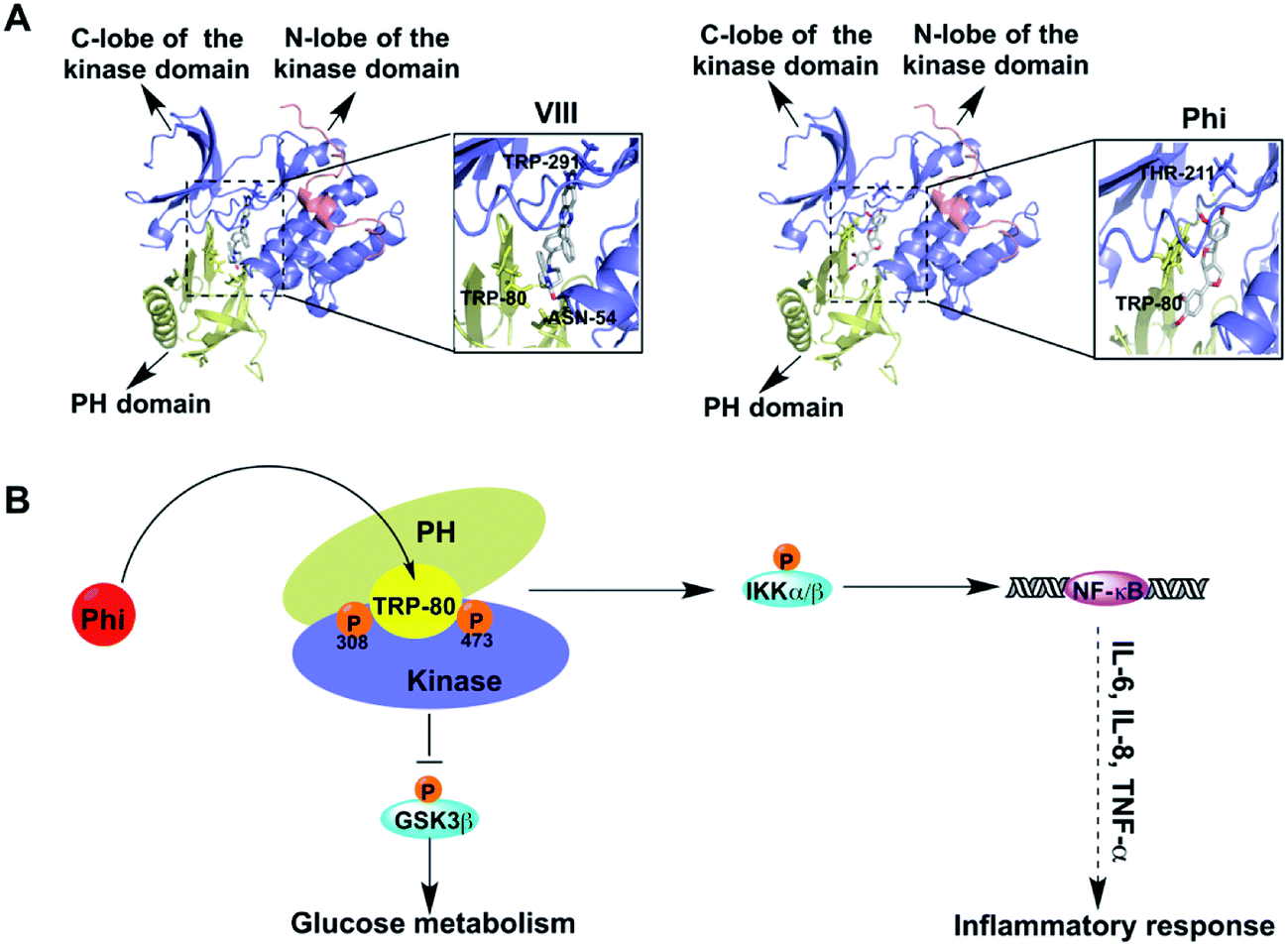 | ||
| Fig. 3 Phi targets an allosteric inhibit pocket on Akt (A) molecular dynamics simulation and docking of Akt with inhibitor VIII (VIII, left panel) and phillygenin (Phi, right panel). (B) Effects of Phi on the anti-inflammatory and glucose metabolism responses are hypothesized to be mediated by the AKT signalling pathway. | ||
Studies have shown that allosteric inhibitors were not competitive with ATP or peptide substrates and are only active against the full-length enzyme, requiring the PH domain for full enzyme inhibition.32–34 Furthermore, unlike ATP-competitive Akt inhibitors, allosteric Akt inhibitors do not result in hyper phosphorylation of Akt at Thr308 and Ser473.35 The inhibition of Thr308 and Ser473 phosphorylation was different, and the mechanism of action of the allosteric inhibitor affected the two sites in a distinct manner. For example, at 5 μmol L−1 AKT inhibitor VIII, the phosphorylation of Thr308 was inhibited by 95%, and Ser473 was inhibited by 70%.31 As shown in Fig. 2B, the levels of p-Akt phosphorylated at both Thr308 and Ser473 were decreased following Phi treatment in a dose-dependent manner, and the inhibition effect on Thr308 phosphorylation was more significant than for Ser473 phosphorylation. Therefore, we speculated Phi may bind to an allosteric binding site rather than competitive with ATP.
Activated Akt, in turn phosphorylates a variety of proteins, including several associated with cell inflammation, cell survival and glucose metabolism pathways, such as IKKα/β, NF-κB and GSK3β (Fig. 3B). The majority of kinase inhibitors have been shown to inhibit kinase signalling pathways by blocking the target kinases' substrate phosphorylation and subsequent downstream pathway components.35 To further confirm the inhibition effect, we determined whether Phi inhibition of Akt activation could recapitulate the physiological function of Akt signalling using a series of experiments.
3.4. Phi reduces inflammation by inhibiting the Akt phosphorylation
Previous studies have demonstrated that Phi inhibits prostaglandin E2 and NO production in LPS-activated RAW 264.7 cells, and also significantly inhibits mouse carrageenan-induced paw edema by intraperitoneal administration.36 However, there is not much evidence to indicate the anti-inflammatory targets of Phi. In the present study, we sought to investigate the anti-inflammatory mechanism of Phi on LPS-induced inflammation in BEAS-2B cells.The Akt signalling pathway is a critical regulator of NF-κB activation, plays an active role in host immune responses and is critical in the pro-inflammatory production of cytokines, such as IL-6 and IL-8.37,38 Here, we investigated the effect of Phi on LPS-induced phosphorylation of Akt, IKKα/β and NF-κB by western blot. The results showed that LPS remarkably induced Akt, IKKα/β and NF-κB phosphorylation within 30 min treatment, and pre-treatment by Phi could significantly reduce these phosphorylation events (Fig. 4C). During inflammatory responses, NF-κB is phosphorylated and translocated into the nucleus, where it induces the expression of genes involved in immune and inflammatory responses. As shown in Fig. 4A, NF-κB was normally sequestered in the cytoplasm (left column), nuclear accumulation of NF-κB was strongly induced by LPS treatment, and pre-treatment of Phi completely abolished the LPS-induced NF-κB activation. We think that the biological effects of Phi could be due to the inhibition of this transcription factor during the inflammatory process. Subsequent experiments showed that Phi could inhibit the production of IL-6 and IL-8 induced by LPS (Fig. 4B). Based on these results, we think the anti-inflammatory effects of Phi may be mediated by down-regulation of Akt. The decreased expression of p-Akt by Phi leads to the suppression of NF-κB and a consequent decrease in the release of pro-inflammatory mediators, such as TNF-α, IL-8 and IL-6.
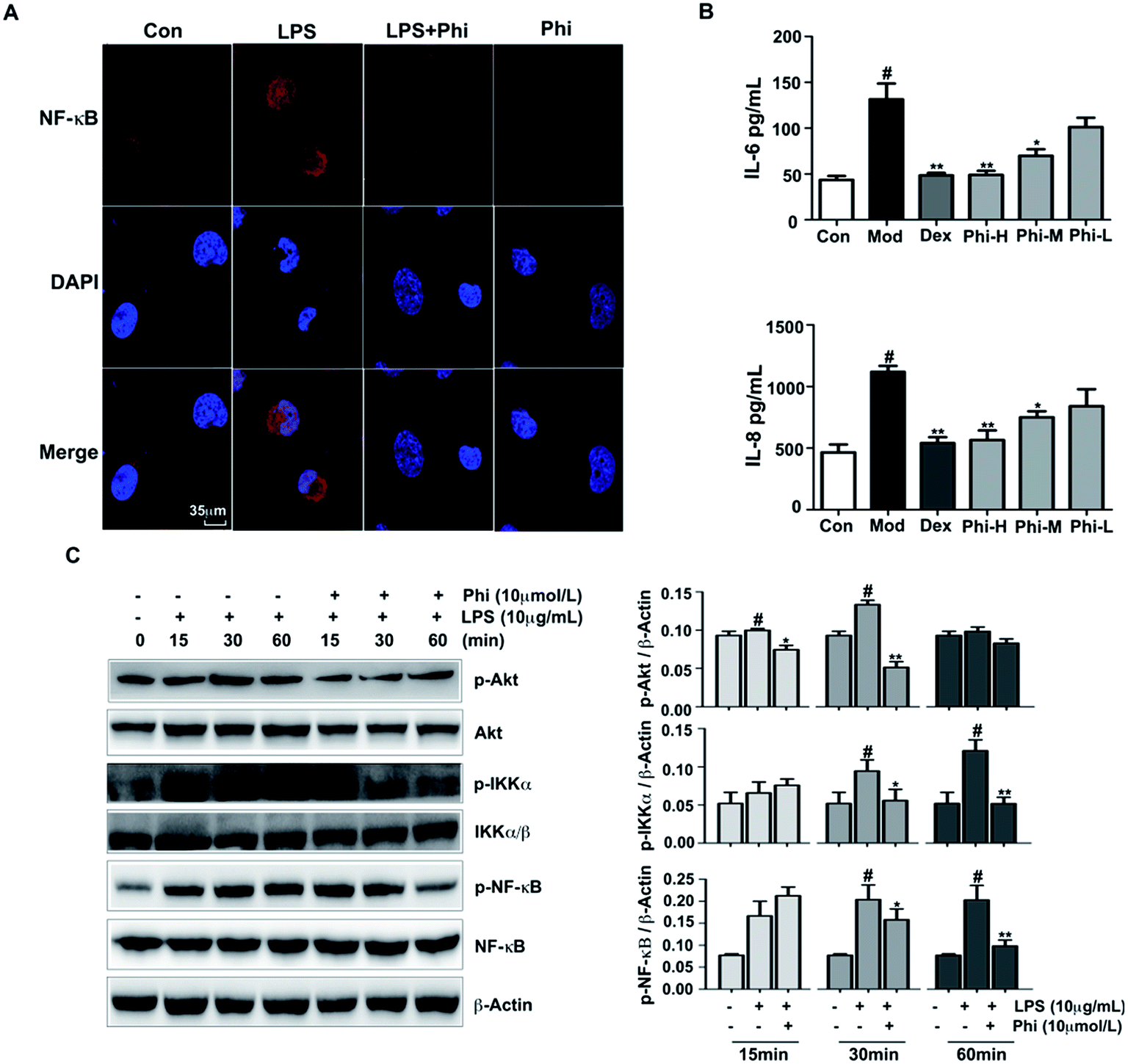 | ||
| Fig. 4 Phi reduces the inflammation by inhibiting Akt activation. BEAS-2B cells were pre-treated with Phi 12 h before LPS treatment. (A) Localization of NF-κB was visualized with fluorescence microscopy after immunofluorescence staining with NF-κB antibody (red). Cells were stained with the DAPI Fluoromount-G® to visualize nuclei (blue). Scale bar 35 μm. (B) BEAS-2B cells were treated with the indicated doses of Phi and Dex for 12 h before LPS treatment for the indicated times. Following 6 h treatment, the supernatants were harvested and amounts of IL-6 (upper histogram) and IL-8 (lower histogram) were measured in the supernatants. The data are shown as the mean ± SD of three independent experiments (*P < 0.05; **P < 0.01 compared to the model group. #P < 0.01 compared to the control group). (C) The expression levels of phosphorylated (P) and total Akt, IKKα/β and NF-κB were assessed by western blot analysis after 15, 30, and 60 min of LPS treatment. β-Actin was used as an internal control. The histograms present the effect of Phi on Akt and the downstream proteins activation by LPS in BEAS-2B cells. The band intensity was quantified by scanning densitometry and standardized with respect to β-actin from five different experiments. Different superscript letters indicate statistically significant differences among groups (*P < 0.05; **P < 0.01 compared to the certain period of model group. #P < 0.01 compared to the certain of control group). The data shown are representative of three independent experiments. | ||
3.5. Phi influences glucose metabolic parameters by inhibiting Akt activation
Glucose metabolism disorder is closely related to many diseases, such as diabetes and obesity. In recent years, Forsythia leaves have been used for weight loss in China as a health food product.39 A few reports have confirmed that Phi can reduce blood lipids in nutritive hyperlipidemia model,40 and also showed a strong lowering blood sugar of diabetic mice.41 However, the specific mechanism of action has not been reported.The Akt signalling pathway is also a critical regulator of GSK3β activation, which plays an active role in glucose metabolism.42 In glucose metabolism, glucose is a critical nutrient with important roles in cellular metabolism, which participates in glycolysis and tricarboxylic acid (TCA) cycle.43 Phosphorylated GSK3β enhances glycogen synthesis and reduces the level of blood glucose.44 Therefore, we examined whether Phi can modulate the glucose metabolism in HEK 293T cells using western blot analyses. Akt and GSK3β expression showed no evident change, but the levels of p-Akt and p-GSK3β decreased following Phi treatment in a dose-dependent manner (Fig. 5A). To confirm this finding, we evaluated the effects of Phi on glucose uptake in HEK 293T cells. We found that Phi could inhibit the consumption of glucose, as shown in Fig. 5B.
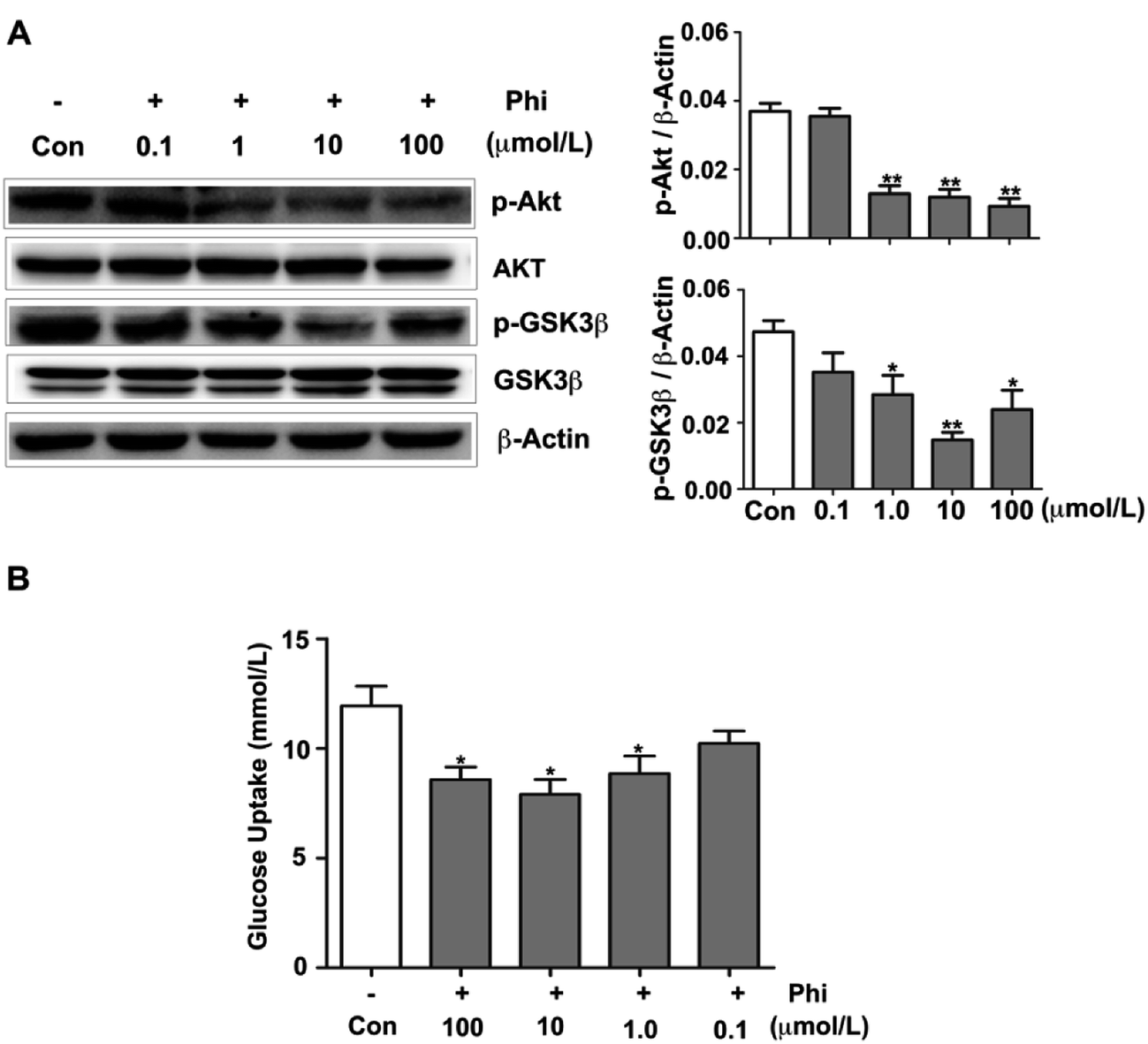 | ||
| Fig. 5 Phi adjusts glucose metabolism by inhibiting Akt activation. (A) Phi modulates the expression of phosphorylation of GSK3β in HEK293T cells. HEK293T cells were serum-starved overnight and then treated with different doses of Phi for 6 h. Phosphorylation of GSK3β and total GSK3β were detected by western blot. β-Actin was used as an internal control. The histograms present the dose-response effect of Phi on GSK3β phosphorylation. The band intensity was quantified by scanning densitometry and standardized with respect to β-actin from three independent experiments. Different superscript letters indicate statistically significant differences among groups (*P < 0.05; **P < 0.01 compared to the control group). (B) The effects of Phi on glucose uptake in HEK293T cells. The data are shown as the mean ± SD of three independent experiments (*P < 0.05 compared to the control group). | ||
4. Conclusion
In conclusion, Phi inhibited Akt kinase activity by specifically targeting an Akt allosteric inhibition pocket. Phosphorylation at Thr308 and Ser473 was reduced at both positions. Moreover, Phi reduced Akt-regulated phosphorylation of IKKα/β and NF-κB, attenuated the production of IL-6, IL-8 in LPS-induced inflammation in BEAS-2B cells, and attenuated p-GSK3β, resulting in reduced glucose uptake by HEK 293T cells. Phi has considerable potential in the treatment or prevention of respiratory and metabolic diseases.Conflicts of interest
The authors have declared no conflicts of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (81473403, 81430095, 81673616), the Natural Science Foundation of Tianjin, China (15JCYBJC28800) and the Science and Technology Program of Tianjin, China (15ZXLCSY00020).References
- H. Qu, Y. Zhang, X. Chai and W. Sun, Bioorg. Chem., 2012, 40, 87–91 CrossRef CAS PubMed.
- P. Zhao, X. Piao, Z. Zeng, P. Li, X. Xu and H. Wang, Anim. Sci. J., 2017, 88, 854–862 CrossRef CAS PubMed.
- H. Qu, Y. Zhang, Y. Wang, B. Li and W. Sun, J. Pharm. Pharmacol., 2008, 60, 261–266 CrossRef CAS PubMed.
- T. Lu, X. L. Piao, Q. Zhang, D. Wang, X. S. Piao and S. W. Kim, Food Chem. Toxicol., 2010, 48, 764–770 CrossRef CAS PubMed.
- L. J. Ni, L. G. Zhang, J. Hou, W. Z. Shi and M. L. Guo, J. Ethnopharmacol., 2009, 124, 79–86 CrossRef CAS PubMed.
- C. Li, Z. H. Yao, Z. F. Qin, J. B. Zhang, R. Y. Cao, Y. Dai and X. S. Yao, Fitoterapia, 2014, 97, 92–97 CrossRef CAS PubMed.
- L. H. Ye, Y. X. Li, C. Peng, X. H. Gong and X. G. Zheng, Eur. J. Drug Metab. Pharmacokinet., 2013, 38, 201–207 CrossRef CAS PubMed.
- P. M. Cheuka, G. Mayoka, P. Mutai and K. Chibale, Molecules, 2017, 22, 58 CrossRef PubMed.
- T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531–541 CrossRef CAS PubMed.
- S. J. Chen and M. C. Cui, Molecules, 2017, 22, 64 Search PubMed.
- M. L. Miller, E. J. Molinelli, J. S. Nair, T. Sheikh, R. Samy, X. Jing, Q. He, A. Korkut, A. M. Crago, S. Singer, G. K. Schwartz and C. Sander, Sci. Signaling, 2013, 6, ra85 Search PubMed.
- L. Huang, Y. Jiang and Y. Chen, Sci. Rep., 2017, 7, 40752 CrossRef CAS PubMed.
- M. Ueda, Chem. Lett., 2012, 41, 658–666 CrossRef CAS.
- F. A. Mandl, V. C. Kirsch, I. Ugur, E. Kunold, J. Vomacka, C. Fetzer, S. Schneider, K. Richter, T. M. Fuchs, I. Antes and S. A. Sieber, Angew. Chem., Int. Ed. Engl., 2016, 55, 14852–14857 CrossRef CAS PubMed.
- C. D. Hein, X. M. Liu and D. Wang, Pharm. Res., 2008, 25, 2216–2230 CrossRef CAS PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- T. Plass, S. Milles, C. Koehler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed. Engl., 2011, 50, 3878–3881 CrossRef CAS PubMed.
- A. Mongis, F. Piller and V. Piller, Bioconjugate Chem., 2017, 28, 1151–1165 CrossRef CAS PubMed.
- S. Sato, Y. Kwon, S. Kamisuki, N. Srivastava, Q. Mao, Y. Kawazoe and M. Uesugi, J. Am. Chem. Soc., 2007, 129, 873–880 CrossRef CAS PubMed.
- X. Liu, S. Ouyang, B. Yu, Y. Liu, K. Huang, J. Gong, S. Zheng, Z. Li, H. Li and H. Jiang, Nucleic Acids Res., 2010, 38, W609–W614 CrossRef CAS PubMed.
- J. Gao, J. Tao, N. Zhang, Y. Liu, M. Jiang, Y. Hou, Q. Wang and G. Bai, Phytother. Res., 2015, 29, 241–250 CrossRef PubMed.
- R. Fang, Q. Cui, J. Sun, X. Duan, X. Ma, W. Wang, B. Cheng, Y. Liu, Y. Hou and G. Bai, Allergy, 2015, 70, 1622–1632 CrossRef CAS PubMed.
- M. H. Han, W. S. Lee, A. Nagappan, S. H. Hong, J. H. Jung, C. Park, H. J. Kim, G. Y. Kim, G. Kim, J. M. Jung, C. H. Ryu, S. C. Shin, S. C. Hong and Y. H. Choi, Phytother. Res., 2016, 30, 1824–1832 CrossRef CAS PubMed.
- K. M. Nicholson and N. G. Anderson, Cell. Signalling, 2002, 14, 381–395 CrossRef CAS PubMed.
- B. D. Manning and L. C. Cantley, Cell, 2007, 129, 1261–1274 CrossRef CAS PubMed.
- D. Kim, H. C. Dan, S. Park, L. Yang, Q. Liu, S. Kaneko, J. Ning, L. He, H. Yang, M. Sun, S. V. Nicosia and J. Q. Cheng, Front. Biosci., Landmark Ed., 2005, 10, 975–987 CrossRef CAS.
- B. Ananthanarayanan, M. Fosbrink, M. Rahdar and J. Zhang, J. Biol. Chem., 2007, 282, 36634–36641 CrossRef CAS PubMed.
- J. Yang, P. Cron, V. Thompson, V. M. Good, D. Hess, B. A. Hemmings and D. Barford, Mol. Cell, 2002, 9, 1227–1240 CrossRef CAS PubMed.
- W. I. Wu, W. C. Voegtli, H. L. Sturgis, F. P. Dizon, G. P. Vigers and B. J. Brandhuber, PLoS One, 2010, 5, e12913 Search PubMed.
- V. Calleja, M. Laguerre and B. Larijani, ChemBioChem, 2009, 2, 11–25 Search PubMed.
- V. Calleja, M. Laguerre, P. J. Parker and B. Larijani, PLoS Biol., 2009, 7, e17 Search PubMed.
- T. A. Yap, L. Yan, A. Patnaik, I. Fearen, D. Olmos, K. Papadopoulos, R. D. Baird, L. Delgado, A. Taylor, L. Lupinacci, R. Riisnaes, L. L. Pope, S. P. Heaton, G. Thomas, M. D. Garrett, D. M. Sullivan, J. S. de Bono and A. W. Tolcher, J. Clin. Oncol., 2011, 29, 4688–4695 CrossRef CAS PubMed.
- M. T. Bilodeau, A. E. Balitza, J. M. Hoffman, P. J. Manley, S. F. Barnett, D. Defeo-Jones, K. Haskell, R. E. Jones, K. Leander, R. G. Robinson, A. M. Smith, H. E. Huber and G. D. Hartman, Bioorg. Med. Chem. Lett., 2008, 18, 3178–3182 CrossRef CAS PubMed.
- C. C. Kumar and V. Madison, Oncogene, 2005, 24, 7493–7501 CrossRef CAS PubMed.
- T. Okuzumi, D. Fiedler, C. Zhang, D. C. Gray, B. Aizenstein, R. Hoffman and K. M. Shokat, Nat. Chem. Biol., 2009, 5, 484–493 CrossRef CAS PubMed.
- H. Lim, J. G. Lee, S. H. Lee, Y. S. Kim and H. P. Kim, J. Ethnopharmacol., 2008, 118, 113–117 CrossRef CAS PubMed.
- H. Kim, N. Banerjee, R. C. Barnes, C. M. Pfent, S. T. Talcott, R. H. Dashwood and S. U. Mertens-Talcott, Mol. Carcinog., 2017, 56, 197–207 CrossRef CAS PubMed.
- W. Zhao, C. Wu, S. Li and X. Chen, Cytokines, 2016, 88, 167–176 CrossRef CAS PubMed.
- Y. Zhao, F. Li, J. Yang, X. An and M. Zhou, Zhongyaocai, 2005, 28, 123–124 Search PubMed.
- W. Kang and J. Wang, Med. Chem. Res., 2010, 19, 617–628 CrossRef CAS.
- Y. Zhang, F. Feng, T. Chen, Z. Li and Q. W. Shen, J. Ethnopharmacol., 2016, 192, 256–263 CrossRef PubMed.
- M. Kuai, Y. Li, X. Sun, Z. Ma, C. Lin, Y. Jing, Y. Lu, Q. Chen, X. Wu, X. Kong and H. Bian, Biomed. Pharmacother., 2016, 84, 1585–1594 CrossRef CAS PubMed.
- Y. M. Woo, Y. Shin, E. J. Lee, S. Lee, S. H. Jeong, H. K. Kong, E. Y. Park, H. K. Kim, J. Han, M. Chang and J. H. Park, PLoS One, 2015, 10, e0132285 Search PubMed.
- E. L. Whiteman, H. Cho and M. J. Birnbaum, Trends Endocrinol. Metab., 2002, 13, 444–451 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |