
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the stability of organolead halide perovskite films through polymer encapsulation†
Barry McKenna a,
Joel R. Troughtonb,
Trystan M. Watsonb and
Rachel C. Evans*ac
a,
Joel R. Troughtonb,
Trystan M. Watsonb and
Rachel C. Evans*ac
aSchool of Chemistry and CRANN, Trinity College, The University of Dublin, Dublin 2, Ireland
bSPECIFIC – College of Engineering, Swansea University, Bay Campus, Swansea, SA1 8EN, UK
cDepartment of Materials Science & Metallurgy, University of Cambridge, UK. E-mail: rce26@cam.ac.uk
First published on 28th June 2017
Abstract
Perovskite solar cells based on organolead halides such as CH3NH3PbX3 (X = Cl, Br, and I) have rapidly established themselves as the frontrunners among emerging photovoltaic technologies. However, their commercial application has been hindered to date in part due to their susceptibility to degradation by UV radiation or heat in the presence of moisture. Herein we investigate the relationship between the physical properties of several polymer encapsulants (poly(methylmethacrylate) (PMMA), ethyl cellulose, polycarbonate and poly(4-methyl-1-pentene)) and their ability to function as barrier layers to improve the stability of CH3NH3PbI3−xClx films under prolonged thermal degradation at 60 °C, 80 °C and 100 °C. In all cases, polymer-coated CH3NH3PbI3−xClx films showed retarded thermal degradation compared to the uncoated films, as indicated by the quantitative decay of the perovskite band edge in the UV/Vis absorption spectrum and the appearance of PbI2 peaks in the powder X-ray diffraction pattern. However, the extent of this reduction was highly dependent on the physical properties of the polymer encapsulant. Notably, PMMA-coated CH3NH3PbI3−xClx films showed no visible signs of degradation to PbI2 after extended heating at 60 °C. However, concomitant studies by epifluorescence microscopy (FM) revealed deterioration of the CH3NH3PbI3−xClx film quality, even in the presence of a polymer-coating, at much shorter heating times (29 h), as evidenced by quenching of the film fluorescence, which was attributed to grain aggregation and the formation of associated non-radiative trap sites. Since grain aggregation occurs on a shorter timescale than chemical degradation to PbI2, this may be the limiting factor in determining the resistance of organolead halide perovskite films to thermal degradation.
1. Introduction
Perovskite solar cells (PSC) have rapidly established themselves as the frontrunners in the field of emerging photovoltaic technologies, with certified power conversion efficiencies (PCEs) of over 20% reported for state-of-the-art devices.1 However, despite high efficiencies and ease of processability through low-cost solution deposition methods, the inherent instability and toxicity of the active organolead halide perovskite layer still presents a significant barrier to market acceptance of PSCs as a commercial product. Moisture, high temperatures and UV exposure are all well-known to result in degradation of the active CH3NH3PbI3 or CH3NH3PbI3−xClx material through the dissolution of methyl ammonium iodide to produce PbI2.2 While PbI2 shows only low solubility in water (Ksp = 9.8 × 10−9 at 25 °C),3 leakage of any quantity of PbI2 into the global water supply is clearly a significant environmental concern and steps must be taken to actively monitor the degradation of the active layer during the manufacturing process, so that any nuisance by-products can be identified and disposed of in a safe and timely manner. Moreover, the encapsulation materials used to protect finished cells must be rigorously tested to ensure that the accidental discharge of lead compounds from both working and legacy devices into the environment is prevented.Significant research has already been undertaken to improve the intrinsic stability of the active perovskite layer, which includes for example, the use of mixed halide perovskites,4 the use of 2D layered perovskite structures,5 the use of formamidinium in place of methylammonium as the organic counterion6 and the use of cross-linking agents.7 To investigate the success of these approaches, elevated temperatures (20–90 °C)8–10 are commonly used to accelerate the degradation of the perovskite material, which can be monitored using a variety of characterisation methods, with varying degrees of ease and success. X-ray diffraction (XRD) has been used extensively to determine the ratio of CH3NH3PbX3 to PbX2 phases in degraded samples from the relative intensities of the peaks in their characteristic diffraction patterns and in response to different conditions/treatments such as: substrates and Cl− doping concentrations;11 the effect of different atmospheres;8,12 effect of temperature;8,9 humidity;13 and moisture.10 X-ray photoelectron spectroscopy (XPS) has also been used to investigate the stability of perovskites to moisture, inert atmospheres, thermal and X-ray beam damage.14 However, such methods are generally not suitable for rapid, real-time analysis and early indication of device degradation on the production line.
Conversion of CH3NH3PbI3 or CH3NH3PbI3−xClx into PbI2 results in a distinctive colour change from brown to yellow, which can be followed using UV/Vis absorption spectroscopy. However, for perovskite thin films in particular, observation of the “true” absorption spectrum is challenging, due to both the high optical density and scattering due to the crystallite size, which results in a spectrum which is largely featureless across the visible range.15 Watson et al. recently reported the spectrothermal evaluation of the thermal annealing step of perovskite formation through a combination of thermogravimetric analysis (TGA), differential scanning calorimetry (DSC) and Fourier transform infrared spectrometry (FTIR).16 This study showed that decomposition of the CH3NH3PbI3−xClx layer did not occur for temperatures less than 150 °C over the timescale investigated (80 min). The same group also demonstrated a method of RGB (red, green, blue) colour analysis to track the degradation of the perovskite layer to PbI2 using time-lapse photography.17 The active CH3NH3PbX3 compound is also photoluminescent and this has been exploited to monitor the layer uniformity of PSCs under operating conditions.18,19 Moreover, Sheng et al. used confocal fluorescence microscopy to observe that the grain size in CH3NH3PbBr3 films increases spontaneously in the presence of moisture and oxygen, and that the larger crystals have a higher number of defects, despite no obvious change in the XRD pattern of the films.20 This increase in defect concentration results in a shorter charge-carrier lifetime, which can negatively impact the efficiency of the perovskite as a light-absorber.
Device encapsulation at the laboratory scale typically involves manual coverage with a glass cover slip and subsequent sealing using an epoxy resin.21–23 While this method is effective for small devices, it is not practical for mass production. Roll-to-roll fabrication will require encapsulation of devices using polymer layers, similar to that demonstrated for organic solar cells.24 With this in mind, a variety of materials have been investigated as device encapsulants, including Teflon,25 Al2O3,2,26 poly(methyl methacrylate) (PMMA) and polycarbonate (PC).10,17 However, while comparison between the performance of different encapsulants is common,10,17 there has been little attempt to clearly identify the key physicochemical characteristics required of the material. This is clearly important since the barrier performance of the encapsulant will depend on a number of factors including the glass transition temperatures (Tg), water vapour transport rate (WVTR), O2 permeability and hydrophobicity.
Recent studies have also investigated the use of polymers as “internal” barrier layers for PSCs. Habisreutinger et al. used polymer composites prepared from a mixture of PMMA or PC, with poly(3-hexylthiophene-2,5-diyl) (P3HT) and carbon nanotubes as a dual barrier-hole transport layer, with the resultant devices displaying improved stability up to 96 h upon heating at 80 °C in air.10 This was achieved by doping the hole conducting P3HT-nanotube composite within the hydrophobic PMMA and PC stock solutions and depositing onto the bare perovskite layer. However, the specific role of temperature and the materials properties of the encapsulation layer were not investigated in detail. Zhao et al. utilised polyethylene glycol (PEG) as a hydrophobic mesostructured layer in PSCs which resulted in improved stability over 300 h at 70% humidity and displayed self-healing behaviour.27 A similar study was also performed using ethyl cellulose (EC) as a mesostructured layer, which led to an increased stability of 6 days at 60% relative humidity, albeit with a decreased device efficiency when compared with the parent device (16.3% to 14.1%).28 Very recently Bella et al. demonstrated a bifunctional encapsulation approach, by coating the back side of the PSC with a hydrophobic photocurable polymer and the front surface with the same polymer doped with a luminophore as a luminescent down-shifting layer.29
The aim of this work is to investigate the effect that the physical properties of a variety of polymer encapsulants have on their ability to act as barrier layers for PSCs. The four polymer encapsulants selected were PMMA, PC, EC and poly(4-methyl-1-pentene) (PMP) (see Fig. S1 ESI† for chemical structures). These polymers were specifically chosen to screen a broad range of values associated with the key physical parameters relevant to encapsulant performance for PSCs, namely oxygen permeability, water vapour transition rate (WVTR), water absorption and glass transition temperature (see Table 1 for values). CH3NH3PbI3−xClx films were coated with polymer layers of known thickness and subjected to accelerated thermal degradation at a series of temperatures to enable the stability of both the active perovskite layer and the polymer barrier to be probed using UV/Vis absorption spectroscopy, powder XRD (PXRD) and epifluorescence microscopy (FM). We will demonstrate that ability of a given polymer to function as an effective barrier layer is highly dependent on its WVTR, and to a lesser extent the glass transition temperature. Moreover, while UV/Vis absorption spectroscopy and PXRD provide quantitative evidence of chemical degradation of the CH3NH3PbI3−xClx film to PbI2, they fail to identify deterioration of the film quality due to grain aggregation, which is easily detected by FM. These results indicate that FM may be a powerful tool for the rapid and non-invasive screening of device viability during the manufacturing process.
2. Experimental
2.1 Materials
Methylamine solution (33 wt% in absolute ethanol), hydroiodic acid (57 wt% in H2O), lead chloride (99.999%), lead iodide (99.999%), titanium diisopropoxide bis(acetylacetonate) (75% in isopropanol), chlorobenzene (99.8%), bis(trifluoromethane)sulfonimide lithium salt (Li-TFSI) (anhydrous 99.8%), 4-tert-butylpyridine (tBP) (96%), tris(2-(1H-pyrazol-1-yl)-4-tert-butylpyridine)cobalt(III) tri[bis(trifluoromethane)sulfonimide] (FK209) (98%), PMMA (MW (av.) 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), EC (48% ethoxyl) and PMP (low molecular weight) were all obtained from Sigma Aldrich. PC (MW (av.) 45000 g mol−1) was obtained from Acros Organics. Dimethyl formamide (DMF) (99.8%), toluene (99.8%), chloroform (99.8%), cyclohexane (99.8%) dimethyl sulfoxide (DMSO) and dichloromethane (DCM) (99.8%) were obtained from Fisher. 2,2′,7,7′-Tetrakis[N,N-di(4-methoxyphenyl)amino]-9,9′-spirobifluorene (spiro-OMeTAD) (99%) was purchased from FMECT. All materials were used as received.
000 g mol−1), EC (48% ethoxyl) and PMP (low molecular weight) were all obtained from Sigma Aldrich. PC (MW (av.) 45000 g mol−1) was obtained from Acros Organics. Dimethyl formamide (DMF) (99.8%), toluene (99.8%), chloroform (99.8%), cyclohexane (99.8%) dimethyl sulfoxide (DMSO) and dichloromethane (DCM) (99.8%) were obtained from Fisher. 2,2′,7,7′-Tetrakis[N,N-di(4-methoxyphenyl)amino]-9,9′-spirobifluorene (spiro-OMeTAD) (99%) was purchased from FMECT. All materials were used as received.
2.2 Fabrication of polymer encapsulated CH3NH3PbI3−xClx films
CH3NH3PbI3−xClx films were prepared following well-established procedures with some minor modifications.30 CH3NH3I was prepared by reacting methylamine solution (33 wt% in ethanol) and HI (57 wt% in H2O) in a 2:1 molar ratio in ethanol. This mixture was stirred at room temperature (RT) under nitrogen for 2 h. The product was isolated by rotary evaporation and filtration before being washed with diethyl ether several times to remove unreacted precursor. A CH3NH3PbI3−xClx stock solution was prepared by mixing CH3NH3I (2.65 mol L−1) and PbCl2 (0.88 mol L−1) in DMF (4 mL). This solution was stirred at RT before being filtered through a 200 nm PTFE syringe filter. CH3NH3PbI3−xClx films were deposited on microscope glass slides by spin coating a fixed volume (400 μL) of the stock solution at 2000 rpm for 45 s, followed by annealing at 100 °C for 60 min.
Polymer coatings of uniform thickness (∼800 nm) were applied directly to the CH3NH3PbI3−xClx films by spin-coating. Due to the varying viscosities of the polymer stock solutions, the spin rates and conditions were carefully controlled to achieve a standard and uniform thickness for all barrier layers. PMMA (560 mg in 4 mL toluene (16.1 wt%)) and PMP (100 mg in 4 mL cyclohexane (3.2 wt%)) films were deposited by spin-coating at 8000 rpm for 60 s, PC (200 mg in 4 mL dichloromethane (3.8 wt%)) and EC (200 mg in 4 mL 80:20 toluene:ethanol (5.9 wt%)) were spun at 2000 and 1000 rpm for 60 s respectively. After spinning, the polymer-coated perovskite layers were placed on a hot plate at 80 °C for 5 min to ensure complete evaporation of the solvent.
2.3 Thermal degradation studies
Thermal degradation of the bare and polymer encapsulated CH3NH3PbI3−xClx was performed under identical ambient conditions (relative humidity 50–70%, indoor lighting) by placing the perovskite films face up onto a hotplate at 60, 80 or 100 °C. The films were only removed briefly at intervals of 24 h for characterisation by the methods outlined below.2.4 Characterisation
UV/Vis absorption spectroscopy was performed using a Lambda 35 (Perkin Elmer) spectrometer. Steady-state photoluminescence (PL) spectroscopy measurements were made at room temperature (RT) on a Fluorolog-3 (Horiba Jobin Yvon) spectrophotometer using the front-face configuration. PL spectra were corrected for the wavelength response of the system using correction factors supplied by the manufacturer. The slit widths were 2 nm for UV/Vis absorption, and 5 nm and 2 nm for excitation and emission monochromators, respectively, for the photoluminescence measurements. PXRD measurements were performed on a Bruker D2 Phaser Powder diffractometer. Samples were exposed to Cu Kα radiation (λ = 1.54 Å) at RT in the range 5–55° 2θ. Fluorescence microscopy images were obtained using a Nikon Labophot epifluorescence microscope fitted with a B2-A filter cube (excitation 450–490 nm, dichromatic mirror with a 500 nm cut-on (longpass), and 515 nm cut-on emission filter (longpass)) and a QImaging QIClick camera. The thicknesses of polymer films were determined using a Filmetrics F20 thin film analyser in contact mode. Contact angle measurements were performed on a First Ten Ångstroms FTA 1000 B-class drop shape instrument with a manual dispenser and analysed using FTA32 software.2.5 Device fabrication and characterisation
Etched FTO glass substrates (NSG Pilkington, TEC7) were cleaned sequentially in detergent, deionised water, acetone and ethanol before undergoing 10 min of O2 plasma treatment. A compact TiO2 layer was deposited on the glass substrates through spray pyrolysis of a 0.2 M solution of titanium diisopropoxide bis(acetylacetonate) in isopropanol at 450 °C. Upon cooling, a mesoporous layer of TiO2 nanoparticles was spin-coated from a 2:7 (w/w) suspension of Dyesol 30NR-D paste in ethanol (4500 rpm for 30 s), followed by sintering at 550 °C for 30 minutes.
A CH3NH3PbI3 perovskite precursor solution was prepared by dissolving 576 mg PbI2, and 199 mg CH3NH3I in a 4:1 (v/v) solution of DMF:DMSO. 100 μL of the perovskite precursor solution was deposited onto the TiO2 films and spin-coated at 4000 rpm for 30 s, with 200 μL of ethyl acetate dripped onto the spinning substrate 10 s prior to the end of the spin-coating process. Perovskite films were annealed at 100 °C for 10 min.
A 70 mM solution of spiro-OMeTAD in chlorobenzene was prepared with dopants including Li-TFSI, tBP and FK209 with molar ratios with respect to the spiro-OMeTAD of 0.5, 3.3 and 0.03, respectively. The hole transport material (HTM) solution was spin-coated onto perovskite films at 4000 rpm for 30 s before 80 nm thick Au contacts were thermally evaporated onto devices.
Current–voltage measurements were performed using both a AAA-rated solar simulator (Oriel Sol3A) calibrated against a KG5-filtered reference diode (Oriel 91150-KG5) and an ABB-rated solar simulator (Abet Technologies 10500) calibrated against a quartz-filtered reference diode (RR 230 O). Solar cells were masked to 0.1 cm2 or 0.28 cm2 and scanned both from forward to reverse bias and vice versa at 100 mV s−1.
3. Results and discussion
3.1 Thermal degradation of pristine and polymer-coated CH3NH3PbI3−xClx films
Fig. 1 shows the change in the optical properties of a CH3NH3PbI3−xClx film (∼500 nm thick) following heating at 60 °C in air. While pristine films are grey-brown in colour, after thermal degradation for 72 h they turn yellow (Fig. 1a). This colour change has previously been assigned to the complete degradation of CH3NH3PbI3−xClx and formation of PbI2.11 This colour change can be monitored quantitatively using UV/Vis absorption spectroscopy (Fig. 1b). Pristine CH3NH3PbI3−xClx films absorb strongly across the UVA/Vis region and exhibit a step-like feature with an onset at ∼750 nm, which corresponds to a bandgap energy of approximately 1.56 eV. Upon thermal degradation, the step-like feature disappears. Pristine CH3NH3PbI3−xClx films are also photoluminescent, exhibiting an emission maximum at approximately 770 nm upon excitation at 500 nm.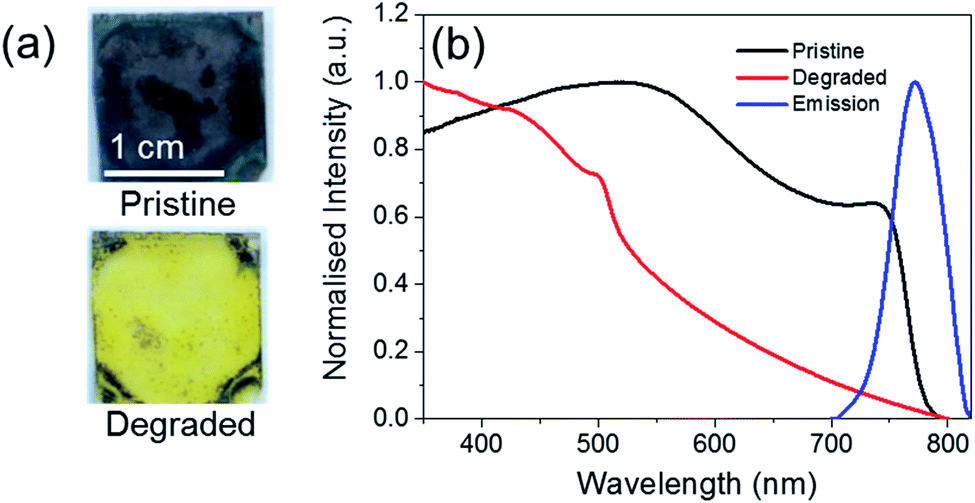 | ||
| Fig. 1 Comparison of the optical properties of a pristine and thermally degraded (60 °C for 72 h in air) CH3NH3PbI3−xClx film (∼500 nm thick). (a) Photograph illustrating the colour change that occurs upon degradation. (b) Normalised UV/Vis absorption spectrum of an uncoated CH3NH3PbI3−xClx film before (black line) and after (red line) thermal degradation. The photoluminescence spectrum (blue line) of the pristine film of CH3NH3PbI3−xClx (λex = 500 nm) is also shown. | ||
Polymer encapsulants were spin-coated as uniform films (∼800 nm thick) onto pristine perovskite films to investigate their potential to act as a barrier to the ambient environment and inhibit the rate of thermal degradation. The PXRD patterns of pristine and coated perovskite films are identical for all polymers screened (Fig. S2, ESI†), indicating that the polymer encapsulant does not alter the crystal structure of the perovskite material. Pristine and coated samples show two intense diffraction peaks at 14° and 28° and a less intense peak at 43°, corresponding to the (110), (220) and (330) planes of the tetragonal perovskite lattice respectively.30 Following extended thermal degradation at 60 °C for 432 h, the PXRD patterns of pristine and coated films reveal degradation of the perovskite layer to differing extents, indicated by the appearance of a new peak at 12.8°, which is characteristic of PbI2 (Fig. 2).13 The uncoated film is completely degraded after this time such that the PXRD pattern corresponds to that of the amorphous glass substrate with only residual PbI2 reflections present. In contrast, the PC-, EC-, and PMP-encapsulated films show some resistance to thermal decomposition and the characteristic reflections of CH3NH3PbI3−xClx are still present in the PXRD pattern, albeit to a significantly lower intensity than those attributed to PbI2. Notably, the PMMA-encapsulated films show remarkable tolerance to thermal degradation, with the PXRD pattern after 432 h heating matching that of the original film, and no discernible PbI2 peaks present.
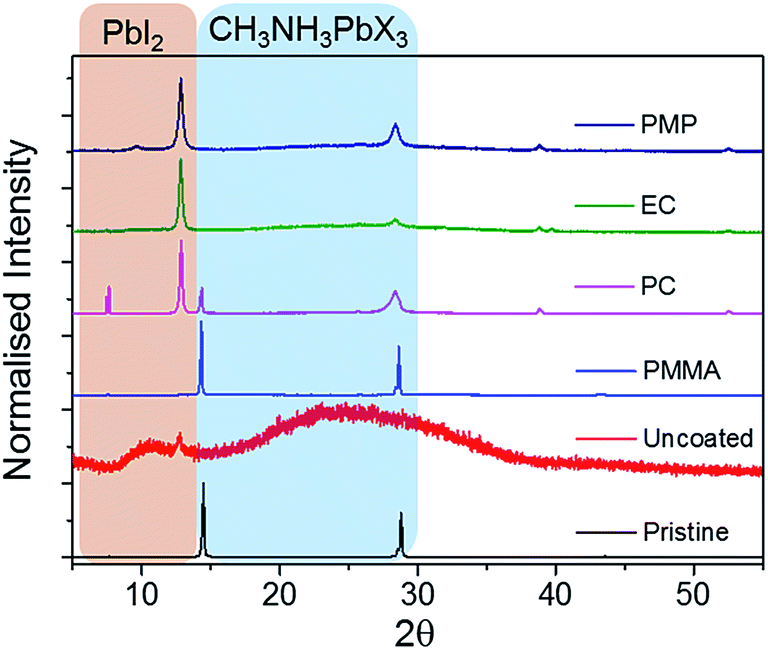 | ||
| Fig. 2 PXRD patterns of uncoated and polymer-encapsulated CH3NH3PbI3−xClx films following thermal degradation in 60 °C in air for 432 h. The PXRD pattern of the pristine uncoated CH3NH3PbI3−xClx film prior to thermal degradation is also shown for comparison (black line). After 432 h, the uncoated film displayed total degradation, such that the observed pattern predominantly represents that of the amorphous glass substrate. | ||
The addition of the polymer coating does, however, induce some changes in the shape and intensity of the UV/Vis absorption spectrum (Fig. S3, ESI†). PMMA- and PC-coated CH3NH3PbI3−xClx films showed a general increase in absorbance at all wavelengths, while EC- and PMP-coated samples showed a decrease in absorbance near the band edge at 750 nm, but an increase in absorbance upon moving towards the higher energy spectral region. This behaviour is likely to be due to partial re-dissolution of the perovskite layer in the polymer stock solution during the spin-coating process, which subsequently self-heals and results in improved coverage of the CH3NH3PbI3−xClx film. This leads to increased absorption in the visible region of the spectrum, due to decreased scattering as the crystal size of the perovskite increases.15 Partial solvation of perovskite layers has been widely investigated to improve surface coverage of the films and thus device efficiencies.36 As each polymer stock solution uses a different solvent, the extent of the improved coverage is determined by the solubility of the perovskite in that solvent. We note that the polymer coatings exhibit negligible absorbance across the visible spectrum (Fig. S4, ESI†) and as such do not contribute to the observed changes. However, the results highlight the challenges associated with measuring the “true” UV/Vis absorption spectrum of organometal halide perovskites.15
3.2 Time-dependent thermal degradation monitored by ex situ UV/Vis absorption spectroscopy
The temporal dependence of the thermal degradation of uncoated and encapsulated perovskite films was initially investigated by UV/Vis absorption spectroscopy. Films were placed on a hot-plate at three degradation temperatures (60, 80 and 100 °C), which were chosen to approximate the range of possible operating temperatures for solar cells in different climates.37 Fig. 3a shows the UV/Vis absorption spectrum of the PMP-coated film as a function of degradation time at 80 °C as a representative example. The step-like feature at 750 nm is observed to decay with increasing degradation time. With the exception of the PMMA-coated film, this trend is observed for all polymer encapsulants and all degradation temperatures, with the only difference being the time take to reach total degradation. The corresponding UV/Vis absorption spectra for all polymer-encapsulated perovskite films at 80 °C can be found in Fig. S5, ESI.†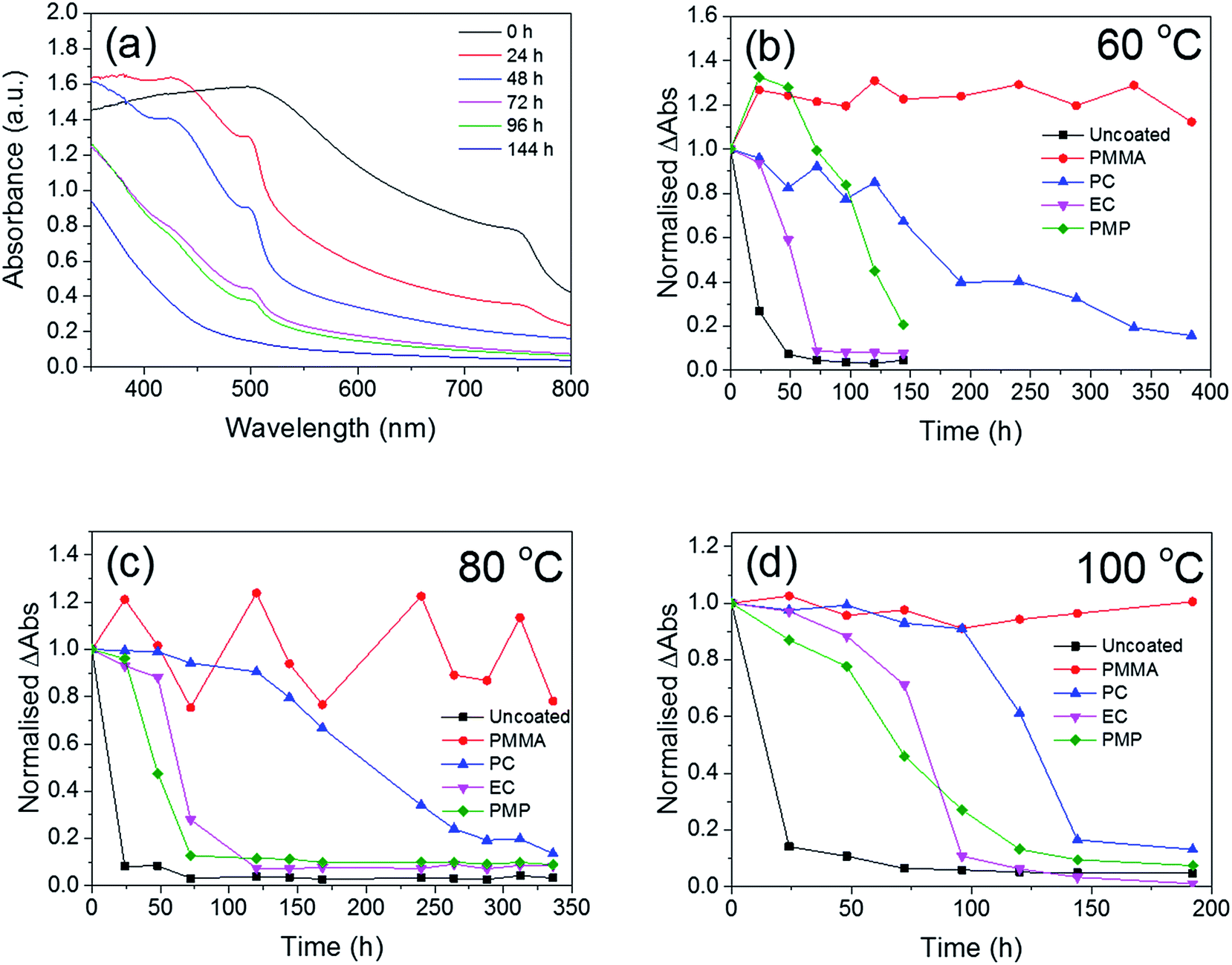 | ||
| Fig. 3 Thermal degradation of pristine and polymer-encapsulated CH3NH3PbI3−xClx films followed by ex situ UV/Vis absorption spectroscopy. (a) UV/Vis absorption spectra of PMP-coated films degraded at 80 °C as a function of time, t. (b) Difference in absorbance between 750 nm and 800 nm (ΔAbs) as a function of time, normalised to t = 0 h for films degraded at 60 °C in air, (c) 80 °C in air and (d) at 100 °C in air. The solid lines serve only to guide the eye. | ||
In order to compare the rate of degradation of the CH3NH3PbI3−xClx polymer-encapsulated films at different temperatures, Fig. 3b–d show the decay in the absorbance at the band edge normalised to time, t = 0. We note that for some samples, the absorbance initially increases between 0–24 h heating, before subsequently decreasing with extended degradation times. This is attributed to both continued annealing of the film and the removal of any unevaporated solvent during the first 24 h, which results in rearrangement of crystallites and improved surface coverage of the film due to moisture present in the solvents.16,36 It can be seen that after 24 h, at all temperatures, the uncoated CH3NH3PbI3−xClx film shows significant loss in absorbance corresponding to near complete degradation, with the additional decrease in absorbance as time progresses representing loss of material from the substrate (Fig. S6, ESI†). As expected, the addition of the polymer encapsulant significantly reduces the rate of degradation, the extent of which is determined by the polymer used. Notably, PMMA-encapsulated layers show no significant degradation at any temperature after 384 h, in good agreement with the PXRD data. For the other polymers, the rate of degradation increases with the degradation temperature. PC is the next best performing encapsulant, with total degradation observed at ∼380 h, 340 h and 190 h at 60 °C, 80 °C and 100 °C, respectively. However, as encapsulants, EC and PMP exhibit a similar performance, but only moderately enhance the thermal stability compared to the uncoated CH3NH3PbI3−xClx film, with the onset of degradation occurring rapidly after 50 h at all temperatures. Fig. 4 summarises the mean times required for complete decay of the band edge for pristine and polymer-coated CH3NH3PbI3−xClx films. At all temperatures, it is apparent that the efficacy of the encapsulant towards inhibiting thermal degradation is related to the water vapour transmission rate (WVTR) of the polymer (see Table 1). PMMA, with the lowest WVTR, vastly outperforms the other polymers, confirming, as expected, that water vapour plays a key role in the thermal degradation mechanism. Habisreutinger et al. showed similar results for a carbon nanotube-doped polymer hole transport layer using PMMA and PC acting as suitable moisture barriers even when placed under running water.10 However, all the encapsulants become less effective at preventing thermal degradation as the temperature is increased. Interestingly, the surface hydrophobicity of the polymer coating appears to have a negligible effect its performance as a barrier layer. Contact angle measurements on polymer films coated on a glass substrate revealed that PMP exhibits the lowest wettability (θc = 105.9°), while the EC surface is highly hydrophilic (θc = 18.5°) (Fig. 5); however, both polymers exhibit similar trends in the thermal degradation studies at all temperatures investigated. It is possible that the low glass transition temperature of PMP (Tg = 29 °C for amorphous PMP, however this can decrease with crystallinity)35 results in the formation of defects in the film, which allow moisture to permeate through facilitating degradation.
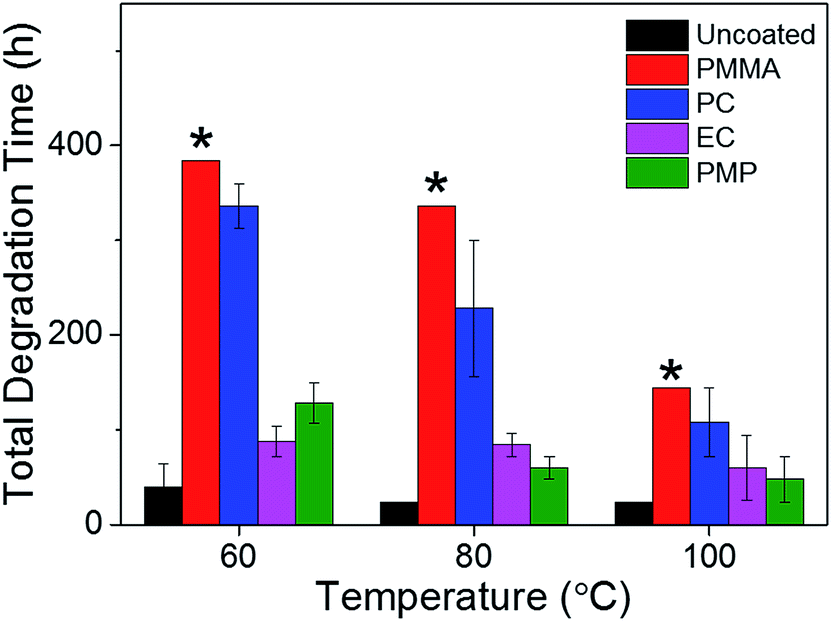 | ||
| Fig. 4 The mean times required for complete degradation of the UV/Vis absorption band edge (750 nm) for pristine and polymer-encapsulated CH3NH3PbI3−xCl3 films as a function of degradation temperature. The error bars represent the standard deviation of three measurements. The * indicates that at that time of characterisation there was still no indication of degradation. | ||
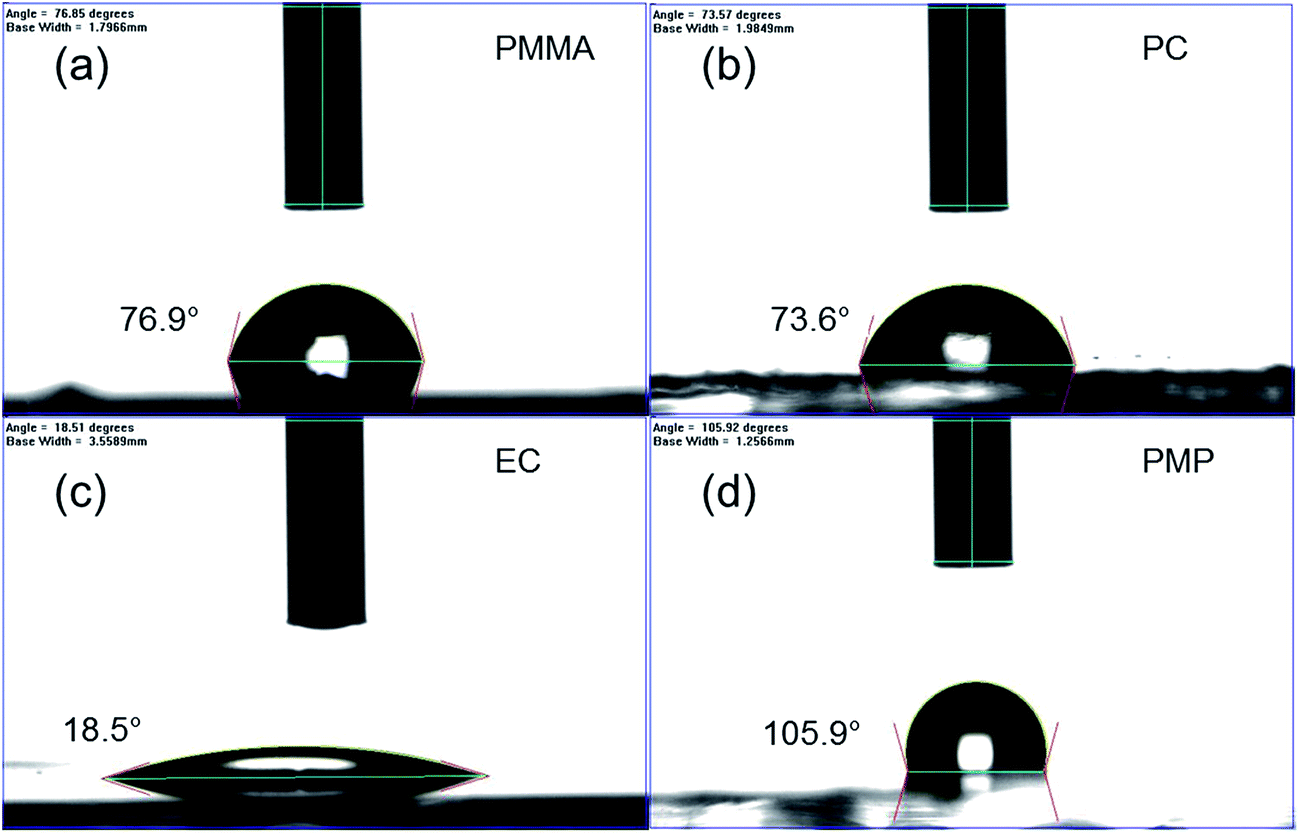 | ||
| Fig. 5 Determination of the surface hydrophobicity of polymer films coated on glass slides using contact angle measurements: (a) PMMA, (b) PC, (c) EC and (d) PMP. | ||
3.3 Real-time evaluation of thermal degradation using epifluorescence microscopy
Fig. 6 shows the FM images of polymer-encapsulated CH3NH3PbI3−xClx films as a function of degradation time at 60 °C. The corresponding images for thermal degradation of the pristine CH3NH3PbI3−xClx film at 60 °C, and polymer-coated CH3NH3PbI3−xClx films at 80 °C and 100 °C can be found in the ESI (Fig. S7–S9†). We note care was taken to limit exposure of the films to the blue excitation source (irradiation time was less than a few seconds) and that the area of the film imaged was also varied to minimise any possible contributions of the light exposure to the degradation of the perovskite layer. | ||
| Fig. 6 Thermal degradation of polymer encapsulated CH3NH3PbI3−xClx films at 60 °C as a function of time monitored by fluorescence microscopy. CH3NH3PbI3−xClx degradation is accompanied by a decrease in emission intensity and hence darkening of the image. (a) PMMA shows negligible degradation after 384 h, (b) PC degrades completely after 96 h, (c) EC shows an onset of degradation after 24 h and (d) PMP shows complete degradation after 72 h. | ||
Prior to heating, the pristine and encapsulated films exhibit a homogeneous red emission that is characteristic of CH3NH3PbI3−xClx, as shown by the fluorescence spectrum in Fig. 1. The addition of the polymer encapsulant has no effect on the fluorescence of the perovskite layer underneath, nor does it affect the observed intensity due to the high optical transparency of all of the polymers in the visible region (Fig. S4, ESI†). Degradation of the CH3NH3PbI3−xClx layer is observed as a darkening of the film and a corresponding decrease in the intensity of the red fluorescence, the rate of which depends on the polymer encapsulant used.
Fluorescence imaging reveals the same trends in the efficacy of the polymer encapsulants at inhibiting thermal degradation at all temperatures. Notably, after heating for 384 h at 60 °C, the red fluorescence of the PMMA-encapsulated film is still present, albeit with a slightly decreased intensity, further supporting its claim as the champion barrier layer (Fig. 4). Notably, FM presents the opportunity to monitor the degradation of the CH3NH3PbI3−xClx layer at significantly shorter timescales than possible by either visual observation or UV/Vis absorption spectroscopy. PC-coated films, for example, showed a significant decrease in the fluorescence intensity after 96 h (Fig. 6b), whereas there was no noteworthy decay in the UV/Vis absorption spectrum until 144 h. EC- and PC-encapsulated films both showed almost complete quenching of fluorescence after 24 h and 72 h respectively, 24 hours before any significant decay was observed in the UV/Vis absorption spectra. Interestingly at elevated temperatures (100 °C), this decrease in the fluorescence intensity is observed for all samples, including those encapsulated in PMMA, after only 48 h, thus indicating that even with an effective barrier layer to the local environment a significant decrease in the fluorescence intensity is observed (Fig. S9, ESI†).
The decrease in the fluorescence intensity can be attributed to a combination of effects, including the inherent degradation of the CH3NH3PbI3−xClx to PbI2 and to the agglomeration of crystal grains under ambient conditions to form larger aggregates.20 Sheng et al. demonstrated for CH3NH3PbBr3 that under storage ambient conditions for 2 weeks spontaneous grain growth was observed, ranging from relatively homogeneous <1 μm grains to variable grains sizes >2 μm, with the larger grains showing less intense emission.20 Spatially-resolved confocal fluorescence-lifetime imaging experiments on pure CH3NH3PbBr3 and CH3NH3PbI3 films have also shown that an increase in grain size leads to a decrease in the PL intensity.20,38 Grain growth has been attributed to the aggregation of smaller grains, essentially resulting in the formation of localised defect sites which enhance the rate of non-radiative recombination.20,38
Epifluorescence imaging does not afford us the spatial resolution required to identify this grain growth. However, while no visual discoloration or quantitative decrease in the UV/Vis absorption spectrum was observed for the PMMA-coated sample following thermal degradation at 100 °C for up to 190 h (Fig. 3d), FM revealed a similar pattern of film darkening to that observed for the other polymer encapsulants (Fig. S9, ESI†), despite no evidence of chemical degradation to PbI2. It seems likely then that the observed darkening of the CH3NH3PbI3−xClx layer is due to spontaneous grain growth (and associated formation of non-radiative trap sites) and that this growth is temperature dependent.
Simple epifluorescence microscopy is therefore a useful tool to observe rapid degradation of CH3NH3PbI3−xClx films through grain agglomeration and/or growth in a non-invasive way before it is observable by UV/Vis absorption spectroscopy or PXRD. In order to observe the lowest time detection limit for the onset of degradation by FM, pristine and EC-coated CH3NH3PbI3−xClx films were degraded at 60 °C and images were recorded at shorter time intervals of 1 h. Fig. 7 shows the images of the samples pre-degradation and when the first instance of film darkening is observed. An increase in fluorescence intensity is observed in the first 2 hours of the stability test, which is attributed to additional thermal annealing.36 Film darkening is observed for the uncoated CH3NH3PbI3−xClx sample after only 8 h, which is reasonable given that both UV/Vis absorption and PXRD measurements show complete degradation of the film to PbI2 within 24 h. For the analogous EC-coated films, significant film darkening was observed by FM after 29 h. In contrast, UV/Vis absorption studies only revealed the onset of thermal degradation after 50 h of heating (Fig. 3). These results demonstrate that FM imaging can be used as a rapid, non-invasive approach to screen the quality of perovskite films and to identify the onset of film degradation before other commonly used techniques.
 | ||
| Fig. 7 Thermal degradation of pristine and EC-encapsulated CH3NH3PbI3−xClx films at 60 °C as a function of time, as monitored by epifluorescence microscopy. (a) The uncoated CH3NH3PbI3−xClx film exhibits significant darkening after 8 h. (b) The EC-encapsulated film exhibits significant darkening after 29 h. After 2 h at 60 °C, the fluorescence of both films is more intense due to the removal of solvent and additional annealing. | ||
In order to confirm this technique is applicable to finished PSC devices, and not simply the perovskite layer itself, a non-encapsulated CH3NH3PbI3 cell was thermally degraded at 100 °C for 14 h to observe if the early onset degradation through crystal growth could (i) be observed by FM and (ii) negatively impacts on the current (I)–voltage (V) performance of the cell. Fig. 8 shows a comparison of the IV curves measured both before and after thermal degradation and the corresponding FM images of the device. Despite significant degradation of the cell power conversion efficiency from 8% to 1%, significant discolouration of the active cell area was not observed. However, this is perhaps unsurprising as the cell was fabricated using an anti-solvent method to promote rapid crystallisation of the active perovskite layer, which results in a significantly more uniform film quality than the previously investigated CH3NH3PbI3−xClx films. Upon heating, however, an increase in the number of pinhole type defects in the perovskite film was observed (Fig. 8b). This supports the results of Soufiani et al. who reported an increase in the number and size of pinhole defects for a CH3NH3PbI3 PSC aged in an inert atmosphere for 2 months.18 The large observed decrease in PCE for the thermally degraded cell here is likely to be due to degradation of the spiro-OMeTAD hole transport layer due to evaporation of the 4-tert-butylpyridine (T > 85 °C)39,40 and a combination of both cell degradation to PbI2 and the formation of pinhole defects. However, the possibility of using FM to rapidly assess the film quality in a non-invasive manner at any stage of the device fabrication process is an attractive option.
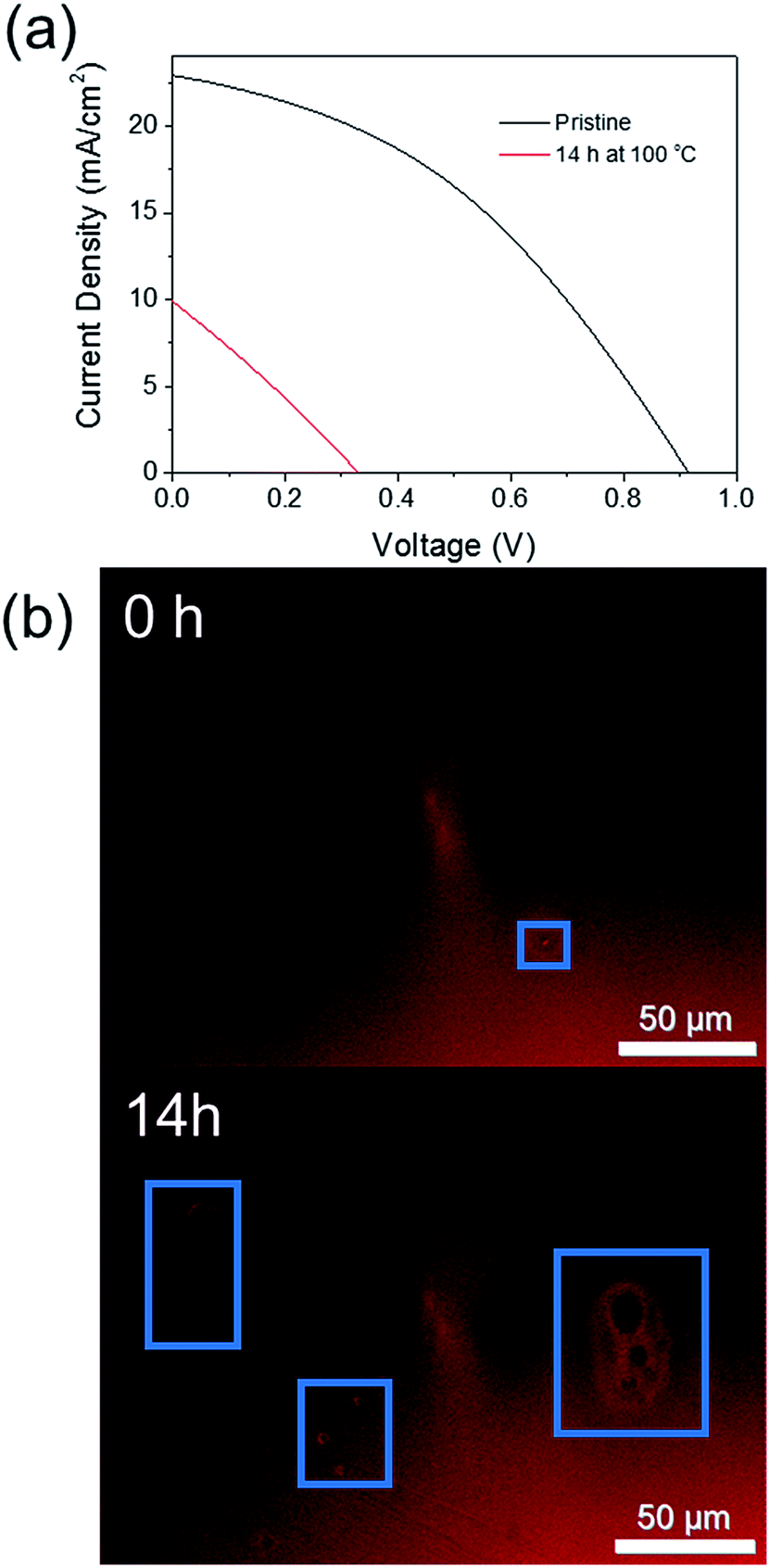 | ||
| Fig. 8 (a) I–V curves of a CH3NH3PbI3 PSC before (black) and after (red) heating at 100 °C for 14 h. (b) Fluorescence microscopy images of the CH3NH3PbI3 PSC before and after heating at 100 °C for 14 h. The blue box highlight the visible pinhole defects. | ||
4. Conclusions
The time-dependent thermal degradation of pristine and polymer-encapsulated CH3NH3PbI3−xClx films at 60 °C, 80 °C and 100 °C has been investigated in detail using a combination of PXRD, UV/Vis absorption spectroscopy and epifluorescence microscopy. PMMA demonstrated the best performance as an encapsulant at all temperatures, extending the lifetime of the CH3NH3PbI3−xClx film from 24 h to >400 h upon continuous heating at 60 °C. PC is the next best performing encapsulant, with total degradation observed at ∼380 h at 60 °C. In contrast, EC and PMP only moderately enhance the thermal stability compared to the uncoated CH3NH3PbI3−xClx film, with the onset of degradation occurring rapidly after 50 h at all temperatures. The results indicate that the physical properties of the polymer must be carefully considered when assessing a material as a potential encapsulant for PSCs. In addition, to a low WVTR, a Tg well above the typical device operating temperatures and/or the ambient conditions is advisable. Through our studies, we have observed that while PXRD and UV/Vis absorption provide quantitative analysis of the degradation of the CH3NH3PbI3−xClx film to PbI2, epifluorescence microscopy provides an indication of the early onset deterioration of the film quality due to grain growth and associated defect formation. This suggests that FM may be an attractive technique for rapid and non-invasive screening of device quality in real-time during the fabrication process.Acknowledgements
This work was supported by the Science Foundation Ireland under Grant No. 12/IP/1608 and the Engineering and Physical Sciences Research Council grant SPACE-Modules EP/M015254/1.References
- M. A. Green, K. Emery, Y. Hishikawa, W. Warta, E. D. Dunlop, D. H. Levi and A. W. Y. Ho-Baillie, Prog. Photovoltaics, 2017, 25, 3–13 Search PubMed.
- G. Niu, W. Li, F. Meng, L. Wang, H. Dong and Y. Qiu, J. Mater. Chem. A, 2014, 2, 705 CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, 84th edn, 2003 Search PubMed.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. Il Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- I. C. Smith, E. T. Hoke, D. Solis-Ibarra, M. D. McGehee and H. I. Karunadasa, Angew. Chem., Int. Ed., 2014, 53, 11232–11235 CrossRef CAS PubMed.
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 982–988 CAS.
- L. Sun, Nat. Chem., 2015, 7, 684–685 CrossRef CAS PubMed.
- B. W. Park, B. Philippe, T. Gustafsson, K. Sveinbjörnsson, A. Hagfeldt, E. M. J. Johansson and G. Boschloo, Chem. Mater., 2014, 26, 4466–4471 CrossRef CAS.
- G. Divitini, S. Cacovich, F. Matteocci, L. Cinà, A. Di Carlo and C. Ducati, Nat. Energy, 2016, 1, 15012 CrossRef CAS.
- S. N. Habisreutinger, T. Leijtens, G. E. Eperon, S. D. Stranks, R. J. Nicholas and H. J. Snaith, Nano Lett., 2014, 14, 5561–5568 CrossRef CAS PubMed.
- P. F. Ndione, W.-J. Yin, K. Zhu, S. H. Wei and J. J. Berry, J. Mater. Chem. A, 2015, 3, 21940–21945 CAS.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D'Haen, L. D'Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca, E. Mosconi, F. De Angelis and H. G. Boyen, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS PubMed.
- B. Philippe, B. W. Park, R. Lindblad, J. Oscarsson, S. Ahmadi, E. M. J. Johansson and H. Rensmo, Chem. Mater., 2015, 27, 1720–1731 CrossRef CAS.
- Y. Tian and I. G. Scheblykin, J. Phys. Chem. Lett., 2015, 6, 3466–3470 CrossRef CAS PubMed.
- A. E. Williams, P. J. Holliman, M. J. Carnie, M. L. Davies, D. A. Worsley and T. M. Watson, J. Mater. Chem. A, 2014, 2, 19338–19346 CAS.
- T. J. Wilderspin, F. De Rossi and T. M. Watson, Sol. Energy, 2016, 139, 426–432 CrossRef CAS.
- A. M. Soufiani, M. J. Y. Tayebjee, S. Meyer, A. Ho-Baillie, J. Sung Yun, R. W. MacQueen, L. Spiccia, M. A. Green and Z. Hameiri, J. Appl. Phys., 2016, 120, 035702 CrossRef.
- M. Okano, M. Endo, A. Wakamiya, M. Yoshita, H. Akiyama and Y. Kanemitsu, Appl. Phys. Express, 2015, 8, 102302 CrossRef.
- R. Sheng, X. Wen, S. Huang, X. Hao, S. Chen, Y. Jiang, X. Deng, M. A. Green and A. W. Y. Ho-Baillie, Nanoscale, 2016, 8, 1926–1931 RSC.
- Y. Han, S. Meyer, Y. Dkhissi, K. Weber, J. M. Pringle, U. Bach, L. Spiccia and Y.-B. Cheng, J. Mater. Chem. A, 2015, 3, 8139–8147 CAS.
- T. Leijtens, G. E. Eperon, S. Pathak, A. Abate, M. M. Lee and H. J. Snaith, Nat. Commun., 2013, 4, 2885 Search PubMed.
- J. W. Lee, D. H. Kim, H. S. Kim, S. W. Seo, S. M. Cho and N. G. Park, Adv. Energy Mater., 2015, 5, 1501310 CrossRef.
- F. C. Krebs, S. A. Gevorgyan and J. Alstrup, J. Mater. Chem., 2009, 19, 5442–5451 RSC.
- I. Hwang, I. Jeong, J. Lee, M. J. Ko and K. Yong, ACS Appl. Mater. Interfaces, 2015, 7, 17330–17336 CAS.
- I. S. Kim and A. B. F. Martinson, J. Mater. Chem. A, 2015, 3, 20092–20096 CAS.
- Y. Zhao, J. Wei, H. Li, Y. Yan, W. Zhou, D. Yu and Q. Zhao, Nat. Commun., 2016, 7, 10228 CrossRef CAS PubMed.
- J. He, C.-F. Ng, K. Young Wong, W. Liu and T. Chen, ChemPlusChem, 2016, 81, 1292–1298 CrossRef CAS.
- F. Bella, G. Griffini, J.-P. Correa-Baena, G. Saracco, M. Grätzel, A. Hagfeldt, S. Turri and C. Gerbaldi, Science, 2016, 354, 203–206 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- Physical properties table, https://static.thermoscientific.com/images/D20826%7E.pdf, accessed December 2016.
- L. W. McKeen, Permeability Properties of Plastics and Elastomers, William Andrew, Norwich, 3rd edn, 2011 Search PubMed.
- J. E. Mark, Polymer Data Handbook, Oxford University Press, New York, 1st edn, 1999 Search PubMed.
- Ethyl Cellulose, http://www.sigmaaldrich.com/catalog/product/aldrich/200689, accessed January 2017.
- B. G. Rånby, K. S. Chan and H. Brumberger, J. Polym. Sci., 1962, 58, 545–552 CrossRef.
- G. E. Eperon, S. N. Habisreutinger, T. Leijtens, B. J. Bruijnaers, J. J. Van, R. A. J. Janssen, A. Petrozza and H. J. Snaith, ACS Nano, 2015, 9, 9380–9393 CrossRef CAS PubMed.
- A. McEvoy, L. Castaner and T. Markvart, Solar Cells: Materials, Manufacture and Operation, Elsevier, Amsterdam, 2nd edn, 2013 Search PubMed.
- M. Yang, Y. Zeng, Z. Li, D. H. Kim, C.-S. Jiang, J. van de Lagemaat and K. Zhu, Phys. Chem. Chem. Phys., 2017, 19, 5043–5050 RSC.
- C. D. Bailie, E. L. Unger, S. M. Zakeeruddin, M. Grätzel and M. D. McGehee, Phys. Chem. Chem. Phys., 2014, 16, 4864–4870 RSC.
- S. Wang, M. Sina, P. Parikh, T. Uekert, B. Shahbazian, A. Devaraj and Y. S. Meng, Nano Lett., 2016, 16, 5594–5600 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical structures of polymer encapsulants; PXRD of bare and polymer encapsulated CH3NH3PbI3−xClx films; photographs of a CH3NH3PbI3−xClx film before and after thermal degradation for 432 h at 60 °C; UV/Vis absorption spectra of CH3NH3PbI3−xClx films before and after polymer coating; UV/Vis transmission spectra of bare polymer films; UV/Vis absorption spectra of bare and polymer CH3NH3PbI3−xClx films degraded at 80 °C as a function of time; FM images of bare, and encapsulated CH3NH3PbI3−xClx films thermally degraded at 80 and 100 °C as a function of time. See DOI: 10.1039/c7ra06002e |
| This journal is © The Royal Society of Chemistry 2017 |