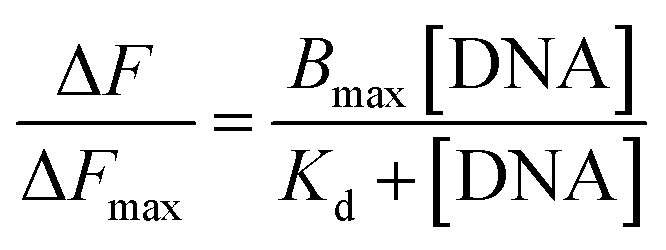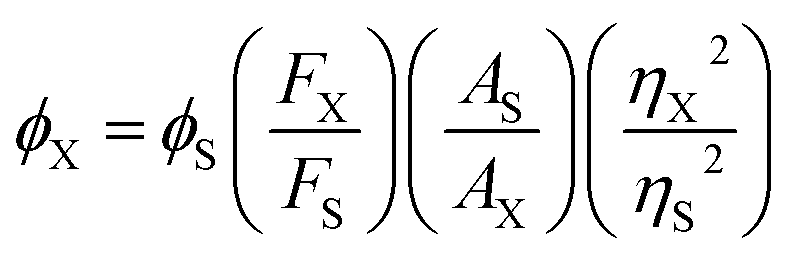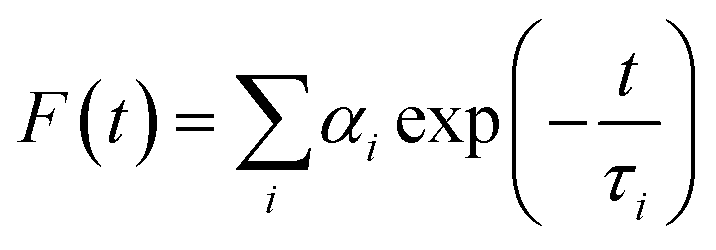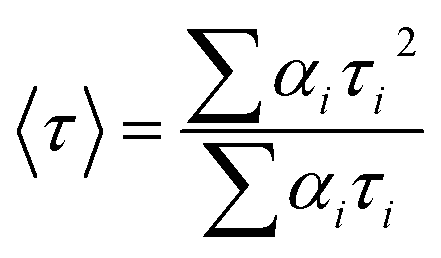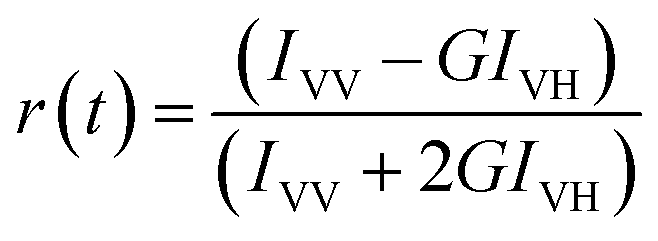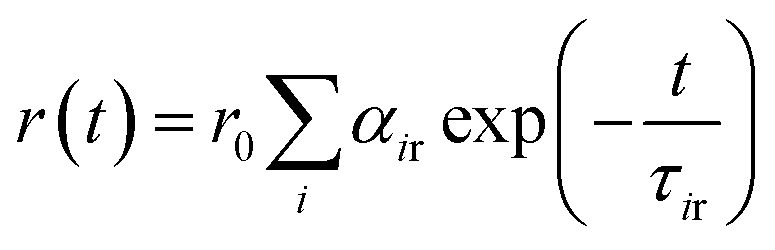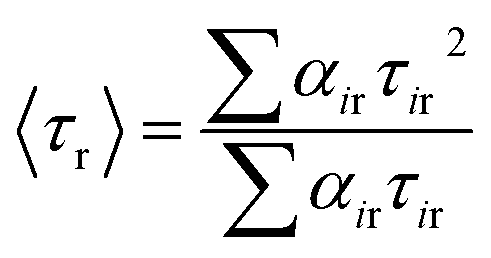DOI:
10.1039/C7RA05930B
(Paper)
RSC Adv., 2017,
7, 37230-37240
Exploring the preferential interaction of quercetin with VEGF promoter G-quadruplex DNA and construction of a pH-dependent DNA-based logic gate†
Received
26th May 2017
, Accepted 21st July 2017
First published on 27th July 2017
Abstract
G-Quadruplex DNA (G4–DNA) is one of the most important non-canonical nucleic acid structures. G4–DNA forming sequences are present in different crucial genomic regions and are abundant in promoter regions of several oncogenes. Therefore, G4–DNA is an important target for anticancer drugs and hence binding interactions between G4–DNA and small molecule ligands are of great importance. Since G4–DNA is a highly polymorphic structure, it is important to identify ligand molecules which preferentially target a particular quadruplex sequence in comparison to other quadruplexes. In the present study, different spectroscopic techniques have been used to explore the interaction of the dietary plant flavonoid quercetin (Que) with various G4–DNA structures (VEGF, c-MYC, c-KIT1, c-KIT2, h-TELO) along with double stranded (duplex) DNA. We found that Que shows preferential interaction with VEGF G4–DNA compared to other G4–DNA structures as well as duplex DNA. This identifies Que as an appropriate natural product based ligand for targeting VEGF G4–DNA. We also observed pH dependent interaction of Que with VEGF G4–DNA, based on which we have designed a complex Boolean logic gate exploiting Que as a sensing molecule.
1. Introduction
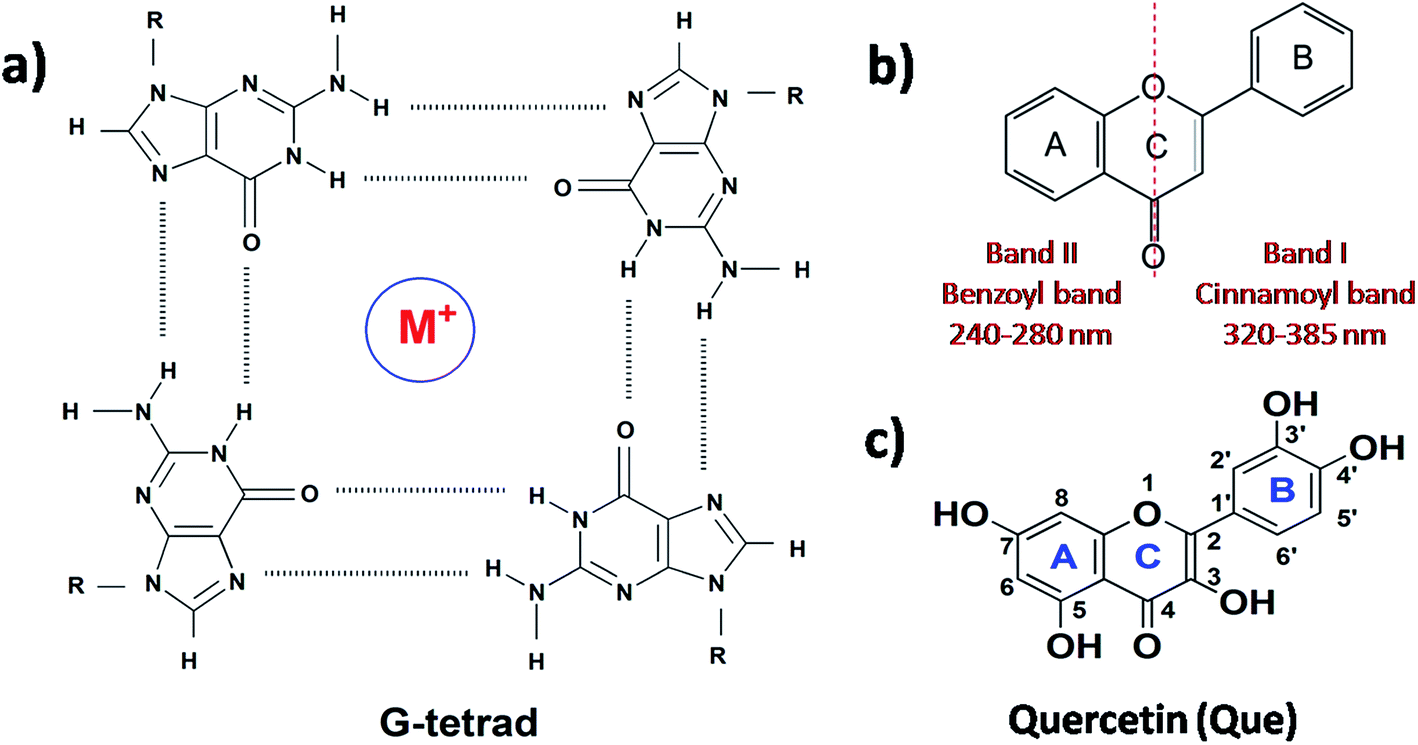
Deoxyribonucleic acid (DNA) is known to adopt a right-handed double helical B-DNA structure, where the two complementary strands are held together by Watson–Crick base pairs. However, a number of alternative non-B-DNA structures have been identified and among them, G-quadruplex (G4) has captivated considerable attention. G4 structures are formed in guanine (G)-rich sequences of DNA that have a high propensity to self-associate into planar G-quartets, which is the building block of G4 structures. G-Quartet is a planar association of four guanosines that are cyclically bound together by Hoogsteen hydrogen bonds. G-Quartets can stack on top of each other to form four-stranded G4 structures. The overall structure is also stabilized by the presence of monovalent cations, such as K+ and Na+ (Fig. 1a).1,2 G4–DNA forming nucleic acid sequences have been found in a number of important regions of genomes such as telomeric ends, as well as in several oncogene promoters, suggesting that G4 structures may play a crucial role in the control of a variety of cellular processes, including telomere maintenance, replication, transcription and translation.3,4 G4 structures have been the focus of enormous interest in recent years because their formation is widely associated with various human diseases such as cancer, neurodegeneration etc.5,6 Therefore, G4–DNA structures are recognized as promising targets for drug binding. G4–DNA structures exhibit extensive structural diversity and polymorphism compared to duplex DNA. In particular, G4–DNA structures exhibit different topologies depending on several factors such as length and sequence of the DNA strands, the number of oligonucleotide strands, the directionality of strands, the angles of the glycosidic bonds, the size and type of intervening loops, and environmental factors, such as cations, molecular crowding etc.7–10 Therefore, drug design must be directed not only to differentiate between G4 and duplex DNA, but also to ensure preferential recognition of particular G4–DNA structure. The quest for G4–DNA ligands, which can discriminate a particular quadruplex over other quadruplexes, is of significant importance in view of their promising prospects for exploiting G4–DNA based therapeutic interventions at the genomic level. Since G4 structures have been reported to be stable and detectable in human and other mammalian cells in vivo,11–13 the identification and sensing of these sequences in cells using natural fluorescent probes have attracted significant interest. Besides interest in their biological significance, G4–DNA structures have provided the basis for the development of DNA-based sensing platforms and biomolecular logic gate construction.14–16 As molecular logic gates detect external inputs such as ions or small biological molecules, the logic gates in themselves can act as sensors for these inputs.
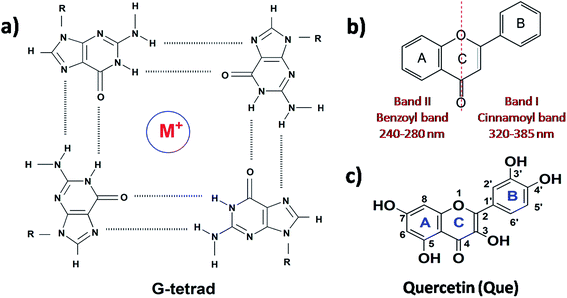 |
| | Fig. 1 Structure of (a) G-tetrad, (b) flavonoid backbone and (c) quercetin. | |
Different classes of small molecules including alkaloids,17 curcuminoids,18 flavonoids,19,20 and acriflavines21 have been investigated as antiparallel and/or mixed-hybrid G4–DNA specific ligand in comparison to double-stranded DNA. Studies on the interactions of alkaloids and flavonoids of different classes (flavone, isoflavone, flavanone and flavan-3-ol) with parallel G4–DNA sequence have also been reported.22,23 The present research has been motivated by our interest in assessing the prospects of plant flavonoids as ligands for promoter G4–DNA structures. Flavonoids are naturally occurring polyphenolic compounds that are found in different citrus fruits, vegetables, seeds etc. and are therefore consumed in considerable amounts through the human diet.24,25 From the chemical context, flavonoids are low molecular weight compounds composed of a three-ring structure with various substitution patterns. This basic structure is comprised of two benzene rings (A and B) linked through a heterocyclic pyran or pyrone (with a double bond) ring (C) in the middle (Fig. 1b).24 Various studies have suggested the protective effects of flavonoids against different diseases such as cardiovascular diseases, cancers, neurodegenerative and other age-related ailments.24,25 The high pharmacological potency and low cytotoxicity exhibited by flavonoids make them viable alternatives to conventional therapeutic drugs. For the present study, we chose quercetin (Que, Fig. 1c) which is the most abundant dietary plant flavonoid, which has been found to possess several beneficial effects on human health. Moreover, Que has remarkable spectroscopic interest because of its unique ‘two color’ fluorescence properties which can be useful for sensing G4–DNA. Free Que exhibits low fluorescence emission in aqueous buffer. However, strong fluorescence signals are observed upon binding to G4–DNA and other target biomolecules.19,26
In the present work, we have made a comparison on the binding of Que with different G4–DNA sequences namely parallel promoter G4–DNA (VEGF, c-MYC, c-KIT1, c-KIT2), hybrid G4–DNA (h-TELO) along with duplex DNA. Our studies reveal that Que exhibits preferential recognition of VEGF promoter G4–DNA in comparison to the other G4–DNA sequences and duplex DNA studied. To our knowledge, this is the first report on the interaction between VEGF G4–DNA and Que. Furthermore, we have designed a complex biomolecular logic gate device based on pH-dependent interaction between Que and VEGF G4–DNA structure. Here, we have employed UV-visible absorption, steady-state and time-resolved fluorescence and circular dichroism (CD) spectroscopy along with isothermal titration calorimetry (ITC) measurement to explore the molecular recognition and interaction scenario and hence the prospect of Que as an extrinsic non-covalent fluorescent marker for VEGF G4–DNA structure.
2. Materials and methods
2.1. Materials and preparation of stock solutions
DNA sequences (Table S1, ESI†) and the flavonoid Que were procured from Sigma Aldrich and were used as obtained. The solvents used were of spectroscopic grade. The desalted oligonucleotides were dissolved in double distilled water and stored at 4 °C. G4–DNA structure was prepared by taking requisite amount of DNA from the main stock to the buffer solution and the resulting solutions were annealed by heating at 95 °C for 5 min. The solutions were then slowly cooled to room temperature and equilibrated overnight at 4 °C. Stock solution of Que was prepared in methanol and the final experimental concentrations of Que were kept on the order of 10−6 M with methanol <1% (v/v). All experiments were carried out using 50 mM KCl, 10 mM KH2PO4, and 1 mM K2EDTA (pH 7.4) at room temperature (25 °C).
2.2. UV-Vis absorption studies
UV-Vis absorption spectra of Que in the absence and presence of DNA were recorded from 230 to 530 nm with Jasco V-630 spectrophotometer (Jasco International Co. Ltd.) using quartz cuvette of 1 cm path length. Experiment was carried out by keeping the concentration of Que constant and was subsequently titrated with increasing concentration of different DNA structures. The spectra were scanned with a scanning speed of 54 nm min−1 and a data interval of 1 nm. The concentrations of DNA and Que during spectrophotometric titrations are mentioned in the relevant discussion.
2.3. Binding stoichiometry of the complexation: Job plot analysis
The continuous variation method (Job plot)27,28 was employed to determine the binding stoichiometry of Que–DNA complexes in each case from fluorescence spectroscopy. The fluorescence intensity of the Que–DNA complex at 540 nm was recorded with concentrations of both DNA and Que being varied while the sum of their concentrations was kept constant. The emission intensity at 540 nm was plotted against the corresponding Que mole fraction and fitted to a Gaussian function using Origin 8.0. The value of mole fraction corresponding to the peak of the Gaussian curve gives the stoichiometry of the complexes.
2.4. Steady-state spectrofluorometric studies
The fluorescence emission spectra were recorded with a Jasco FP-8500 fluorescence spectrometer (Jasco International Co. Ltd.) using quartz cuvette of 1 cm path length from 490 to 650 nm (only tautomer emission) at room temperature. During emission spectral measurements both the excitation and emission spectral bandwidths were set to 5 nm and the excitation wavelength for Que was set at 370 nm. Experiment was carried out by keeping the concentration of Que constant and was subsequently titrated with increasing concentrations of different DNA structures. Emission spectra were recorded with a scanning speed of 100 nm min−1 and a response time of 50 ms. The binding constant (Ka = 1/Kd) was calculated from the plot of ΔF/ΔFmax at 540 nm versus concentration of DNA using the following equation,20
where Kd is the dissociation constant, ΔF is the change in emission spectrum at 540 nm after each addition of DNA, ΔFmax is the change in emission spectrum when Que is totally bound to DNA, and Bmax is the maximum specific binding, in the same units as ΔF/ΔFmax.
2.5. Quantum yield calculation
The fluorescence quantum yields were calculated relative to quinine sulphate in 0.1 N H2SO4 (ϕ = 0.54) using the formula,29
where FX and FS are the integrated area under the fluorescence emission profiles of the sample and standard solutions, respectively, and, AX and AS are the corresponding absorbance values at the excitation wavelength and η denotes the refractive index of the solvent medium.
2.6. Time-resolved fluorescence studies
Fluorescence lifetimes were measured by the Time Correlated Single Photon Counting (TCSPC) technique on a FluoroCube-01-NL spectrometer (Horiba Jobin Yvon). Laser-diode was used as an excitation source and Que was excited at 375 nm. The instrument response function (IRF) was measured at the excitation wavelength using Ludox (colloidal silica) as the scatterer. The full width at half-maximum (fwhm) of the IRF was 108 ps. To optimize the signal-to-noise ratio, 5000 photon counts were collected in the peak channel. The signals were collected at the magic angle of 54.7° to eliminate any considerable contribution from polarisation artifacts.29 The emission decay data at 540 nm were analyzed using the software, DAS-6 (Decay Analysis Software 6), provided with the TCSPC instrument and was fitted with a multi-exponential decay function:29
where F(t) is the fluorescence intensity at time t, and αi and τi are the pre-exponential factor and decay time respectively, corresponding to the ith decay. The goodness of the fits was evaluated by χ2 criteria and visual inspection of the residuals of the fitted function to the data. The mean (average) fluorescence lifetimes (〈τ〉) were calculated using the following equation:29
For time-resolved fluorescence anisotropy decay measurements, the polarized fluorescence decays of the parallel (IVV) and perpendicular (IVH) emission polarizations with respect to the vertical excitation polarization were first collected at the emission maxima of the fluorophore. The anisotropy decay function r(t) was constructed from these IVV and IVH decays using the following equation:29
in which
G is the correction factor for the detector sensitivity of the instrument. Anisotropy decay functions were also deconvoluted on DAS-6 decay analysis software using a similar multi-exponential model using the equation:
29in which
αir is the pre-exponential factor corresponding to the
ith rotational time constant (
τir), and
r0 is the limiting anisotropy at time
t = 0. Mean (average) rotational correlation times (〈
τr〉) were calculated using the following equation:
29
2.7. Circular dichroism (CD) spectroscopic studies
Circular dichroism (CD) spectra were recorded on a JASCO J-815 spectropolarimeter (Jasco International Co. Ltd.) using a cuvette of 1 mm path length at room temperature. Each spectrum was the average of three scans, in which the scan range was from 200 to 350 nm with an interval of 1 nm at a scan rate of 100 nm min−1. The concentration of DNA was kept constant while varying the concentration of Que. The concentrations of DNA and Que during CD measurements are mentioned in the relevant discussion. The corresponding buffer blanks were used to correct background.
2.8. Isothermal titration calorimetric (ITC) studies
ITC experiments were performed on a GE Microcal ITC 200, (Northampton, USA) microcalorimeter. All samples and buffer were degassed prior to titration to ensure that no bubble formation takes place. Preannealed DNA (300 μM) was injected into a fixed volume of Que (25 μM) after an interval of 240 s. A corresponding blank experiment was employed by injecting an equal volume of DNA into the buffer solution. The heat associated with the blank experiment was subtracted from the heat change associated with the Que–DNA complex, to reveal the actual heat change related to the formation of the Que–DNA complex. The isotherms were analyzed using in-built Origin 7.0 software. The data points obtained best fitted the ‘one set of sites’ model. Equilibrium constant (Ka), change in enthalpy (ΔH), and change in entropy (ΔS) associated with Que–DNA complex formation were evaluated after fitting the isotherms. The Gibbs free energy (ΔG) was calculated using the equation: ΔG = ΔH − TΔS.
3. Results and discussion
3.1. UV-visible absorption studies
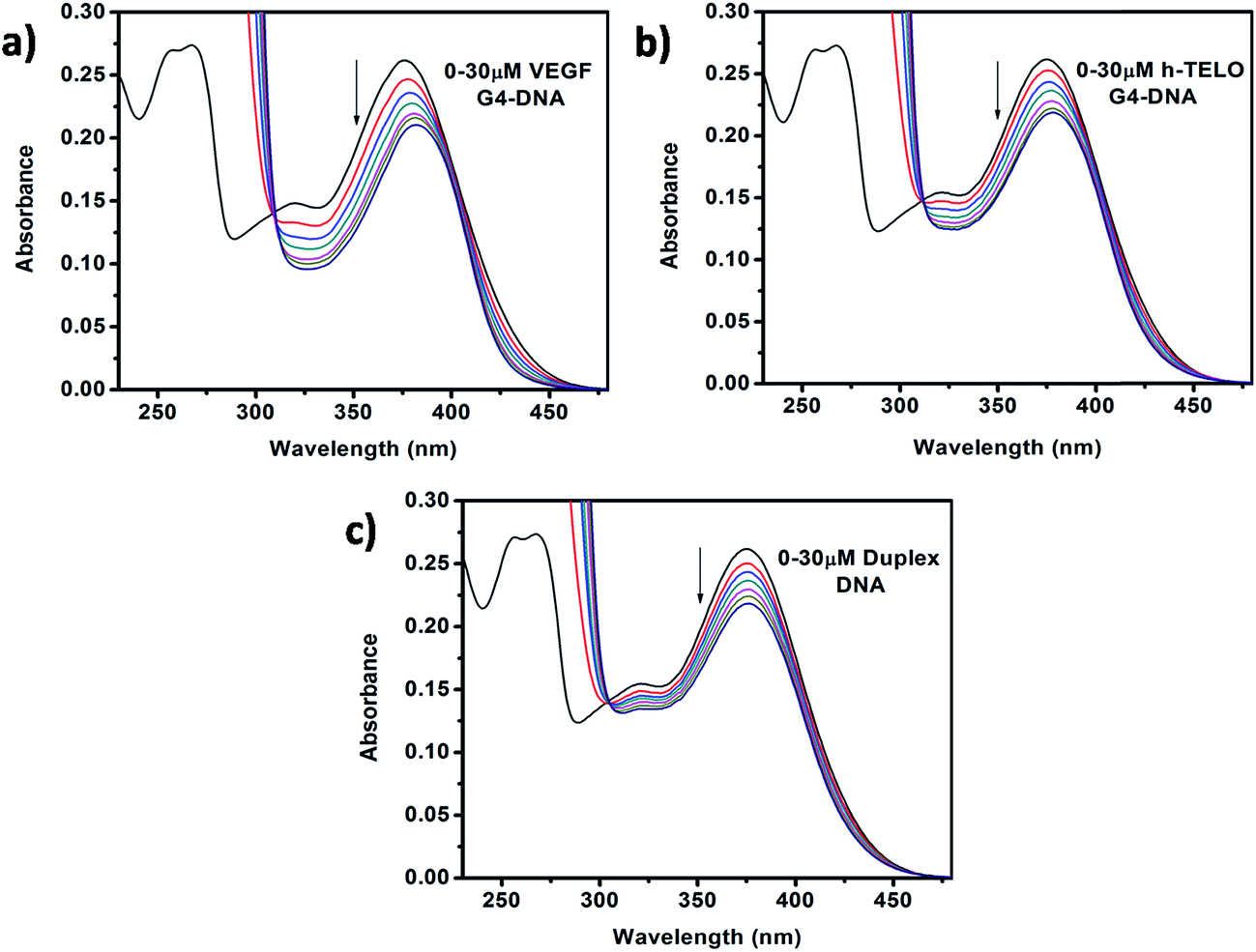
The absorption profile of Que shows absorption maxima at 375 nm (band I) and 260 nm (band II) which is in good agreement with previous report.30 Band I, located in the wavelength range of 300–400 nm, is attributed to the light absorption of the cinnamoyl system (B + C ring), and band II located in the wavelength range of 240–300 nm is related to the absorption of the benzoyl system (A + C ring) (Fig. 1b).31 Upon addition of DNA to Que solution in aqueous buffer, the absorption spectra of Que show a decrement in absorbance (hypochromism) along with a bathochromic (red) shift of the absorption maximum (Fig. 2 and S1, ESI†), indicating that Que senses a hydrophobic environment within the DNA matrix where it is protected against the polar aqueous environment.21 The extent of hypochromism as well as bathochromism differs significantly depending on the DNA structures and it is greater for the VEGF G4–DNA compared to the duplex DNA and other G4–DNA structures that have been studied (Table 1). The observed bathochromic effect arises due to the coupling of the empty π antibonding orbital (π*) of Que with the π bonding orbital of the DNA base pairs and form a π–π* conjugation resulting in the decrease of the π → π* transition energy, which causes a red shift in the absorption band. Meanwhile, the empty π*-orbital of Que is partially filled with electrons and thus decreases the transition probability, which causes hypochromism because the lower the probability, the smaller the molar absorption coefficient.32 As reported earlier, the intercalative binding of small molecules to DNA is characterized by large changes in the absorbance (hypochromism ≥ 35%) and an appreciable shift in the absorption spectrum (red shift Δλ ≥ 15 nm) while outside binders display smaller red shifts (Δλ ≤ 8 nm).33 Therefore, our results indicate that an external binding mode exists between Que and DNA structures. However, in the present context we observed that the DNA-induced absorption spectral changes of Que lack an isosbestic point, indicating that 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry is not maintained. It indicates that more than one type of interactions may be present during the binding process. Hence, we have not employed the absorption study for the determination of the binding constant.34,35 We have also performed continuous variation method (Job plot) to determine the binding stoichiometry of Que–DNA complexes and we found that 1:1 binding stoichiometry is not maintained, which corroborates our spectrophotometric results (Fig. S2, ESI†).
:1 binding stoichiometry is not maintained. It indicates that more than one type of interactions may be present during the binding process. Hence, we have not employed the absorption study for the determination of the binding constant.34,35 We have also performed continuous variation method (Job plot) to determine the binding stoichiometry of Que–DNA complexes and we found that 1:1 binding stoichiometry is not maintained, which corroborates our spectrophotometric results (Fig. S2, ESI†).
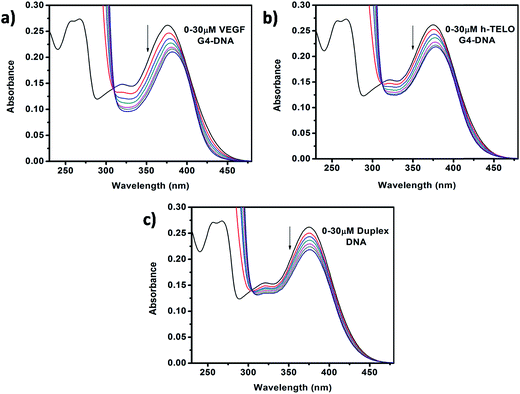 |
| | Fig. 2 Absorption spectra of Que (15 μM) in the absence and presence of successive additions of (a) VEGF G4–DNA, (b) h-TELO G4–DNA, and (c) duplex DNA. | |
Table 1 Spectral and binding parameters of interactions between Que and different DNA structures obtained from absorption and steady-state fluorescence studies
| Sample |
Absorption studies |
Fluorescence studies |
| Hypochromicity (%) |
Red shift (nm) |
Fluorescence enhancement (fold) |
Binding constant (M−1) |
Quantum yield (×10−3) |
| Que + VEGF |
22.2 |
6 |
8.46 |
9.40 × 104 |
1.62 |
| Que + c-MYC |
19.8 |
6 |
2.29 |
4.20 × 104 |
0.45 |
| Que + c-KIT1 |
18.9 |
5 |
2.93 |
3.10 × 104 |
0.59 |
| Que + c-KIT2 |
19.8 |
3 |
2.69 |
3.05 × 104 |
0.53 |
| Que + h-TELO |
16.8 |
3 |
2.12 |
4.04 × 104 |
0.42 |
| Que + duplex |
16.5 |
1 |
1.53 |
2.20 × 104 |
0.27 |
3.2. Steady-state spectrofluorometric studies
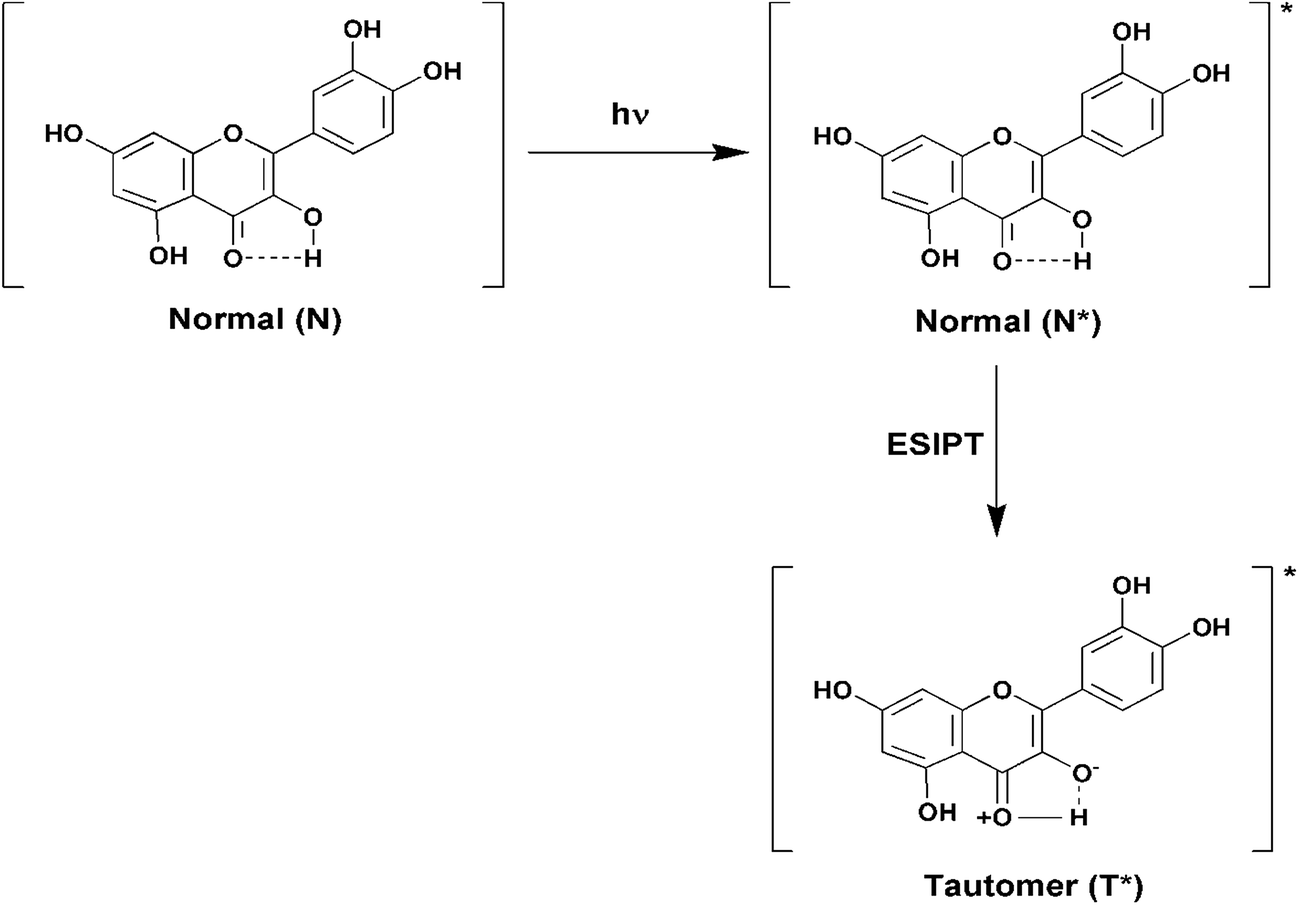
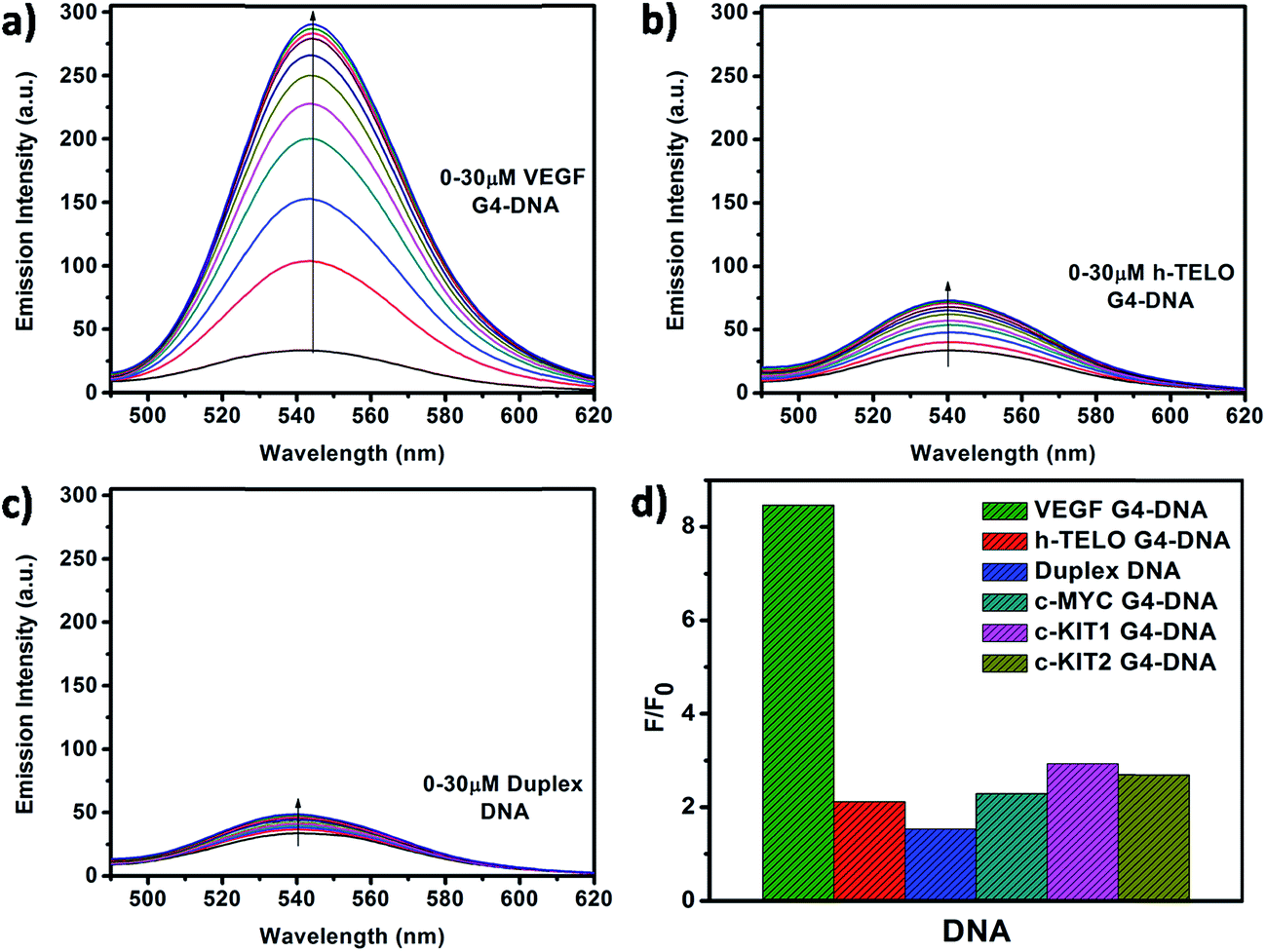
Fluorescence emission spectra of Que were monitored in the absence and presence of increasing concentrations of different forms of DNA. In aqueous buffer free Que exhibits very low fluorescence intensity with emission maximum at ∼540 nm corresponding to the photoinduced tautomer species of Que. It is known that flavonols which have a C(3)–OH group adjacent to the C(4)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O readily undergo photoinduced excited state intramolecular proton transfer (ESIPT), exhibiting large Stokes shifted, high fluorescence yield tautomer emission in non-polar environment (Fig. 3).36 In Que both 5-OH and 3-OH groups are present together and they are located adjacent to the C(4)O group (Fig. 1c). The C(4)O⋯HO–C(5) hydrogen bond interferes with the C(4)O⋯HO–C(3) hydrogen bond which hinders ESIPT process, resulting in low tautomer fluorescence emission in free states of Que. Binding of Que to biomolecular targets disrupts the internal hydrogen bond involving the 5-OH group. Upon photo-excitation of the bound Que, the ESIPT process across the C(4)O⋯HO–C(3) hydrogen bond is facilitated, which leads to activation of pronounced fluorescence from the intrinsically weakly fluorescent Que. The binding environment drastically modulates the ESIPT process and related photophysical parameters of fluorescence.19,26,36 In the present study, we observed that in the presence of DNA there is a significant enhancement in the tautomer fluorescence emission of Que, which clearly depicts the binding interaction between Que and DNA. Further, we observed that Que has preferential affinity towards VEGF G4–DNA, resulting in ∼9-fold enhancement in the tautomer fluorescence intensity. This enhancement of fluorescence is much higher compared to duplex and other G4–DNA structures (Table 1, Fig. 4 and S3, ESI†). This observation thus indicates that Que molecules are predominantly located in a less polar and less protic environment within the VEGF G4–DNA matrix which makes non-radiative deactivation processes less operative and favors the ESIPT process of Que resulting in enhanced tautomer emission. We also estimated the binding constant (Kb) values for different DNA structures by plotting the Que fluorescence changes with concentration of DNA and we found that the binding affinity of Que is much higher for VEGF G4–DNA compared to other DNA structures (Table 1 and Fig. S4, ESI†). Furthermore, we determined fluorescence quantum yield (ϕ) values of Que in free and DNA-bound conditions. We found that quantum yield of free Que is very low (0.16 × 10−3) in aqueous buffer which is consistent with previous studies.37 When Que binds to VEGF G4–DNA the quantum yield becomes ∼10-fold, relative to the value for the free fluorophore in aqueous buffer solution. This dramatic enhancement in quantum yield can be explained as being due to the C(4)O⋯HO–C(5) hydrogen bond being disrupted and C(4)O⋯HO–C(3) hydrogen bond being facilitated, thus promoting the ESIPT process and activating Que fluorescence.19,26,36 Among the various DNA structures we examined, the highest quantum yield enhancement is observed for VEGF G4–DNA environment (Table 1).
O readily undergo photoinduced excited state intramolecular proton transfer (ESIPT), exhibiting large Stokes shifted, high fluorescence yield tautomer emission in non-polar environment (Fig. 3).36 In Que both 5-OH and 3-OH groups are present together and they are located adjacent to the C(4)O group (Fig. 1c). The C(4)O⋯HO–C(5) hydrogen bond interferes with the C(4)O⋯HO–C(3) hydrogen bond which hinders ESIPT process, resulting in low tautomer fluorescence emission in free states of Que. Binding of Que to biomolecular targets disrupts the internal hydrogen bond involving the 5-OH group. Upon photo-excitation of the bound Que, the ESIPT process across the C(4)O⋯HO–C(3) hydrogen bond is facilitated, which leads to activation of pronounced fluorescence from the intrinsically weakly fluorescent Que. The binding environment drastically modulates the ESIPT process and related photophysical parameters of fluorescence.19,26,36 In the present study, we observed that in the presence of DNA there is a significant enhancement in the tautomer fluorescence emission of Que, which clearly depicts the binding interaction between Que and DNA. Further, we observed that Que has preferential affinity towards VEGF G4–DNA, resulting in ∼9-fold enhancement in the tautomer fluorescence intensity. This enhancement of fluorescence is much higher compared to duplex and other G4–DNA structures (Table 1, Fig. 4 and S3, ESI†). This observation thus indicates that Que molecules are predominantly located in a less polar and less protic environment within the VEGF G4–DNA matrix which makes non-radiative deactivation processes less operative and favors the ESIPT process of Que resulting in enhanced tautomer emission. We also estimated the binding constant (Kb) values for different DNA structures by plotting the Que fluorescence changes with concentration of DNA and we found that the binding affinity of Que is much higher for VEGF G4–DNA compared to other DNA structures (Table 1 and Fig. S4, ESI†). Furthermore, we determined fluorescence quantum yield (ϕ) values of Que in free and DNA-bound conditions. We found that quantum yield of free Que is very low (0.16 × 10−3) in aqueous buffer which is consistent with previous studies.37 When Que binds to VEGF G4–DNA the quantum yield becomes ∼10-fold, relative to the value for the free fluorophore in aqueous buffer solution. This dramatic enhancement in quantum yield can be explained as being due to the C(4)O⋯HO–C(5) hydrogen bond being disrupted and C(4)O⋯HO–C(3) hydrogen bond being facilitated, thus promoting the ESIPT process and activating Que fluorescence.19,26,36 Among the various DNA structures we examined, the highest quantum yield enhancement is observed for VEGF G4–DNA environment (Table 1).
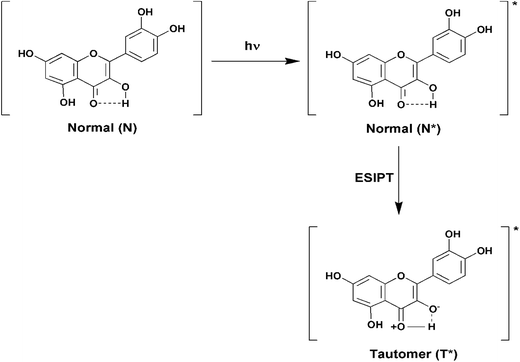 |
| | Fig. 3 Schematic diagram depicting the ground and excited (denoted by an asterisk (*)) states of normal and tautomer forms of Que. | |
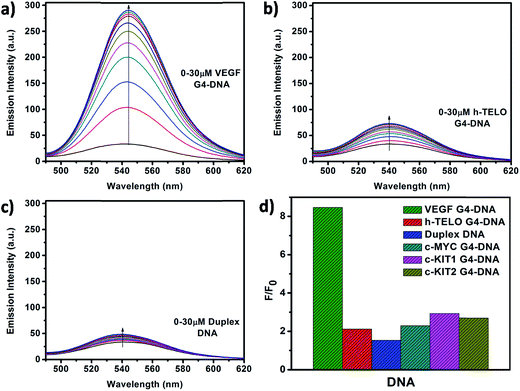 |
| | Fig. 4 Fluorescence emission spectra of Que (10 μM) with increasing concentrations of (a) VEGF G4–DNA, (b) h-TELO G4–DNA, and (c) duplex DNA. Figure (d) represents the values of F/F0 at 540 nm for Que in different DNA environments. (λex = 370 nm). | |
3.3. Time-resolved fluorescence studies
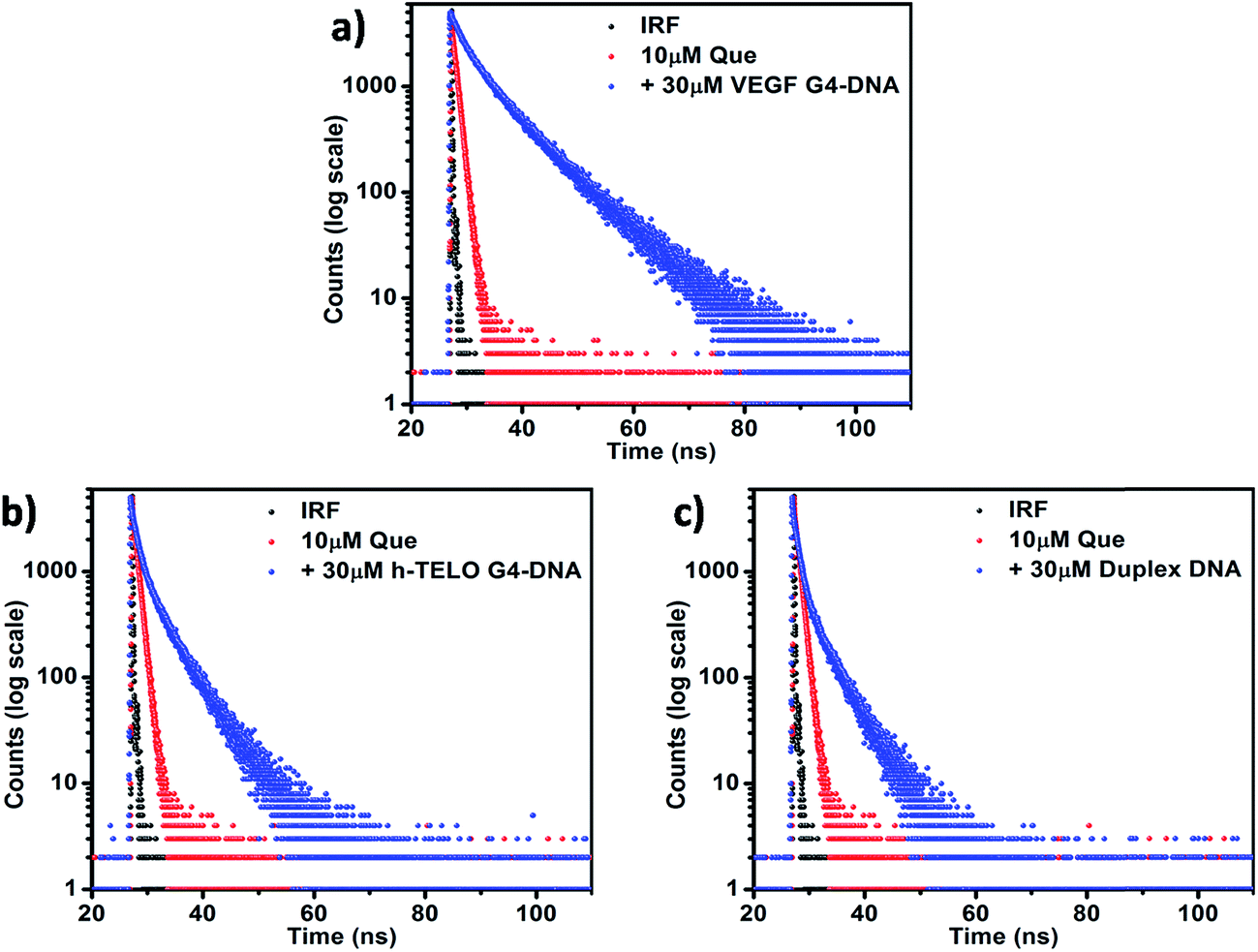
Fluorescence lifetime measurement helps in monitoring the excited state interactions between fluorophores and DNA. In aqueous buffer, Que exhibits a bi-exponential decay pattern, consisting of a fast component (0.02 ns) ascribed to the free ligand (having a contribution of ∼91%) and another slower component (0.86 ns) attributed to the solvated cluster of the ligand (having a contribution of ∼9% only). The time-resolved fluorescence decay of the DNA-bound Que exhibits a complex tri-exponential pattern with three distinct lifetime components. Such multi-exponential decay of fluorophore molecules in the presence of DNA has been previously reported in the literature.38,39 The first component τ1 having the highest contribution may be attributed to the free (i.e. unbound) Que, which shows no appreciable change in magnitude but a decrement in amplitude in the presence of DNA (Table 2). The third component τ3, which shows a large increment in magnitude coupled with an increment in the amplitude in the presence of DNA, is predicted to be the outcome of the binding interaction between Que and DNA (Table 2). The second component τ2 having an intermediate lifetime is of special interest. The fact that its magnitude is significantly larger than that of the free Que in solution implies that the corresponding species is an outcome of a different binding phenomenon between the Que and DNA (Table 2). The presence of this component is also consistent with the absence of an isosbestic point in the absorption spectra indicating more than one mode of binding. Moreover if we consider the average fluorescence lifetime (〈τ〉) as a reliable parameter for exploring the interaction between Que and DNA, it is observed that, 〈τ〉 of Que increases in the presence of DNA molecules which confirms the occurrence of a binding interaction. The reduced polarity around Que in DNA environment is also reflected in a perceptible increment in the fluorescence lifetime. When Que molecule is trapped within the hydrophobic regions of DNA, lesser accessibility to water leads to a suppression of the nonradiative decay channels, thereby producing an enhancement of the lifetime of the bound Que.38 The observed 〈τ〉 of Que in VEGF G4–DNA environment (6.98 ns) is much higher than that of other G4–DNA structures as well as duplex DNA, which further corroborates that Que experiences more hydrophobicity in VEGF G4–DNA environment compared to other DNA structures (Table 2, Fig. 5 and S5, ESI†).
Table 2 Fluorescence decay parameters of Que tautomer species in aqueous buffer and in the presence of different DNA structures
| Sample |
τ1 (ns) |
α1 |
τ2 (ns) |
α2 |
τ3 (ns) |
α3 |
〈τ〉 (ns) |
χ2 |
| Que |
0.02 |
0.91 |
0.86 |
0.09 |
|
|
0.70 |
1.06 |
| Que + VEGF |
0.08 |
0.24 |
3.36 |
0.44 |
8.90 |
0.32 |
6.98 |
1.04 |
| Que + c-MYC |
0.03 |
0.81 |
1.19 |
0.11 |
4.24 |
0.08 |
3.23 |
1.04 |
| Que + c-KIT1 |
0.05 |
0.59 |
1.50 |
0.26 |
4.35 |
0.15 |
3.19 |
1.06 |
| Que + c-KIT2 |
0.08 |
0.55 |
1.38 |
0.39 |
6.20 |
0.06 |
3.20 |
1.16 |
| Que + h-TELO |
0.03 |
0.80 |
1.12 |
0.15 |
4.98 |
0.05 |
3.24 |
1.10 |
| Que + duplex |
0.02 |
0.86 |
0.95 |
0.11 |
4.51 |
0.03 |
2.76 |
0.99 |
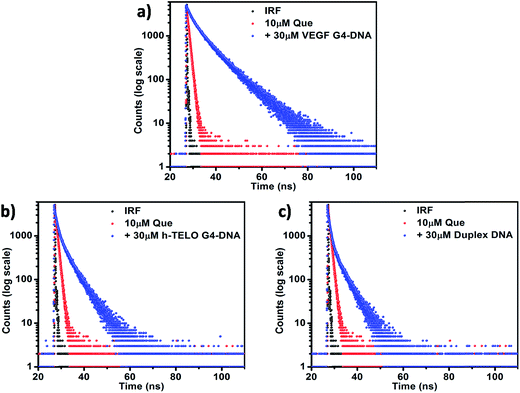 |
| | Fig. 5 Fluorescence lifetime decay profiles of 10 μM Que in aqueous buffer and in the presence of (a) VEGF G4–DNA, (b) h-TELO G4–DNA, and (c) duplex DNA. [DNA] = 30 μM; (λex = 375 nm and λem = 540 nm). | |
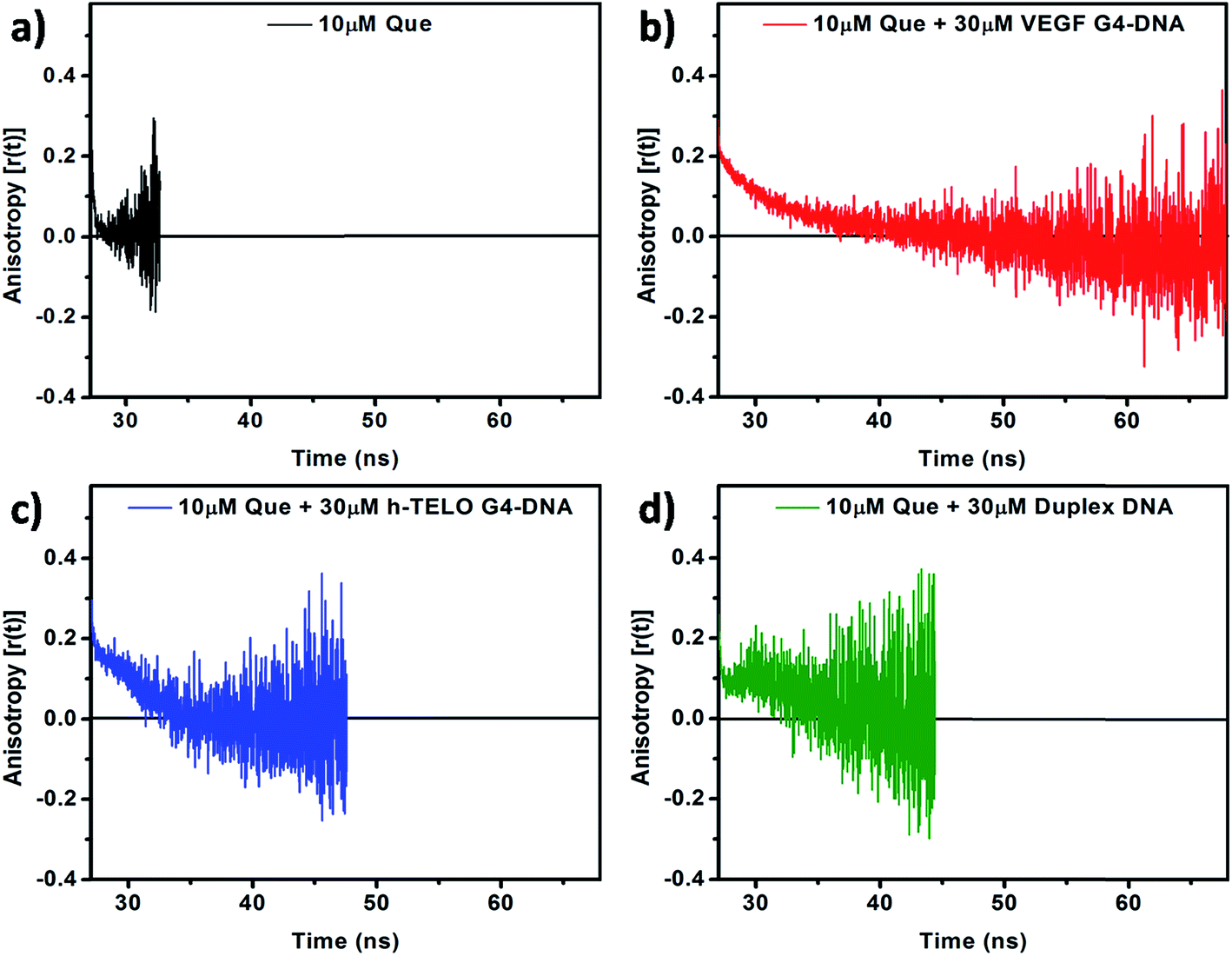
To obtain critical insight into the motional rigidity of the fluorophore in the DNA environment, time-resolved fluorescence anisotropy decay studies of Que in aqueous buffer and in the presence of DNA have been performed. Time-resolved fluorescence anisotropy profile of Que in the presence of DNA is found to be quite interesting. In the absence of DNA, Que exhibits single-exponential anisotropy decay with an average rotational correlation time (〈τr〉) of 0.18 ns (Fig. 6a and Table 3). Rotational diffusion of unbound Que is fast which leads to the rapid decay in anisotropy. We found that in the VEGF G4–DNA environment the decay follows a biexponential pattern consisting of a fast component (0.13 ns) representing the free Que (with a contribution of only 20%) and another slower dominating component (4.89 ns) attributed to the DNA bound Que (having a contribution of 80%), with the 〈τr〉 being 4.86 ns (Fig. 6b and Table 3). As Que binds to the VEGF G4–DNA its rotational freedom is restricted leading to slow decay in anisotropy. However, in the presence of h-TELO G4–DNA and duplex DNA, anisotropy decays displayed “dip-rise-dip” kind of profiles, indicating that in addition to the short and long anisotropy decay components, a growth component at intermediate time is also observed (Fig. 6c and d). The observed 〈τr〉 of Que in VEGF G4–DNA matrix is appreciably higher compared to c-MYC (4.12 ns) and c-KIT1 (3.26 ns) G4–DNA (Table 3 and Fig. S6, ESI†). Again, in c-KIT2 G4–DNA environment, anisotropy decays also displayed “dip-rise-dip” kind of profiles similar to h-TELO G4–DNA and duplex DNA (Fig. S6, ESI†). Such anisotropy profiles may be explained as follows: upon excitation, the fluorescence anisotropy quickly decreases at first because the fraction of free Que decays faster. With time, the DNA-bound fraction increases and the anisotropy recovers; and finally it shows a slow decay because only DNA-bound Que form remains in the excited state. This type of decay pattern is characteristic for associated anisotropy decays.38,40 Similar kind of “dip-rise-dip” pattern was observed in our previous report with another plant flavonoid fisetin in duplex DNA environment.41 This kind of time-resolved anisotropy behavior can be explained with the help of a two-component anisotropy equation as suggested by Ludescher et al.42
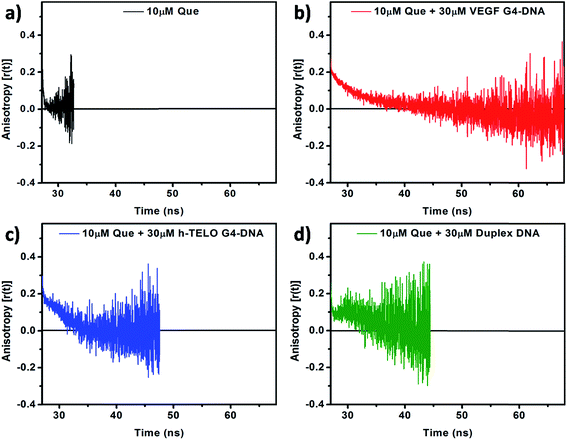 |
| | Fig. 6 Time-resolved fluorescence anisotropy decay profiles of 10 μM Que in (a) aqueous buffer and in the presence of (b) VEGF G4–DNA, (c) h-TELO G4–DNA, and (d) duplex DNA. [DNA] = 30 μM; (λex = 375 nm and λem = 540 nm). | |
Table 3 Fluorescence anisotropy decay parameters of Que tautomer species in aqueous buffer and in the presence of different G4–DNA structures
| Sample |
τ1 (ns) |
α1 |
τ2 (ns) |
α2 |
〈τr〉 (ns) |
χ2 |
| Que |
0.18 |
1 |
|
|
0.18 |
1.01 |
| Que + VEGF |
0.13 |
0.20 |
4.89 |
0.80 |
4.86 |
1.15 |
| Que + c-MYC |
0.11 |
0.36 |
4.18 |
0.64 |
4.12 |
0.99 |
| Que + c-KIT1 |
0.13 |
0.28 |
3.31 |
0.72 |
3.26 |
1.08 |
3.4. Circular dichroism (CD) studies
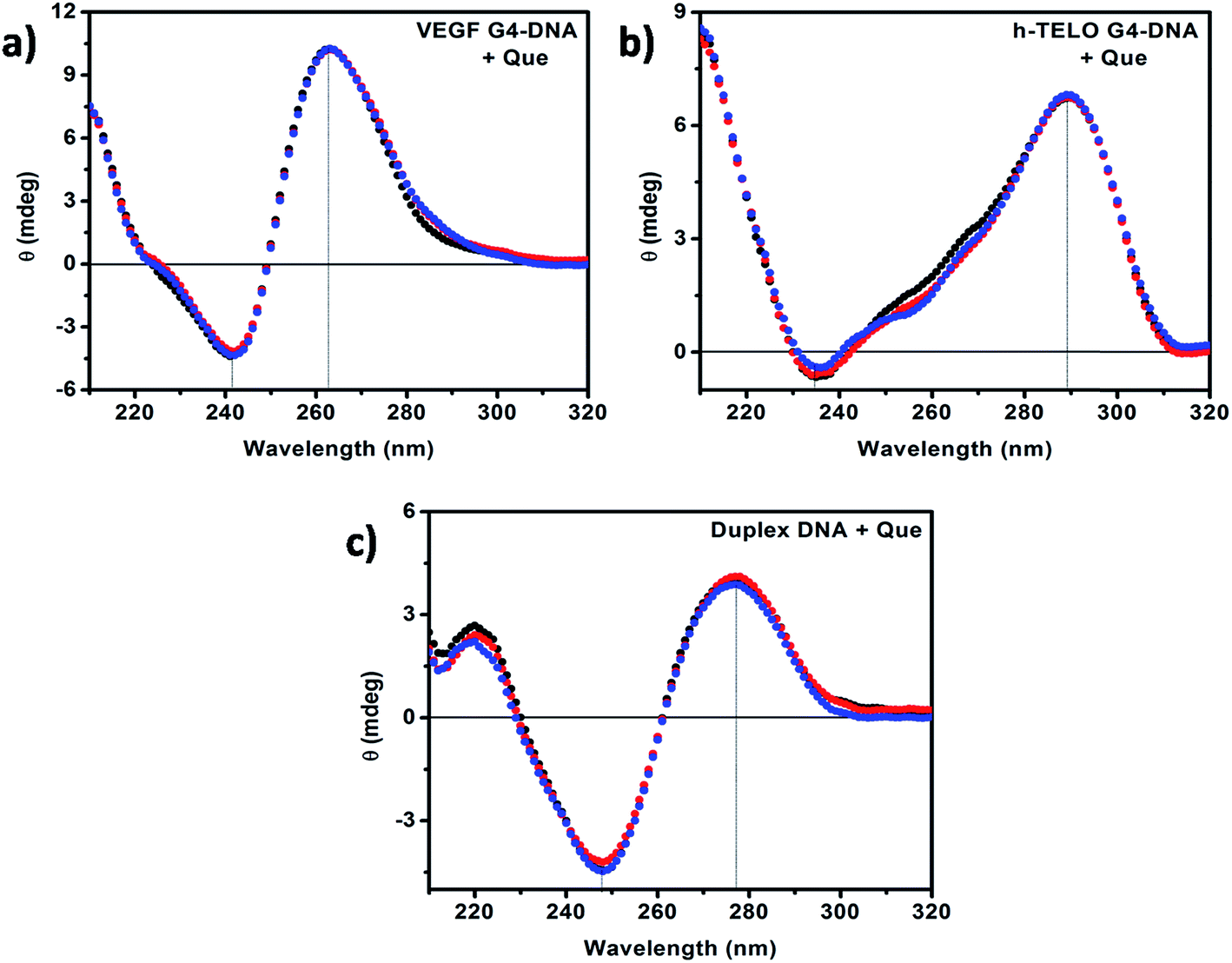
CD has been used to investigate the conformational changes in DNA structure during interactions with Que. Promoter G4–DNA structures such as VEGF, c-MYC, c-KIT1 and c-KIT2 exhibit a positive band at ∼260 nm and a negative band at ∼240 nm indicating the formation of parallel G4–DNA structure while the CD spectrum of the h-TELO G4–DNA exhibits a positive band at ∼290 nm, and a small negative peak at ∼235 nm with a small hump at ∼260 nm suggesting the formation of hybrid G4–DNA that have both parallel and antiparallel characteristics.43,44 The CD spectrum of the duplex DNA consists of two major peaks, a positive band at ∼275 nm and a negative band at ∼245 nm, which are characteristic of double stranded B-DNA.35,38 It has been reported earlier that the CD spectra show almost no change in the case of external binding such as groove binding and electrostatic binding, whereas intercalative binding affects both the positive and negative bands.35,45 In our present studies, with the addition of Que we observed no remarkable change in the position of the peaks and band intensities irrespective of DNA structure (Fig. 7 and S7, ESI†). Therefore, from the CD spectroscopic analyses, we can infer that there is no significant perturbation in the secondary structures of the DNA molecules in the presence of Que. The CD data also suggest the presence of external mode of binding (i.e. binding in the loop or groove region) between Que and DNA structures rather than strong intercalative binding. This is consistent with the absorption spectroscopic studies discussed earlier.
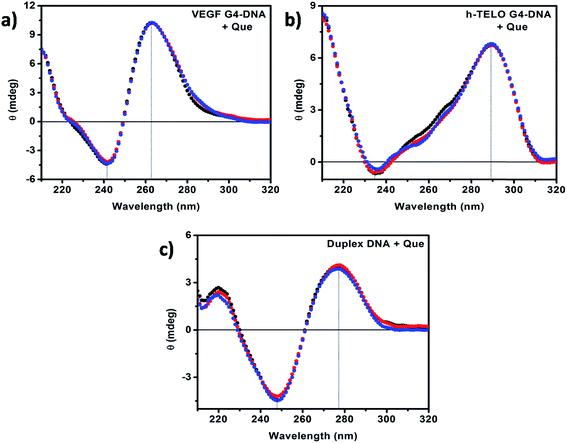 |
| | Fig. 7 CD spectra of (a) VEGF G4–DNA, (b) h-TELO G4–DNA, and (c) duplex DNA in the absence (black) and in the presence of 50 μM (red) and 100 μM (blue) Que, respectively. [DNA] = 20 μM. | |
3.5. Isothermal titration calorimetric (ITC) studies
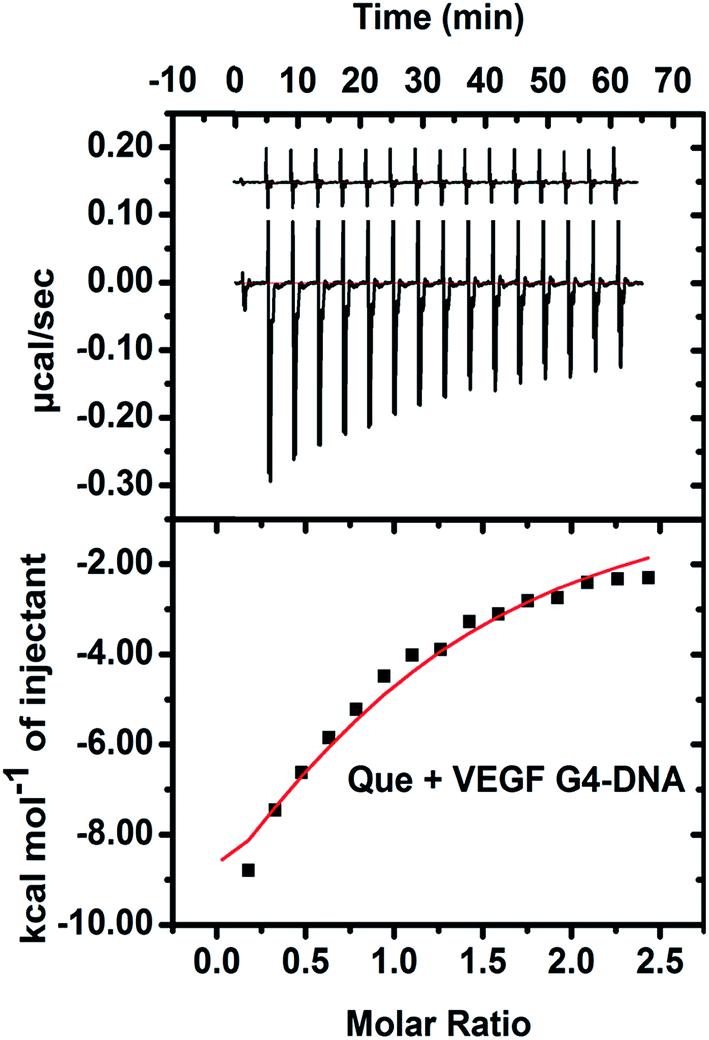
The energetics of binding between Que and VEGF G4–DNA was studied by isothermal titration calorimetry (ITC). The negative peaks in the ITC thermogram reveal that the binding reaction is exothermic (Fig. 8). The enthalpy change (ΔH is −17.9 kcal mol−1) for the association process is more negative than the entropy change (TΔS is −0.98 kcal mol−1), implying the binding process to be enthalpy driven. The Gibbs free energy change (ΔG is −16.92 kcal mol−1) is also negative, indicating the binding reaction to be spontaneous. The ITC data of Que and VEGF G4–DNA complexation yielded a Kb value of 3.8 × 104 M−1 which is of the same order as obtained from fluorometric studies. In the case of h-TELO G4–DNA, duplex and other promoter G4–DNA structures, ITC data were erratic due to much less interaction with Que (data not shown).
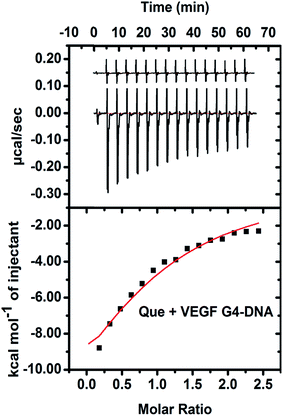 |
| | Fig. 8 ITC profile generated from the interaction between VEGF G4–DNA with Que. Upper panel shows the isothermal plot of Que and VEGF G4–DNA complex formation and the lower panel represents the integrated binding isotherm generated from integration of peak area as a function of molar ratio. The solid line (red) represents the best fit data using the ‘one site binding model’. | |
3.6. Construction of DNA-based molecular logic gate
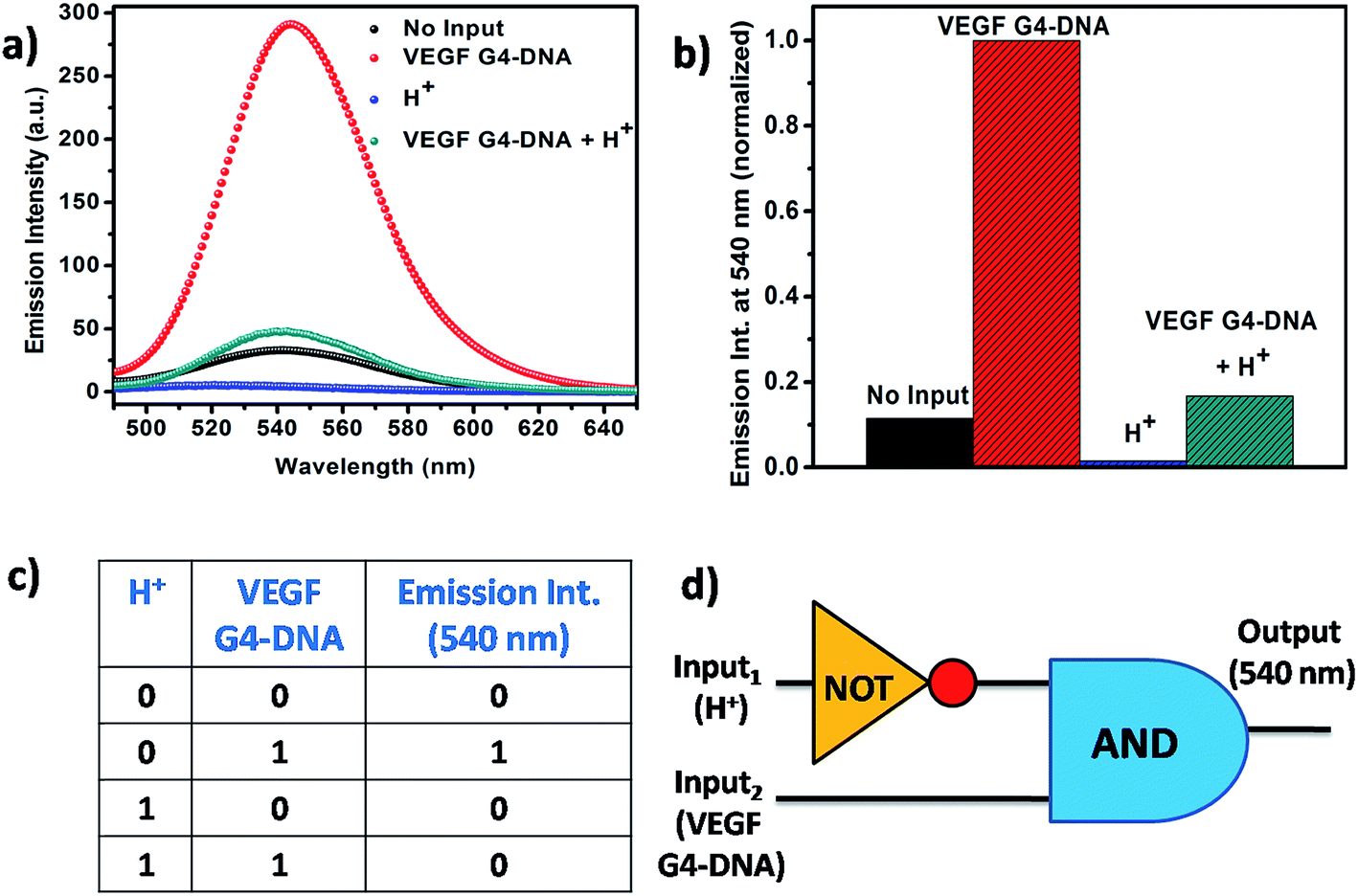
From the context of application of Que, we have designed a logic gate on the basis of the noncovalent interaction of Que with the VEGF G4–DNA at two different pH values pH 7.4 and 5.4. VEGF G4–DNA and the pH of the solution are taken as the inputs and the fluorescence intensity of Que at 540 nm is chosen as the output. The threshold value for the construction of logic gate is set at Que fluorescence intensity of 60 a.u. At pH 7.4 [input (0, 0)], free Que has a low fluorescence signal (output taken as 0) but in the presence of VEGF G4–DNA at pH 7.4 [input (0, 1)], binding of Que to VEGF G4–DNA enhances the intensity of Que (output taken as 1). When pH of the solution is decreased to 5.4 [input (1, 0)], the fluorescence intensity of free Que is very low (output taken as 0). However, when VEGF G4–DNA is added and the pH of the system is adjusted to 5.4, this input system (1, 1) gives rise to a low fluorescence signal (output taken as 0). The reason behind this effect is probably the structure of VEGF G4–DNA at pH 5.4 has been distorted and consequently the binding of Que with VEGF G4–DNA is quite less which in turn is reflected by very low increase of Que fluorescence. Out of the four cases, since input system (0, 1) gives rise to the most intensified fluorescence signal, it is taken as 1 and all other intensities are kept as 0 with respect to it. Subsequent analysis of the normalized fluorescence according to Boolean logic leads to the formation of a complex logic gate system based on NOT and AND gate (Fig. 9).
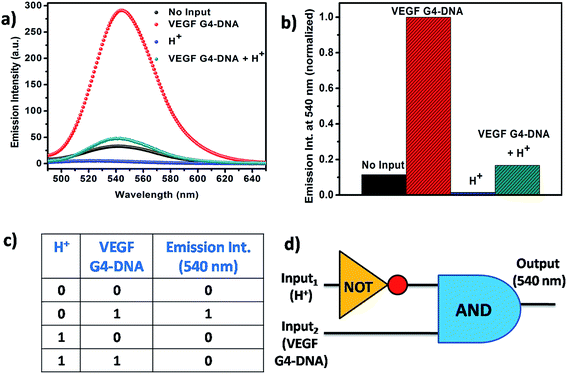 |
| | Fig. 9 (a) Fluorescence emission spectra of Que in the presence of different inputs. [Que] = 10 μM and [VEGF G4–DNA] = 30 μM. (b) Normalized fluorescence intensity at 540 nm in the presence of different inputs. (c) Truth table of the Boolean logic gate. (d) Scheme of the Boolean logic gate. | |
4. Conclusions
G4–DNA structures have now emerged as a new class of molecular target for anticancer drugs and therefore, it is important to investigate interactions between small molecule ligands and G4–DNA, towards quest for G4–DNA targeting drugs. Various studies suggest that there is no general rule that explains the interaction between ligands and G4–DNA, and a sequence-dependent interaction could also be important. Among various such small molecules, Que, a bioactive plant flavonoid, has immense biological potential. In this article, we report an investigation on the interaction of Que with different promoter G4–DNA, telomeric G4–DNA and duplex DNA structures using different spectroscopic techniques. UV-visible absorption studies show that the binding of Que with G4–DNA does not involve intercalation, instead presumably external mode of binding occurs between Que and G4–DNA. Upon addition of increasing amounts of VEGF G4–DNA to Que, larger spectral shifts and decrement in absorbance were observed compared to other G4–DNA as well as duplex DNA, suggesting a greater extent of interaction of Que with VEGF G4–DNA than that of other DNA structures. Steady-state fluorometric studies reveal that the binding affinity of Que and VEGF G4–DNA is much higher compared to other G4–DNA structures and duplex DNA. We also observed that the enhancement of the fluorescence emission intensity (quantified by quantum yield measurements) of Que upon DNA binding is much higher in VEGF G4–DNA environment which is also an indication of strong binding interaction. From time-resolved fluorescence decay studies, we observed an increase in the average lifetime (〈τ〉) of Que in DNA environments, but the value is much higher in VEGF G4–DNA than that of other DNA structures which also indicates the preferential binding interaction of Que with VEGF G4–DNA. Time-resolved anisotropy measurements also support our findings, showing higher rotational correlation time (〈τr〉) of Que in VEGF G4–DNA environment indicating higher motional restriction of Que in VEGF G4–DNA matrix. CD results reveal that the DNA structures remain unperturbed upon binding with Que and also the minimal change in band intensities indicates that external mode of binding is present instead of intercalative binding. Binding energetics between Que and VEGF G4–DNA was evaluated using ITC and we found that the binding process is exothermic and enthalpy driven. Therefore, from the above observations we can conclude that Que shows preferential binding interaction towards VEGF G4–DNA as compared to telomeric G4–DNA, duplex DNA and other promoter G4–DNA structures (c-MYC, c-KIT1 and c-KIT2). This indicates that binding of Que with different G4–DNA structures is strongly modulated by the DNA base sequences, with a remarkably preferential interaction with VEGF G4–DNA. In this connection, it is noteworthy that although VEGF G4–DNA, h-TELO G4–DNA and duplex DNA adopt different secondary structures, other promoter G4–DNAs such as c-MYC, c-KIT1 and c-KIT2 adopt a structure similar to that of VEGF G4–DNA. However, the bases (which are mainly cytosine, Table S1, ESI†) present in the intervening region (loop) between the G-tracts of VEGF G4–DNA differs from other DNA sequences that have been studied here. Therefore, despite the fact that the promoter G4–DNAs VEGF, c-MYC, c-KIT1 and c-KIT2 possess similar structure, probably because of the difference in the intervening sequences of G-tract, Que interacts more strongly with VEGF G4–DNA. Que bound in the loop region is presumably partially shielded from the bulk aqueous environment, which can explain the dramatically enhanced tautomer fluorescence of Que bound to VEGF G4–DNA. We have also found that the interaction between Que and VEGF G4–DNA is pH dependent and based on this property we have designed a complex biomolecular logic gate device. Furthermore, it is evident that the interaction between Que and VEGF G4–DNA can be used as a sensor for pH detection. To the best of our knowledge, based on the present study, we report here for the first time the preferential interaction of Que with VEGF G4–DNA manifested by remarkably large increase in steady-state as well as time-resolved fluorescence emission parameters of Que, in sharp contrast with other G4–DNA structures and duplex DNA, where only modest changes are noted. Finally, the construction of G4–DNA based logic gate sensing platform with Que and VEGF G4–DNA in different pH environments signifies a further application of Que, based on its interesting and exquisitely sensitive fluorescence.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work has been funded by Department of Science and Technology (DST), Govt of India (DST INSPIRE Grant: DST/INSPIRE/04/2014/000217). Prof. P. K. Sengupta gratefully acknowledges UGC, India, for the award of an Emeritus Fellowship.
References
- T. Simonsson, Biol. Chem., 2001, 382, 621–628 CrossRef CAS PubMed.
- J. L. Huppert, Chem. Soc. Rev., 2008, 37, 1375–1384 RSC.
- M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev. Genet., 2012, 13, 770–780 CrossRef CAS PubMed.
- D. Yang and K. Okamoto, Future Med. Chem., 2010, 2, 619–646 CrossRef CAS PubMed.
- Y. Wu and R. M. Brosh Jr, FEBS J., 2010, 277, 3470–3488 CrossRef CAS PubMed.
- R. Simone, P. Fratta, S. Neidle, G. N. Parkinson and A. M. Isaacs, FEBS Lett., 2015, 589, 1653–1668 CrossRef CAS PubMed.
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402–5415 CrossRef CAS PubMed.
- A. Guedin, J. Gros, P. Alberti and J. L. Mergny, Nucleic Acids Res., 2010, 38, 7858–7868 CrossRef CAS PubMed.
- N. Smargiasso, F. Rosu, W. Hsia, P. Colson, E. S. Baker, M. T. Bowers, E. De Pauw and V. Gabelica, J. Am. Chem. Soc., 2008, 130, 10208–10216 CrossRef CAS PubMed.
- A. Arora and S. Maiti, J. Phys. Chem. B, 2009, 113, 8784–8792 CrossRef CAS PubMed.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef CAS PubMed.
- A. Henderson, Y. Wu, Y. C. Huang, E. A. Chavez, J. Platt, F. B. Johnson, R. M. Brosh Jr, D. Sen and P. M. Lansdorp, Nucleic Acids Res., 2014, 42, 860–869 CrossRef CAS PubMed.
- E. Y. Lam, D. Beraldi, D. Tannahill and S. Balasubramanian, Nat. Commun., 2013, 4, 1796–1803 CrossRef PubMed.
- H. Chen, H. Sun, X. Zhang, X. Sun, Y. Shi and Y. Tang, RSC Adv., 2015, 5, 1730–1734 RSC.
- Y. F. Huo, L. N. Zhu, X. Y. Li, G. M. Han and D. M. Kong, Sens. Actuators, B, 2016, 237, 179–189 CrossRef CAS.
- S. Bhowmik, R. N. Das, B. Parasar and J. Dash, Chem. Commun., 2013, 49, 1817–1819 RSC.
- P. Mandal, M. Bhattacharya and J. Chowdhury, RSC Adv., 2016, 6, 109846–109856 RSC.
- N. S. Jha, S. Mishra, A. S. Mamidi, A. Mishra, S. K. Jha and A. Surolia, RSC Adv., 2016, 6, 7474–7487 RSC.
- B. Sengupta, S. M. Reilly, D. E. Davis Jr, K. Harris, R. M. Wadkins, D. Ward, D. Gholar and C. Hampton, J. Phys. Chem. B, 2015, 119, 2546–2556 CrossRef CAS PubMed.
- S. Mondal, J. Jana, P. Sengupta, S. Jana and S. Chatterjee, Mol. BioSyst., 2016, 12, 2506–2518 RSC.
- V. Kumar, A. Sengupta, K. Gavvala, R. K. Koninti and P. Hazra, J. Phys. Chem. B, 2014, 118, 11090–11099 CrossRef CAS PubMed.
- N. Xu, H. Yang, M. Cui, F. Song, Z. Liu and S. Liu, J. Mass Spectrom., 2012, 47, 694–700 CrossRef CAS PubMed.
- N. Xu, H. Yang, M. Cui, F. Song, Z. Liu and S. Liu, Chin. J. Chem., 2012, 30, 1433–1438 CrossRef CAS.
- E. Middleton Jr, C. Kandaswami and T. C. Theoharides, Pharmacol. Rev., 2000, 52, 673–751 CAS.
- L. H. Yao, Y. M. Jiang, J. Shi, F. A. Tomas-Barberan, N. Datta, R. Singanusong and S. S. Chen, Plant Foods Hum. Nutr., 2004, 59, 113–122 CrossRef CAS PubMed.
- B. Sengupta and P. K. Sengupta, Biochem. Biophys. Res. Commun., 2002, 299, 400–403 CrossRef CAS PubMed.
- P. Job, Ann. Chim., 1928, 9, 113–203 CAS.
- C. Y. Huang, Methods Enzymol., 1982, 87, 509–525 CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 Search PubMed.
- Z. Jurasekova, C. Domingo, J. V. Garcia-Ramos and S. Sanchez-Cortes, Phys. Chem. Chem. Phys., 2014, 16, 12802–12811 RSC.
- L. Huang and D. Q. Yu, Application of Ultraviolet Spectroscopy in Organic Chemistry, Science Press, Beijing, 2000 Search PubMed.
- G. Zhang, J. Guo, J. Pan, X. Chen and J. Wang, J. Mol. Struct., 2009, 923, 114–119 CrossRef CAS.
- Y. Sun, F. Ji, R. Liu, J. Lin, Q. Xu and C. Gao, J. Lumin., 2012, 132, 507–512 CrossRef CAS.
- P. Ghosh and P. Purkayastha, RSC Adv., 2014, 4, 22442–22448 RSC.
- M. A. Husain, S. U. Rehman, H. M. Ishqi, T. Sarwar and M. Tabish, RSC Adv., 2015, 5, 64335–64345 RSC.
- P. K. Sengupta, in Reviews in Fluorescence 2016, ed. C. D. Geddes, Springer International Publishing AG, Switzerland, 1st edn, 2017, vol. 10, ch. 4, pp. 45–70 Search PubMed.
- R. Simkovitch and D. Huppert, J. Phys. Chem. B, 2015, 119, 10244–10251 CrossRef CAS PubMed.
- A. Ganguly, S. Ghosh and N. Guchhait, Phys. Chem. Chem. Phys., 2015, 17, 483–492 RSC.
- S. Ghosh, D. Banik, A. Roy, N. Kundu, J. Kuchlyan and N. Sarkar, Phys. Chem. Chem. Phys., 2014, 16, 25024–25038 RSC.
- A. Ganguly, B. K. Paul, S. Ghosh, S. Dalapati and N. Guchhait, Phys. Chem. Chem. Phys., 2014, 16, 8465–8475 RSC.
- S. Bhattacharjee, S. Chakraborty, P. K. Sengupta and S. Bhowmik, J. Phys. Chem. B, 2016, 120, 8942–8952 CrossRef CAS PubMed.
- R. D. Ludescher, L. Peting, S. Hudson and B. Hudson, Biophys. Chem., 1987, 28, 59–75 CrossRef CAS PubMed.
- M. Vorlickova, I. Kejnovska, J. Sagi, D. Renciuk, K. Bednarova, J. Motlova and J. Kypr, Methods, 2012, 57, 64–75 CrossRef CAS PubMed.
- A. Ambrus, D. Chen, J. Dai, T. Bialis, R. A. Jones and D. Yang, Nucleic Acids Res., 2006, 34, 2723–2735 CrossRef CAS PubMed.
- A. Manna and S. Chakravorti, J. Phys. Chem. B, 2012, 116, 5226–5233 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05930b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Pradeep K. Sengupta
,
Pradeep K. Sengupta