
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile method for cellular N-glycomic profiling by matrix-assisted laser desorption/ionization mass spectrometry†
Wenjie Gao‡
 ,
Yanhua Jiang‡,
Zhihui Zhang‡,
Yifang Zhang,
Yanyan Liu,
Yanhong Zhou and
Xin Liu*
,
Yanhua Jiang‡,
Zhihui Zhang‡,
Yifang Zhang,
Yanyan Liu,
Yanhong Zhou and
Xin Liu*
The Key Laboratory for Biomedical Photonics of MOE at Wuhan National Laboratory for Optoelectronics-Hubei Bioinformatics & Molecular Imaging Key Laboratory, Systems Biology Theme, Department of Biomedical Engineering, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan 430074, China. E-mail: xliu@mail.hust.edu.cn; Tel: +86-27-87793180
First published on 20th July 2017
Abstract
Conventional protocols for cellular N-glycan profiling often need several time-consuming and labor-intensive steps, and a large amount of material (5–20 × 106 cells) is required. In this study, an optimized co-derivatization method was applied for convenient, rapid and highly-sensitive analysis of cellular N-glycan. Taking human cervical carcinoma cells (HeLa) as a model, the required amount for comprehensive cellular N-glycans analysis was reduced down to an original cell number of 105 and the total analysis time was decreased from several days to several hours. Good reproducibility of the method was also obtained with the CV averaging to less than 13.1%. In addition, compared to permethylation derivatization, more than 5-fold sensitivity improvement was achieved and more concise and clear mass spectra were obtained with the new developed method. As a preliminary study, this method was also successfully applied to reveal the difference in cellular N-glycan profiling of two heterogeneous live cell lines, human normal liver cells (L-02) and human hepatocyte carcinoma cells (HepG2), showing great potential for high-throughput analysis of N-glycans from limited cell samples, such as primary cell lines, cells obtained from cell sorting and small size cancer specimens.
1 Introduction
Protein glycosylation is one of the most common and complicated co- or post-translational modifications. Almost all membrane and secreted proteins of mammals,1 as well as numerous cytoplasm and nucleus proteins2–4 are glycosylated. It has been well documented that N-glycans play crucial roles in many biological processes such as cell growth and development,5,6 immune recognition,7–9 cell–cell communication,10 infectivity of many pathogenic bacteria11,12 and most viruses.13,14 Therefore, comprehensive structural characterization of cellular glycan is of great important to gain a better understanding of glycan functions in cells, and to provide deeper insight into pathological processes that occur in diseases, which could contribute to progress in early diagnosis and targeted treatment.Sample preparation is an important step for the analysis of cellular N-glycan profiling. The conventional protocol used for the cellular N-glycan profiling includes the following steps: (1) cell lysis; (2) protein extraction; (3) deglycosylation and glycan enrichment; and/or (4) derivatization.15 However, efficient analysis of cellular N-glycans was often hampered by both the requirement of large amount samples and tedious sample preparation. Hence, many researchers have developed some convenient and efficient methods for the N-glycan profiling of cellular or membrane glycoproteins. Rahman et al. have reported a sample preparation method for mammalian cells N-glycan profiling based on using filter-aided removal of detergent.16 Hamouda et al. have used trypsin to release glycopeptides from cell surfaces and then for the analysis of membrane glycoprotein N-glycan profiling.17 However, traditional deglycosylation used in these studies was still time-consuming. Additionally, even though permethylation has been shown to increase the glycan abundance but are not useful with larger glycans due to incomplete reaction dispersing the glycan signal across several different m/z channels.18 Lattová et al. have reported an efficient method for N-glycan analysis which involved one-step organic extraction in prior to Peptide-N-Glycosidase F (PNGase F) deglycosylation.19 Phenyl-hydrazine (PHN) was used for derivatization to achieve the high sensitive analysis. However, neuraminidase was employed to remove sialic acid residues to inhibit in-and post-source decay of sialylated glycans during MALDI-MS analysis, which resulted in the incomplete cellular N-glycan profiling. Moreover, polyhexose contaminations from cytoplasm could decrease the signal intensities of other glycan peaks and increase the difficulties in data interpretation when the above derivatization strategies were applied. More recently, Holst et al. have applied an optimized, high-throughput membrane-based enzymatic glycan release for cellular N-glycan analysis with sialic linkage specific carboxyl derivatization method.20 However, this method has some disadvantages, such as the instability of the generated lactones and byproducts of the derivatives.21
Up to now, matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) has been widely used in glycan analysis for the advantages of impurity tolerance, simplicity and ease of operation.22 However, the use of a matrix introduces a number of problems into the MALDI-MS analysis and one major limitation is the unsatisfying variability of signal intensities between different desorption spots across the preparation.23 Therefore, an often time-consuming search for “sweet spots” is necessary in order to yield good signal-to-noise ratios or an analyte signal at all. In addition, the resulting lack of shot-to-shot reproducibility mostly limited the use of MALDI-MS for quantification experiments. The main reason for low shot-to-shot reproducibility surely resulted from inhomogeneous segregation of analytes and matrix molecules over different areas of the preparation during drying and co-crystallization.
Previously, we reported a novel strategy to rapid and sensitive analysis of N-glycans by MALDI-MS using permanent charge derivatization and methylamidation.24 Rapid deglycosylation was achieved using enzyme-friendly detergents assisted method with DDM as the detergents, which significantly reduced sample digestion time to several minutes. The corresponding glycosylamines of N-glycans can be easily labeled by a permanent charge derivatization reagent, TMPP-Ac-OSu. Additionally, the neutralization of sialic acid by methylamidation was also employed to avoid dissociation of sialic acid moiety during MALDI-TOF analysis of sialoglycans. This developed dual derivatization method could dramatically improve the sensitivity and give the opportunity to simultaneous analysis of neutral glycans and sialoglycans when the sample amounts is highly limited.
In this paper, an optimized co-derivatization method was applied to achieve the cellular N-glycan analysis with reduction of cell numbers and preparation time. With the new developed method, the required amount for cellular N-glycans analysis was reduced down to 105 and total sample preparation time was decreased from several days to several hours. We also found that using α-cyano-4-hydroxycinnamic acid (CHCA) as matrix could obtained homogeneous crystal produced on the steel plate with better mass spectra and improved sensitivity and resolution when it was compared to 2,5-dihydroxybenzoic acid (DHB). As a result, almost 60 glycans were detected with only 105 cells using HeLa cell as a model. In addition, two heterogeneous live cell lines, HepG2 and L-02 were successfully applied to validate our modified method and significant difference of the cellular N-glycan profiling was observed between these two cell lines. Therefore, the new developed method has the potential to be a powerful tool for the cellular N-glycan analysis and biomarker discovery with limited cell samples.
2 Materials and methods
2.1 Chemicals and reagents
Tris(2,4,6-trimethoxyphenyl)-phosphine (TMPP), bromoacetic acid N-hydroxysuccinimide ester, anhydrous dimethyl sulfoxide (DMSO), methylamine hydrochloride, (7-azabenzotriazol-1-yloxy) tripyrrolidinophosphonium hexafluorophosphate (PyAOP), microcrystalline cellulose (MCC), 2,5-dihydroxybenzoic acid (DHB), α-cyano-4-hydroxycinnamic acid (CHCA), phenylmethylsulfonyl fluoride (PMSF), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were purchased from Sigma Aldrich (St. Louis, MO, U.S.A.). PNGase F was purchased from LCP Biomed (Lianyungang, China). Acetonitrile (ACN) and methanol (HPLC grade) were purchased from Merck KGaA (Darmstadt, Germany). The empty cartridges and frits were purchased from Tianjin Bonna-Agela Technologies Inc. (Tianjin, China). Trifluoroacetic acid (TFA), Tris base, chloroform, ethanol (HPLC grade) and N-dodecyl β-D-maltoside (DDM) were purchased from Aladdin Industrial Inc. (Shanghai, China). All the reaction solutions were prepared with water purified by the Direct-Q system (Millipore, Bedford, MA).2.2 Cell culture
Human cervical carcinoma cell line (HeLa) and human hepatocyte carcinoma cell line (HepG2) were obtained from the American Type Culture Collection (Bethesda, MD). Human normal liver cell line (L-02) was a gift from Dr Shuai Xia. All the above cell lines were cultured in Dulbecco's Modified Eagle Medium (DMEM), supplemented with 10% (v/v) fetal bovine serum (FBS), 100 U mL−1 penicillin and streptomycin. All cells were maintained in a T-25 flask with 5 mL at 37 °C incubator with 5% CO2 atmosphere until they reached approximately 90% confluence. Cells were detached with trypsin and washed with cold D-PBS by centrifugation for 3 times. When necessary, 10 μL of the cell suspension was removed from for counting to determine the total number of cells used. After third washing and pipetting off the supernatant, the pelleted cells were directly for lysis or stored at −20 °C.2.3 Cell lysis and protein extraction
Collected cells (usually 5 × 105) were dissolved in an ice-cold lysis buffer (200 μL) containing 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.2% SDS, 0.5 mM PMSF and were incubated at 4 °C for 15 min. Then the solution was ultrasonicated at intervals of 3 s for 1 min, with 3 s pause between each treatment. The cell lysate was centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 5 min and the supernatant was carefully collected and used for further protein extraction. The following two protocols were applied for protein extraction: (1) chloroform–methanol–water (CMW) method. 600 μL of methanol, 150 μL of chloroform and 400 μL of pure water were added in turn and all vortex briefly. The mixture was centrifuged for 5 min at 13000g, then carefully removed and discard as much of the upper aqueous phase as possible while leaving the interface layer containing the protein precipitate intact. 450 μL of methanol was added to the tube and vortex briefly and centrifuged for 5 min at 13000g to pellet the protein. (2) ethanol extraction. 800 μL of ethanol was added to the lysis buffer and left in the freezer for 2 h. After centrifugation (5 min at 13000g), the supernatant was carefully removed and the protein pellet was left to air-dry.
000g for 5 min and the supernatant was carefully collected and used for further protein extraction. The following two protocols were applied for protein extraction: (1) chloroform–methanol–water (CMW) method. 600 μL of methanol, 150 μL of chloroform and 400 μL of pure water were added in turn and all vortex briefly. The mixture was centrifuged for 5 min at 13000g, then carefully removed and discard as much of the upper aqueous phase as possible while leaving the interface layer containing the protein precipitate intact. 450 μL of methanol was added to the tube and vortex briefly and centrifuged for 5 min at 13000g to pellet the protein. (2) ethanol extraction. 800 μL of ethanol was added to the lysis buffer and left in the freezer for 2 h. After centrifugation (5 min at 13000g), the supernatant was carefully removed and the protein pellet was left to air-dry.
2.4 Synthesis of TMPP-Ac-OSu
TMPP-Ac-OSu was synthesized by a protocol as previously reported.25 Briefly, 0.1 mmol (23.6 mg) bromoacetic acid N-hydroxysuccinimide ester was dissolved in 1 mL of toluene and 0.1 mmol (53.3 mg) Tris(2,4,6-trimethoxyphenyl) phosphine in 1 mL of toluene was added. A white precipitate was formed immediately. After standing at room temperature for 0.5 h, the mixture was diluted with 4 mL of hexane, filtered and washed on the filter with hexane. Drying in a desiccator over CaCl2 overnight gave pure TMPP-Ac-OSu bromide as a white crystalline powder. It should be noted that experiments were carried out under a hood because of the use of toluene.2.5 Rapid digestion of cellular glycoproteins and TMPP-Ac labeling
The rapid digestion of cellular glycoproteins and TMPP-Ac labeling were performed as previously reported with small modification.24 Briefly, crude cellular proteins sample was dissolved in 100 μL digestion buffer containing 50 mM HEPES (pH 8.0), 1% N-dodecyl β-D-maltoside (DDM), 20 mM dithiothreitol (DTT), 0.2% sodium dodecyl sulfate (SDS). Then the solution was incubated at 100 °C for 2 min, and was subsequently cooled to 50 °C. 1.0 μL of PNGase F (5 units) was added to the mixture, and incubated at 50 °C for 5 min. Following enzymatic deglycosylation, enzyme was deactivated at 100 °C for 2 min. For the TMPP-Ac labeling, a freshly prepared solution of 20 mM TMPP-Ac-OSu in acetonitrile (100 μL) was added to the reaction tube. The mixture was agitated vigorously for 30 s and incubated at 40 °C for 20 min. Purification was performed by an HILIC method26 with some modifications using MCC SPE as follows: the MCC cartridge was first washed with 3.0 mL of water to prevent contamination by cellulose-derived materials into the eluent and then equilibrated with 5.0 mL binding solution of ACN/H2O/TFA (85:15:0.1, v/v/v). The reaction mixture was diluted in 500 μL binding solution and then applied to the cartridge. The cartridge was washed with 1.0 mL binding solution for five times to remove the excessive derivative reagents and other impurities. Finally, the derivatives were eluted with 1.5 mL of ethanol/H2O (1:1, v/v) and dried by a rotary concentrator.
2.6 Methylamidation of the cellular sialoglycans
Cellular sialoglycans were derivatized with methylamidation after TMPP-Ac labeling following a previously reported method.27 Briefly, dried samples were dissolved in 20 μL DMSO containing 1 M methylamine hydrochloride and 0.5 M N-methylmorpholine, followed by addition of 20 μL PyAOP (50 mM in DMSO) solution. The reaction mixture was incubated at room temperature for 30 min and purified according to the HILIC method as described above.2.7 MALDI-TOF MS analysis
All of the MALDI-TOF-MS were acquired using 5800 MALDI-MS (SCIEX, Concord, Canada) equipped with a 355 nm Nd:YAG laser in the reflector positive mode. DHB and CHCA matrix were prepared in 50% ACN aqueous solution with a final concentration of 10 mg mL−1. To suppress potassium adduct formation in the mass spectra of the permethylated glycans, 10 mM sodium acetate aqueous solution was applied to dissolve the sample, which resulted in yielding almost exclusively [M + Na]+ species in reflectron positive mode. Samples of 0.5 μL mixed with 0.5 μL freshly prepared matrix were directly loaded onto the stainless steel MALDI plate and allowed to dry in a gentle stream of warm air. A total of 1000 laser shots were employed in each sample spot. The MS data processing was further performed by DataExplorer 4.0 (AB SCIEX, Concord, Canada). The glycan annotation was used the consortium for functional glycomics (CFG) as the model system.28 The cartoons of possible structures of glycans were adapted from Glycoworkbench.292.8 MS/MS analysis of glycans using NanoLC-ESI-MS
The derivatized glycans were investigated by a NanoLC Ultra System (Eksigent, U.S.A.) equipped with a trap column (150 μm I.D. × 10 mm long; C18, 5 μm; Proteomics Front, China) and a separation column (75 μm I.D. × 100 mm long; C18, 5 μm; Proteomics Front, China). The mobile phases consisted of 0.1% formic acid in 5% acetonitrile (solvent A) and 0.1% formic acid in 95% acetonitrile (solvent B). The following gradient program was used at a flow rate of 300 nL min−1: t = 0 min, 5% solvent B; t = 5 min, 30% solvent B; t = 15 min, 80% solvent B; t = 20 min, 95% solvent B; t = 21 min, 5% solvent B; t = 25 min, 5% solvent B. MS was operated in positive-ion mode with mass range of 500–2000 m/z. MS/MS was acquired in the information dependent acquisition (IDA) mode for the 20 most abundant precursor ions with mass range of 100–2000 m/z. Data were processed with PeakView 1.2 software (SCIEX, USA).3 Results and discussion
Current protocols for the analysis of cellular N-glycans often require large amount samples (5–20 million cells) and the total sample preparation procedure was time-consuming and tedious. The main objective of this study was to develop a convenient, rapid and sensitive method for the analysis of cellular N-glycans. As shown in Fig. 1, the general routine of our method was using rapid deglycosylation and TMPP-Ac/methylamidation instead of the traditional overnight digestion and permethylation.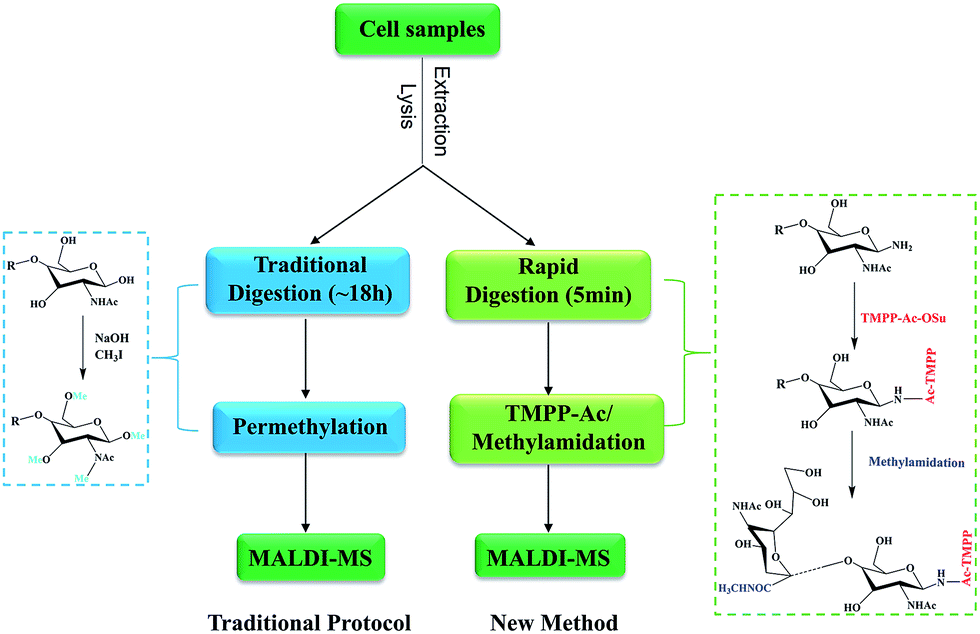 | ||
| Fig. 1 Workflow of the conventional strategy and our method for the analysis of N-glycans from cell lines. The conventional strategy using overnight deglycosylation and permethylation and our method using rapid digestion and TMPP-Ac/Methylamidation. | ||
3.1 Optimization of cellular protein extraction
Requirement of total cellular proteins was essential for the total cellular N-glycan analysis. Therefore, it is critical to effectively extract cellular proteins from complicated cell lysate. Several strategies for the cellular protein extraction have been reported, such as chloroform/methanol/water (CMW), ethanol and acetone.15,30,31 These methods could give different results based on how well they extract proteins. In this study, CMW and ethanol were evaluated. As shown in Fig. S1,† these two protein extraction methods resulted in a very consistent N-glycan profiling. However, use of CMW showed higher signal intensities, which might result from the better protein recovery.19 In addition, less impurity peaks were observed in the low m/z range of mass spectrum by using CMW. As a result, the choice of extraction method was CMW for further experiments.3.2 Choice of matrix
Several matrices have been employed for the analysis of N-glycans by MALDI-MS, such as DHB.32 However, one major drawback in the application of MALDI-MS using DHB as the matrix is the formation of inhomogeneous needle-shaped crystals, which resulted in variability of signal intensities in the same or between different desorption spots across the preparation.23 In addition, the resulting lack of shot-to-shot reproducibility mostly limits the use of MALDI-MS for the relative quantification experiments.As shown in Fig. S2a and b,† two matrices, DHB and CHCA, were used to compare the performance for the analysis of derivatized glycans in details. The S/N and resolution of major glycan peaks were summarized in Table S1.† Compared with DHB as the matrix, most of the peaks obtained two or more fold sensitivity improvement using CHCA for derivatized glycans. These results showed that CHCA obtained superior MS spectrum for TMPP-Ac derivatized glycans, which may result from the addition of a permanent charge.33 In addition, using the CHCA as the matrix could improve the resolution of glycan peaks. For example, the resolution of m/z 3636 was 6536 when DHB was used as the matrix, while the resolution was improved to 11391 when CHCA was used (embedded in Fig. S2a and b†). The photograph of different matrix for dried-droplet preparation was presented in Fig. S2c.† Moreover, the crystallization was homogeneous and glycans were detected throughout the entire matrix-analyte dried spot when CHCA was used. However, the crystallization using DHB was inhomogeneous with many needle-shaped crystals. Using CHCA as the matrix, searching for “hot spots” could be omitted and it might be used for quantification analysis of glycans because of the homogeneous crystals. Taken together, CHCA was the preferred matrix for the derivatized glycans analysis in this study.
3.3 N-Glycan profiling of HeLa cells
Permethylation was a commonly used method for the derivatization of N-glycans, which could enhance the ionization efficiency and improve the detection sensitivity by increasing the hydrophobicity of the glycans.34,35 Therefore, many researchers have used permethylation for the analysis of cellular or membrane N-glycans.15–17 However, it appears less satisfactory to small samples analysis due to oxidative degradation and peeling reaction during the derivatization reaction. In addition, complete permethylation of large glycans is still challenging.36 Moreover, polyhexose contaminations from cytoplasm were commonly observed in the mass spectra, which could decrease the signal intensities of other glycan peaks and increase the difficulties in data interpretation.18As shown in Fig. 2a, more than 60 N-glycan structures were obtained using 5 × 105 HeLa cells with the new developed method. However, less than 30 N-glycans and several polyhexose contaminations were detected using same cells with permethylation (Fig. 2a). More importantly, TMPP-Ac labeling was only occurred to the glycosylamines that produced by rapid deglycosylation, while the polyhexose contaminations could be hided due to the difference of ionization efficiency. Consequently, more concise and clear mass spectra was achieved with the new developed method. Moreover, the N-glycan profiling of HeLa cell using our method was in good agreement with previously reported article.15 The detailed N-glycan profile of HeLa cell lines was presented in Table S2.†
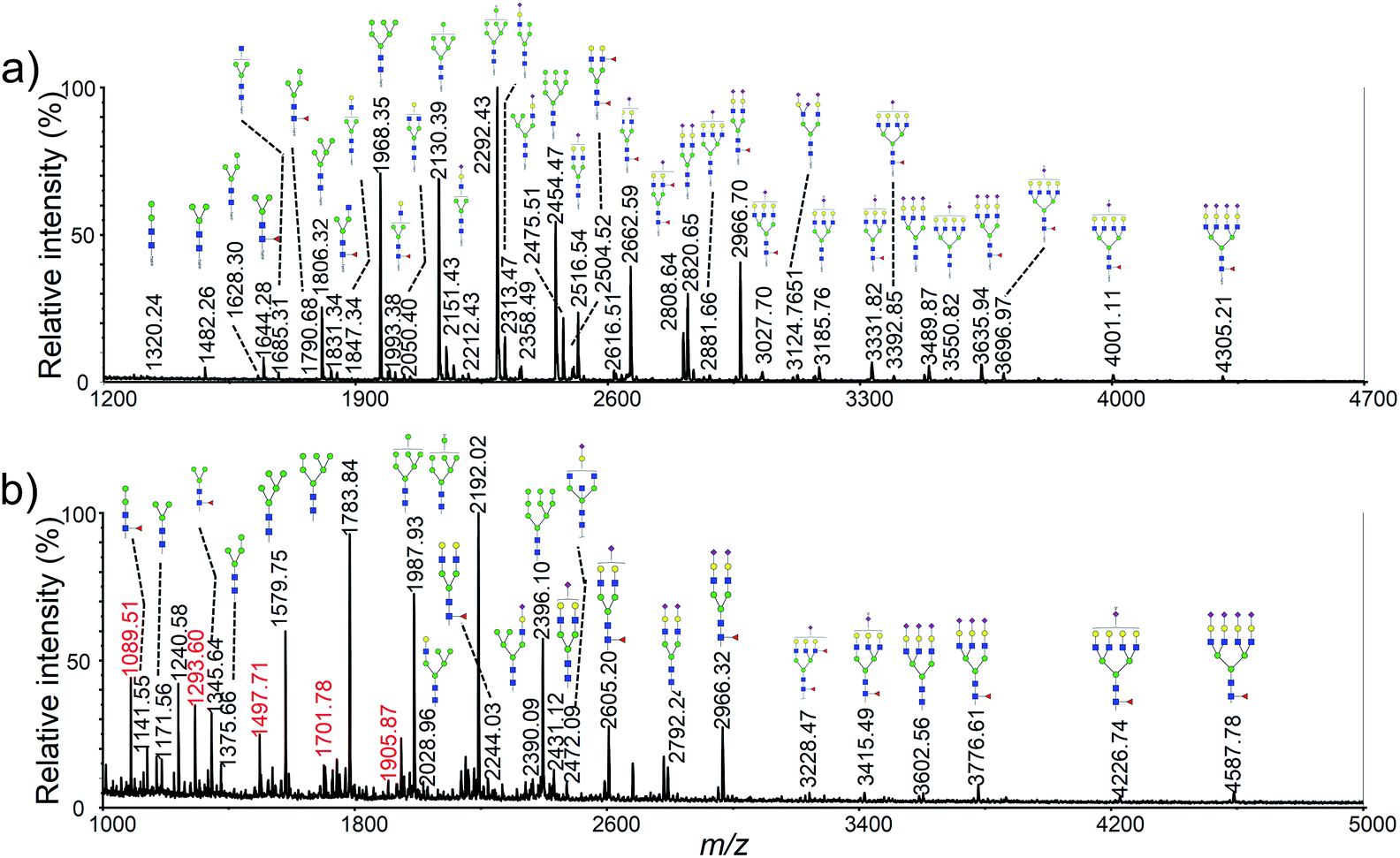 | ||
| Fig. 2 Typical MALDI-TOF mass spectra of HeLa cell N-glycan profiling corresponding to 5 × 105 cells with different sample preparations: (a) optimized method with co-derivatization; (b) traditional method with permethylation. Peaks with m/z values in red color in (b) correspond to polyhexose contaminations. | ||
It should be pointed out that several glycan moieties with a mass shift of 16 Da were also observed, such as 2516.95 vs. 2532.95 and 2821.10 vs. 2837.12. Previous article has reported that human embryonic stem cells could generate nonhuman sialic acid Neu5Gc as they were typically cultured with animal-derived “serum replacement”.37 We supposed that the abnormal glycans might be Neu5Gc containing glycans generated by the cells using the fetal bovine serum in the cell culture medium. NanoLC-ESI-MS/MS was used to confirm the unusual glycan moieties observed in this study. As shown in Fig. 3, several sets of diagnostic ions between the Neu5Ac and the Neu5Gc containing glycans was shown in a typical MS/MS spectra. For example, m/z 287 (Neu5Ac–H2O), 305 (Neu5Ac), 670 (Hex1HexNAc1Neu5Ac1), corresponded to fragment ions from the Neu5Ac containing glycan moiety of Hex5HexNAc4NeuAc2, and m/z 303 (Neu5Gc–H2O), 321 (Neu5Gc), 686 (Hex1HexNAc1Neu5Gc1), corresponded to fragment ions from the Neu5Gc containing glycan moiety of Hex5HexNAc4NeuAc1NeuGc1. To our knowledge, it was the first time that observed the Neu5Gc containing glycans from humanized cells.
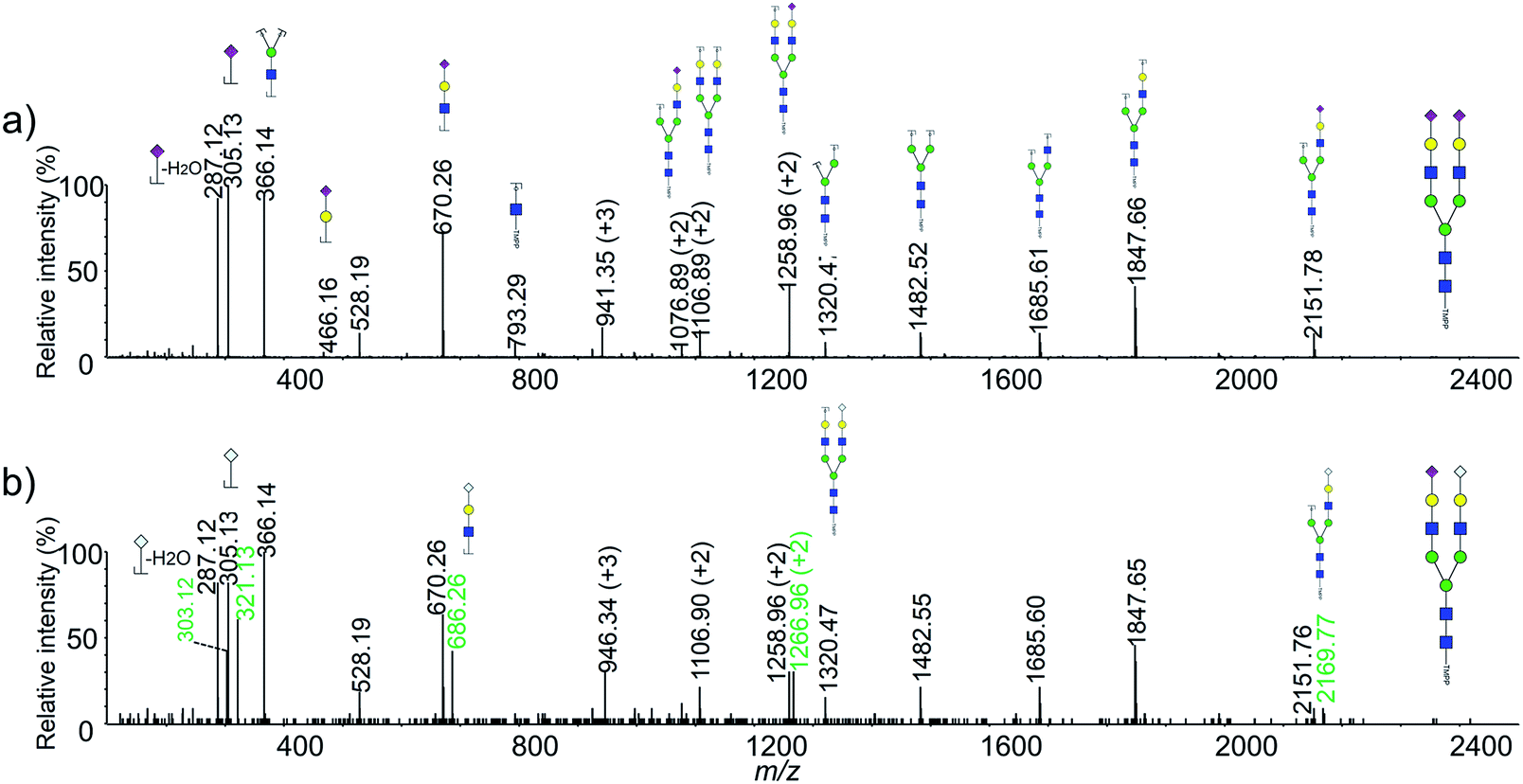 | ||
| Fig. 3 Typical MS/MS spectra of the derivatized N-glycans from HeLa cell line. (a) NanoLC-ESI-MS/MS spectrum of m/z 941.35 [M + 3H]3+ with the composition of Hex5HexNAc4NeuAc2; (b) NanoLC-ESI-MS/MS spectrum of m/z 946.34 [M + 3H]3+ with the composition Hex5HexNAc4NeuAc1NeuGc1. | ||
3.4 Detection limit and reproducibility of the method
To determine detection limit of our method, using HeLa cell as a model, three different original cell numbers, 5 × 105, 1 × 105 and 2 × 104 was used (Fig. 4). As shown in Fig. 4b, more than 30 glycan were observed using only 1 × 105 cells. Clearly, more than 5-fold sensitivity enhancement was obtained using our method when it was compared to permethylation (Fig. 2b vs. 4b). Moreover, nearly all the high-abundant glycans moieties were detected with only original cell number of 2 × 104. It should also be noted that the sample amount loaded onto the steel plate was 1/20 of the original sample amount.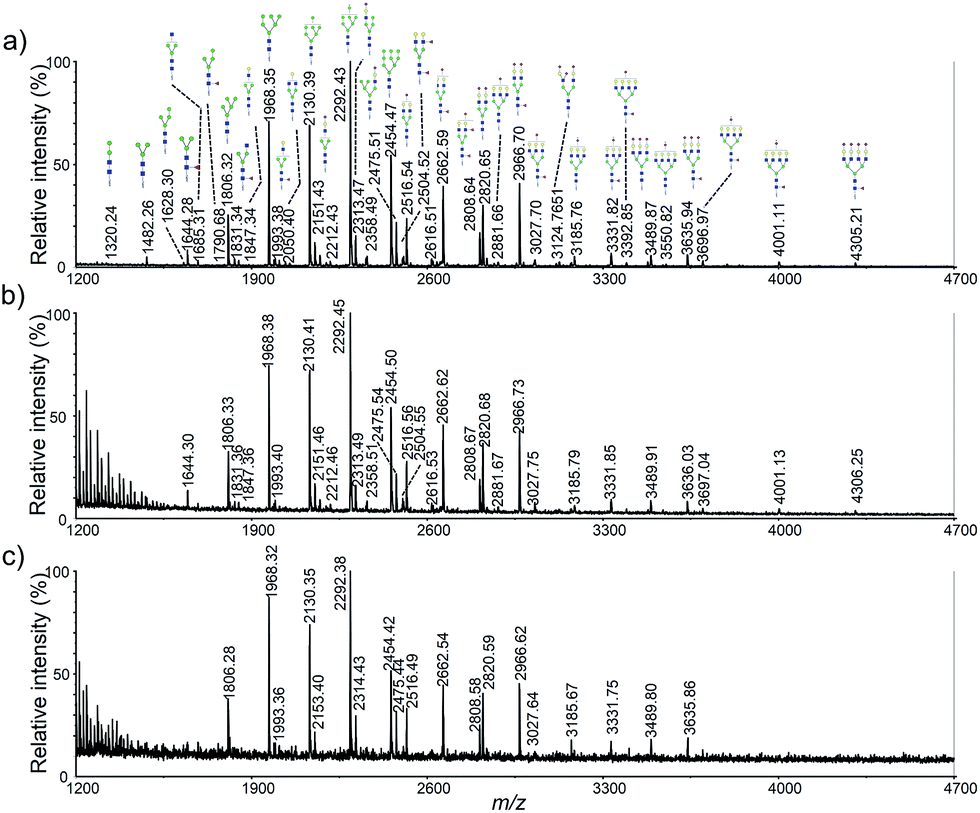 | ||
| Fig. 4 MALDI-TOF mass spectra of HeLa cell N-glycan profiling corresponding to different original cells: (a) 5 × 105; (b) 1 × 105; (c) 2 × 104. | ||
In order to assess the reproducibility of the optimized method, three independent experiences using HeLa cells were carried out (Fig. S3†). Glycan signals were integrated, normalized to the sum of intensities, and the relative signals and standard deviations were calculated. Analysis of the 27 most abundant glycan moieties found in HeLa cells revealed high repeatability within one batch, as well as between three batches prepared and measured on different days (with averaging coefficient of variation (CV) less than 13.1%). Average relative intensity values for the highest peak (H8N2) were around 20.5% (SD ± 0.38%) across all measurements, with CV averaging to 3.03%. Analysis of all assigned glycan moieties shows low variability even for values below 0.1% of the cumulative distribution.
3.5 Application of the method to the comparison of L-02 and HepG2 cells
To further demonstrate the application of our method, a relative quantitative comparison of a human normal liver cell line (L-02) and a human hepatocyte carcinoma cell line (HepG2) was performed. Three biological replicates of these two cell lines were carried out. Representative cellular N-glycan profiling of L-02 and HepG2 is presented in Fig. 5a and b. Most of the observed glycans were in consistent with known mammalian glycans structures, including high mannose, complex, and hybrid type structures.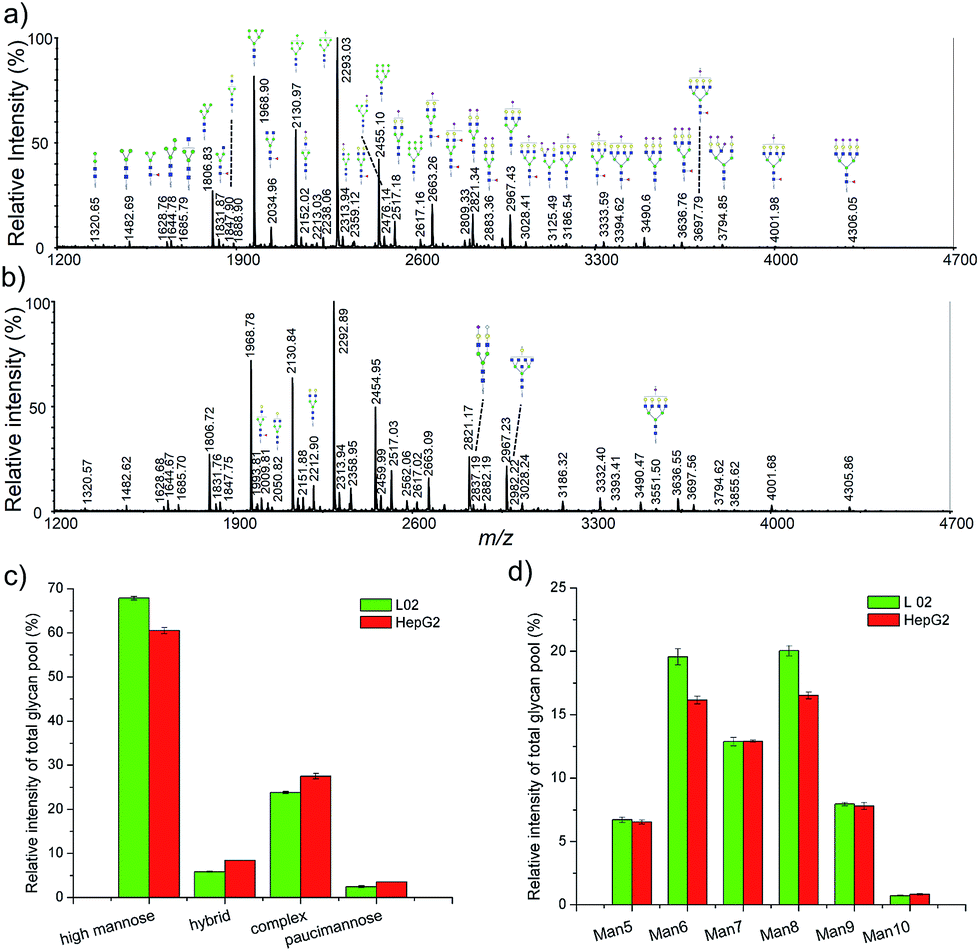 | ||
| Fig. 5 MALDI-TOF mass spectra of L-02 and HepG2 cell N-glycan profiling and the differential of cellular N-glycans across the two cell lines. (a) A typical mass spectrum of N-glycans from L-02 cells; (b) a typical mass spectrum of N-glycans from HepG2 cells; (c) distribution of four major types, i.e., high mannose, hybrid, complex and paucimannose; (d) distribution of the different types of high-mannose (Man5–10) structures. | ||
In order to identify major differences in the glycan profiles between these two cell lines, classification of the detected N-glycans into the main structural N-glycan types was applied. The N-glycan structures were categorized into four major types, high mannose, hybrid, complex, and paucimannose (Fig. 5c). High mannose structures (Man5–10GlcNAc2) was the most abundant type of glycans in both L-02 and HepG2 cell lines, showing relative abundance of 67.86 ± 0.42% and 60.52 ± 0.67%, respectively. Complex-type N-glycans comprised 23.78 ± 0.24% of L-02 signals and 27.52 ± 0.61% of HepG2 signals. Hybrid and paucimannose-typed N-glycans were less abundant in all these two cell lines. Fig. 5d shows a detailed distribution of different types of high mannose structures. The relative abundance of high mannose structures between L-02 and HepG2 were nearly the same except Man6GlcNAc2 and Man8GlcNAc2. These results showed that the N-glycan profiles of L-02 and HepG2 were characteristic differences.
4 Conclusion
In summary, an optimized co-derivatization method was presented for rapid analysis of cellular N-glycan profiling with limited cell amounts. The general routine of our method was using rapid deglycosylation and TMPP-Ac/methylamidation instead of the traditional overnight digestion and permethylation. Consequently, the required amount for cellular N-glycans analysis was reduced down to 105 (using HeLa cells as a model) and the total sample preparation time was decreased from several days to several hours. In addition, compared to permethylation derivatization, more than 5-fold sensitivity improvement was achieved and more concise and clear mass spectra was obtained with our method. Moreover, two heterogeneous live cell lines, human hepatocyte carcinoma cells (HepG2) and human normal liver cells (L-02), were also applied to validate the new developed method. Results shown that it was able to reveal the difference in cellular N-glycan profiling of these two cell lines. Finally, due to rapid, high-sensitive and robust of the method, it could be used as a powerful analytical tool for the reliable detection and quantification of N-glycans from limited cellular samples, and could be enable researcher to discover marker oligosaccharides characteristically observed in various disease such as cancer and congenital disorder.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (20905027).References
- R. Apweiler, H. Hermjakob and N. Sharon, Biochim. Biophys. Acta, 1999, 1473, 4–8 CrossRef CAS.
- L. Wells, K. Vosseller and G. W. Hart, Science, 2001, 291, 2376–2378 CrossRef CAS PubMed.
- R. G. Spiro, Glycobiology, 2002, 12, 43r–56r CrossRef CAS PubMed.
- G. W. Hart, M. P. Housley and C. Slawson, Nature, 2007, 446, 1017–1022 CrossRef CAS PubMed.
- R. S. Haltiwanger and J. B. Lowe, Annu. Rev. Biochem., 2004, 73, 491–537 CrossRef CAS PubMed.
- K. Aoki, M. Perlman, J. M. Lim, R. Cantu, L. Wells and M. Tiemeyer, J. Biol. Chem., 2007, 282, 9127–9142 CrossRef CAS PubMed.
- P. M. Rudd, T. Elliott, P. Cresswell, I. A. Wilson and R. A. Dwek, Science, 2001, 291, 2370–2376 CrossRef CAS PubMed.
- J. D. Marth and P. K. Grewal, Nat. Rev. Immunol., 2008, 8, 874–887 CrossRef CAS PubMed.
- Y. van Kooyk and G. A. Rabinovich, Nat. Immunol., 2008, 9, 593–601 CrossRef CAS PubMed.
- N. Haines and K. D. Irvine, Nat. Rev. Mol. Cell Biol., 2003, 4, 786–797 CrossRef CAS PubMed.
- C. J. Day, E. N. Tran, E. A. Semchenko, G. Tram, L. E. Hartley-Tassell, P. S. K. Ng, R. M. King, R. Ulanovsky, S. McAtamney, M. A. Apicella, J. Tiralongo, R. Morona, V. Korolik and M. P. Jennings, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E7266–E7275 CrossRef CAS PubMed.
- D. Park, N. Arabyan, C. C. Williams, T. Song, A. Mitra, B. C. Weimer, E. Maverakis and C. B. Lebrilla, Mol. Cell. Proteomics, 2016, 15, 3653–3664 CAS.
- P. C. Chen, P. K. Chuang, C. H. Chen, Y. T. Chan, J. R. Chen, S. W. Lin, C. Ma, T. L. Hsu and C. H. Wong, ACS Chem. Biol., 2014, 9, 1437–1443 CrossRef CAS PubMed.
- L. J. Stroh and T. Stehle, Annu. Rev. Virol., 2014, 1, 285–306 CrossRef PubMed.
- N. Fujitani, J. Furukawa, K. Araki, T. Fujioka, Y. Takegawa, J. Piao, T. Nishioka, T. Tamura, T. Nikaido, M. Ito, Y. Nakamura and Y. Shinohara, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2105–2110 CrossRef CAS PubMed.
- S. Abdul Rahman, E. Bergstrom, C. J. Watson, K. M. Wilson, D. A. Ashford, J. R. Thomas, D. Ungar and J. E. Thomas-Oates, J. Proteome Res., 2014, 13, 1167–1176 CrossRef CAS PubMed.
- H. Hamouda, M. Kaup, M. Ullah, M. Berger, V. Sandig, R. Tauber and V. Blanchard, J. Proteome Res., 2014, 13, 6144–6151 CrossRef CAS PubMed.
- S. H. Walker, B. N. Papas, D. L. Comins and D. C. Muddiman, Anal. Chem., 2010, 82, 6636–6642 CrossRef CAS PubMed.
- E. Lattova, J. Bryant, J. Skrickova, Z. Zdrahal and M. Popovic, J. Proteome Res., 2016, 15, 2777–2786 CrossRef CAS PubMed.
- S. Holst, A. J. Deuss, G. W. van Pelt, S. J. van Vliet, J. J. Garcia-Vallejo, C. A. Koeleman, A. M. Deelder, W. E. Mesker, R. A. Tollenaar, Y. Rombouts and M. Wuhrer, Mol. Cell. Proteomics, 2016, 15, 124–140 CAS.
- H. Li, W. Gao, X. Feng, B. F. Liu and X. Liu, Anal. Chim. Acta, 2016, 924, 77–85 CrossRef CAS PubMed.
- D. J. Harvey, Mass Spectrom. Rev., 2015, 34, 268–422 CrossRef CAS PubMed.
- M. Mank, B. Stahl and G. Boehm, Anal. Chem., 2004, 76, 2938–2950 CrossRef CAS PubMed.
- W. Gao, H. Li, Y. Liu, Y. Liu, X. Feng, B. F. Liu and X. Liu, Talanta, 2016, 161, 554–559 CrossRef CAS PubMed.
- Z. H. Huang, T. Shen, J. Wu, D. A. Gage and J. T. Watson, Anal. Biochem., 1999, 268, 305–317 CrossRef CAS PubMed.
- Q. W. Zhang, H. H. Li, X. J. Feng, B. F. Liu and X. Liu, PLoS One, 2014, 9, e94232 Search PubMed.
- X. Liu, H. Qiu, R. K. Lee, W. Chen and J. Li, Anal. Chem., 2010, 82, 8300–8306 CrossRef CAS PubMed.
- R. Raman, M. Venkataraman, S. Ramakrishnan, W. Lang, S. Raguram and R. Sasisekharan, Glycobiology, 2006, 16, 82r–90r CrossRef CAS PubMed.
- A. Ceroni, K. Maass, H. Geyer, R. Geyer, A. Dell and S. M. Haslam, J. Proteome Res., 2008, 7, 1650–1659 CrossRef CAS PubMed.
- D. Wang, X. Zhou, L. Wang, S. H. Wang and X. L. Sun, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 944, 75–81 CrossRef CAS PubMed.
- H. M. Park, M. P. Hwang, Y. W. Kim, K. J. Kim, J. M. Jin, Y. H. Kim, Y. H. Yang, K. H. Lee and Y. G. Kim, Carbohydr. Res., 2015, 413, 5–11 CrossRef CAS PubMed.
- Y. Fukuyama, Mass Spectrom., 2015, 4, A0037 CrossRef PubMed.
- X. Liu, G. Zhang, K. Chan and J. Li, Chem. Commun., 2010, 46, 7424–7426 RSC.
- P. Kang, Y. Mechref, I. Klouckova and M. V. Novotny, Rapid Commun. Mass Spectrom., 2005, 19, 3421–3428 CrossRef CAS PubMed.
- P. Kang, Y. Mechref and M. V. Novotny, Rapid Commun. Mass Spectrom., 2008, 22, 721–734 CrossRef CAS PubMed.
- W. R. Alley Jr, M. Madera, Y. Mechref and M. V. Novotny, Anal. Chem., 2010, 82, 5095–5106 CrossRef PubMed.
- M. J. Martin, A. Muotri, F. Gage and A. Varki, Nat. Med., 2005, 11, 228–232 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06071h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |