
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient production of (R)-3-TBDMSO glutaric acid methyl monoester by manipulating the substrate pocket of Pseudozyma antarctica lipase B†
Jing Wu‡
 ab,
Hongjiang Wang‡bc,
Bin Yangbc,
Wei Songbc,
Chenchen Liangab and
Liming Liu*bc
ab,
Hongjiang Wang‡bc,
Bin Yangbc,
Wei Songbc,
Chenchen Liangab and
Liming Liu*bc
aSchool of Pharmaceutical Sciences, Jiangnan University, 1800 Lihu Road, Wuxi, Jiangsu 214122, China
bThe Key Laboratory of Industrial Biotechnology, Ministry of Education, Jiangnan University, 1800 Lihu Road, Wuxi, Jiangsu 214122, China. E-mail: mingll@jiangnan.edu.cn; Fax: +86-510-85197875; Tel: +86-510-85197875
cState Key Laboratory of Food Science and Technology, Jiangnan University, 1800 Lihu Road, Wuxi, Jiangsu 214122, China
First published on 3rd August 2017
Abstract
Optically pure (R)-3-substituted glutaric acid methyl monoesters are multifunctional chiral building blocks used in the pharmaceutical industry. In the current study, a combined in silico/mutagenesis approach was used to improve the performance of Pseudozyma antarctica lipase B (CALB) as a biocatalyst in the asymmetric synthesis of (R)-3-t-butyl-dimethyl-silyloxy (TBDMSO) glutaric acid methyl monoester (R-J6). Candidate amino acids that likely affected the enantioselectivity of CALB were identified by substrate structure analysis. Mutant variants were screened in silico; CALB enantioselectivity was reversed and enhanced based on molecular docking analyses, followed by reshaping of the substrate pocket. EF5 CALB variant, generated by semi-rational design and selected by high-throughput screening, exhibited high R-selectivity with an eeR (enantiomeric excess) value of 85%, while the wild-type (WT) CALB showed S-selectivity; the kcat/KM of EF5 towards R-J6 increased 14-fold, from 0.59 to 8.29 mM−1 s−1. Compared with WT CALB, the affinity of EF5 for 3-TBDMSO glutaric anhydride increased 2.31-fold. By optimizing the fermentation conditions for the yeast host for protein production and enzyme immobilization conditions, the hydrolytic activity of EF5 was increased to 2401.5 ± 5.3 U mL−1 and 2706.7 ± 11.4 U g−1, respectively. The yield of R-J6 generated by the EF5 variant in non-aqueous media increased to 55 ± 1.6 g L−1, with an eeR value of 98.5%. Semi-rational design was hence successfully employed to generate gram quantities of (R)-3-substituted glutaric acid monoesters with enormous potential and high ee.
Introduction
Optically pure (R)-3-substituted glutaric acid monoesters ((R)-J6) are attractive building blocks for the synthesis of a number of pharmaceutically important compounds, including pitavastatin, fluvastatin, atorvastatin, and rosuvastatin. Among these, rosuvastatin inhibits hydroxymethylglutaryl-CoA reductase and has few side-effects.1 (R)-J6 can be obtained by four approaches: chemical synthesis,2 hydrolysis of dialkyl-3-substituted glutaric acids using hydrolases,3 kinetic resolution of racemates,4,5 and desymmetrization of prochiral compounds.6 To date, industrial (R)-J6 is mainly prepared by chemical synthesis;7 (S)-1-phenethylamin is used to catalyze the asymmetric reduction of 3-substituted glutaric acid monoesters, with a space-time yield of 13.6 g L−1 h−1, low yield (54.9%), and a low enantiomeric excess (ee; 80%) at −78 °C.Fortunately, biocatalysis provides an attractive alternative to chemical synthesis and is environmentally friendly. R,S-J6 may be prepared using pig liver esterase and Novozym 435.8,9 The space-time yield of such a reaction is high (≥4.5 g L−1 h−1) but the selectivity for the (R)-isomer is low (Table 1). α-Chymotrypsin is also employed in (R)-J6 preparation, hydrolyzing the dialkyl-3-substituted glutaric acid with a high eeR of 97%.10 However, this approach comprises six steps, starting with diethyl-3-t-butyl-dimethyl-silyloxy (TBDMSO) glutaric acid (conversion, 65.4%; isolated yield, 53.2%), and the 3-substituent group of the substrate appreciably affects the catalytic efficiency and enantioselectivity of the enzyme.10 Thus, α-chymotrypsin performs poorly with TBDMSO as the 3-substituent group of the substrate. Furthermore, the process entails high production costs, and two high-selectivity biocatalysts (α-chymotrypsin and cephalosporin C acetyl esterase) are required.10 Moreover, α-chymotrypsin exhibits low activity and selectivity during R-J6 preparation in an organic solvent, as described by us in a previous study.9
| Catalyst | Substrate load (g L−1) | Time (h) | Catalyst load (g L−1) | Yielda (%) | Temp. (°C)/strategy | ee (%)/config. | STYb (g L−1 h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Note: This value refers to the isolation yield.b STY, space-time yield (g L−1 h−1).c NG, not given.d CS, chemical synthesis.e PS, previous study.f H, hydrolysis.g AS, asymmetric synthesis. | ||||||||
| (S)-1-Phenethylamin | 45 | 2 | 44 | 54.9 | −78 (CS)d | 80 (R) | 13.60 | 2, 7 and 28 |
| Novozym 435 | 45 | 64 | 20 | 92.6 | 30 (H)f | 95.6 (R) | 0.654 | 29 |
| PLE | 50 | 6 | 7.5 | 76 | NGc (H)f | 22 (S) | 6.330 | 3 and 30 |
| α-Chymotrypsin | 50 | 48 | 20 | 56 | NGc (H)f | 97 (R) | 0.677 | 3 and 10 |
| Novozym 435 | 52 | 24 | 48 | 93 | 35 (AS)g | 98 (S) | 2.030 | 29 |
| Novozym 435 | 200 | 24 | 50 | 52.6 | 30 (AS)g | <22 | 4.800 | 9 (PS)e |
| EF5 mutant | 60 | 48 | 80 | 92.1 | 5 (AS)g | 98.5 (R) | 1.152 | This work |
With a theoretical yield close to 100%, enzymatic desymmetrization of prochiral compounds is currently recognized as an efficient route for the preparation of optically pure compounds. Therein, the hydrophobic substrates are dissolved in organic solvents and the desired products are subsequently easily isolated, purified, and dried.11,12 Therefore, the research focus has shifted from chemical synthesis to the desymmetrization of 3-substituted glutaric acid anhydrides by biocatalysts.13 Literature search, however, revealed a lack of one type of enzyme that would meet the industry requirements; the reactions catalyzed by the available biocatalysts are characterized by low titers, low yields, and low ee values.14 Pseudozyma antarctica lipase B (CALB) was previously used for the development of a biocatalytic process for R-J6 production; high titer and yield of R-J6 were indeed achieved (117.19 g L−1 and 58.6%, respectively), but the ee value was very low (<22%) because of the enzyme's preference for the synthesis of (S)-isomers.9 The aim of this study was to generate a high-performance CALB-based biocatalyst using a semi-rational protein design to facilitate R-J6 production.
Materials and methods
Chemicals
3-TBDMSO glutaric anhydride was purchased from Henan Yuchen Fine Co., Ltd. (Henan, China). Hexane and iso-propanol (high-performance liquid chromatography grade) were purchased from Fisher Chemical (Fairlawn, NJ, USA); other chemicals and solvents (analytical grade) were obtained from local suppliers. An R-J6 standard and racemic 3-TBDMSO glutaric acid methyl monoester were gifts from Chanyoo Pharmatech Co., Ltd. (Nantong, China). All other chemicals were of analytical grade and obtained from companies in China. A Daicel Chiralpak AD-H column (4.6 × 250 mm) was purchased from Daicel Chiral Technologies Co., Ltd. (Shanghai, China).Bacterial strains, plasmids, and enzymes
The calB gene (GeneBank accession no. Z30645.1) was amplified by polymerase chain reaction (PCR) from the chromosome of P. antarctica JCM3941 purchased from the Japan Collection of Microorganisms (JCM; RIKEN, Saitama, Japan). A fragment of the gene (ca. 1029 bp) was amplified using the following primers: 5′-CGCTCGAGAAAATGAGACTACCTTCCGGTTCGGACC-3′ (forward) and 5′-TGCTCTAGATTAGGGGGTGACGATGCCG-3′ (reverse; the underlined regions are XhoI and XbaI restriction sites, respectively). A recombinant plasmid vector pMD19-T-CALB was constructed and used as a parental vector for the generation of enzyme variants.15 Escherichia coli JM109 [recA supE hsR Δ(lac-pro)] was used as the host for plasmid amplification. Pichia pastoris GS115 strain (Invitrogen, Carlsbad, CA, USA) served as the host for CALB production. The expression vector pGAPZαA was purchased from Novagen (Madison, WI, USA). LATaq DNA polymerase, PrimeSTAR DNA polymerase, and restriction enzymes were from TakaRa (Dalian, China). AvrII (Thermo Fisher Scientific, Waltham, MA, USA) was used to linearize the recombinant plasmids. E. coli was incubated in lysogeny broth (LB) medium at 37 °C, 200 rpm, for 24 h. P. pastoris GS115 and P. antarctica JCM3941 were inoculated into yeast extract peptone dextrose (YPD) medium (10 g L−1 of yeast extract, 20 g L−1 of peptone, and 20 g L−1 of glucose), and grown at 30 °C on a rotary shaker (200 rpm) for 2 d.The design and construction of CALB variants
CALB amino acid sequences were downloaded from the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/genbank/), for a multiple sequence alignment using Geneious software (http://www.geneious.com, Biomatters Ltd., Auckland, New Zealand). The predicted structures of CALB were downloaded from the Protein Data Bank (PDB ID, 1TCA; (http://www.rcsb.org-/pdb/explore/explore.do?structureId=1TCA) and the structures of the variants were obtained by homology modeling.16 The three-dimensional structures of the enantiomer products [(R)-J6 and (3S)-TBDMSO glutaric acid methyl monoester (S-J6)] were generated using the ChemOffice Ultra 11.0 program (Cambridge Soft Corporation, Cambridge, MA, USA). All generated structures were then minimized using the CHARMM force field.17 Only ligand molecules were considered to be flexible during the docking simulation, and only the free energy of their best orientation was used to compute the docking free energy. The cavity volume of the redesigned mutants was calculated by the Molegro Virtual Docker software.18For site-directed mutagenesis, candidate amino acids, excluding the catalytic triad (Ser105, His224, and Asp187), were changed to ones that occurred with high frequency in the homologous sequence (ESI Table S1†). Introduction of specific amino acid substitutions into CALB was performed using primer pairs listed in ESI Table S2.†19,20 PCR products were digested by DpnI (Thermo Fisher Scientific) and used to transform E. coli JM109; the transformants were selected on LB agar plates containing 100 μg mL−1 of ampicillin. The introduced mutations were confirmed by DNA sequencing (Sangon Biotech, Shanghai, China), and the mutagenized sequences were inserted into the pGAPZαA vector. The pGAPZαA-CALB-variant plasmids were linearized by AvrII and introduced into the host, P. pastoris GS115, by electroporation.21
CALB variant library expression in P. pastoris and biocatalyst preparation
Degenerate NNN, NDK, and NDT codons were used to create CALB variant libraries based on the results of docking analysis; the primers used for the semi-saturation/saturation mutagenesis of A141S-A283V are shown in ESI Table S3.† The prepared plasmid libraries were introduced into P. pastoris GS115 by electroporation,22 the cells were then incubated at 30 °C in YPD medium (1 mL) for 2 h, which was followed by plating on YPD agar plates containing zeocin (100 μg mL−1), the plates were then incubated at 30 °C for 3 d. Single colonies were picked, inoculated into conical, deep 96-well plates, and incubated for 3 d, with shaking at 200 rpm, at 29 °C. Each well contained YPD (800 μL) supplemented with zeocin (100 μg mL−1). Clones were verified by PCR using a pair of validation primers (pGAP-forward, 5′-TCCCTATTTCAATCAATTGAA-3′; and 3′-AOX1-Rev, 5′-GCAAATGGCATTCTGACATCC-3′). After gene expression, the yeast cells were pelleted by centrifugation (2500 × g, 10 min). The supernatant was harvested by aspiration and used directly for optical screening. Master plates were stored at −70 °C prior to further analysis or cultivation. The best-performing CALB variants were purified on a nickel-affinity column (Ni Sepharose 6 Fast Flow, Amersham Biosciences, Freiburg, Germany), and prepared for kinetic analysis.20 The enzyme solution was immobilized on D101 macroporous resin (Shanghai Hualing Resin Factory, Shanghai, China) according to the manufacturer's guidelines and the pellets were freeze-dried.Hydrolytic activity determinations and enantioselectivity screening
The hydrolytic activity assay was performed by monitoring the amount of released p-nitrophenol during the hydrolysis of 10 mM p-nitrophenyl butyrate (p-NPB) in 50 mM Tris–HCl buffer (pH 8.0) at 410 nm and 30 °C.22 One unit of hydrolytic activity (1 U) was defined as the amount of enzyme that generated 1 μmol of p-nitrophenol per min. The protein concentration was determined using Bradford's method, with bovine serum albumin as the standard.23The screening buffer [120 μL; 100 mM potassium phosphate, 10% (v/v) methanol, and 4% (v/v) Triton X-100, pH 7.4] and (R or S)-methyl mandelate (10 μL; 100 mM in methanol) were premixed as screening buffer I. The yeast library supernatant (20 μL) and screening buffer I (150 μL) were dispensed into a microtiter plate, and incubated at 30 °C for 15 min, following which the absorbance was measured at 655 nm. Individual enantiomers of the substrate were hydrolyzed in parallel reactions.
Characterization of variant function and analytical methods
Immobilized CALB variants were characterized using a model esterification reaction for (R)-J6 synthesis; the reaction was conducted in 10 mL capped flasks containing methyl tert-butyl ether and equal concentrations of substrates. The mixtures were incubated in batches for 36 h on an orbital shaker (200 rpm) at 5 °C, and the system water activity was adjusted by using appropriate mesh sieves.6,24 The products were analyzed using a high-performance liquid chromatograph (Waters E2695, Waters Corporation, Milford, MA, USA) equipped with an AD-H column and an ultraviolet detector (Waters Corporation). The mobile phase consisted of 94% hexane and 6% iso-propanol with 0.01% (v/v) acetic acid, filtered through a 0.22 μm membrane before use. Samples (10 μL) were injected into the chromatograph (25 °C detection temperature and 1 mL min−1 flow rate); the detection took 12 min. The retention times of R-J6 and S-J6 were 6.1 and 6.6 min, respectively; racemic 3-TBDMSO glutaric acid methyl monoester (racemic J6) served as an internal standard. The eeR and eeS values were defined as follows: eeR = (R − S)/(R + S) × 100% and eeS = (S − R)/(R + S) × 100%, wherein R and S represent the concentrations of (R)-J6 and (S)-J6, respectively.Results and discussion
Potential CALB derivatives with improved enantioselectivity
To select the key amino acid residues that pronouncedly affect CALB enantioselectivity, the docking energies of different 3-hydroxy protecting groups on wild-type (WT) CALB were first compared in Table 2. The highest and lowest docking energy values were −124.33 kcal mol−1 (TBDPSO–) and −72.92 kcal mol−1 (OH–), respectively. Specific amino acid residues were identified by analyzing the interaction (H-bonds and salt bridges) of these structures [(R)-TBDPSO-J6, (R)-OH-J6, and R-J6] with WT CALB. Thr42, Ile189, Asp134, Thr138, Gln157, and Gln106 were selected as candidate residues that appeared twice and interacted with WT CALB via an H-bond or a salt bridge. Similarly, 3-TBDMSO glutaric acid monoalkyl esters with different alcohol moieties as ligands were docked into WT CALB (Table 3); amino acid residues Ala141, Leu278, Trp104, Val190, Ser47, Ala282, and Ile285 were selected. Collectively, 13 amino acid residues were selected as candidate residues to be substituted.| Protecting group | R-Isomer structure | Docking energy (kcal mol−1) | Related amino acidsa |
|---|---|---|---|
| a Note: Related amino acids are the ones interacting with the R-isomer structure via H bond or salt bridge, excluding the catalytic triad (Ser105, His224, and Asp187). | |||
| OH– | 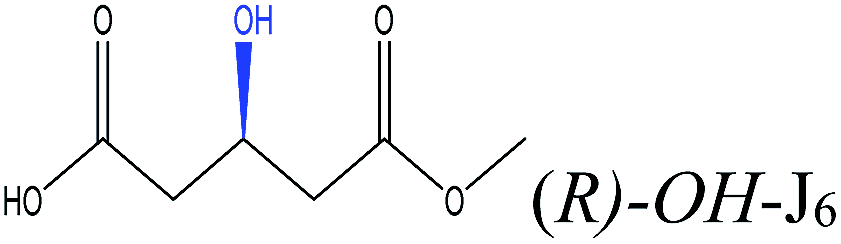 |
−72.92 | Gln106 Gln157 Thr138 Asp134 Thr42 |
| Acetyl– | 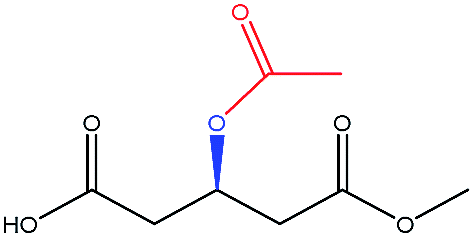 |
−76.34 | — |
| TBDMSO– | 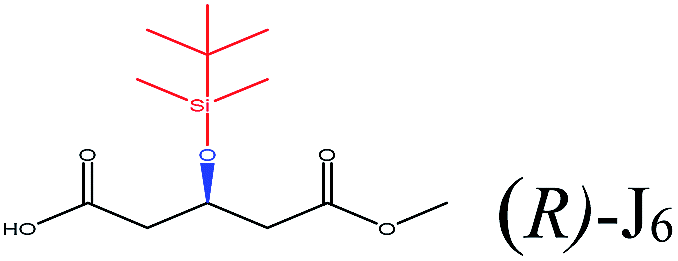 |
−96.99 | Asp134 Gln106 Gln157 Val154 Ile189 |
| Benzoyl– | 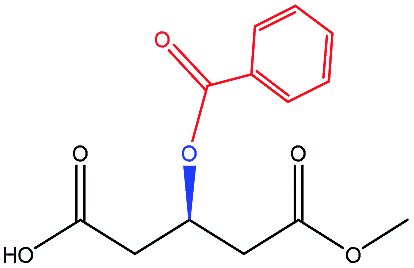 |
−86.48 | — |
| TIPSO– | 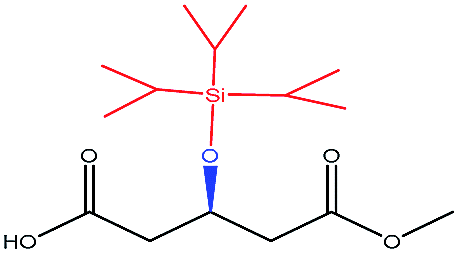 |
−104.20 | — |
| TBDPSO– | 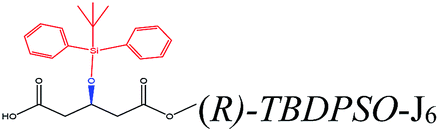 |
−124.33 | Ile285 Thr42 Trp104 Ile189 Thr138 Leu140 |
| TMSO– | 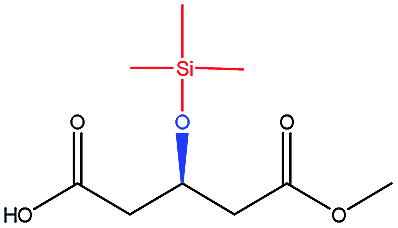 |
−86.98 | — |
| Alcohol moiety | Compound structure | Docking energy (kcal mol−1) | Related amino acidsa |
|---|---|---|---|
| a Note: Related amino acids are the ones interacting with the R-isomer structure via H bond or salt bridge, excluding the catalytic triad (Ser105, His224, and Asp187). | |||
| EtOH |  |
−89.34 | — |
| Methanol | 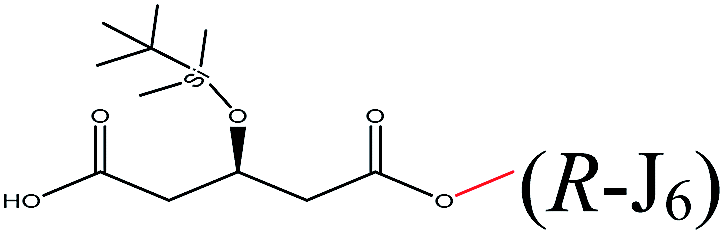 |
−96.19 | Asp134 Gln106 Gln157 Val154 Ile189 Val190 Lue278 Thr40 |
| n-Propanol | 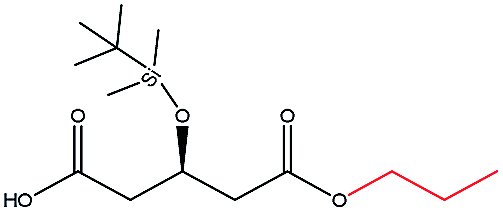 |
−101.67 | — |
| i-Propanol | 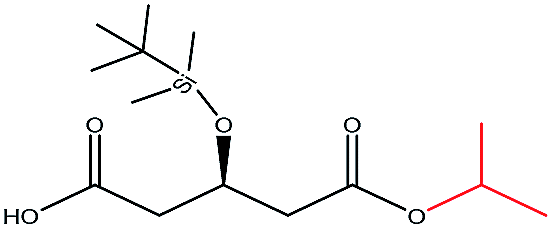 |
−90.25 | — |
| n-Butanol | 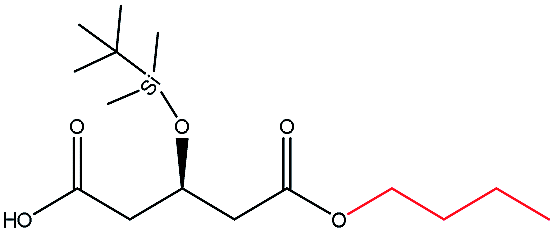 |
−104.40 | Gln106 Thr40 Gln157 Ala282 Ile189 Val190 Leu278 Ser47 |
| t-Butanol | 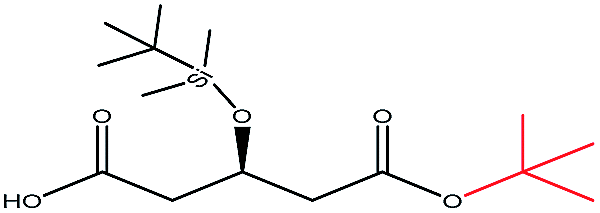 |
−90.44 | — |
| i-Butanol | 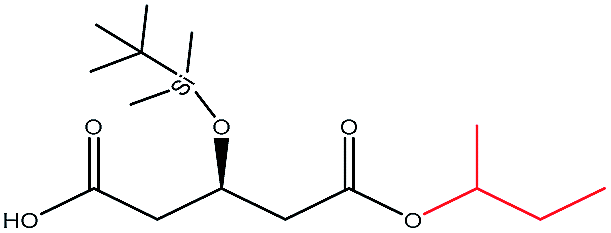 |
−86.97 | — |
| Phenylmethanol | 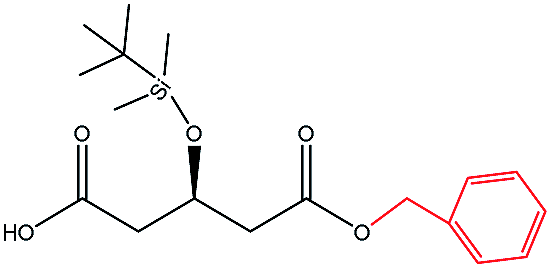 |
−115.07 | Ala141 Ile285 Ile189 Trp104 Leu140 Gln157 Ala282 Asp134 Ser47 |
| 2-Phenylethanol | 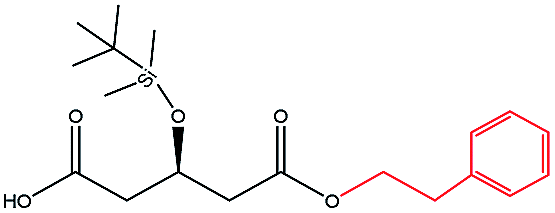 |
−86.71 | — |
| 1-Phenylethanol | 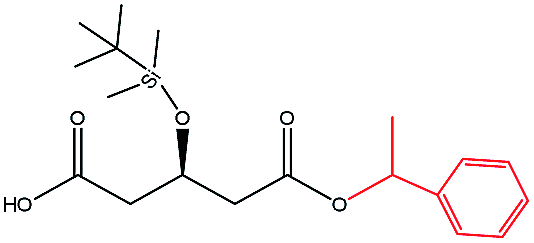 |
−83.25 | Ala141 Ile285 Ile189 Thr40 Val154 |
Enhancing the enantioselectivity of CALB by manipulating the substrate pocket
The enantioselectivity of CALB is affected by the affinity, and the substrate pocket.25,26 Hence, to increase the affinity of CALB for the R-isomer, the enantiomer products R-J6 and S-J6 were docked in silico into thirteen potential enzyme variants (the acceptors); the respective docking energies of (R)-J6 and (S)-J6 are listed in ESI Table S4.† The ratios of the docking energies of (R)-J6 to (S)-J6 (ER/ES) were then calculated and used as an index to compare the potential variants in silico: the higher of ER/ES value, the higher the R-selectivity. Consequently, mutants S47N (ER/ES = 1.138) and A141S (ER/ES = 1.119), with the highest ER/ES values, were identified. Of the two, the R-J6 docking energy was lower for A141S (−92.01 kcal mol−1), i.e., the mutant exhibited a higher affinity for R-J6, and was chosen for further analysis. The titer and ee values of R-J6 were 4.4 ± 0.3 g L−1 and −63%, respectively, for WT CALB; and 10.1 ± 0.6 g L−1 and 17.6%, respectively, for the A141S mutant.To manipulate the substrate pocket by altering the its cavity volume, the amino acid residue A282 and the adjacent amino acids, A281 and A283, which limit the size of the substrate pocket,27 were substituted. The cavity volumes for the variants were calculated using the Molegro Virtual Docker software18 (Table 4); when A281 was changed to A281E, A281V, or A281G, the ER/ES decreased to 1.051, 0.976, and 0.953, respectively; the cavity volume decreased to 57.344, 55.088, and 68.608 Å3, respectively; and the M value [M = (ER/ES) × cavity per volume] was 0.0183, 0.0177, and 0.0139 Å−3, respectively. Similarly, when A282 was changed to A282G, A282L, or A282N, the M value was 0.0185, 0.0080, and 0.0140 Å−3, respectively. When A283 was changed to A283V, A283L, or A283S, the M value was 0.0205, 0.0171, and 0.0155 Å−3, respectively.
| Variant | ER/ESa | Cavity volume (Å3) | Mc (Å−3) |
|---|---|---|---|
| a Note: ER/ES represents the ratio of the docking energy of the R-isomer to the S-isomer.b WT, wild-type CALB.c M = (ER/ES) × (cavity volume − 1). | |||
| WTb | 0.975 | 99.840 | 0.0098 |
| A141S | 1.119 | 66.560 | 0.0168 |
| A141S-A281E | 1.051 | 57.344 | 0.0183 |
| A141S-A281V | 0.976 | 55.088 | 0.0177 |
| A141S-A281G | 0.953 | 68.608 | 0.0139 |
| A141S-A282G | 1.006 | 54.272 | 0.0185 |
| A141S-A282L | 0.958 | 119.296 | 0.0080 |
| A141S-A282N | 0.839 | 59.940 | 0.0140 |
| A141S-A283V | 1.083 | 52.736 | 0.0205 |
| A141S-A283L | 0.952 | 55.808 | 0.0171 |
| A141S-A283S | 0.867 | 55.808 | 0.0155 |
Among these, the mutant A141S-A283V had the highest M value (0.0205 Å−3), indicating the highest R-selectivity. This protein was then overproduced in P. pastoris; its hydrolytic activity and enantioselectivity were determined as 16.06 U mL−1 and 60% (eeR value), respectively, and were greater by 47.8% and 56.8%, respectively, than the corresponding parameters of the A141S mutant (ESI Fig. S2†). Furthermore, the protein structure of the A141S-A283V mutant has changed from an α-helix to β-sheet, decreasing the distance between R-J6 and Ser105 (from 4.7 Å to 3.3 Å, compared with the WT CALB) (Fig. 1B and C).
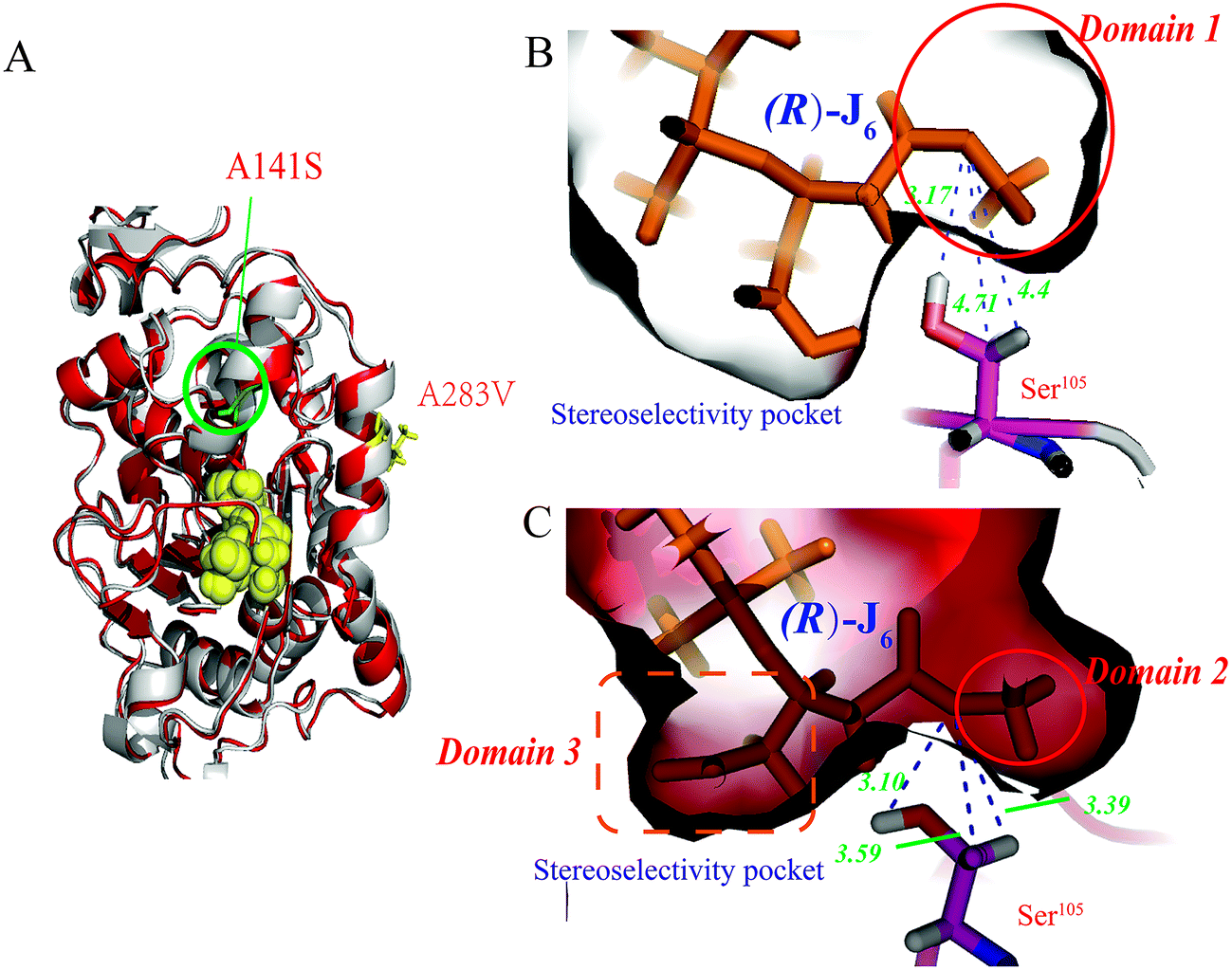 | ||
| Fig. 1 Comparison of the A141S-A283V mutant and WT CALB. (A) Structural comparison between the A141S-A283V mutant and WT CALB. The structures of the A141S-A283V mutant and WT CALB are shown in red and blue, respectively, and the catalytic triad (S105, D187, and H224) is in yellow. Models of the active site for WT CALB (C) and the A141S-A283V mutant (B). In both cases, (R)-3-TBDMSO glutaric acid methyl monoester [(R)-J6] is covalently bound to the enzyme. Hydrogen bonds are indicated with black lines. Ser105 is represented in stick form, where the C, H, N, S, and O atoms are in magenta, white, blue, yellow, and red, respectively. | ||
Construction and characterization of the EF5 mutant
Compared with WT CALB, the volume of the stereoselectivity pocket of the A141S-A283V variant obviously decreased (domains 1 and 2 in Fig. 1). Hence, to further increase the enantioselectivity of CALB, potential amino acid candidates close to domains 2 or 3 (Fig. 1C) were selected to further reshape the substrate pocket. Val190, Ile189, Asp134, and Thr138, all located in domain 3, which accommodates the free carboxyl group of R-J6; and Leu278, Ala282, Ser47, and Thr42, located in the hydrophobic tunnel (domain 2), were all selected for modification (Fig. 2). A combinational mutagenesis approach was employed to further increase the eeR value of the A141S-A283V mutant. A141S-A283V variants were obtained and further screened using (R)-methyl mandelate and (S)-methyl mandelate as substrates in 96-well microtiter plate assays. In this screening procedure, the R/S value [the relative hydrolytic activity with (R)-methyl mandelate to that with (S)-methyl mandelate] was used to assess the enantioselectivity of the variants. Among the variants, the highest R/S value (18.94) was observed for EF5 (A141S-A283V-T138S-S47K). The enantioselectivity of this variant was then evaluated in non-aqueous media, as shown in Fig. 2C; the eeR value (85%) indicated high R-selectivity.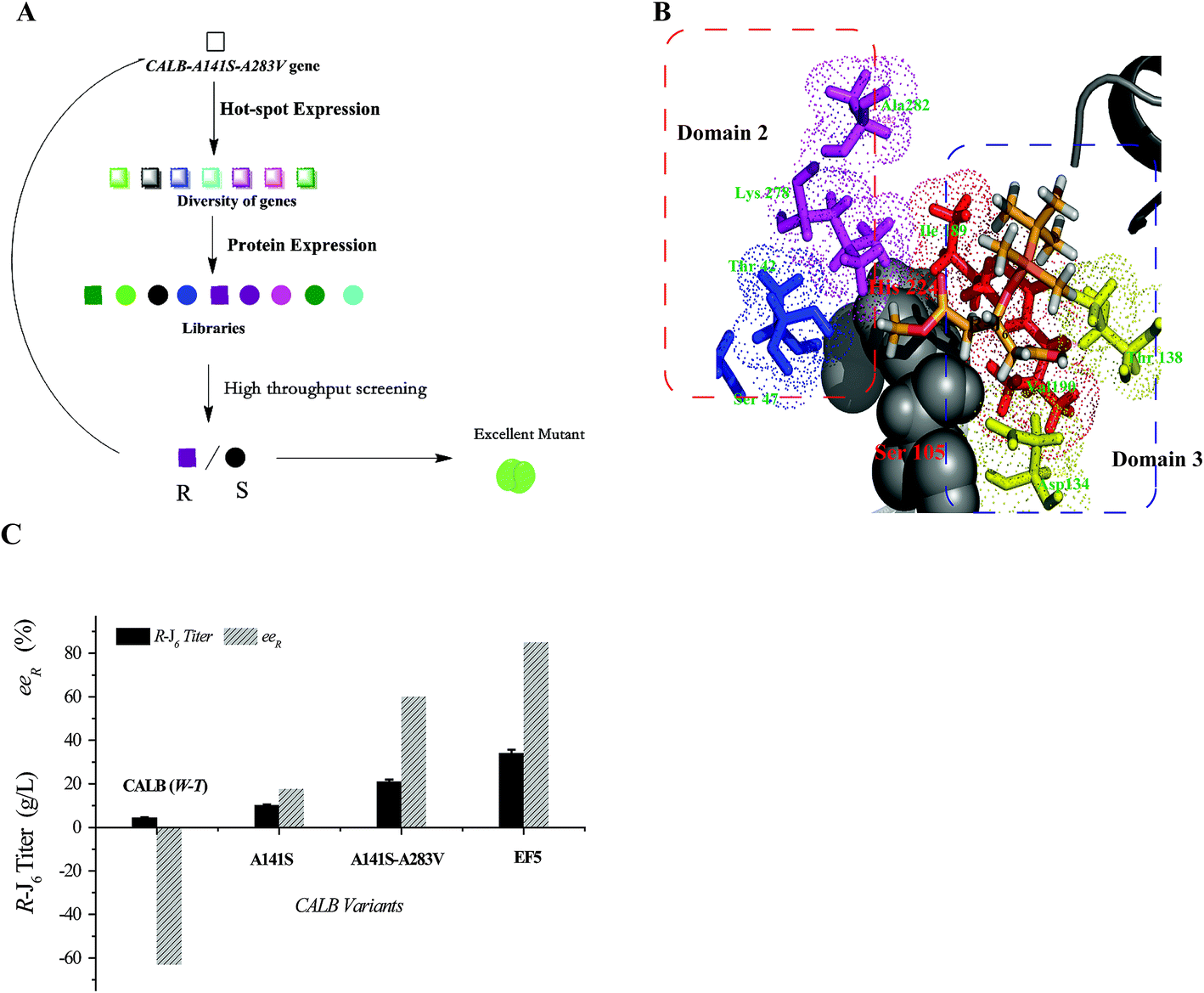 | ||
| Fig. 2 Generation of highly R-selective variants. (A) Amino acids selected for the combinational mutagenesis. (B) Domain 3 (Val190, Ile189, Asp134, and Thr138) and domain 2 (Leu278, Ala282, Ser47, and Thr42). (C) (R)-J6 production by CALB variants. | ||
Kinetic parameters of the EF5 variant are presented in Table 5. For the R-J6 substrate, the KM, kcat, and kcat/KM values of the EF5 variant were better (−90.45%, 34.42%, and 1305.09%, respectively) than the corresponding values of WT CALB; higher (−53.49%, 16.43%, and 151.21%, respectively) than the corresponding values of the A141S variant; and higher (−50.82%, 5.31%, and 114.77%, respectively) than the corresponding values of the A141S-A283V variant. For the p-NPB substrate, the KM, kcat, and kcat/KM values of the EF5 variant were different (785.37%, −33.68%, and −92.50%), respectively, than the corresponding values of WT CALB; higher (295.42%, −30.33%, and −82.40%, respectively) than the corresponding values of the A141S variant; and higher (13.58%, −18.88%, and −28.68%, respectively) than the corresponding values of the A141S-A283V variant. The optimum reaction pH and temperature of the EF5 variant were 8.0 and 65 °C, respectively (Fig. 3).
| Enzyme | p-NPB | (R)-J6 | ||||
|---|---|---|---|---|---|---|
| KM | kcat | kcat/KM | KM | kcat | kcat/KM | |
| (mM) | (s−1) | (mM−1 s−1) | (mM) | (s−1) | (mM−1 s−1) | |
| a Note: Linewear–Burk plots were plotted to determine Km and Vmax of CalB variants. All measurements were taken in triplicate to evaluate the standard deviation. | ||||||
| WT | 2.05 ± 0.05 | 26.57 ± 1.74 | 12.94 | 6.28 ± 0.61 | 3.69 ± 0.05 | 0.59 |
| A141S | 4.59 ± 0.16 | 25.29 ± 1.48 | 5.51 | 1.29 ± 0.10 | 4.26 ± 0.08 | 3.30 |
| A141S-A283V | 15.98 ± 1.52 | 21.72 ± 1.16 | 1.36 | 1.22 ± 0.08 | 4.71 ± 0.09 | 3.86 |
| EF5 | 18.15 ± 2.09 | 17.62 ± 0.76 | 0.97 | 0.60 ± 0.02 | 4.96 ± 0.11 | 8.29 |
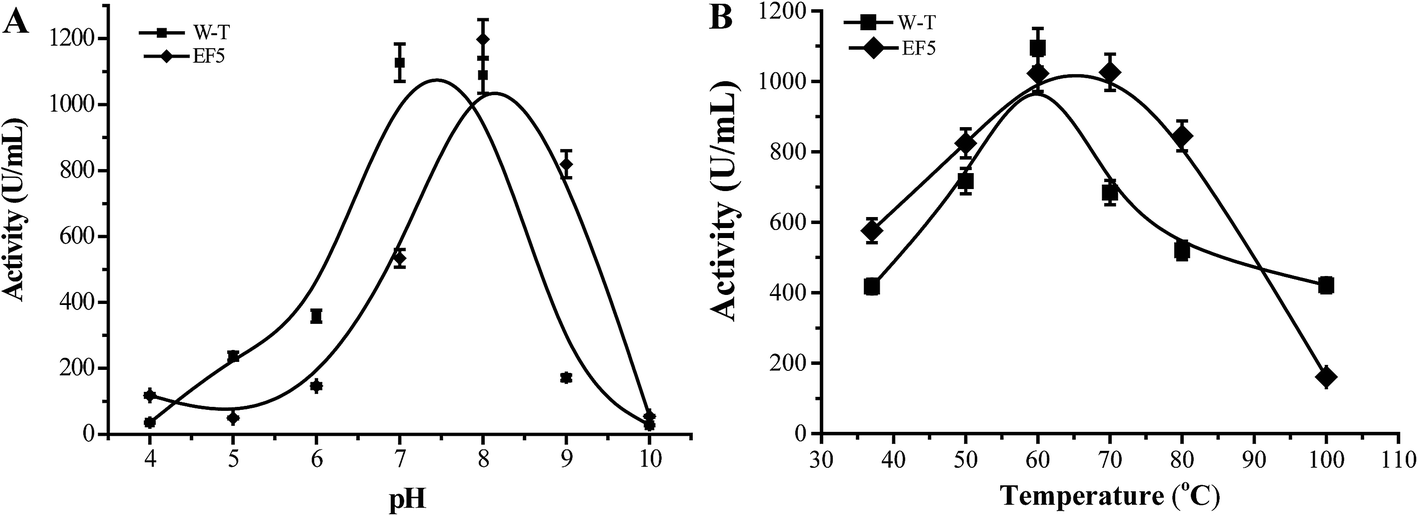 | ||
| Fig. 3 Effect of pH (A) and temperature (B) on the activity of EF5 variant. | ||
Production of (R)-J6 by EF5
The effect of glucose concentration, glucose feeding, pH, and temperature on the specific activity of EF5 variant were then investigated (ESI Fig. S3 and S4†). Under optimum fermentation conditions (pH 6.0, 29 °C, glucose as the inducer, and glucose feeding concentration of 25 g L−1), the activity of EF5 variant was 1114 ± 8.7 U mL−1. The activity of the variant was further increased to 2401.5 ± 5.3 U mL−1 by increasing the gene copy number upon the addition of 800 ng mL−1 zeocin. When the enzyme variant was immobilized under optimal conditions [macroporous resin D101 as the carrier, 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/w) ratio of the enzyme solution to the carrier, pH 8.2, 37 °C, 5 h adsorption time], the activity of immobilized EF5 increased to 2706.7 ± 11.4 U g−1 (ESI Fig. S5†).
:1 (v/w) ratio of the enzyme solution to the carrier, pH 8.2, 37 °C, 5 h adsorption time], the activity of immobilized EF5 increased to 2706.7 ± 11.4 U g−1 (ESI Fig. S5†).
The conditions for the enzymatic transformation of 3-TBDMSO glutaric anhydride to (R)-J6 by the EF5 variant were then investigated. The titer of (R)-J6 increased but the enantioselectivity of the immobilized EF5 catalyst decreased with increasing temperature; at temperature below 5 °C, the obtained (R)-J6 titer was 9.56 ± 1.7 g L−1, with the eeR value of 95.6% (Fig. 4A). As shown in Fig. 4B, the titer of R-J6 increased but the enantioselectivity of EF5 decreased with the molar ratio of 3.0; the obtained R-J6 titer was 20.2 ± 1.7 g L−1, with the eeR value of 69.7%. With increasing EF5 variant concentrations, the titer and ee value of R-J6 increased. When 80 g L−1 of the immobilized EF5 variant was used in a 10 L reaction system, the titer and ee values of R-J6 reached 55.28 ± 1.6 g L−1 and 98.5%, respectively (Fig. 5); in comparison, the ee value was 60% during S-J6 preparation with WT CALB (Fig. 2C). These results indicated that an excess of EF5 biocatalyst is required to obtain a high-optical purity product.
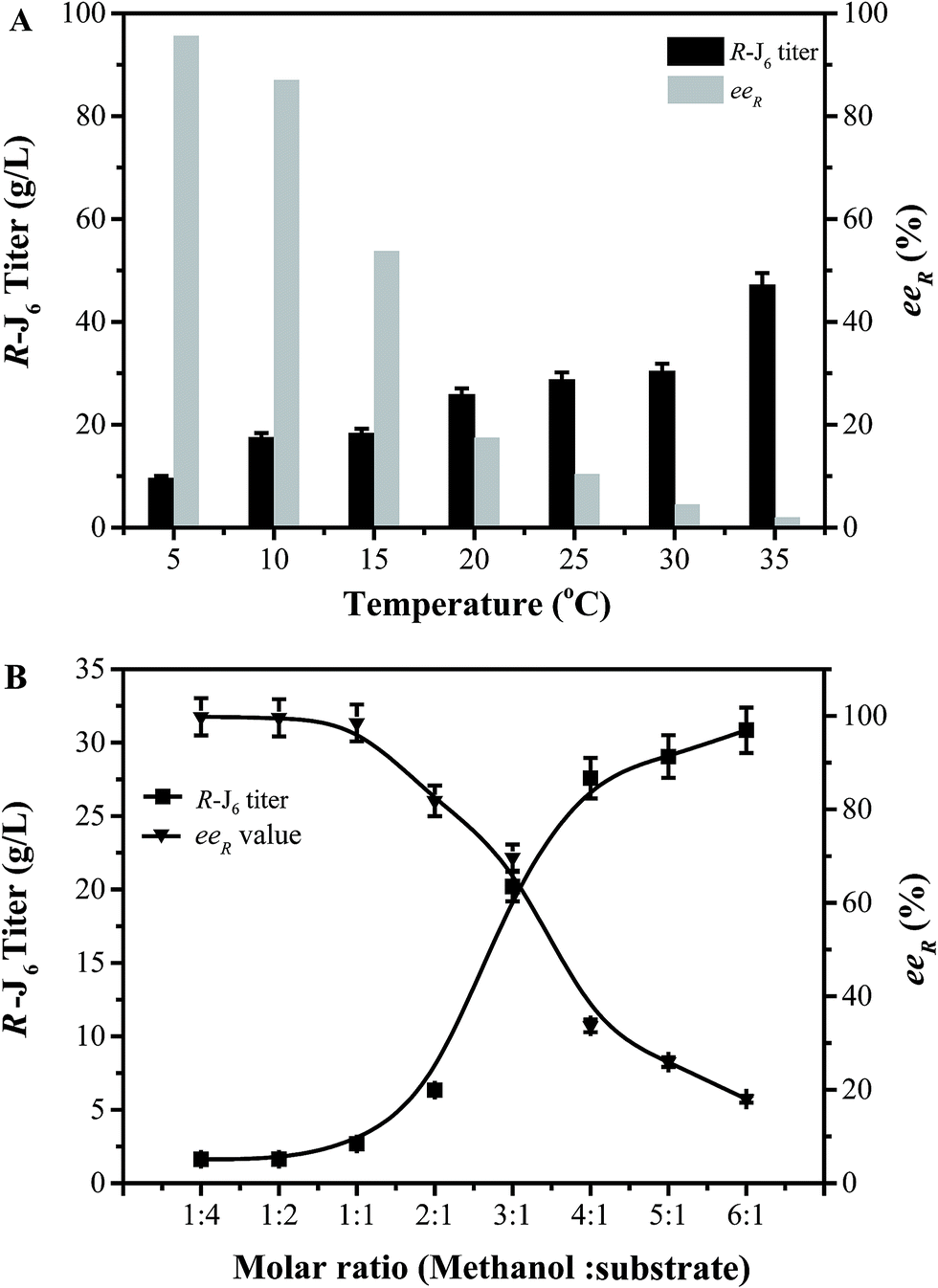 | ||
| Fig. 4 Optimization of the conversion conditions. (A) Effect of reaction temperature on R-J6 production. (B) Effect of methanol concentration on R-J6 production by immobilized EF5. | ||
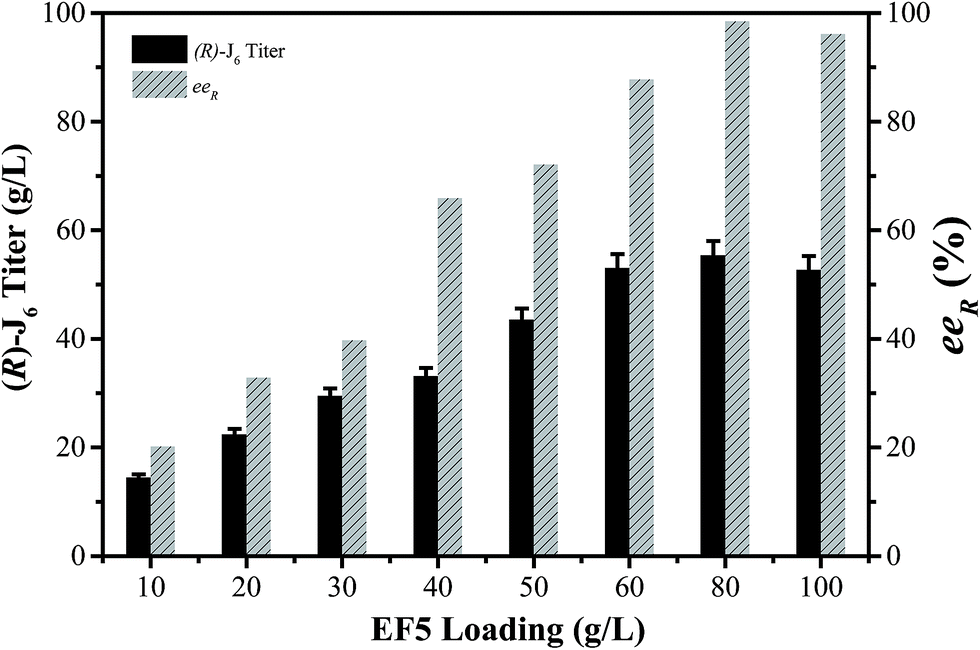 | ||
| Fig. 5 Effect of EF5 load on (R)-J6 synthesis. Reaction conditions: 60 g L−1 of 3-TBDMSO glutaric anhydride (substrate) in MTBE, 5 °C, 200 rpm; methanol:substrate = 2:1, methanol was added at the beginning of the reaction; an appropriate amount of molecular sieves as a desiccant was added to the system to adjust the water activity. | ||
Conclusions
In summary, in the current study, the eeR value of R-J6 was increased by manipulating the substrate pocket of CALB using a semi-rational design strategy. The key amino acid candidates Ala141, Ala283, Thr138, and Ser47 were identified by analyzing the relationship between WT CALB and the target product structure (the 3-substituent group and alcohol moiety). The A141S-A283V mutant was generated after in silico screening, and displayed R-selectivity (with eeR of 60%) during asymmetric alcoholysis of 3-TBDMSO glutaric anhydride. The eeR value was further increased to 85% in a reaction catalyzed by the EF5 mutant, which was generated by combining mutagenesis with high-throughput screening. The kcat/KM value of the EF5 mutant toward R-J6 increased 14-fold compared with WT CALB. Upon optimization of the fermentation conditions and increase the gene copy number, the activity of EF5 increased to 2401.5 ± 5.3 U mL−1; after immobilization, EF5 activity increased to 2706.7 ± 11.4 U g−1. By optimizing the enzymatic reaction system, the R-J6 titer increased to 55.28 ± 1.6 g L−1, with the ee value of 98.5%. These results indicate that the employed semi-rational design strategy is a promising approach for redesigning other biocatalysts and it has a great potential for the production of R-J6 on an industrial scale.Conflict of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant no. 21422602) and the Fundamental Research Funds for the Central Universities (grant no. JUSRP116022).References
- J. W. Blasetto, E. A. Stein, W. V. Brown, R. Chitra and A. Raza, Am. J. Cardiol., 2003, 91, 3–10 CrossRef.
- T. Konoike and Y. Araki, J. Org. Chem., 1994, 59, 7849–7854 CrossRef CAS.
- E. E. Jacobsen, B. H. Hoff, A. R. Moen and T. Anthonsen, J. Mol. Catal. B: Enzym., 2003, 21, 55–58 CrossRef CAS.
- F. Wang, S. Hou, Q. Wang, P. Wang, J. Liu, B. Yang and Y. Wang, Adv. Appl. Microbiol., 2015, 5, 493 CrossRef CAS.
- E. Rogalska, C. Cudrey, F. Ferrato and R. Verger, Chirality, 1993, 5, 24–30 CrossRef CAS PubMed.
- W. M. Liu, Y. Hu, Y. Zhang, Y. Ma and H. Huang, Biotechnol. Bioprocess Eng., 2014, 19, 449–455 CrossRef CAS.
- M. Wolberg, W. Hummel, C. Wandrey and M. Muller, Angew. Chem., Int. Ed., 2000, 39, 4306–4308 CrossRef CAS.
- M. López-García, I. Alfonso and V. Gotor, Tetrahedron: Asymmetry, 2003, 14, 603–609 CrossRef.
- H. J. Wang, Z. B. A. Li, X. X. Yu, R. D. Chen, X. L. Chenabc and L. M. Liu, RSC Adv., 2015, 5, 75160–75166 RSC.
- R. Metzner, W. Hummel, F. Wetterich, B. Konig and H. Groger, Org. Process Res. Dev., 2015, 19, 635–638 CrossRef CAS.
- E. Garcia-Urdiales, I. Alfonso and V. Gotor, Chem. Rev., 2011, 111, Pr110–Pr180 CrossRef PubMed.
- J. M. Palomo and Z. Cabrera, Curr. Org. Synth., 2012, 9, 791–805 CrossRef CAS.
- A. Fryszkowska, M. Komar, D. Koszelewski and R. Ostaszewski, Tetrahedron: Asymmetry, 2005, 16, 2475–2485 CrossRef CAS.
- A. Fryszkowska, M. Komar, D. Koszelewski and R. Ostaszewski, Tetrahedron: Asymmetry, 2006, 17, 961–966 CrossRef CAS.
- J. K. Yang, L. Y. Liu, J. H. Dai and Q. Li, PLoS One, 2013, 8, e53939 CAS.
- M. Biasini, S. Bienert, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer, T. Gallo Cassarino, M. Bertoni, L. Bordoli and T. Schwede, Nucleic Acids Res., 2014, 42, W252–W258 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes and I. Vorobyov, J. Comput. Chem., 2010, 31, 671–690 CAS.
- D. H. Priscilla, D. Roy, A. Suresh, V. Kumar and K. Thirumurugan, Chem.-Biol. Interact., 2014, 210, 77–85 CrossRef CAS PubMed.
- X. D. Kong, S. Yuan, L. Li, S. Chen, J. H. Xu and J. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15717–15722 CrossRef CAS PubMed.
- X. J. Li, R. C. Zheng, H. Y. Ma and Y. G. Zheng, Appl. Microbiol. Biotechnol., 2014, 98, 2473–2483 CrossRef CAS PubMed.
- A. K. Vadhana, P. Samuel, R. M. Berin, J. Krishna, K. Kamatchi and S. Meenakshisundaram, Enzyme Microb. Technol., 2013, 52, 177–183 CrossRef CAS PubMed.
- R. Margesin, G. Feller, M. Hammerle, U. Stegner and F. Schinner, Biotechnol. Lett., 2002, 24, 27–33 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- B. Wang, J. Liu, X. L. Tang, C. Cheng, J. L. Gu, L. Y. Dai and H. W. Yu, Tetrahedron Lett., 2010, 51, 309–312 CrossRef CAS.
- E. Corey and M. C. Noe, J. Am. Chem. Soc., 1996, 118, 319–329 CrossRef CAS.
- F. Hou, T. Miyakawa, M. Kataoka, D. Takeshita, S. Kumashiro, A. Uzura, N. Urano, K. Nagata, S. Shimizu and M. Tanokura, Biochem. Biophys. Res. Commun., 2014, 446, 911–915 CrossRef CAS PubMed.
- R. T. Otto, H. Scheib, U. T. Bornscheuer, J. Pleiss, C. Syldatk and R. D. Schmid, J. Mol. Catal. B: Enzym., 2000, 8, 201–211 CrossRef CAS.
- T. Rosen, M. Watanabe and C. H. Heathcock, J. Org. Chem., 1984, 49, 3657–3659 CrossRef CAS.
- I. Høegh, S. Patkar, T. Halkier and M. T. Hansen, Can. J. Bot., 1995, 73, 869–875 CrossRef.
- L. Novak, J. Rohály, L. Poppe, G. Hornyánszky, P. Kolonits, I. Zelei, I. Fehér, J. Fekete, É. Szabó and U. Záhorszky, Liebigs Ann. Chem., 1992, 1992, 145–157 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06016e |
| ‡ The co-first authors. |
| This journal is © The Royal Society of Chemistry 2017 |