DOI:
10.1039/C7RA06107B
(Paper)
RSC Adv., 2017,
7, 42986-42996
Long non-coding RNA NEAT1 contributes to docetaxel resistance of prostate cancer through inducing RET expression by sponging miR-34a
Received
1st June 2017
, Accepted 29th August 2017
First published on 5th September 2017
Abstract
Background: Nuclear paraspeckle assembly transcript 1 (NEAT1) was demonstrated to serve as a carcinogenic long non-coding RNA (lncRNA) in multiple tumors including prostate cancer (PC). However, the potential biological role of NEAT1 in docetaxel resistance of PC and the underlying mechanism are largely unknown. Methods: Expressions of NEAT1 and miR-34a in PC tissues and cells were detected by qRT-PCR. The IC50 value of docetaxel and apoptosis in PC cells were examined by CCK-8 assay and flow cytometry analysis, respectively. Bioinformatics software and a luciferase reporter assay were used to predict and confirm the potential targets of miR-34a. A tumor xenograft assay was employed to verify the effect of NEAT1 on docetaxel resistance of PC cells in vivo. Results: NEAT1 was upregulated and miR-34a was downregulated in PC tissues, parental PC cells and docetaxel-resistant PC cells. NEAT1 knockdown improved sensitivity to docetaxel in docetaxel-resistant PC cells. In addition, NEAT1 functioned as a ceRNA of miR-34a in docetaxel-resistant PC cells to positively regulate the expression of RET (rearranged during transfection). Moreover, miR-34a inhibition partially reversed NETA1-knockdown-induced sensitivity to docetaxel in docetaxel-resistant PC cells. NEAT1 overexpression partially overturned RET-knockdown-mediated sensitivity to docetaxel in docetaxel-resistant PC cells. Furthermore, NEAT knockdown enhanced docetaxel sensitivity of docetaxel-resistant PC cells in vivo. Conclusion: NEAT1 contributed to docetaxel resistance of PC through inducing RET expression by sponging miR-34a, implying that targeting the NEAT1/miR-34a/RET axis may be a potential application in improving chemotherapy of PC.
1. Introduction
Prostate cancer (PC) is currently the most commonly diagnosed non-cutaneous malignancy and remains one of the primary causes of cancer-related mortality in men in the developed world.1 Despite great efforts in early diagnosis and treatments, PC continues to pose a significant public health burden, with more than 220![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 newly confirmed cases and an estimated 27000 deaths yearly in the United States.2 Although androgen deprivation therapy (ADT) is initially effective for many patients with PC, most patients ultimately evolve into castration-resistant prostate cancer (CRPC) at the advanced stages.3 Docetaxel, an important chemotherapy option, has been believed to exhibit modest survival benefits for patients with CRPC.4 However, resistance formation still remains a challenging and pervasive problem for the success of chemotherapy.5,6 Therefore, to develop effective therapeutic strategies to overcome this drug resistance, it is imperative to elucidate molecular mechanisms involved in the development of resistance to docetaxel in PC patients.
000 newly confirmed cases and an estimated 27000 deaths yearly in the United States.2 Although androgen deprivation therapy (ADT) is initially effective for many patients with PC, most patients ultimately evolve into castration-resistant prostate cancer (CRPC) at the advanced stages.3 Docetaxel, an important chemotherapy option, has been believed to exhibit modest survival benefits for patients with CRPC.4 However, resistance formation still remains a challenging and pervasive problem for the success of chemotherapy.5,6 Therefore, to develop effective therapeutic strategies to overcome this drug resistance, it is imperative to elucidate molecular mechanisms involved in the development of resistance to docetaxel in PC patients.
The human genome sequence data indicates that only 2% of mammalian genome is able to encode proteins, while the majority of the transcripts are referred to as the non-coding RNAs, such as long non-coding RNAs (lncRNAs).7 LncRNAs, a group of transcripts with more than 200 nucleotides and lack of protein-coding ability, modulate gene expression at the transcriptional, post-transcriptional and epigenetic levels.8,9 There is increasing evidence that these differently expressed lncRNAs may be involved in tumorigenesis, tumor progression and metastasis, highlighting the emerging roles of lncRNAs as diagnostic and prognostic biomarkers in various types of cancer.10–12 LncRNAs have attracted broad attention in cancer research for its differential expression in multiple tumors.13 Nuclear paraspeckle assembly transcript 1 (NEAT1), a nuclear lncRNA with two isoforms (3.7 kb NEAT1-1 and 23 kb NEAT1-2), was found to be a core architectural component of nuclear paraspeckle structure.14,15 Aberrant expression of NEAT1 was observed to play a significant role in tumorigenesis of a wide variety of cancers, including acute promyelocytic leukemia, breast cancer, non-small cell lung cancer, and glioma.15–18 NEAT1 was also demonstrated to be significantly overexpressed in PC and promote PC tumorigenesis.19 However, the potential biological role of NEAT1 in docetaxel resistance of PC cells and the underlying mechanism are largely unknown.
microRNAs (miRNAs) are endogenous, small non-coding RNAs which post-transcriptionally regulate gene expression by binding to the 3′-untranslated region (3′-UTR) of mRNAs. miRNAs are reported to act as oncogenes or tumor suppressors to participate in the regulation of a wide range of critical pathological processes of cancers.20 There is a growing body of evidence indicating the involvement of miRNAs in chemoresistance of human cancers, including PC.21,22 miR-34a was revealed to be markedly downregulated in PC cells and ectopic miR-34a expression inhibited cell growth and attenuated chemoresistance to camptothecin in PC cells.23 Recently, a novel regulatory circuit has been proposed in which lncRNAs function as competing endogenous RNAs (ceRNAs) to sponge miRNAs, thereby derepressing miRNA target genes at the posttranscriptional level.24–26 On the contrary, miRNAs could also target lncRNAs for degradation through Argonaute 2 (Ago2)-mediated pathway.27 However, whether NEAT1 could interact with miR-34a to regulate docetaxel activity of PC cells remains to be investigated.
In our study, we reported that knockdown of NEAT1 overcomed docetaxel resistance of PC cells by upregulating rearranged during transfection (RET) proto-oncogene mediated by miR-34a. Our study provided a novel evidence for the cross-talk between NEAT1, miR-34a and RET, elucidating a potential molecular mechanism involved in PC chemoresistance.
2. Materials and methods
2.1. Patient tissue samples
Fresh PC specimens and matched adjacent normal tissues were collected from 30 patients with PC who were treated with radical prostatectomy at Huaihe Hospital of Henan University between June 2013 and November 2015. Without any preoperative treatment such as chemotherapy or radiotherapy, all 30 patients were diagnosed with prostate cancer postoperatively by histopathology. These tissue samples were immediately frozen and stored in liquid nitrogen for subsequent analysis. These experimental protocols were approved by the local ethics committee of Huaihe Hospital and written informed consent was obtained from all patients. This study was conducted in accordance with the Declaration of Helsinki Principles.
2.2. Cell culture
Human PC cell lines (PC3 and DU-145) and normal human prostate epithelial cells (PrEC) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). These cells were cultivated in Roswell Park Memorial Institute (RPMI) 1640 medium (HyClone, GE Healthcare Life Science, Logan, USA) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA), 100 μg mL−1 streptomycin and 100 U mL−1 penicillin (Invitrogen) in a 5% CO2 humidified atmosphere at 37 °C. Docetaxel-resistant PC cell lines (PC3-DR and DU-145-DR) were generated from parental PC3 and DU-145 cells by exposing them to an initial dose of 5 nmol L−1 docetaxel (Sigma, St. Louis, MO, USA). Then the survivable cells were continuously cultured with gradually increasing doses of docetaxel in the culture medium until the final concentration of 100 nmol L−1.
2.3. Transfection
Small interfering RNA (siRNA) targeting NEAT1 (si-NEAT1), siRNA scrambled control (si-con), miR-34a mimic (miR-34a), mimic scrambled control (miR-con) miR-34a inhibitor (anti-miR-34a), inhibitor control (anti-miR-con), siRNA against RET (si-RET) and corresponding scrambled control (si-con) were purchased from Genepharma (Shanghai, China). The full-length sequence of NEAT1 was amplified and sub-cloned into pcDNA3.0 empty control (Invitrogen), named as pcDNA-NEAT1. Cell transfection was performed using the Lipofectamine 2000 (Invitrogen) when the cell fusion rate reached 70%. Cells were collected 48 h after transfection and subjected to further analysis.
2.4. Lentivirus construction, package and infection
Short hairpin RNA (shRNA) targeting NEAT1 and its matched controls were cloned into the lentiviral expression vector pLV-H1tetO-GFP-Puro (Biosettia, San Diego, CA, USA), generating sh-NEAT1 and sh-con. Then the constructed lentivirus plasmids were cotransfected with PG-P3-RRE, PG-P2-REV and PG-P1-VSVG into 293T cells. At 72 h post-transfection, PC3-DR cells were infected with the collected lentiviruses, followed by being screened with puromycin for almost 7 days in order to obtain stable lentivirus-transfected cells.
2.5. Quantitative real-time RT-PCR (qRT-PCR)
Total RNA samples were extracted from PC tissues or cells using Trizol (Invitrogen). 1 μg of total of RNA was reverse-transcribed to complementary DNA (cDNA) using random primers (10 μM) and high capacity cDNA Reverse Transcription Kit (Applied Biosystems, Boston, MA, USA). The expression of NEAT1 was quantified using SYBR Green PCR Kit (Qiagen, Valencia, CA, USA) and normalized to GAPDH. The expression of miR-34a was determined by TaqMan miRNA assays (Applied Biosystems, Foster City, CA, USA) and U6 small nuclear RNA (U6 snRNA) was used as the internal control. RT-PCR analysis was conducted using an Applied Biosystems 7500 Real-Time PCR Systems (Applied Biosystems) and relative gene expression was calculated according to the 2−ΔΔct method. The primer sequences were presented as blow: NEAT1 forward, 5′-CTTCCTCCCTTTAACTTATCCATTCAC-3′, NEAT1 reverse, 5′-CTCTTCCTCCACCATTACCAACAATAC-3′; GAPDH forward, 5′-AGAAGGCTGGGGCTCATTTG-3′, GAPDH reverse, 5′-AGGGGCCATCCACAGTCTTC-3′; miR-34a forward, 5′-GTGCAGGGTCCGAGGT-3′, reverse, 5′-GCCGCTGGCAGTGTCTTAGCTG-3′; U6 forward, 5′-CTCGCTTCGGCAGCACA-3′, reverse, 5′-AACGCTTCACGAATTTGCGT-3′.
2.6. Western blot
Total protein (20 μg) from cell lysate were loaded onto 10% SDS-PAGE and then electrophoretically transferred to PVDF membranes. After blocked in 5% non-fat milk at room temperature for 2 h, the membranes were probed overnight with primary antibodies RET and β-actin (Cell Signaling Technology, Danvers, MA, USA) at 4 °C overnight, followed by incubation with HRP-coupled goat anti-rabbit secondary antibody.
2.7. Docetaxel resistance assay
Docetaxel resistance was assessed using a Cell-Counting Kit 8 (CCK-8) (Dojindo, Kumamoto, Japan) as described by the manufacturer. The PC cells were plated in 96-well plates at a density of 8 × 103 per well 48 h post-transfection and then were treated with various concentrations of docetaxel (0.32 to 100 μM) for 48 h. Subsequently, 100 μL of complete medium containing 10 μL of CCK-8 solution was added for another incubation of 2 h at 37 °C. The absorbance value was determined at 450 nm using the Infinite M200 spectrophotometer (Bio-Rad Laboratories). Half maximal inhibitory concentration (IC50) values of docetaxel were calculated by the GraphPad Prism software.
2.8. Apoptosis analysis using flow cytometry
Apoptotic rate of cells was examined with Annexin-V-FITC apoptosis detection kit (BD Biosciences, San Jose, CA, USA). Briefly, 1 × 106 treated PC cells were harvested, washed with ice-cold PBS, and incubated with Annexin V-FITC and propidium iodide (PI) for 10 min in the dark. FACScan Flow Cytometer (BD Biosciences) was applied to test apoptotic event.
2.9. Luciferase reporter assay
The 3′UTR of RET mRNA containing the putative wild-type or corresponding mutated miR-34a binding sites or NEAT1 fragment containing the predicted wild-type or mutated miR-34a binding sites was synthesized and subcloned into the downstream of luciferase gene in the pmiR-GLO reporter vector (Promega, Madison, WI, USA), namely pmiR-GLO-RET-3′UTR-WT (RET-WT), pmiR-GLO-RET-3′UTR-MUT (RET-MUT), pmiR-GLO-NEAT1-WT (NEAT1-WT), and pmiR-GLO-NEAT1-MUT (NEAT1-MUT). For the luciferase reporter assay, 293T, PC3-DR and DU-145-DR cells were cultured in 96-well plates and cotransfected with miR-34a or miR-con and reporter plasmids (RET-WT, RET-MUT, NEAT1-WT, or NEAT1-MUT) using Lipofectamine 2000 (Invitrogen). Luciferase activity was measured using the dual-luciferase assay system (Promega) 48 h post-transfection.
2.10. In vivo tumor xenograft model
The procedures involving animal experiments were conducted with the approval of the Ethics Committee of Huaihe Hospital and in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and Animal Welfare Act. About 1 × 107 logarithmically growing PC3-DR cells stably transfected with sh-NEAT1 or sh-con were subcutaneously injected into the right hind flank of female nude mice from the Animal Center of Guangzhou Province (Guangdong, China). At 7 days after the injection, mice were administered intraperitoneally with 10 mg kg−1 docetaxel or same volume of PBS every 3 days. Tumor growth was measured every 3 days using a caliper rule according to the formula: volume = (width2 × length)/2. The mice were euthanized at 28 days post-inoculation, and the xenografted tumors were excised and weighed. The resected xenografted tumors were subjected to qRT-PCR and western blot analyses.
2.11. Statistical analyses
All results are expressed as mean ± standard deviation (SD). Statistical analysis was performed using SPSS software version 18.0 (SPSS, Inc., Chicago, IL, USA) with Student's t-test or one-way ANOVA. P values less than 0.05 were considered to indicate a statistically significant difference.
3. Results
3.1. NEAT1 was upregulated and miR-34a was downregulated in PC tissues and cells
To explore the biological functions of NEAT1 and miR-34a in docetaxel resistance of PC, we initially detected the expressions of NEAT1 and miR-34a in 30 PC tissues and adjacent normal tissues by qRT-PCR. The results showed that NEAT1 expression was significantly higher (Fig. 1A) and miR-34a expression was dramatically lower (Fig. 1B) in PC tissues than that in adjacent normal tissues. Furthermore, we analyzed the expressions of NEAT1 and miR-34a in parental PC cells (PC3 and DU-145), docetaxel-resistant PC cells (PC3-DR and DU-145-DR), and normal human prostate epithelial cells (PrEC) by qRT-PCR. As compared with PrEC cells, NEAT1 was exceptionally upregulated in PC3 (Fig. 1C) and DU-145 (Fig. 1D) cells, and it was much higher in PC3-DR and DU-145-DR cells than that in PC3 and DU-145 cells. On the contrary, miR-34a expression was obviously decreased in PC3 (Fig. 1E) and DU-145 (Fig. 1F) cells compared with PrEC cells, and it was much lower in PC3-DR and DU-145-DR cells than that in PC3 and DU-145 cells. These results demonstrated the involvement of NEAT1 and miR-34a in docetaxel resistance in PC cells.
 |
| | Fig. 1 NEAT1 was upregulated and miR-34a was downregulated in PC tissues and cells. Expressions of NEAT1 (A) and miR-34a (B) in 30 PC tissues and paired adjacent normal tissues. Expressions of NEAT1 (C and D) and miR-34a (E and F) in parental PC cells (PC3 and DU-145), docetaxel-resistant PC cells (PC3-DR and DU-145-DR), and normal human prostate epithelial cells PrEC. *P < 0.05. | |
3.2. NEAT1 knockdown improved sensitivity to docetaxel in docetaxel-resistant PC cells
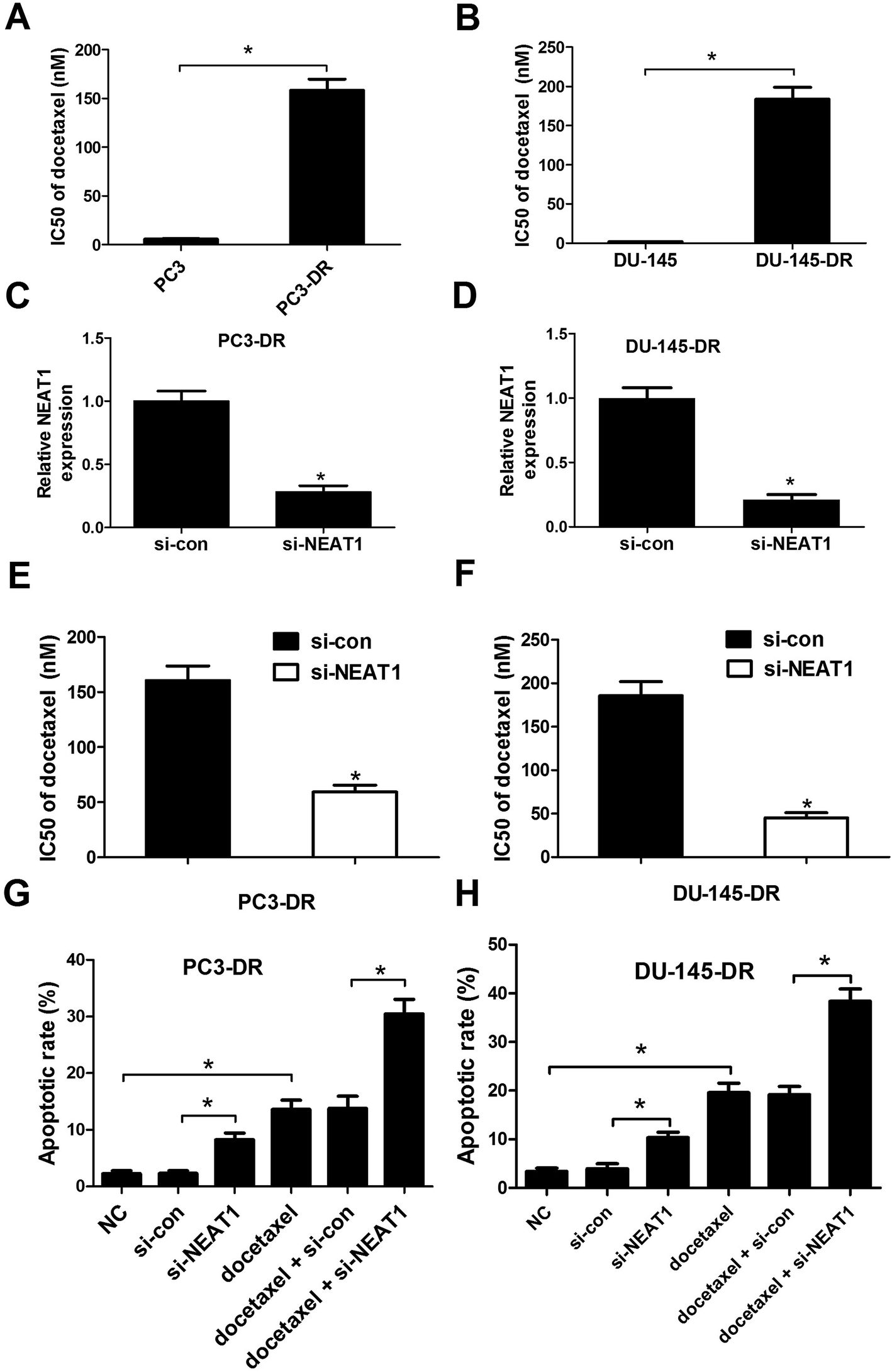
We firstly evaluated the docetaxel resistance of PC3-DR and DU-145-DR cells by IC50 value of docetaxel using CCK-8 assay. As shown in Fig. 2A and B, IC50 value of docetaxel in PC3-DR and DU-145-DR cells was strikingly higher than PC3 and DU-145 cells, demonstrating the docetaxel resistance of PC3-DR and DU-145-DR cells. To explore the effect of NEAT1 on docetaxel resistance of PC cells, loss-of-function experiments were employed by transfecting si-NEAT1 or si-con into PC3-DR and DU-145-DR cells. qRT-PCR was used to verify the transfection efficiency and the results exhibited that si-NEAT1 transfection led to a significant decrease of NEAT1 expression in PC3-DR (Fig. 2C) and DU-145-DR (Fig. 2D) cells. Next, the effect of NEAT1 knockdown on docetaxel resistance of PC3-DR and DU-145-DR cells was investigated. The results indicated that introduction of si-NEAT1 significantly reduced IC50 value of docetaxel in PC3-DR (Fig. 2E) and DU-145-DR (Fig. 2F) cells compared to si-con-transfected cells. The effect of NEAT1 knockdown on docetaxel-induced apoptosis was further explored by flow cytometry analysis. As presented in Fig. 2G and H, NEAT1 knockdown or docetaxel treatment effectively induced apoptosis, while combination treatment with si-NEAT and docetaxel dramatically aggravated docetaxel-induced apoptosis in PC3-DR and DU-145-DR cells. Collectively, these data suggested that NEAT1 knockdown increased the sensitivity to docetaxel in docetaxel-resistant PC cells.
 |
| | Fig. 2 NEAT1 knockdown sensitized docetaxel-resistant PC cells to docetaxel. (A and B) IC50 value of docetaxel was detected by CCK-8 assay in PC3, DU-145, PC3-DR and DU-145-DR cells. qRT-PCR analysis was perform to determine the expression level of NEAT1 in PC3-DR (C) and DU-145-DR (D) cells transfected with si-NEAT or si-con. CCK-8 assay was employed to measure IC50 value of docetaxel after PC3-DR (E) and DU-145-DR (F) cells transfected with si-NEAT1 or si-con were treated with various concentrations of docetaxel (0.32 to 100 μM) for 48 h. Flow cytometry analysis was applied to evaluate apoptosis after PC3-DR (G) and DU-145-DR (H) cells introduced with si-NEAT1 or si-con were exposed to 30 μM docetaxel for 48 h. *P < 0.05. | |
3.3. NEAT1 interacted with miR-34a in docetaxel-resistant PC cells
Increasing of publications reported that lncRNAs contain motif with complementary sequence to miRNAs. Therefore, Starbase v.2.0 was used to predict potential miRNAs that directly interacted with NEAT1. As a result, miR-34a was predicted to contain the complementary binding sites with NEAT1 (Fig. 3A). To confirm whether NEAT1 could interact with miR-34a, luciferase reporter assay was performed. As expected, ectopic expression of miR-34a significantly suppressed the luciferase activity of NEAT1-WT reporter in PC3-DR (Fig. 3B) and DU-145-DR (Fig. 3C) cells, but did not impair the luciferase activity of NEAT1-MUT reporter, suggesting that miR-34a could directly bind to NEAT1 at the recognized sites. Furthermore, qRT-PCR was conducted to assess the real regulatory effect of NEAT1 on miR-34a expression in PC3-DR and DU-145-DR cells. Our data demonstrated that miR-34a expression was markedly decreased in pcDNA-NEAT1-transfected PC3-DR and DU-145-DR cells (Fig. 3D), while miR-34a expression was apparently increased in si-NEAT-transfected PC3-DR and DU-145-DR cells (Fig. 3E). Therefore, we concluded that NEAT1 could interact with miR-34a and suppress miR-34a expression in docetaxel-resistant PC cells.
 |
| | Fig. 3 Reciprocal repression between NEAT1 and miR-34a in docetaxel-resistant PC cells. (A) Diagram of the predicted wild type or mutated miR-34a recognization region in NEAT1. Luciferase reporter assay was conducted to measure the luciferase activities in PC3-DR (B) and DU-145-DR (C) cells cotransfected with miR-34a or miR-con and NEAT1-WT or NEAT1-MUT. qRT-PCR analysis was applied to quantify the expression of miR-34a in PC3-DR (D) and DU-145-DR (E) cells after treatment with pcDNA3.0, pcDNA-NEAT1, si-con, or si-NEAT1. *P < 0.05. | |
3.4. NEAT1 knockdown enhanced sensitivity to docetaxel by sponging miR-34a in docetaxel-resistant PC cells
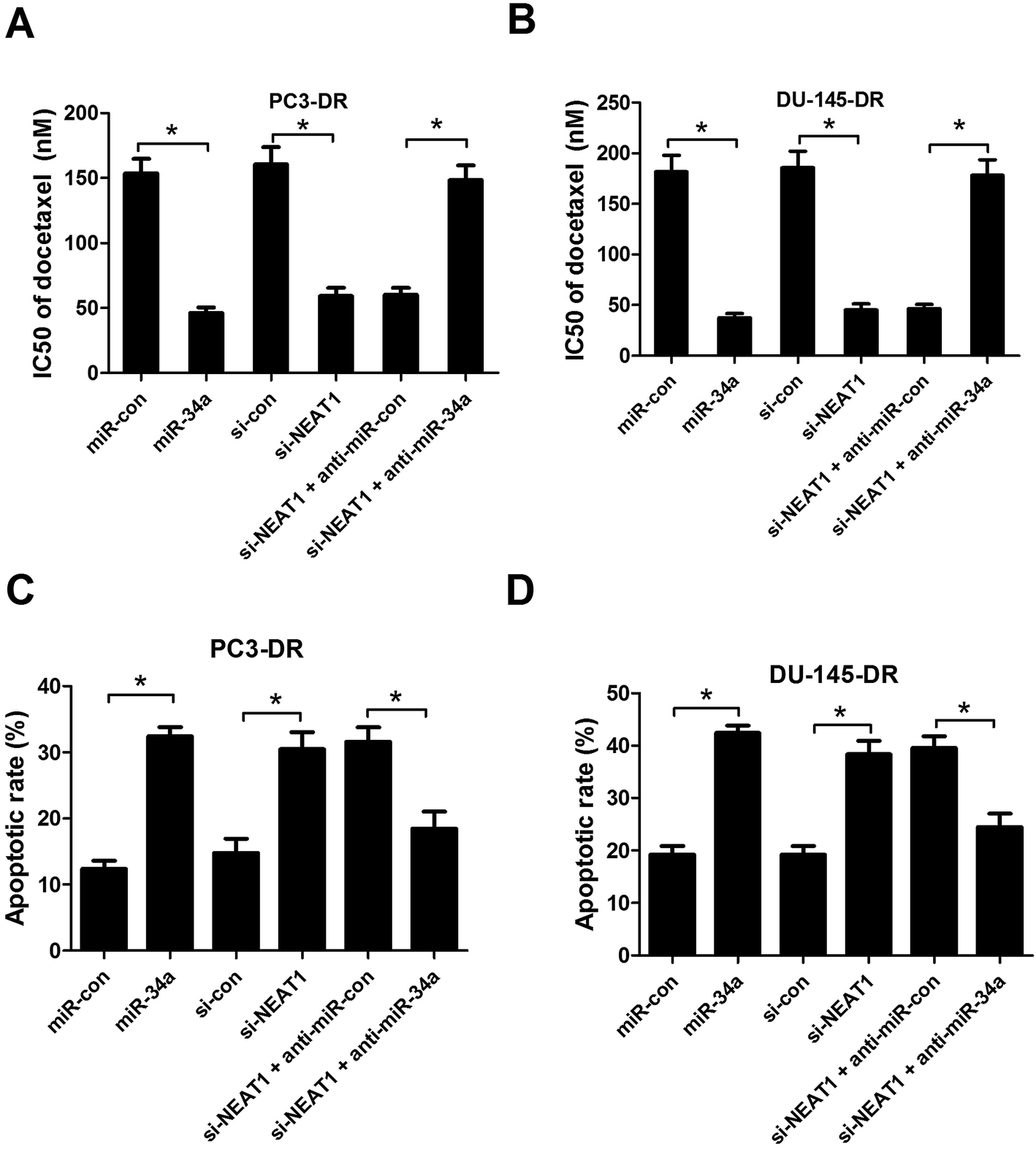
To investigate whether the involvement of NEAT1 in docetaxel resistance was mediated by miR-34a in docetaxel-resistant PC cells, PC3-DR and DU-145-DR cells were introduced with miR-34a, miR-con, si-con, si-NEAT1, si-NEAT1 + anti-miR-con, si-NEAT1 + anti-miR-34a, and then docetaxel resistance assay and flow cytometry analysis were performed. As illustrated in Fig. 4A and B, forced expression of miR-34a or NEAT1 knockdown led to a marked decrease of IC50 value of docetaxel in PC3-DR and DU-145-DR cells, whereas anti-miR-34a transfection strikingly abolished NEAT1-knockdown-mediated reduction in IC50 value of docetaxel. Meanwhile, our results revealed that exogenous expression of miR-34a or NEAT1 knockdown obviously induced apoptosis of PC3-DR (Fig. 4C) and DU-145-DR (Fig. 4D) cells, while miR-34a inhibition significantly reversed NEAT1-knockdown-induced apoptosis, as demonstrated by flow cytometry analysis. Taken together, these data revealed that NEAT1 knockdown enhanced sensitivity to docetaxel by downregulating miR-34a in docetaxel-resistant PC cells.
 |
| | Fig. 4 NEAT1 knockdown enhanced sensitivity to docetaxel by sponging miR-34a in docetaxel-resistant PC cells. PC3-DR and DU-145-DR cells were transfected with miR-con, miR-34a, si-con, si-NEAT1, si-NEAT1 + anti-miR-con, or si-NEAT1 + anti-miR-34a. CCK-8 assay was conducted to measure IC50 value of docetaxel in transfected PC3-DR (A) and DU-145-DR (B) cells. Flow cytometry analysis was carried out to examine cell apoptosis in transfected PC3-DR (C) and DU-145-DR (D) cells. *P < 0.05. | |
3.5. NEAT1 positively regulated RET expression by functioning as a ceRNA of miR-34a
To further explored the molecular mechanism by which NEAT1 and miR-34a exerted their regulatory effect on docetaxel resistance of PC cells, targetscan (http://www.targetscan.org) and miRanda (http://www.microrna.org) algorithms were employed to predict the potential targets of miR-34a. Target prediction analysis revealed that RET was a potential target of miR-34a (Fig. 5A). Subsequently, luciferase reporter assay was performed to confirm the direct binding between miR-34a and RET. 293T, PC3-DR and DU-145-DR cells were cotransfected with constructed luciferase reporter plasmids containing the wild-type or mutated miR-34a binding sites in the 3′UTR of RET mRNA and miR-34a, miR-con, miR-34a + pcDNA3.0, or miR-34a + pcDNA-NEAT1. The results revealed that increased expression of miR-34a significantly reduced the luciferase activity of RET-WT reporter in 293T (Fig. 5B), PC3-DR and DU-145-DR (Fig. 5C) cells, while co-treatment with miR-34a and pcDNA-NEAT1 markedly reversed this effect. However, the luciferase activity of RET-MUT reporter was not affected by any treatment. We further analyzed the protein level of RET by western blot in PC3-DR and DU-145-DR cells transfected with si-con, si-NEAT1, miR-con, miR-34a, miR-34a + pcDNA3.0, or miR-34a + pcDNA-NEAT1. As shown in Fig. 5D and E, NEAT1 knockdown or miR-34a overexpression dramatically reduced the protein level of RET in PC3-DR and DU-145-DR cells, but ectopic expression of NETA1 effectively restored miR-34a-mediated reduction of RET protein level. Collectively, these data indicated that NEAT1 positively regulated RET expression by sponging miR-34a.
 |
| | Fig. 5 NEAT1 upregulated RET expression by acting as a ceRNA of miR-34a. (A) The predicted miR-34a binding sequences in the 3′UTR of RET mRNA or the corresponding mutant type. Luciferase reporter assay was performed to detect luciferase activity after 293T (B), PC3-DR (C) and DU-145-DR (C) cells were cotransfected with RET-WT or RET-MUT and miR-34a, miR-con, miR-34a + pcDNA3.0, or miR-34a + pcDNA-NEAT1. Western blot was carried out to determine the protein level of PC3-DR (D) and DU-145-DR (E) cells transfected with si-con, si-NEAT1, miR-con, miR-34a, miR-34a + pcDNA3.0, or miR-34a + pcDNA-NEAT1. *P < 0.05. | |
3.6. NEAT1 overexpression reversed RET-knockdown-induced sensitivity to docetaxel in docetaxel-resistant PC cells
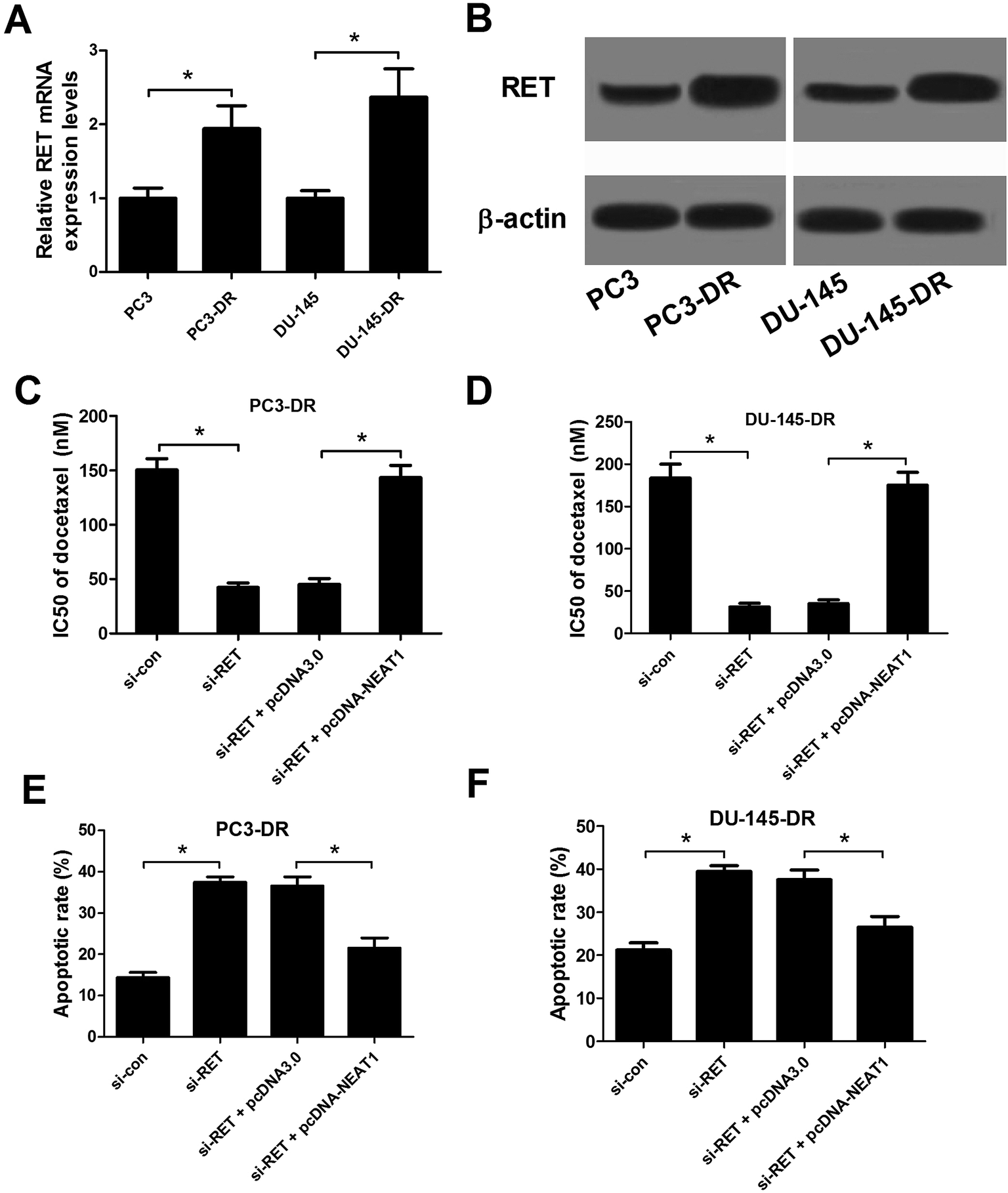
To explore the role of RET in docetaxel resistance of PC, the expression of RET in parental PC cells (PC3 and DU-145) and docetaxel-resistant PC cells (PC3-DR and DU-145-DR) was initially detected by qRT-PCR and western blot. As shown in Fig. 6A and B, RET expression at both mRNA and protein levels was markedly increased in PC3-DR and DU-145-DR cells in comparison with PC3 and DU-145 cells. Due to the positive effect of NEAT1 on RET expression, we investigated the regulatory role of NEAT1 on RET's function in docetaxel-resistant PC cells. Docetaxel resistance assay and flow cytometry analysis were conducted in PC3-DR and DU-145-DR cells treated with si-con, si-RET, si-RET + pcDNA3.0, or si-RET + pcDNA-NEAT1. CCK-8 assay implicated that IC50 value of docetaxel was remarkably decreased in RET-knockdown-PC3-DR (Fig. 6C) and DU-145-DR (Fig. 6D) cells, suggesting that RET knockdown dramatically improved sensitivity to docetaxel in docetaxel-resistant PC cells. Nevertheless, NEAT1 overexpression greatly relieved the reduction in IC50 value of docetaxel by RET knockdown. Flow cytometry analysis indicated that RET knockdown significantly promoted apoptosis of PC3-DR (Fig. 6E) and DU-145-DR (Fig. 6F) cells in contrast to si-con-transfected cells, whereas cotransfection with si-RET and pcDNA-NETA1 obviously inhibited si-RET-induced apoptosis of PC3-DR and DU-145-DR cells compared to si-RET + pcDNA3.0 group. Thus, these data demonstrated that NEAT1 overexpression abated RET-knockdown-induced sensitivity to docetaxel in docetaxel-resistant PC cells.
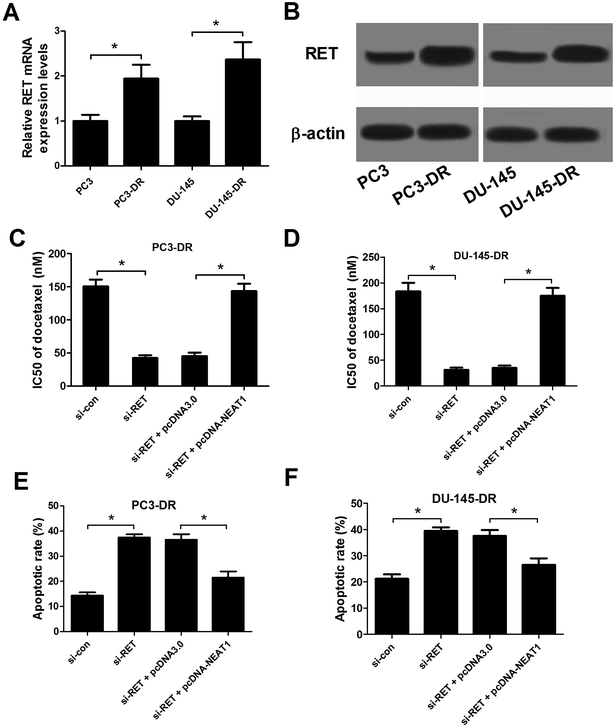 |
| | Fig. 6 NEAT1 overexpression abrogated RET-knockdown-induced sensitivity to docetaxel in docetaxel-resistant PC cells. RET expressions at mRNA and protein levels in parental PC cells (PC3 and DU-145) and docetaxel-resistant PC cells (PC3-DR and DU-145-DR) were determined by qRT-PCR (A) and western blot (B). PC3-DR and DU-145-DR cells were transfected with si-con, si-RET, si-RET + pcDNA3.0, or si-RET + pcDNA-NEAT1. The effect of RET knockdown or combined with NEAT1 overexpression on IC50 value of docetaxel was examined using CCK-8 assay in transfected PC3-DR (C) and DU-145-DR (D) cells. The effect of RET knockdown or combined with NEAT1 overexpression on apoptosis of transfected PC3-DR (E) and DU-145-DR (F) cells was explored by flow cytometry analysis. *P < 0.05. | |
3.7. NEAT1 knockdown enhanced sensitivity to docetaxel of docetaxel-resistant PC cells through suppressing RET expression by sponging miR-34a in vivo
To further confirm the biological function of 1 in docetaxel resistance of PC cells in vivo, a xenograft nude mice model was established. PC3-DR cells stably transfected with sh-NEAT1 or sh-con were subcutaneously injected into nude mice. At 7 days after the inoculation, mice were administered intraperitoneally with 10 mg kg−1 docetaxel or same volume of PBS every 3 days. As presented in Fig. 7A and B, NEAT1 knockdown or docetaxel treatment significantly impeded tumor growth including tumor volume and weight, however, cotreatment with NEAT1 and docetaxel aggravated docetaxel-induced inhibition on tumor growth, indicating that NEAT1 knockdown enhanced sensitivity to docetaxel of docetaxel-resistant PC cells in vivo. In addition, we estimated the effects of NEAT1 on the expression of miR-34a and the protein level of RET in vivo. Consistently with the in vitro results, transfection with sh-NEAT1 significantly promoted miR-34a expression (Fig. 7C) and suppressed RET level (Fig. 7D) compared with control group in the resected tumors in the presence of docetaxel, as demonstrated by qRT-PCR and western blot, suggesting that NEAT1 knockdown suppressed RET by sponging miR-34a in vivo.
 |
| | Fig. 7 NEAT1 knockdown enhanced sensitivity to docetaxel of docetaxel-resistant PC cells in vivo. PC3-DR cells transfected with sh-NEAT1 or sh-con were subcutaneously injected into nude mice. At 7 days post-inoculation, mice were administered intraperitoneally with 10 mg kg−1 docetaxel or same volume of PBS every 3 days. (A) Tumor growth was measured every 3 days using a caliper rule. (B) Xenografted tumor masses were excised and weighed at 28 days after implantation. (C) qRT-PCR analysis of miR-34a expression in the resected xenografted tumors. (D) Western blot analysis of RET protein level in the resected xenografted tumors. *P < 0.05. | |
4. Discussion
Aberrant expression of lncRNAs is involved in the development of drug resistance of various tumors and provides new insights into cancer treatment and drug resistance.28,29 Nevertheless, the biological function of lncRNA in drug resistance of PC cells remains largely unclear. Elucidating the detailed molecular mechanism by which lncRNAs function in the drug resistance of human malignancies would contribute to the development of lncRNA-mediated therapeutics against cancers that respond poorly to chemotherapy. In the current study, we demonstrated that NEAT1 was aberrantly upregulated in docetaxel-resistant PC cells. Functional and mechanistic analyses revealed that NEAT1 knockdown improved docetaxel sensitivity of docetaxel-resistant PC cells, which was partially credited to its ability to serve as a ceRNA of miR-34a, resulting in derepression of RET.
According to previous studies, NEAT1 functioned as a crucial transcriptional regulator for numerous genes, some of which are implicated in cancer progression.30 Anomalous expression of NEAT1 has been reported in several human malignancies and exerts oncogenic activity in breast cancer,31 hepatocellular carcinoma,32 thyroid carcinoma,33 PC,19 and pancreatic cancer.34 More importantly, previous studies demonstrated the involvement of NEAT1 in chemoresistance in several tumors. For example, NEAT1 expression was found to be repressed in leukemia cells and overexpression of NEAT1 alleviated multidrug resistance in leukemia cells through inhibition of ATP-binding cassette G2.35 NEAT1 was demonstrated to function as a ceRNA of miR-98-5p to upregulate copper transporter 1 (CTR1), thus enhancing cisplatin sensitivity in lung cancer cells.36 Additionally, NETA1 was reported to be highly expressed in gastric cancer tissues and cells, moreover, NEAT1 knockdown depressed chemotherapy resistance to adriamycin and promoted adriamycin-induced apoptosis in gastric cancer.37 Consistent with the previous study,19 our study confirmed the upregulation of NEAT1 in PC tissues and cell lines. Also, higher expression of NEAT1 in docetaxel-resistant PC cells was observed, suggesting that NEAT1 may be involved in docetaxel resistance of PC cells. Loss-of-function experiments demonstrated that NEAT1 knockdown significantly improved sensitivity to docetaxel in vitro and in vivo. Our results provided new evidence in the current literature linking drug resistance and NEAT1 in PC.
The molecular mechanism by which NEAT1 mediated sensitivity to docetaxel in docetaxel-resistant PC cells remains to be thoroughly elucidated. Increasing evidence has demonstrated that interactions between lncRNAs and miRNAs are involved in many pathological processes of a wide range of human carcinomas.38 In this case, lncRNAs are implicated in the ceRNA regulatory system and may serve as ceRNAs or molecular sponges to suppress the expressions and activities of miRNAs, thus leading to derepression of miRNA targets.39 According to a previous study, NEAT1 functioned as a ceRNA for miR-377-3p, antagonized its functions and led to the derepression of its endogenous targets E2F transcription factor 3 (E2F3), which played an oncogenic role in promoting non-small cell lung cancer progression.40 In nasopharyngeal carcinoma, NEAT1 overexpression upregulated zinc finger E-box binding homeobox 1 (ZEB1) by negatively regulating miR-204 expression.41 NEAT1 was found to be able to regulate cyclin-dependent kinase 6 (CDK6) in laryngeal squamous cell cancer, which was mediated by miR-107.42 In the present study, we figured out the underlying molecular mechanism of NEAT1 involved in docetaxel resistance in PC cells, by functioning as a miRNA sponge to regulate miR-34a. miR-34a has been reported to mediate drug resistance in various cancers, such as colorectal cancer,43 diffuse large B-cell lymphoma,44 bladder cancer,45 and PC.22,46 Consistently, our study demonstrated that miR-34a was significantly downregulated in PC tissues, parental PC cell lines, as well as docetaxel-resistant PC cell lines, and enforced expression of miR-34a enhanced sensitivity to docetaxel in docetaxel-resistant PC cells. In addition, we discovered that NEAT1 functioned as a molecular sponge of miR-34a to positively regulate miR-34a target RET. Proto-oncogene RET encodes a receptor tyrosine kinase (RTK) which plays an important role in cell proliferation, migration, and differentiation in several human malignancies.47 Our study revealed that RET was upregulated in docetaxel-resistant PC cells compared with parental PC cells, suggesting that RET may be involved in docetaxel resistance of PC. Moreover, miR-34a inhibition partially reversed NETA1-knockdown-induced sensitivity to docetaxel in docetaxel-resistant PC cells, while NEAT1 overexpression abrogated RET-knockdown-triggered sensitivity to docetaxel in docetaxel-resistant PC cells in vitro. Furthermore, in vivo experiment demonstrated that NEAT1 knockdown suggesting that NETA1 knockdown significantly improved sensitivity to docetaxel by inhibiting RET level and promoting miR-34a expression in vivo. NEAT1 regulated sensitivity to docetaxel in docetaxel-resistant PC cells through inducing RET expression by sponging miR-34a in vitro and in vivo.
5. Conclusions
In conclusion, these results demonstrated that NEAT1 was upregulated and miR-34a was downregulated in docetaxel-resistant PC cells. Furthermore, mechanistic analyses revealed that NEAT1 contributed to docetaxel resistance in docetaxel-resistant PC cells through inducing RET expression by sponging miR-34a, shedding light on the vital role of NEAT1/miR-34a/RET asix in PC chemoresistance. Therefore, NEAT1 may be used as an effective chemotherapeutic target in the clinical diagnosis and treatment of PC.
Authors' contribution
Chaoyang Zhu and Yang Li designed the experiment. Xin Tian and Guangwei Zhang performed the experiment, analyzed the data, and wrote the manuscript. Hui Zhao and Chaoyang Zhu supervised the study and reviewed the manuscript.
Conflicts of interest
There is no conflict of interest regarding the publication of this paper.
References
- R. Siegel, J. Ma, Z. Zou and A. Jemal, Ca-Cancer J. Clin., 2014, 64, 9–29 CrossRef PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 5–29 CrossRef PubMed.
- M. Gaztañaga and J. Crook, J. Natl. Compr. Cancer Network, 2012, 10, 1088–1095 CrossRef.
- D. P. Petrylak, C. M. Tangen, M. H. Hussain, P. N. Lara Jr, J. A. Jones, M. E. Taplin, P. A. Burch, D. Berry, C. Moinpour, M. Kohli, M. C. Benson, E. J. Small, D. Raghavan and E. D. Crawford, N. Engl. J. Med., 2004, 351, 1513–1520 CrossRef CAS PubMed.
- C. Rolfo, F. Passiglia, M. Castiglia, L. E. Raez, P. Germonpre, I. Gil-Bazo, K. Zwaenepoel, W. A. De, G. Bronte and A. Russo, Transl. Lung Cancer Res., 2014, 3, 250–261 CAS.
- K. Komura, S. H. Jeong, K. Hinohara, F. Qu, X. Wang, M. Hiraki, H. Azuma, G. M. Lee, P. W. Kantoff and C. J. Sweeney, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6259–6264 CrossRef CAS PubMed.
- S. Djebali, C. A. Davis, A. Merkel, A. Dobin, T. Lassmann, A. M. Mortazavi, A. Tanzer, J. Lagarde, W. Lin and F. Schlesinger, Nature, 2012, 489, 101–108 CrossRef CAS PubMed.
- J. L. Rinn and H. Y. Chang, Annu. Rev. Biochem., 2012, 81, 145–166 CrossRef CAS PubMed.
- S. Geisler and J. Coller, Nat. Rev. Mol. Cell Biol., 2013, 14, 699–712 CrossRef CAS PubMed.
- M. Zhou, Y. Sun, Y. Sun, W. Xu, Z. Zhang, H. Zhao, Z. Zhong and J. Sun, Oncotarget, 2016, 7, 32433–32448 Search PubMed.
- M. Zhou, H. Zhao, W. Xu, S. Bao, L. Cheng and J. Sun, Mol. Cancer, 2017, 16, 16 CrossRef PubMed.
- M. Zhou, Z. Diao, X. Yue, Y. Chen, H. Zhao, L. Cheng and J. Sun, Oncotarget, 2016, 7, 56383–56394 Search PubMed.
- J. R. Prensner and A. M. Chinnaiyan, Cancer Discovery, 2011, 1, 391–407 CrossRef CAS PubMed.
- T. Naganuma and T. Hirose, RNA Biol., 2013, 10, 456–461 CrossRef CAS PubMed.
- C. Zeng, Y. Xu, L. Xu, X. Yu, J. Cheng, L. Yang, S. Chen and Y. Li, BMC Cancer, 2014, 14, 693 CrossRef PubMed.
- L. J. Pan, T. F. Zhong, R. X. Tang, P. Li, Y. W. Dang, S. N. Huang and G. Chen, Asian Pac. J. Cancer Prev., 2015, 16, 2851–2855 CrossRef PubMed.
- H. Ke, L. Zhao, X. Feng, H. Xu, L. Zou, Q. Yang, X. Su, L. Peng and B. Jiao, Gene Regul. Syst. Biol., 2016, 10, 11–17 Search PubMed.
- L. Zhen, L. Yun-Hui, D. Hong-Yu, M. Jun and Y. Yi-Long, Tumor Biol., 2016, 37, 673–683 CrossRef PubMed.
- D. Chakravarty, A. Sboner, S. S. Nair, E. Giannopoulou, R. Li, S. Hennig, J. M. Mosquera, J. Pauwels, K. Park, M. Kossai, T. Y. MacDonald, J. Fontugne, N. Erho, I. A. Vergara, M. Ghadessi, E. Davicioni, R. B. Jenkins, N. Palanisamy, Z. Chen, S. Nakagawa, T. Hirose, N. H. Bander, H. Beltran, A. H. Fox, O. Elemento and M. A. Rubin, Nat. Commun., 2014, 5, 5383 CrossRef CAS PubMed.
- M. V. Iorio and C. M. Croce, Carcinogenesis, 2012, 33, 1126–1133 CrossRef CAS PubMed.
- M. I. Aslam, M. Patel, B. Singh, J. S. Jameson and J. H. Pringle, J. Transl. Med., 2012, 10, 128 CrossRef CAS PubMed.
- E. Kopczynska, Contemp. Oncol., 2015, 19, 423–427 Search PubMed.
- Y. Fujita, K. Kojima, N. Hamada, R. Ohhashi, Y. Akao, Y. Nozawa, T. Deguchi and M. Ito, Biochem. Biophys. Res. Commun., 2008, 377, 114–119 CrossRef CAS PubMed.
- J. H. Yoon, K. Abdelmohsen and M. Gorospe, Semin. Cell Dev. Biol., 2014, 34, 9–14 CrossRef CAS PubMed.
- Z. Dong, A. Zhang, S. Liu, F. Lu, Y. Guo, G. Zhang, F. Xu, Y. Shi, S. Shen, J. Liang and W. Guo, Mol. Cancer Res., 2017, 15, 800–810 CrossRef CAS PubMed.
- M. Zhou, X. Wang, H. Shi, L. Cheng, Z. Wang, H. Zhao, L. Yang and J. Sun, Oncotarget, 2016, 7, 12598–12611 Search PubMed.
- J. Saakshi, B. Deeksha, L. M. Kumar, S. Sridhar and S. Vinod, PLoS One, 2013, 8, e53823 Search PubMed.
- J. Liu, L. Wan, K. Lu, M. Sun, X. Pan, P. Zhang, B. Lu, G. Liu and Z. Wang, PLoS One, 2015, 10, e0114586 Search PubMed.
- C. Shang, Y. Guo, J. Zhang and B. Huang, Cancer Chemother. Pharmacol., 2016, 77, 1061–1067 CrossRef CAS PubMed.
- C. M. Clemson, J. N. Hutchinson, S. A. Sara, A. W. Ensminger, A. H. Fox, A. Chess and J. B. Lawrence, Mol. Cell, 2009, 33, 717–726 CrossRef CAS PubMed.
- M. Zhang, W. B. Wu, Z. W. Wang and X. H. Wang, Eur. Rev. Med. Pharmacol. Sci., 2017, 21, 1020–1026 CAS.
- Y. Mang, L. Li, J. Ran, S. Zhang, J. Liu, L. Li, Y. Chen, J. Liu, Y. Gao and G. Ren, OncoTargets Ther., 2017, 10, 1003–1016 CrossRef PubMed.
- J. H. Li, S. Q. Zhang, X. G. Qiu, S. J. Zhang, S. H. Zheng and D. H. Zhang, Internet J. Oncol., 2017, 50, 708–716 CrossRef PubMed.
- B. Huang, C. Liu, Q. Wu, J. Zhang, Q. Min, T. Sheng, X. Wang and Y. Zou, Biochem. Biophys. Res. Commun., 2017, 482, 828–834 CrossRef CAS PubMed.
- C. Gao, J. Zhang, Q. Wang and C. Ren, Oncol. Lett., 2016, 12, 1051–1057 Search PubMed.
- P. Jiang, X. Wu, X. Wang, W. Huang and Q. Feng, Oncotarget, 2016, 7, 43337–43351 Search PubMed.
- J. Zhang, B. Zhao, X. Chen, Z. Wang, H. Xu and B. Huang, Pathol. Oncol. Res., 2017, 1–5 Search PubMed.
- S. Ergun and S. Oztuzcu, Tumor Biol., 2015, 36, 3129–3136 CrossRef CAS PubMed.
- Y. Tay, J. Rinn and P. P. Pandolfi, Nature, 2014, 505, 344–352 CrossRef CAS PubMed.
- C. Sun, S. Li, F. Zhang, Y. Xi, L. Wang, Y. Bi and D. Li, Oncotarget, 2016, 7, 51784–51814 Search PubMed.
- Y. Lu, T. Li, G. Wei, L. Liu, Q. Chen, L. Xu, K. Zhang, D. Zeng and R. Liao, Tumor Biol., 2016, 37, 11733–11741 CrossRef CAS PubMed.
- P. Wang, T. Wu, H. Zhou, Q. Jin, G. He, H. Yu, L. Xuan, X. Wang, L. Tian, Y. Sun, M. Liu and L. Qu, J. Exp. Clin. Cancer Res., 2016, 35, 22 CrossRef PubMed.
- C. Sun, F. J. Wang, H. G. Zhang, X. Z. Xu, R. C. Jia, L. Yao and P. F. Qiao, World J. Gastroenterol., 2017, 23, 1816–1827 CrossRef PubMed.
- S. C. Marques, B. Ranjbar, M. B. Laursen, S. Falgreen, A. E. Bilgrau, J. S. Bodker, L. K. Jorgensen, M. N. Primo, A. Schmitz, M. S. Ettrup, H. E. Johnsen, M. Bogsted, J. G. Mikkelsen and K. Dybkaer, Exp. Hematol., 2016, 44, 238–246 CrossRef CAS PubMed.
- H. Li, G. Yu, R. Shi, B. Lang, X. Chen, D. Xia, H. Xiao, X. Guo, W. Guan, Z. Ye, W. Xiao and H. Xu, Mol. Cancer, 2014, 13, 8 CrossRef PubMed.
- K. Kojima, Y. Fujita, Y. Nozawa, T. Deguchi and M. Ito, Prostate, 2010, 70, 1501–1512 CrossRef CAS PubMed.
- C. Eng, J. Clin. Oncol., 1999, 17, 380–393 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a