
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Syntheses, characterisation and solid-state study of alkali and ammonium BArF salts†
Lucas Carreras a,
Laura Roviraa,
Mónica Vaqueroa,
Ignasi Mona,
Eddy Martina,
Jordi Benet-Buchholza and
Anton Vidal-Ferran*ab
a,
Laura Roviraa,
Mónica Vaqueroa,
Ignasi Mona,
Eddy Martina,
Jordi Benet-Buchholza and
Anton Vidal-Ferran*ab
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: avidal@iciq.cat
bICREA, Pg. Lluís Companys 23, 08010 Barcelona, Spain
First published on 27th June 2017
Abstract
A new synthetic protocol for synthesising a number of BArF derivatives has been developed. Single crystal X-ray analysis of an array of alkali metal and ammonium salts has allowed the determination of the coordination sphere and/or the map of short contacts of the positively charged atoms. The increasing number of coordination bonds and/or short contacts between the alkali metal cation and the surrounding atoms has been rationalised in terms of the size of the alkali metal centre. It has also been demonstrated that an increase in the number of coordination bonds and/or short contacts translates into longer M–F distances. In the case of the ammonium BArF salts, the N–B distances are shorter than the M–B distances in the alkali metal BArF salts, indicating stronger interactions between the cationic nitrogen and the anionic boron than those between the boron and the alkali metal centres. Finally, a study of the structures of alkali metal hydrated and THF-solvated BArF salts showed that the interactions between the metal centre and the surrounding atoms depend not only on the size of the alkali metal centre but also on the occupancy of the first coordination sphere.
Introduction
The term weakly coordinating anion (WCA) appeared in the literature in the 1990s and was used to describe anions that interacted weakly with cations.1 This qualifying term was used in the past for counterions such as [ClO4]−, [PF6]−, [SbF6]−, [AsF6]−, [BF4]−, [CF3SO3]−, carborane anions (i.e., [HCB11H11−nXn]− with X = halogen) and [AlCl4]−, since these anions proved to have a weakly coordinating character in aqueous solutions due to the delocalisation of their negative charge over all their groups or atoms.A wide variety of WCAs are known2 and of these, borate-based anions (i.e. [BPh4]− and its derivatives)3 deserve special mention. The [BPh4]− anion was used as counterion in Fe-based olefin polymerisation catalysts.4 The BPh4 group is prone to hydrolysis and sensitive to decomposition.2 All these drawbacks were solved by attaching fluorine-containing substituents to the phenyl groups.
Several fluorine-containing derivatives of [BPh4]− have been developed (i.e., [B(C6F5)4]− (ref. 3a), [B(3,5-(CF3)2C6H3)4]− (ref. 3e) and [B(CF3)4]− (ref. 3j)). The tetrakis[3,5-bis(trifluoromethyl)phenyl]borate anion ([B(3,5-(CF3)2C6H3)4]−, hereafter BArF)3e has gained significance within this group of fluorine-containing anions because of its stability and weak interaction with cations.
New and important applications for the BArF anion have been reported in recent years and have led to classical anions being replaced.3b–d,5–13 The BArF anion has been used in organic transformations as phase transfer catalyst,3b–d Lewis acid catalyst,5,6 in electrochemistry,7 in lithium batteries,8 as a component of ionic liquids9 and as a remover of lanthanide ions.10 Regarding applications in enantioselective catalysis, the BArF anion has been widely used as a counterion in Ir-mediated hydrogenations,11 and as regulation agent or additive for supramolecular catalysts.12,13
The synthesis of NaBArF was first reported by the Kobayashi group, who carried out the addition of arylmagnesium derivatives to boron reagents.3b–e Bergman and co-workers14 subsequently described more efficient synthetic protocols that avoided the use of magnesium metal as reagent. Analogous synthetic protocols have been developed for LiBArF3h,13c and KBArF.3h,15 It has also been demonstrated that RbBArF12d or CsBArF12d can be efficiently prepared by displacing the sodium cation in NaBArF with rubidium or cesium salts.16 The fact that RbBArF and CsBArF have lower solubility in water than NaBArF is the driving force behind the exchange of the cations bonded with the BArF moiety. Our group has pioneered the syntheses of a number of enantiopure ammonium BArF salts by displacement of the chloride with BArF anions in the corresponding amine hydrochlorides.12b,d
Though alkali metal and some ammonium-based BArF salts were known in the literature, their structures in the solid-state had not been systematically determined and studied.17 Moreover, neither had systematic characterisation data of these compounds in solution ever been reported.18 Thus, we report herein our efforts to develop efficient methods for the preparation of a number of BArF salts and to characterise in the solid-state and in solution an array of alkali [i.e., LiBArF (1), NaBArF (2), KBArF (3), RbBArF (4) and CsBArF (5)] and ammonium BArF derivatives (structures 6–8, see Fig. 1). The present article provides valuable information on the spatial atomic arrangement, distances and geometries together with the coordination bonds and/or short contacts of the positively charged atoms in the solid-state.
 | ||
| Fig. 1 Ammonium BArF derivatives. | ||
Experimental
For the general information on the preparation methods for the BArF salts, see the ESI.† The spectroscopic data of the BArF salts prepared using known methods can also be found in the ESI.†Lithium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate etherate, (LiBArF·(EtO2)2)
Lithium bis(trimethylsilyl)amide (0.212 g, 1.23 mmol) was dissolved in 30 mL of diethyl ether. This solution was added dropwise over a solution of HBArF·(EtO2)2 (ref. 19) (1.24 g, 1.23 mmol) in 40 mL of diethyl ether at −20 °C. After stirring overnight at −10 °C, the solvent was concentrated under reduced pressure to dryness. The solid was dissolved in 6 mL of diethyl ether and the solution was cooled at −78 °C. Hexane (20 mL) was slowly added over this solution to precipitate the desired product. The solvent was filtered-off via cannula and dried under reduced pressure to afford the product LiBArF·(Et2O)2 as a white solid. Isolated 1.11 g, 89% yield. 1H NMR (CD2Cl2, 400 MHz): δ 7.72 (br s, 8H), 7.57 (br s, 4H), 3.69 (q, J = 7.1 Hz, 8H), 1.25 (t, J = 7.1 Hz, 12H) ppm. 13C{1H} NMR (CD2Cl2, 126 MHz): δ 162.2 (q, JC–B = 50 Hz), 135.2, 129.3 (qm, JC–F = 32 Hz), 125.0 (q, JC–F = 272 Hz), 117.9 (m), 67.0, 14.7 ppm. 19F{1H} NMR (C4D8O, 376 MHz): δ −63.2 (24F) ppm. 11B{1H} NMR (C4D8O, 128 MHz): δ −8.6 ppm. 7Li NMR (D2O, 194 MHz): δ −0.4 ppm. IR (neat, cm−1): 1624, 1611, 1354, 1274, 1111, 1101. Mp = 145–148 °C. Content in Li by ICP-AES: calcd 0.68%, found 0.67%. IR and 1H NMR data were in agreement with those previously reported for LiBArF·4H2O.3hPotassium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate, (KBArF)
Potassium bis(trimethylsilyl)amide (0.18 g, 0.85 mmol) was dissolved in 50 mL of diethyl ether. This solution was added dropwise over a solution of HBArF·(EtO2)2 (0.86 g, 0.85 mmol) in 30 mL of diethyl ether at −20 °C. After stirring overnight at −20 °C, the solvent was concentrated under reduced pressure to dryness. The solid was dissolved in 6 mL of diethyl ether and the solution was cooled at −78 °C. Hexane (20 mL) was slowly added over this solution to precipitate the desired product. The solvent was filtered-off via cannula and dried under reduced pressure to afford the product KBArF as a white solid. Isolated 0.61 g, 80% yield. 1H NMR (C4D8O, 400 MHz): δ 7.79 (br s, 8H), 7.58 (br s, 4H) ppm. 13C{1H} NMR (C4D8O, 126 MHz): δ 162.8 (q, JC–B = 50 Hz), 135.6, 130.0 (qm, JC–F = 32 Hz), 125.5 (q, JC–F = 272 Hz), 118.2 (m) ppm. 19F{1H} NMR (C4D8O, 376 MHz): δ −63.2 (24F) ppm. 11B{1H} NMR (C4D8O, 128 MHz): δ −8.6 ppm. IR (neat, cm−1): 1612, 1357, 1281, 1187, 1124. Mp ≥ 350 °C.20 Content in K by ICP-OES: calcd 4.33%, found 4.40%. IR and 1H NMR data were in agreement with those previously reported.15(R)-(1-Phenylethyl)ammonium tetrakis[3,5-bis(trifluoromethyl)-phenyl]borate, (7)
NaBArF (0.28 g, 0.32 mmol) was mixed with (R)-(1-phenylethyl)ammonium hydrochloride21 (0.05 g, 0.32 mmol) in 5 mL of Et2O and allowed to react overnight. Filtration and evaporation of the solvent yielded the target compound as a white solid (0.30 g, 95% yield). 1H NMR (CD2Cl2, 400 MHz): δ 7.73 (br s, 8H), 7.58–7.52 (m, 7H), 7.44–7.39 (m, 2H), 5.91 (br s, 3H), 4.90 (q, J = 6.9 Hz, 1H), 1.91 (d, J = 6.9 Hz, 3H) ppm. 13C{1H} NMR (CD2Cl2, 100 MHz): δ 162.2 (q, JC–B = 49 Hz), 135.2, 133.7, 132.1, 130.9, 129.3 (qm, JC–F = 31 Hz), 126.7, 125.1 (q, JC–F = 272 Hz), 117.9 (m), 56.9, 20.1 ppm. 19F{1H} NMR (CD2Cl2, 376 MHz): δ −63.3 (24F) ppm. 11B{1H} NMR (CD2Cl2, 128 MHz): δ −8.6 ppm. IR (neat, cm−1): 3683, 3606, 3323, 3281, 2857, 1612, 1492, 1353, 1275, 1098. Mp = 149–154 °C. [α]25D +1.0 (c 1.03, CH3OH). MS (ESI): m/z calcd for C8H12N: 122.1; found: 122.1 [M − BArF]+. Elemental analysis calcd (%) for C41H24BF24N: C 48.75, H 2.45, N 1.42; found: C 48.12, H 2.49, N 1.49.Details on the determination of single crystal X-ray diffraction structures and general structural comments were included in the ESI† of this article.
Results and discussion
Syntheses of alkali metal and ammonium BArF derivatives
Though the preparation of LiBArF and KBArF was known in the literature,3h,13c,15 these syntheses require the use of organomagnesium intermediates and aqueous work-ups. Thus, we envisaged that these compounds could alternatively be prepared via an acid–base reaction (Scheme 1) between the corresponding acid of BArF (9) and the corresponding alkali metal bis(trimethylsilyl)amide (i.e., LiN(SiMe3)2 or KN(SiMe3)2, respectively) in a low boiling point organic solvent.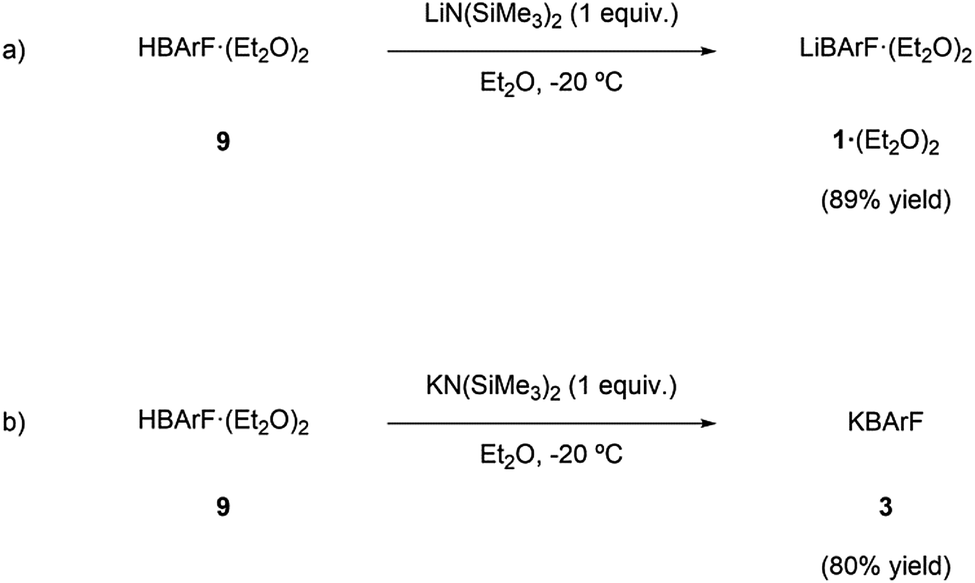 | ||
| Scheme 1 Syntheses of (a) LiBArF·(Et2O)2 and (b) KBArF. | ||
The low boiling point of the liquid components of the mixture would facilitate the isolation of the desired compound by evaporation in vacuo. This synthetic procedure was followed and LiBArF·(Et2O)2 and KBArF were isolated in high yield. It should be noted that this preparation method does not require separating the target compound from other salts.
The preparation of RbBArF, CsBArF and the ammonium BArF derivatives is well documented and relies on a counteranion exchange reaction around the alkali metal or ammonium groups.12d The difference in the solubility of the target BArF derivative and the by-product of the reaction (i.e. the salt arising from the combination of the cation initially bound to the BArF-containing reagent and the anion initially bound to the alkali metal or ammonium groups) constitutes the driving force of the reaction and shifts the equilibrium. The preparation methods for the required starting materials and BArF salts have been detailed in the ESI.†
Characterisation in solution of alkali metal and ammonium BArF derivatives
The whole array of BArF salts was characterised using standard spectroscopic techniques (IR and 1H, 7Li, 11B, 13C, 19F and 133Cs NMR spectroscopy). 19F{1H} and 11B{1H} NMR spectra showed only a singlet for the signals of the BArF anion with very small differences in the chemical shifts (see Table 1). 1H was in agreement with the proposed structures and 13C{1H} NMR showed the expected set of signals for the four equivalent 3,5-bis(trifluoromethyl)phenyl groups and the corresponding organic moieties in the case of the ammonium BArF salts. IR spectroscopy also confirmed the structure of the BArF salts with intense bands in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching (ca. 1600–1650 cm−1) and C–F stretching (ca. 1100–1350 cm−1) vibration regions. Intense bands in the N+–H stretching region (ca. 2700–3000 cm−1, broad) were also observed for the ammonium BArF salts.
C stretching (ca. 1600–1650 cm−1) and C–F stretching (ca. 1100–1350 cm−1) vibration regions. Intense bands in the N+–H stretching region (ca. 2700–3000 cm−1, broad) were also observed for the ammonium BArF salts.
| Entry | Compound | 11B{1H}a | 19F{1H}b |
|---|---|---|---|
| a 11B{1H} chemical shifts are quoted in ppm relative to trimethyl borate (TMB) in C4D8O.b 19F{1H} chemical shifts are quoted in ppm relative to trifluoroacetic acid (TFA) in C4D8O. | |||
| 1 | LiBArF·(Et2O)2 | −8.6 | −63.2 |
| 2 | NaBArF | −8.6 | −63.1 |
| 3 | KBArF | −8.6 | −63.2 |
| 4 | RbBArF | −8.5 | −63.3 |
| 5 | CsBArF | −8.6 | −63.1 |
| 6 | 6 | −8.5 | −63.0 |
| 7 | 7 | −8.5 | −63.3 |
| 8 | 8 | −8.6 | −63.3 |
Crystal structure description and analysis
One of the key structural features of the BArF anion is that its negative charge is distributed over its skeleton, in which the electronegative fluorine atoms play a major role in accommodating the single negative charge of the boron.The distribution of this single negative charge over a multitude of atoms renders the BArF anion both weakly nucleophilic and basic, which poses questions about the coordination sphere (CS) of the cationic species and about the number of weak interactions (or short contacts) required for the single positive charge of the alkali metal or ammonium cation to be compensated. Regarding the alkali metal BArF salts, we reasoned that the different charge densities in the alkali metal cationic series (i.e., Li+, Na+, K+, Rb+ and Cs+) arising from the differences in the cationic radii would translate into a different number of interactions between the fluorine atoms or solvate molecules and the cation in each alkali metal BArF derivative. In terms of the ammonium BArF derivatives, we reasoned that the size and topology of the backbones of the substituents at nitrogen would affect the number of interactions between the electronegative atoms present in the BArF anion or solvate molecules and the nitrogen cation. We considered that determining the coordination spheres, the map of short contacts and the space-charge compensation map for the positively charged atom in a number of alkali metal BArF and ammonium BArF derivatives would be highly useful for understanding the role of these interesting derivatives in catalytic processes,3b–d,5,6,11–13 electrochemistry7,8 and other applications.9,10 Thus, we turned our attention to obtaining single crystals suitable for X-ray analysis for the whole series of BArF derivatives and to performing the corresponding structural studies by X-ray analysis. Single crystals suitable for X-ray analysis were obtained for the whole series of alkali metal BArF salts and the ammonium derivatives studied. Molecules of solvate were incorporated into the crystal structure in some structures (i.e., LiBArF·4THF, NaBArF·6THF, CsBArF·7THF and CsBArF·DCM). Moreover, residual water molecules from the solvent were incorporated into the crystal structure in some cases (i.e., LiBArF, NaBArF, KBArF, RbBArF and 7).
| Entry | Distance | LiBArF | NaBArF | KBArF | RbBArF | CsBArF |
|---|---|---|---|---|---|---|
| a (′) the prime code corresponds to disordered atoms. | ||||||
| 1 | M–B | 7.509(4) | 7.546(1) | 7.891(4) | 7.994(9) | 8.025(3) |
| 2 | M–X | O1 – 1.874(9) | O1 – 2.193(5) | O1 – 2.814(9) | O1 – 2.9392(8) | Cl1S – 3.807(3) |
| 3 | O2 – 1.930(8) | O2 – 2.274(17) | ||||
| 4 | O2′ – 1.856(15) | O2′ – 2.258(7) | ||||
| 5 | M–F | F4 – 2.287(3) | F4 – 2.395(2) | F3 – 2.873(18) | F1′ – 3.160(3) | F2′ – 3.063(19) |
| 6 | F4′ – 3.490(4) | F4′ – 2.995(11) | F3′ – 2.615(4) | F3 – 2.997(2) | F3 – 3.070(11) | |
| 7 | F4′′ – 2.520(2) | F4 – 2.792(2) | F3′ – 3.040(2) | F4 – 3.671(6) | ||
| 8 | F4′ – 2.728(6) | F4 – 2.925(18) | F5 – 3.282(8) | |||
| 9 | F6′ – 3.042(10) | |||||
 | ||
| Fig. 2 Crystal structure of the alkali metal BArF derivatives. Some phenyl rings from the BArF unit have been omitted for the sake of clarity. Colour scheme: C: black, H: blue, B: orange, F: green, Li: violet, O: red; Na/K/Rb/Cs: purple. Atomic displacement ellipsoids are drawn at a 50% probability level. For LiBArF: octahedral (A) and trigonal bipyramidal (B) geometries, in which fluorine atoms F4A, F4B and F4C are symmetry equivalent to F4. For NaBArF: octahedral (C) and trigonal bipyramidal (D) geometries, in which fluorine atoms F4, F4A, F4B and F4C are symmetry equivalent. For KBArF: see structure (E) for the geometry, in which fluorine atoms F3D, F3E, F3F and F3G are symmetry equivalent to F3 and F4A, F4B and F4C to F4. For RbBArF: see structure (F) for the geometry, in which fluorine atoms F3D, F3E, F3F and F3G are symmetry equivalent to F3 and F4A, F4B and F4C to F4. For CsBArF: see structure (G) for the geometry, in which fluorine atoms F3D, F3E, F3F, F3G, F4A, F4B, F4C, F5A, F5B and F5C are symmetry equivalent to F3, F4 and F5, respectively. | ||
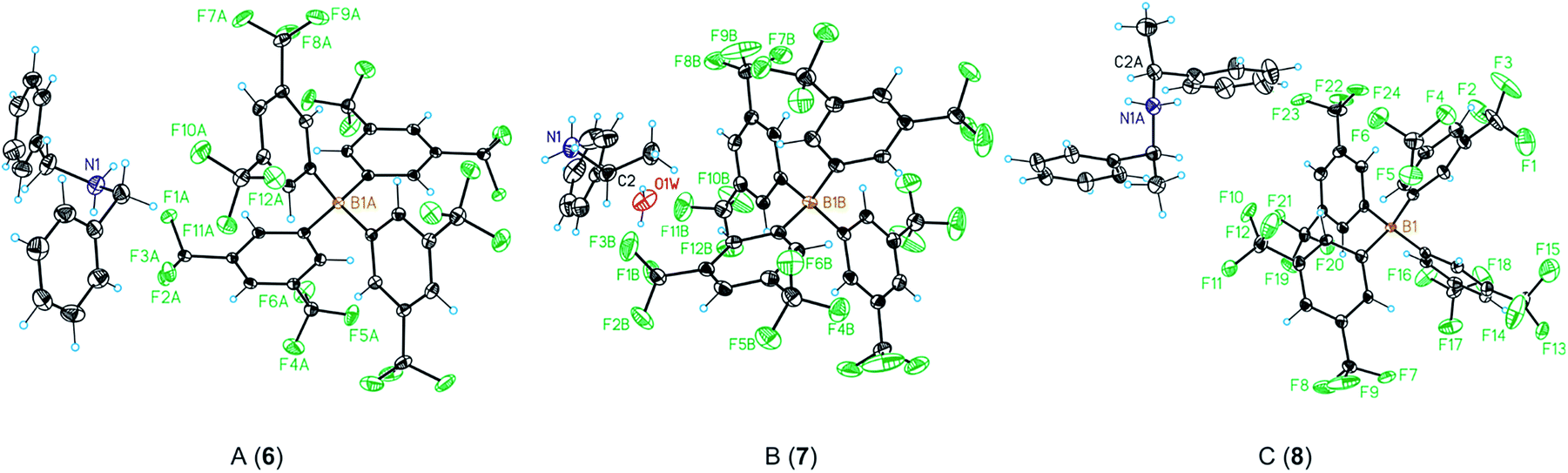 | ||
| Fig. 3 Crystal structure of the ammonium BArF derivatives 6–8 ((A) structure of 6; (B) structure of 7; (C) structure of 8). Colour scheme: C: black, H: blue, B: orange, F: green, N: purple, O: red. Atomic displacement ellipsoids are drawn at a 50% probability level. | ||
| Entry | Distance | 6 | 7 | 8 |
|---|---|---|---|---|
| 1 | N–B | 6.724(2) | 7.477(3) | 6.778(6) |
| 2 | N–F | 3.058(2) | 2.959(3) | 2.995(5) |
| 3 | N–O | — | 2.705(4) | — |
 | ||
| Fig. 4 Crystal structure of alkali metal BArF solvates ((A) structure of LiBArF·4THF; (B) structure of NaBArF·6THF; (C) structure of CsBArF·7THF). Colour scheme: C: black, H: blue, B: orange, F: green, Li: violet, Na: purple, Cs: light green, O: red. Atomic displacement ellipsoids are drawn at a 50% probability level. | ||
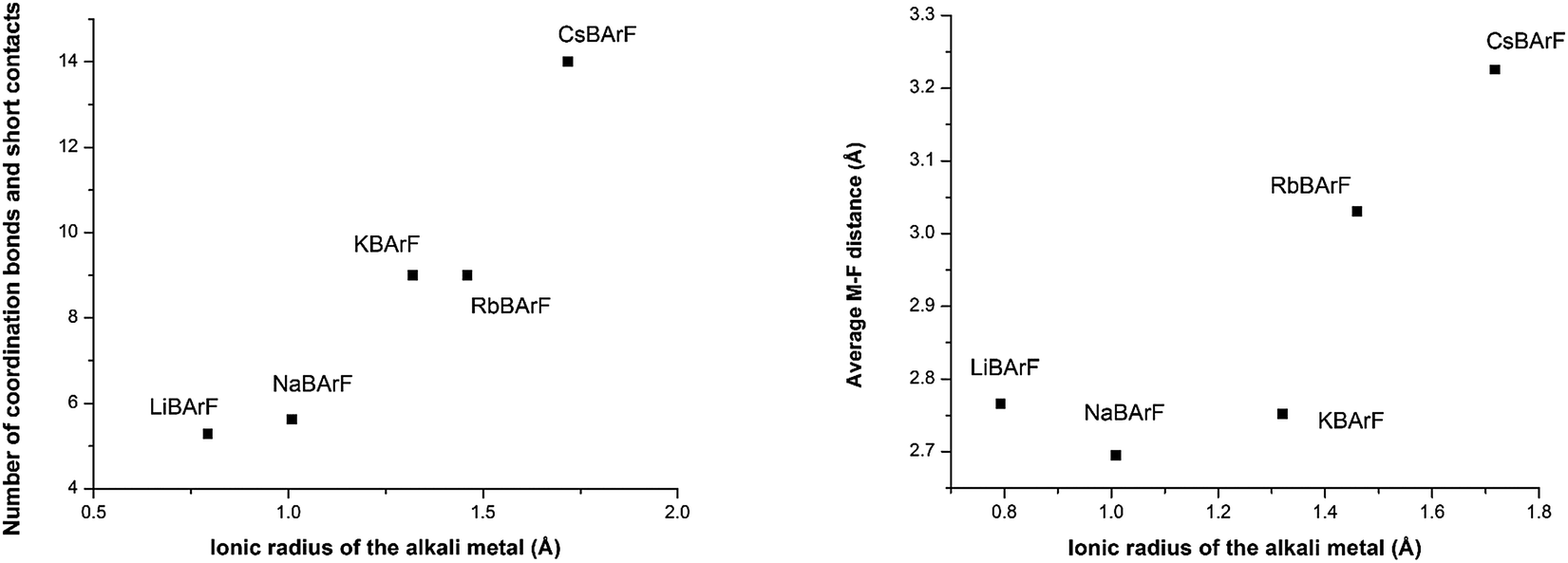 | ||
| Fig. 5 (Left) The number coordination bonds or/and short contacts around the cationic metal alkali atom compared with the ionic radius of the metal, (right) the average distance between the fluorine atoms and the cationic metal alkali atom compared with the ionic radius of the metal. | ||
Conclusions
A new synthetic protocol for the efficient preparation of lithium and potassium BArF has been developed by reaction between HBArF·(Et2O)2 and the corresponding lithium or potassium bis(trimethylsilyl)amide. This new synthesis constitutes an unreported, short, efficient and practical means of synthesising LiBArF and KBArF. The method paves the way for the preparation of new metal BArF derivatives. Moreover, a number of alkali metal and ammonium BArF derivatives have been fully characterised both in solution and in the solid-state by using standard spectroscopic techniques.Single crystals suitable for X-ray analysis for an array of structurally diverse BArF derivatives were obtained, thus yielding unreported structural details of the structures of KBArF, RbBArF, CsBArF, 6, 7 and 8. The increasing number of coordination bonds and/or short contacts between the alkali metal cation and the surrounding atoms has been rationalised in terms of the size of the alkali metal centre. A study of the structures of alkali metal hydrated and THF-solvates (i.e., LiBArF·4H2O, LiBArF·4THF, NaBArF·6THF and CsBArF·7THF) showed that the interactions between the metal centre and the surrounding atoms depend not only on the size of the alkali metal centre but also on its solvation.
The detailed structural information in the solid-state provided herein regarding an array of alkali metal and ammonium BArF derivatives may well allow a better understanding of the role of BArF derivatives in catalysis, electrochemistry and supramolecular processes and broaden their applicability in future processes of interest.
Acknowledgements
The authors would like to thank MINECO (CTQ2014-60256-P and Severo Ochoa Excellence Accreditation 2014–2018 SEV-2013-0319) and the ICIQ Foundation for financial support. L. C. thanks MINECO for a FPI-SO pre-doctoral fellowship. L. R. and I. M. thank ICIQ for pre-doctoral fellowships. Dr M. V. is grateful for the financial support received from the CELLEX Foundation.Notes and references
- S. H. Strauss, Chem. Rev., 1993, 93, 927–942 CrossRef CAS.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
- (a) A. G. Massey and A. J. Park, J. Organomet. Chem., 1964, 2, 245–250 CrossRef CAS; (b) H. Kobayashi, T. Sonoda, H. Iwamoto and M. Yoshimura, Chem. Lett., 1981, 579–580 CrossRef CAS; (c) H. Kobayashi, T. Sonoda and H. Iwamoto, Chem. Lett., 1982, 1185–1186 CrossRef CAS; (d) H. Iwamoto, M. Yoshimura, T. Sonoda and H. Kobayashi, Bull. Chem. Soc. Jpn., 1983, 56, 796–801 CrossRef CAS; (e) H. Nishida, N. Takada, M. Yoshimura, T. Sonoda and H. Kobayashi, Bull. Chem. Soc. Jpn., 1984, 57, 2600–2604 CrossRef CAS; (f) K. Fujiki, M. Kashiwagi, H. Miyamoto and A. Sonoda, J. Fluorine Chem., 1992, 57, 307–321 CrossRef CAS; (g) K. Seppelt, Angew. Chem., Int. Ed. Engl., 1993, 32, 1025–1027 CrossRef; (h) J. H. Golden, P. F. Mutolo, E. B. Lobkovsky and F. J. DiSalvo, Inorg. Chem., 1994, 33, 5374–5375 CrossRef CAS; (i) K. Fujiki, J. Ichikawa, H. Kobayashi, A. Sonoda and T. Sonoda, J. Fluorine Chem., 2000, 102, 293–300 CrossRef CAS; (j) E. Bernhardt, G. Henkel, H. Willner, G. Pawelke and H. Burger, Chem.–Eur. J., 2001, 7, 4696–4705 CrossRef CAS PubMed.
- R. F. Jordan, Adv. Organomet. Chem., 1991, 32, 325–387 CrossRef CAS.
- K. Fujiki, S.-Y. Ikeda, H. Kobayashi, A. Mori, A. Nagira, J. Nie, T. Sonoda and Y. Yagupolskii, Chem. Lett., 2000, 62–63 CrossRef CAS.
- S. Aldridge, C. Jones, T. Gans-Eichler, A. Stasch, D. L. Kays, N. D. Coombs and D. J. Willock, Angew. Chem., Int. Ed., 2006, 45, 6118–6122 CrossRef CAS PubMed.
- F. Barrière, N. Camire, W. E. Geiger, U. T. Mueller-Westerhoff and R. Sanders, J. Am. Chem. Soc., 2002, 124, 7262–7263 CrossRef.
- F. Kita, H. Sakata, S. Sinomoto, A. Kawakami, H. Kamizori, T. Sonoda, H. Nagashima, J. Nie, N. V. Pavlenko and Y. L. Yagupolskii, J. Power Sources, 2000, 90, 27–32 CrossRef CAS.
- A. Boesmann, G. Franciò, E. Janssen, M. Solinas, W. Leitner and P. Wasserscheid, Angew. Chem., Int. Ed., 2001, 40, 2697–2699 CrossRef CAS.
- H. Suzuki, H. Naganawa and S. Tachimori, Phys. Chem. Chem. Phys., 2003, 5, 726–733 RSC.
- (a) A. Pfaltz, J. Blankenstein, R. Hilgraf, E. Hormann, S. McIntyre, F. Menges, M. Schonleber, S. P. Smidt, B. Wustenberg and N. Zimmermann, Adv. Synth. Catal., 2003, 345, 33–44 CrossRef CAS; (b) X. Cui and K. Burgess, Chem. Rev., 2005, 105, 3272–3296 CrossRef CAS PubMed.
- (a) I. Mon, D. A. Jose and A. Vidal-Ferran, Chem.–Eur. J., 2013, 19, 2720–2725 CrossRef CAS PubMed; (b) A. Vidal-Ferran, I. Mon, A. Bauzá, A. Frontera and L. Rovira, Chem.–Eur. J., 2015, 21, 11417–11426 CrossRef CAS PubMed; (c) L. Rovira, M. Vaquero and A. Vidal-Ferran, J. Org. Chem., 2015, 80, 10397–10403 CrossRef CAS PubMed; (d) H. Fernández-Pérez, I. Mon, A. Frontera and A. Vidal-Ferran, Tetrahedron, 2015, 71, 4490–4494 CrossRef; (e) L. Rovira, H. Fernández-Pérez and A. Vidal-Ferran, Organometallics, 2016, 35, 528–533 CrossRef CAS.
- (a) Y. Li, B. Ma, Y. He, F. Zhang and Q.-H. Fan, Chem.–Asian J., 2010, 5, 2454–2458 CrossRef CAS PubMed; (b) F.-T. Song, G.-H. Ouyang, Y. Li, Y.-M. He and Q.-H. Fan, Eur. J. Org. Chem., 2014, 6713–6719 CrossRef CAS; (c) Z. Cai, D. Xiao and L. H. Do, J. Am. Chem. Soc., 2015, 137, 15501–15510 CrossRef CAS PubMed; (d) X.-C. Zhang, Y.-H. Hu, C.-F. Chen, Q. Fang, L.-Y. Yang, Y.-B. Lu, L.-J. Xie, J. Wu, S. Li and W. Fang, Chem. Sci., 2016, 7, 4594–4599 RSC.
- N. A. Yakelis and R. G. Bergman, Organometallics, 2005, 24, 3579–3581 CrossRef CAS PubMed.
- W. E. Buschmann, J. S. Miller, K. Bowman-James and C. N. Miller, Inorg. Synth., 2002, 33, 83–91 CAS.
- Though seminal pieces of work on alkali metal BArF salts indicate that these compounds can be prepared by cation exchange (see ref. 3e, 3h and 5), detailed preparation methods and isolated yields are not provided.
- This statement is made on the basis of the structures checked in the CCDC† database. A query was made in the CCDC database. The reponse to this query provided all the crystal structures including the [BArF]− sub-structure (2060 results) submitted to the CCDC until February 2017 {CSD version 5.38 [November 2016, update 2 (February 2017)]}. For CsBArF, only one crystal structure was found (CCDC ref. code FUPBOJ) coordinated to a thioether macrocycle. For RbBArF, two crystal structures were found (CCDC ref. codes FUPBID and OJAMIX) coordinated to a thioether and an aza-macrocycle respectively. For KBArF, six crystal structures were found (CCDC ref. codes: FUPBEZ and FUPCEA (coordinated to a thioether macrocyle), FUPCOK (coordinated to a selenoether macrocyle), OJAMET and OJAMOD (coordinated to an aza-macrocycle), and ZUZVEX (coordinated to a polyether chain)). For LiBArF, eight crystal structures were found (CCDC ref. codes: FUNZUL and FUPCAW (coordinated to a thioether macrocyle), OJAMUJ and OJEBAI (coordinated to an aza-macrocycle), AGOBUV, UQOQOI and YUCGAG (coordinated to a C60 fullerene), and finally a tetrahydrated structure for LiBArF, ref. code: YEMZUL). For NaBArF, 26 crystal structures were found, but only one relevant structure was found (NaBArF·2H2O, CCDC ref. code: RENPIK). None of the ammonium-based BArF salts studied had been indexed by the time of the query.
- On the contrary, solution and in the solid-state characterisation of alkali metal BArF salts coordinated to a variety of ligands has been recently reported: (a) J. Dyke, W. Levason, M. E. Light, D. Pugh, G. Reid, H. Bhakhoa, P. Ramasami and L. Rhyman, Dalton Trans., 2015, 44, 13853–13866 RSC; (b) M. J. D. Champion, W. Levason, D. Pugh and G. Reid, Dalton Trans., 2015, 44, 18748–18759 RSC.
- M. Brookhart, B. Grant and A. F. Volpe Jr, Organometallics, 1992, 11, 3920–3922 CrossRef CAS.
- The upper temperature limit of the melting point apparatus was 350 °C.
- (R)-(1-Phenylethyl)ammonium hydrochloride was synthesised following the reported procedure: K. J. Halloran, A. Comelly, Z. Chen and S. Krishnan, US Pat., WO2011053835A1, 2011.
- Interestingly LiBArF crystallised also as a tetrahydrate (LiBArF·4H2O). This specific structure has been reported previously (see ref. 3h) but the crystallographic study was performed at 173 K. The crystallographic data obtained at 100 K in the present work for LiBArF·4H2O were used for comparison with the LiBArF structures measured under identical conditions. In the case of the LiBArF·4H2O, the lithium atom is coordinated to four H2O molecules in a tetrahedral fashion, and Li–F and B–Li distances are longer in the tetrahydrate LiBArF·4H2O than in LiBArF-A or LiBArF-B (see ESI†).
- C.-T. Chang, C.-L. Chen, Y.-H. Liu, S.-M. Peng, P.-T. Chou and S.-T. Liu, Inorg. Chem., 2006, 45, 7590–7592 CrossRef CAS PubMed.
- Pörschke et al. have reported a sixteen-coordinated cesium centre. For example, see: D. Pollak, R. Goddard and K.-R. Pörschke, J. Am. Chem. Soc., 2016, 138, 9444–9451 CrossRef CAS PubMed.
- A. E. H. Wheatley, Chem. Soc. Rev., 2001, 30, 265–273 RSC.
- The measured LiBArF·4THF sample was a multicomponent crystal formed by a minimum of two crystals (ratio 51
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49). Additionally only a limited number of reflections could be collected. Due to these difficulties, the data obtained is less precise.
:49). Additionally only a limited number of reflections could be collected. Due to these difficulties, the data obtained is less precise. - The CsBArF·7THF sample was an inversion twin and presented high disorder in the solvent molecules and CF3− units. As a result, the bond lengths for a reduced number of C–C bonds are less precise.
- P. F. Lang and B. C. Smith, Dalton Trans., 2010, 39, 7786–7791 RSC.
- (a) H. Plenio, Chem. Rev., 1997, 97, 3363–3384 CrossRef CAS PubMed; (b) H. Plenio, ChemBioChem, 2004, 5, 650–655 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic and crystallographic data. CCDC 1546291–1546302. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra05928k |
| This journal is © The Royal Society of Chemistry 2017 |