
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of structure–activity relationships of ginsenosides against amyloid β induced pathological behaviours in transgenic Caenorhabditis elegans†
Mu
Zhang‡
 ,
Fei
Qian‡
,
Qingling
Liu
,
Cheng
Qian
,
Pyone Myat
Thu
,
Yanyan
Wang
,
Zu-Guo
Zheng
,
Hua
Yang
,
Ping
Li
* and
Xiaojun
Xu
*
,
Fei
Qian‡
,
Qingling
Liu
,
Cheng
Qian
,
Pyone Myat
Thu
,
Yanyan
Wang
,
Zu-Guo
Zheng
,
Hua
Yang
,
Ping
Li
* and
Xiaojun
Xu
*
State Key Laboratory of Natural Medicines, China Pharmaceutical University, No. 24 Tongjia Lane, Nanjing 210009, Jiangsu, China. E-mail: xiaojunxu@cpu.edu.cn; liping@cpu.edu.cn; Fax: +86 25 83271379; Tel: +86 25 83271379
First published on 16th August 2017
Abstract
Amyloid β (Aβ) induced toxicity has been postulated to initiate synaptic loss and subsequent neuronal degeneration in Alzheimer's disease (AD). The activities of ginsenosides against AD are widely reported, however, a systematic in vivo study comparing the effects of different ginsenosides on Aβ induced toxicity and cognitive impairment has not been described. In addition, the correlation between molecular structures and their anti-Aβ activities remains uncovered. In the present study, the in vivo anti-Aβ effects of 17 ginsenosides were explored using three transgenic Caenorhabditis elegans (C. elegans) models that exhibits pathological behaviors associated with Aβ. We found that (1) ginsenoside Rc, Rd, 20(S)-Rg3, Compound K (CK) and Rg1 could significantly reduce Aβ deposits, (2) ginsenoside Rc, Rd, CK, Rk3 and Rg1 inhibit Aβ induced paralysis, and (3) ginsenosides 20(S)-Rg3, CK and Rk3 prevented Aβ induced defects in associative learning capacity and showed antioxidative activity both in vitro and in vivo. The structure–activity relationships of 17 ginsenosides were investigated by comparing their anti-Aβ effects in C. elegans models. Results showed that the protopanaxadiol-type has higher activities than the protopanaxatriol-type or oleanane-type; the anti-Aβ effects of ginsenosides are inversely related to the sugar numbers. The results of the present study gain insight into the in vivo protective effects of ginsenosides against Aβ induced toxicity, and provide useful information for the clinical use of ginsenosides.
1. Introduction
Alzheimer's disease (AD) is an age-related neurodegenerative disease and is the most common form of dementia.1 Abnormal production of amyloid β (Aβ) peptide and deposition of plaques in the brain are the primary events in AD pathogenesis.2 The 42-amino acid Aβ fragment, Aβ42 peptide, is the primary constituent of plaque3 and regarded as the main toxic fragment in AD.4Ginseng is one of the most widely used dietary supplements and herbal medicines in the world.5 Asian ginseng (Panax ginseng), American ginseng (Panax quinquefolius) and notoginseng (Panax notoginseng) are the three most generally used ginseng herbs.6 The extraction of ginseng, total saponins of ginseng and specific ginsenosides have been used for improving cognitive function in AD.7,8 Ginsenosides are regarded as the major bioactive components of ginseng,9,10 and can be classified into several categories according to their structure. The protopanaxadiol (PPD)-type saponins with sugar moieties at the C-3 and/or C-20, the protopanaxatriol (PPT)-type saponins with sugar moieties attached to the C-6 and/or at C-20, and the oleanane-type with a nonsteroidal structure.11 Some rare ginsenosides are generated from naturally occurring types through heating,6,12 enzymatic,13 microbial14 or chemical treatment.12 Several ginsenosides, including Rb1,15−17 Rg1,18 Rg2,19 Rg3,20 Rd,21−24 Re,25 Rh1,26 Rh2 (ref. 27) and CK,28 have been reported to have potential benefits to improve cognition through (1) regulating the synaptic plasticity,16–18,28 (2) inhibiting neuroinflammation,20,25 (3) enhancing the production of neurotrophic factors,24 (4) regulating the homeostasis of Aβ,23,27 (5) promoting the release of acetylcholine from hippocampus15 and other mechanisms. While to our best knowledge, there is no systematic in vivo study comparing the effects of different ginsenosides on Aβ induced toxicity and cognitive impairment. In addition, the correlation between structures and anti-Aβ activities still has not been fully elucidated.
Since AD is a complex neurodegenerative disease, the cultured mammalian cell models that lack the organism complexity involved in AD are not suitable to fully evaluate the anti-AD activity of candidate compounds. On the other hand, the use of a transgenic mice model of AD for pharmacological evaluation and mechanistic studies is time and labor consuming. Simple invertebrate models of neurodegenerative diseases offer experimental advantages for addressing basic cellular processes that are conserved among all animals.29 In the present study, by using several transgenic AD Caenorhabditis elegans (C. elegans) models,30 we explored the in vivo anti-Aβ effects of 17 ginsenosides (Fig. 5, Tables 2–4), and discussed the possible structure–activity relationship based on types of aglycones and sugar moieties.
2. Materials and method
2.1 Reagents
Reference ginsenosides Rb1, Rb2, Rb3, Rc, Rd, Re, 20(S)–Rh1, Rg1, Rg2, 20(S)-Rg3, Rg6, Rk3, Ro, CK, PPD, PPT and oleanolic acid (OA) were purchased from Must Biological Technology Co. Ltd. (Chengdu, China). Diacetyl and donepezil hydrochloride (Dop) were purchased from Tokyo Chemical Industry (Tokyo, Japan). Thioflavine T (ThT), sodium azide, vitamin E (VitE) and ethanol were purchased from Sigma-Aldrich (St. Louis, MO, USA). Stock solution of all candidate drugs were made with DMSO (Sigma-Aldrich, St. Louis, MO, USA), and diluted by M9 buffer to final concentrations in C. elegans experiments, or diluted by cell culture medium in cell treatment. The final concentration of DMSO did not exceed 0.1%. Diacetyl was prepared immediately before to any experiments by dilution in ethanol. Sodium azide was distilled in water to obtain a final concentration of 1 M.312.2 C. elegans strains
C. elegans strains used in this study are listed in Table 1. CL4176 (smg-1ts [myo-3/Aβ1–42 long 3′-untranslated region (UTR)]), CL802 (smg-1(cc546) I; rol-6(su1006) II), CL2006 (dvIs2[pCL12(unc-54/human Aβ1–42 minigene) + pRF4]), CL2122 (dvIs15[(pPD30.38) unc-54(vector) + (pCL26) mtl-2:GFP]) were obtained from Caenorhabditis Genetics Center (University of Minnesota, Minneapolis, MN). CL2241 (smg-1(cc546ts); dvls29(pCL45(snb-1/SP:Aβ1–42 minigene/long 3′ UTR) + pRF4 (rol-6(su1006)),30 CL2109 (dvIs[pDF1 (unc-54/SP:Aβ1–42 dimer minigene + pRF4 (rol-6(su1006))) are kind gifts from Professor Christopher D. Link (Institute for Behavioral Genetics, University of Colorado).| Strains | Transgene | Expression | Phenotype |
|---|---|---|---|
| CL802 | myo-3 | Control for CL4176 | Wt. movement |
| CL4176 | myo-3/Aβ1–42 | Inducible muscle | Rapid paralysis |
| CL2109 | unc-54/Aβ dimer | Control for CL2006 | No Aβ formation |
| CL2006 | unc-54/Aβ1–42 | Constitutive muscle | Progressive paralysis |
| CL2122 | mtl-2/GFP | Control for CL2241 | Wt. movement |
| CL2241 | snb-1/Aβ1–42 | Inducible neuronal | Wt. movement |
2.3 C. elegans maintenance and treatment
Nematodes were cultured in temperature controlled incubator. All strains were maintained at 16 °C, and propagated on solid nematode growth medium (NGM) seeded with E. coli (OP50) as a food source.32 To prepare age-synchronized populations, nematodes reaching reproductive maturity at 3 days of age were transferred to fresh NGM plates and allowed to lay eggs for 4 h. The synchronized eggs were cultured on fresh NGM plates. All drug treatments were performed in 34.8 × 10 mm Petri plates containing NGM agar. Placed 500 μL of compound solution onto each plate and distributed over the surface of NGM agar. Allowed solution to dry overnight at room temperature and followed by adding 100 μL of E. coli (OP50) onto the center of plate. The worms were fed with the drugs either from 72 h of age or starting from the egg.2.4 Fluorescence staining of Aβ in C. elegans
Methoxy-X04 (MedChem Express, USA) is a Congo-red derivative and has been widely used for in vivo imaging of Aβ plaques.33 In this study, methoxy-X04 was used for fluorescence staining of Aβ deposit in C. elegans. Transgenic worms, CL2006 and its control strain CL2109, were synchronized and plated on NGM plate with OP50 at 16 °C for 72 h.34 Then worms were transferred into NGM plates, which contain vehicle (DMSO, 0.1%) or compounds solution for 48 h. Live worms were collected and incubated in methoxy-X04 solution (1 mM in 10 mM Tris, pH 8.0) for four hours at room temperature. After staining, worms were subsequently transferred to seeded NGM plates for 12 h to allow destaining via normal metabolism. Stained worms were mounted and photographed by using fluorescence microscope (Nikon, Japan). The quantitative analysis of Aβ deposits was performed as described before.352.5 Paralysis assay
Paralysis assay was performed as described before.36 Briefly, Petri plates (34.8 × 10 mm) containing NGM agar were supplemented with vehicle (0.1% DMSO) or compounds solution. The strain CL4176 and CL802 maintained at 16 °C were egg-synchronized onto NGM plates containing vehicle or drug. Transgene expression was induced by upshifting the culture temperature from 16 °C to 25 °C at the 36 h after egg laying (third-stage larvae). Culture temperature was kept at 25 °C until the end of the paralysis assay. Nematodes were scored as paralyzed if they failed to move during observation and exhibited ‘halos’ of cleared bacteria around their heads (indication of insufficient body movement to access food).37 Paralysis was scored at 2 h intervals.2.6 Odorant preference assay
Chemotaxis assay was performed as described before31 with slightly modified. CL2241 and its control strain CL2122 were treated with or without Dop, VitE or ginsenosides starting from egg at 16 °C for 36 h, and then the culture temperature was upshifted to 23 °C to induce expression of Aβ and incubated for another 36 h. Worms were then collected, washed with M9 buffer38 three times, and transferred to NGM plates without food for 2 h. To condition the worms, 2 μL of odorant (0.01% diacetyl in 95% ethanol) was placed in the lid of the plates and then the plates were inverted. Naïve worms were starved in the absence of diacetyl, synchronously. At the end of starvation, all worms were subjected to chemotaxis assay.39 Assays were performed in 100 mm plates containing 1.9% agar, 1 mM CaCl2, 1 mM MgSO4, and 25 mM phosphate buffer, pH 6.0. For chemotaxis assay, 1 μL of diacetyl was placed at 2.5 cm from the center of the plate on one side, and 1 μL of 95% ethanol was added on the opposite. Twenty worms were quickly transferred to the center of the plate, then an additional 1 μL of odorant or 95% ethanol along with 1 μL of 1 M sodium azide were added to the original spot. Assay plates were incubated at 20 °C for 1 h and chemotaxis index (CI) was scored.Chemotaxis index is defined as follows: (number of worms at the attractant spot − number of worms at the control spot)/total number of worm
2.7 Oxidative free radicals assay
Intracellular oxidative free radicals (ROS) in C. elegans and cells were measured using 2,7-dichlorofluorescein diacetate (DCF-DA) (Beyotime, Shanghai, China) as described before.35 For in vitro assay, SH-SY5Ysw cells were pretreated with vehicle (DMSO, 0.1%) or indicated compounds for 48 h, then stimulated for transgene Aβ expression by addition of 1 μM butyric acid (BA) (Sigma, St. Louis, MO, USA) for 24 h. For in vivo assay, age synchronized groups of CL4176 and CL802 were treated with compounds as mentioned before. After 36 h long temperature upshift, at least 60 worms from each group were collected into 100 μL of PBS with 1% Tween 20 (PBST). Worms were subjected to homogenization and sonication to break up the outer cuticle. Both cell and worm samples were incubated with DCF-DA solution (50 μM in PBS) at 37 °C for 30 min. Fluorescence was measured by microplate fluorescence reader with the excitation at 485 nm and emission at 530 nm.2.8 Cell culture
All cells were cultured at 37 °C in a humid incubator with 5% CO2. The human neuroblastoma cell line SH-SY5Y was purchased from American Type Culture Collection (Manassas, VA, USA) and grown in Dulbecco's modified Eagles Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Gibco, New York, USA). The transgenic cell line SH-SY5Ysw cells that stably expressing amyloid β protein precursor (APP) with the Swedish mutation were purchased from Velox Pharmaceutics, Inc. (VLXSTC0002, Changzhou, China) and grown in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin (Gibco, New York, USA) and 200 μg mL−1 G418 (Invitrogen, California, USA).40The laboratory animal facility in the animal center has been accredited by Association for Assessment and Accreditation of Laboratory Animal Care International. All experiments and animal care in this study were conducted in accordance with the Provision and General Recommendation of Chinese Experimental Animals Administration Legislation and approved by the Science and Technology Department of Jiangsu Province (SYXK (SU) 2016-0011). Primary neurons were isolated as described before.41 Briefly, Pregnant Sprague Dawley rat were sacrificed and E18 embryos were removed from abdomen. Cerebral cortices were separated and placed into ice cold Hank's balanced salt solution. After the scalp and skull were peeled away, cortex tissue was digested by 0.05% trypsin–EDTA (Gibco, New York, USA) for 10 min at 37 °C. The digestion was stopped by addition of Neurobasal medium containing 0.1 mg mL−1 DNase I (Gibco, New York, USA). Digested tissues were centrifuged at 1500 rpm for 5 min at 4 °C after gentle trituration and resuspended in a Neurobasal culture medium supplemented with 2% B27 (Invitrogen, Grand Island, NY) and 0.5 mM GlutaMax (Life Technologies, USA), and then filtered through a 70 μm nylon cell restrainer. Cells were diluted to 1 × 106 cells mL−1 and seeded into poly-L-lysine (Beyotime, Shanghai, China) coated plates.
2.9 Cell viability assay
Assessment of primary cortical neurons viability was performed by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide] (Sigma, St. Louis, MO, USA) assay on day in vitro (DIV) 8. Stock solutions of ginsenosides in DMSO were diluted to final concentration and administrate to cells for 24 h. Then, MTT (5 mg ml−1 in PBS) were added to culture medium in each wells to make the final concentration at 0.5 mg ml−1 and incubated for another 4 h. The medium was removed and 200 μL of DMSO was added to dissolve the formazan crystals. Absorbance values were measured at 490 nm.2.10 Statistical analysis
Data were presented as mean ± standard deviation (SD). Difference between two groups was analyzed for statistical significance by independent Student's t test. Difference between three or more groups was using the one-way ANOVA followed by Dunnett's test. p < 0.05 was considered significant.3. Results
3.1 Effects of ginsenosides on Aβ deposits in AD C. elegans
The Aβ42 peptides are highly fibrillogenic and readily aggregate. They could assemble into different forms including monomer, oligomer, fibrillar and finally made the insoluble plaques.2 To validate whether ginsenosides affect Aβ deposits formation in vivo, the transgenic C. elegans line CL2006 worms in which human Aβ42 peptide is expressed in muscle cells and form deposits42 were treated with Dop, ThT or ginsenosides. CL2109 was used as a genetic background control strain of CL2006. Worms were subjected to Aβ staining using methoxy-X04 at the end of drug treatment. The number of Aβ deposits was scored in the worm's head region, which is separated from the rest of the body by the pharyngeal bulb (Fig. 1Ba and b). As shown in Fig. 2Ac, Aβ deposits (white arrowheads) were detected in the CL2006 worms but not in CL2109 (Fig. 1Bb) as observed before.43 Dop and ThT were served as positive control in this assay. Dop is an inhibitor of acetylcholinesterase and widely used in clinical treatment of mild to moderate AD. ThT could bind to Aβ and ameliorate Aβ induced toxicity in C. elegans through regulating the protein homeostasis network.37 The number of Aβ deposits was significantly reduced in worms treated with ThT (Fig. 1Be), Rc (Fig. 1Bi), Rd (Fig. 1Bj), Rg3 (Fig. 1Bk), CK (Fig. 1Bl) and Rg1 (Fig. 1Bo). The average number of Aβ deposits from 25 worms in each group is shown in Fig. 2C.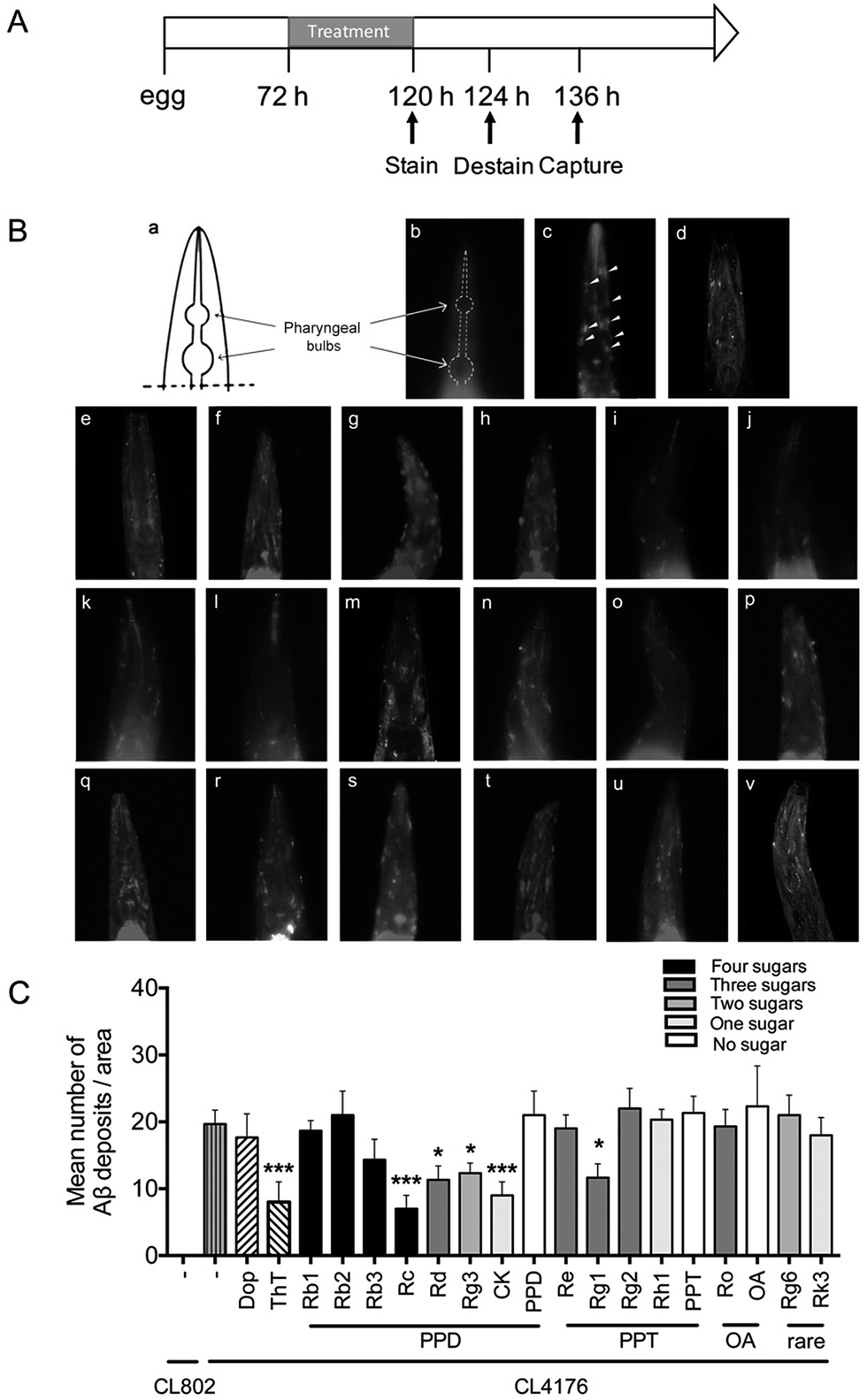 | ||
| Fig. 1 Effects of ginsenosides on Aβ deposits in transgenic C. elegans. (A) An illustrative diagram indicating the time duration of treatment and detectable Aβ deposits stain. Synchronized CL2006 or CL2109 were placed at 16 °C for 72 h, and then treated with vehicle (0.1% DMSO), Dop (50 μM), ThT (50 μM) or ginsenosides (50 μM) for 48 h. The worms were then stained with Methoxy-X04 (1 mM in 10 mM Tris, pH 8.0) for 4 h at room temperature. After staining, worms were subsequently returned to seeded NGM plates for 12 h to allow destaining via normal metabolism. Stained worms were mounted and photographed by using fluorescence microscope. (B) The schematic diagram indicates position of pharyngeal bulbs (a) and representative images of fluorescence in CL2109 (b), CL2006 treated with vehicle (0.1% DMSO) (c), Dop (d), ThT (e) or ginsenoside Rb1 (f), Rb2 (g), Rb3 (h), Rc (i), Rd (j), Rg3 (k), CK (l), PPD (m), Re (n), Rg1 (o), Rg2 (p), Rg2 (q), Rh1 (r), PPT (s), Ro (s), OA (t), Rg6 (u) and Rk3 (v) at 50 μM in M9 buffer. White arrows indicate Aβ deposits in head region. (C) Quantitative analysis of Aβ deposits in the transgenic C. elegans CL2006 treated with different ginsenosides for 48 h. The quantity is expressed as mean number of Aβ deposits/anterior area of worm (n = 25 for each analysis). Error bars indicate SD. ###p < 0.001, ***p < 0.001 compared with CL2006 vehicle treated group. | ||
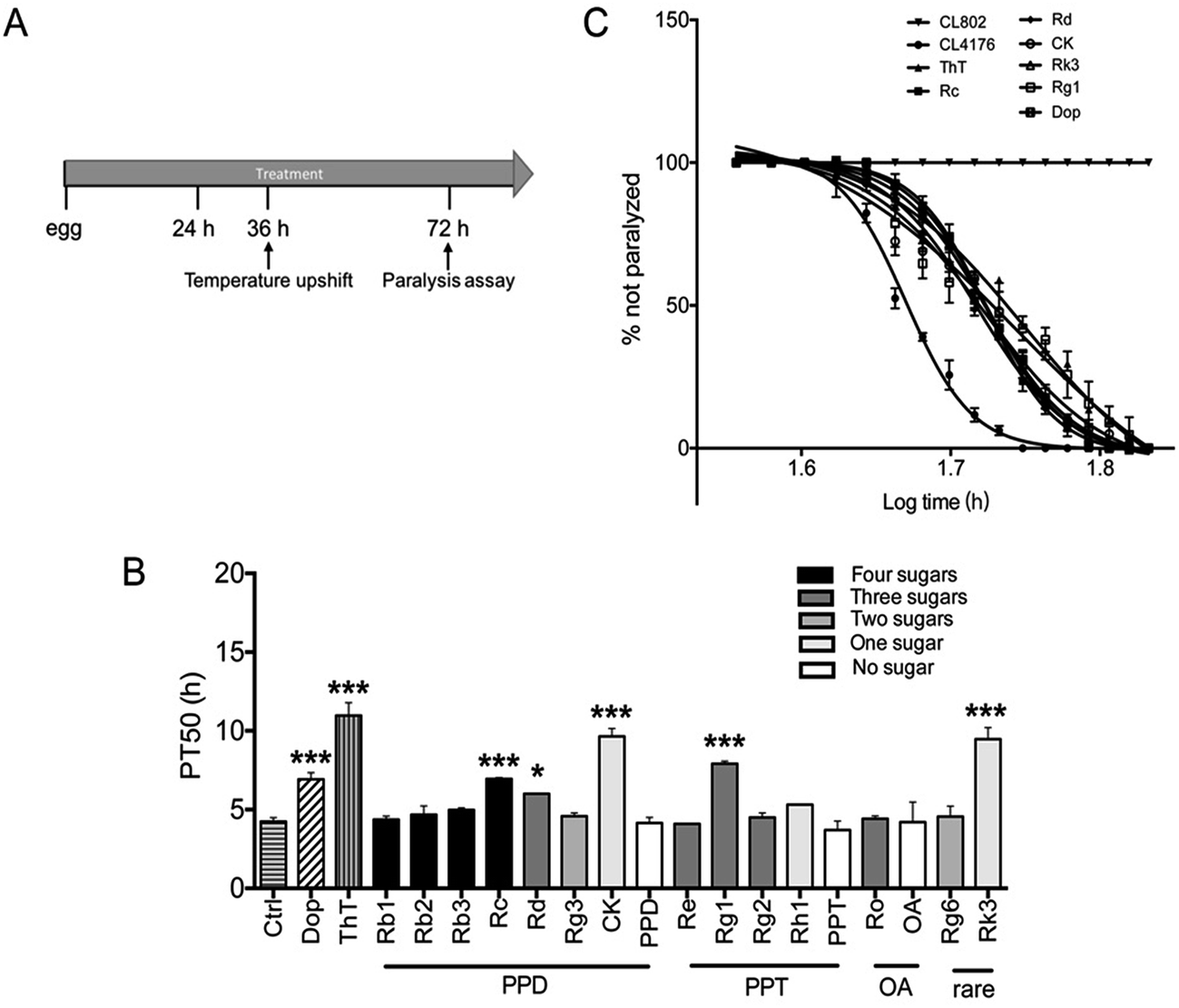 | ||
| Fig. 2 Effects of ginsenosides on Aβ induced paralysis in the transgenic C. elegans (A) an illustrative diagram indicating the time duration of treatment and paralysis assays in CL4176 worms treated with different compounds (50 μM). Synchronized eggs of CL802 or CL4176 worms were maintained at 16 °C on the NGM plates. CL4176 worms were treated with vehicle (0.1% DMSO), Dop (50 μM), ThT (50 μM) or ginsenosides (50 μM) for 36 h. Then the temperature was increased to 25 °C to induce Aβ expression. Paralysis was scored at 2 h intervals. (B) The paralysis assays were quantified for mean time duration at which 50% worms were paralyzed (PT50) from the transgenic worms fed with or without the drugs. Values of p were obtained from 6 independent assays for the worms compared with control group (treated with vehicle). Error bars indicate SD. ###p < 0.001, *p < 0.05, ***p < 0.001 compared with Ctrl group. (C) Time course of paralysis assays in CL802 or CL4176 fed with different drugs. Paralysis in the transgenic strain CL4176 is attributable to Aβ expression compared with the control strain CL802, which does not express Aβ transgene. Data are expressed as percentage of nonparalyzed worms from at least three independent assays of 100 worms in each experiment. | ||
3.2 Effects of ginsenosides on Aβ induced paralysis in the transgenic C. elegans
To determine whether ginsenosides specifically protects against Aβ induced toxicity in vivo, ginsenosides or aglycones were administrated to CL4176, the transgenic C. elegans line in which human Aβ42 peptide expressed in muscle cells and induce an Aβ dependent paralysis phenotype.44 The process of paralysis assay was described in Fig. 2A. Synchronized eggs of CL4176 and CL802 (used as genetic background control strain of CL4176) were placed on NGM plates containing vehicle, Dop (50 μM), ThT (50 μM) or different ginsenosides (50 μM) for 36 h, followed by upshifting temperature from 16 °C to 25 °C to induce Aβ42 expression. PT50 is defined as the time interval from the onset of paralysis to 50% of the worms were paralyzed45 and used for quantitative analysis. As shown in Fig. 2B and C, ThT treatment significantly delay Aβ induced paralysis which was consistent with the data reported previously,37 similarly, Dop. Rc, Rd, CK, Rg1 and Rk3 exhibited a significant delay of paralysis (Fig. 2B and C).3.3 Effects of ginsenosides on neuronal Aβ expression induced defect in associative learning capacity
Transgenic worm in which human Aβ42 peptide is expressed in neuronal cells was described and used for researching the influences of Aβ on neurological functions.31 The chemotaxis response in C. elegans is related to activation of several sensory neurons and interneurons to stimulate the motor neurons.46 This innate behavior was paired with a conditioned stimulant and can be utilized to assess associative learning ability.31 Chemotaxis index (CI) value is defined as the fraction of worms that are able to arrive at the location of the attractants (diacetyl in this study),47 and is used to investigate the effects of ginsenosides on the performance of chemotaxis behavior in the present study. The transgenic strain CL2241 that has inducible expression of human Aβ42 in the neurons30 was used for investigating the effects of ginsenosides on neuronal Aβ expression induced defect in associative learning capacity. Synchronized eggs of CL2241 and CL2122 (used as genetic background control strain of CL2241) were placed on NGM plates containing vehicle (0.1% DMSO) or different compounds for 36 h, followed by upshifting the temperature from 16 °C to 23 °C to induce Aβ42 expression for another 36 h before odorant assay (Fig. 3A). Worms from the same treated plates were distributed equally into conditioned group or naïve group. Conditioned worms were starved for 2 h in the presence of diacetyl. Naïve worms were starved simultaneously in the absence of odorant. Then both groups were subjected to odorant preference assay for 1 h (Fig. 3B).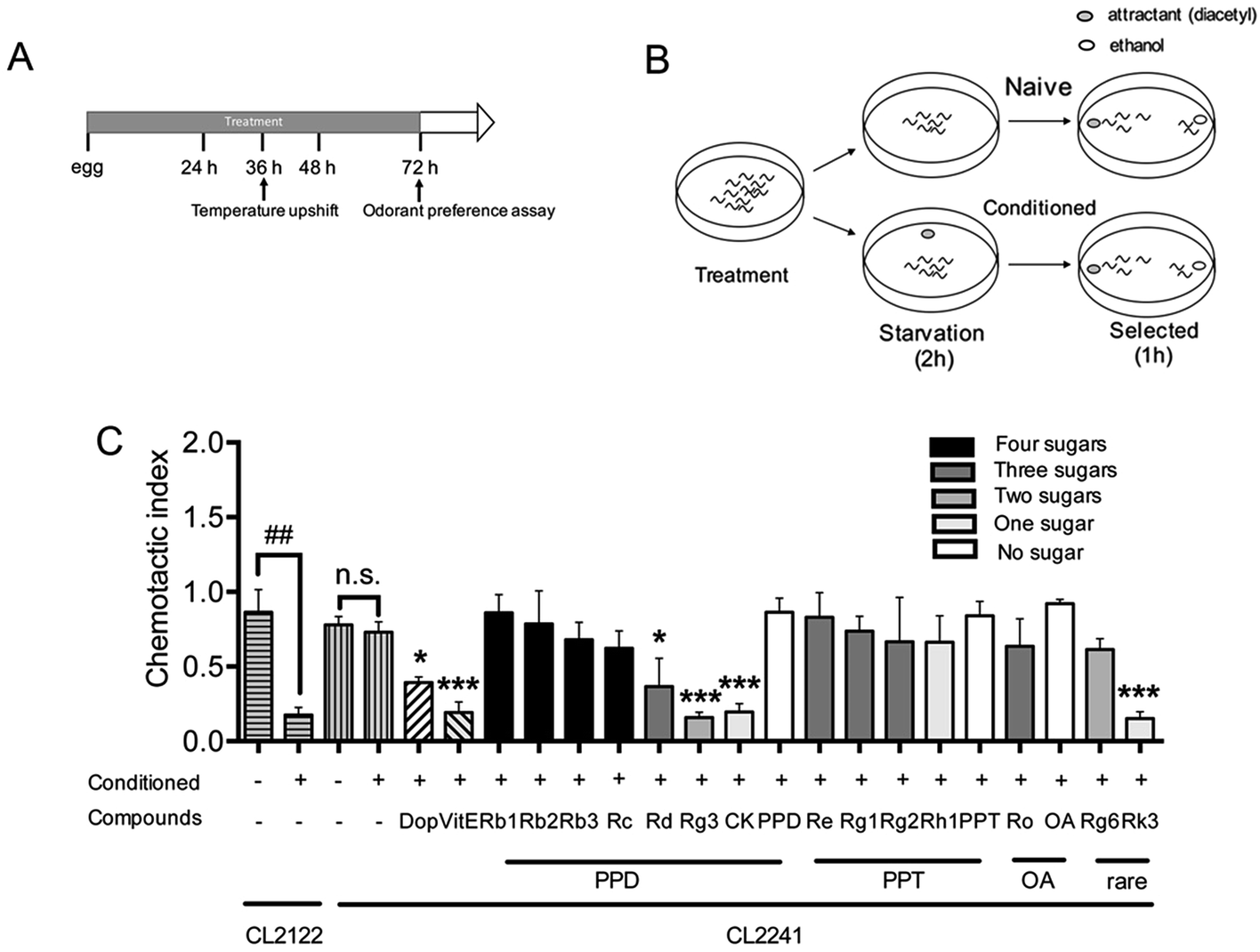 | ||
| Fig. 3 Effects of ginsenosides on neuronal Aβ expression induced defect in associative learning capacity (A) an illustrative diagram indicating the time duration of treatment and paralysis assays in CL2122 or CL2241 worms. (B) Schematic illustration of odorant preference associative learning assay. At the end of drug treatment (Dop, 50 μM; ThT, 50 μM; or ginsenosides, 50 μM), worms were starved for 2 h either in the presence (conditioned) or absence (naïve) of the odorant diacetyl and subsequently subjected to a chemotaxis assay. (C) The chemotaxis index in the control strain (CL2122) and the transgenic strain (CL2241) were scored as mentioned in method part. Each experiment was performed in triplicate. Error bars represent the standard error. ##p < 0.01. *p < 0.05, ***p < 0.001 compared with relevant naïve group. | ||
As shown in Fig. 3C, both CL2241 and CL2122 naïve worms preferred diacetyl though the transgenic worms displayed a slightly lower basal affinity for the diacetyl, indicating that the chemotaxis behavior of CL2241 worms was not impaired. After conditioned, CI of CL2122 worms was significantly decreased, while the CL2241 worms still prefer the attractants. These results indicated that CL2241 worms failed to associate the nutrient deprived environment with the environmental cue. CI of conditioned CL2241 worms were significantly decreased after Dop, VitE, Rg3, CK and Rk3 treatment, but not other ginsenosides or aglycones. These results indicated that Dop, VitE, Rg3, CK and Rk3 could ameliorate Aβ induced neurotoxicity.
3.4 Effect of ginsenosides on the production of ROS
Numerous evidences have shown that oxidative stress is associated with Aβ induced toxicity in AD48. We first tested the effects of ginsenosides on the production of ROS in transgenic muscle expression Aβ strain CL4176 and its control strain CL802. VitE is known to be an antoxidant and used in AD treatment.49 As shown in Fig. 4A, the untreated CL4176 worms exhibited increased levels of ROS compared with CL802 worms. Dop, VitE, Rg3, CK and Rk3 treatment before the induction of Aβ expression significantly prevent the elevation of ROS levels in CL4176 worms. We further found that Rg3, CK and Rk3 could attenuate the intracellular levels of ROS in CL4176 worms in a does-dependent manner (Fig. 4B).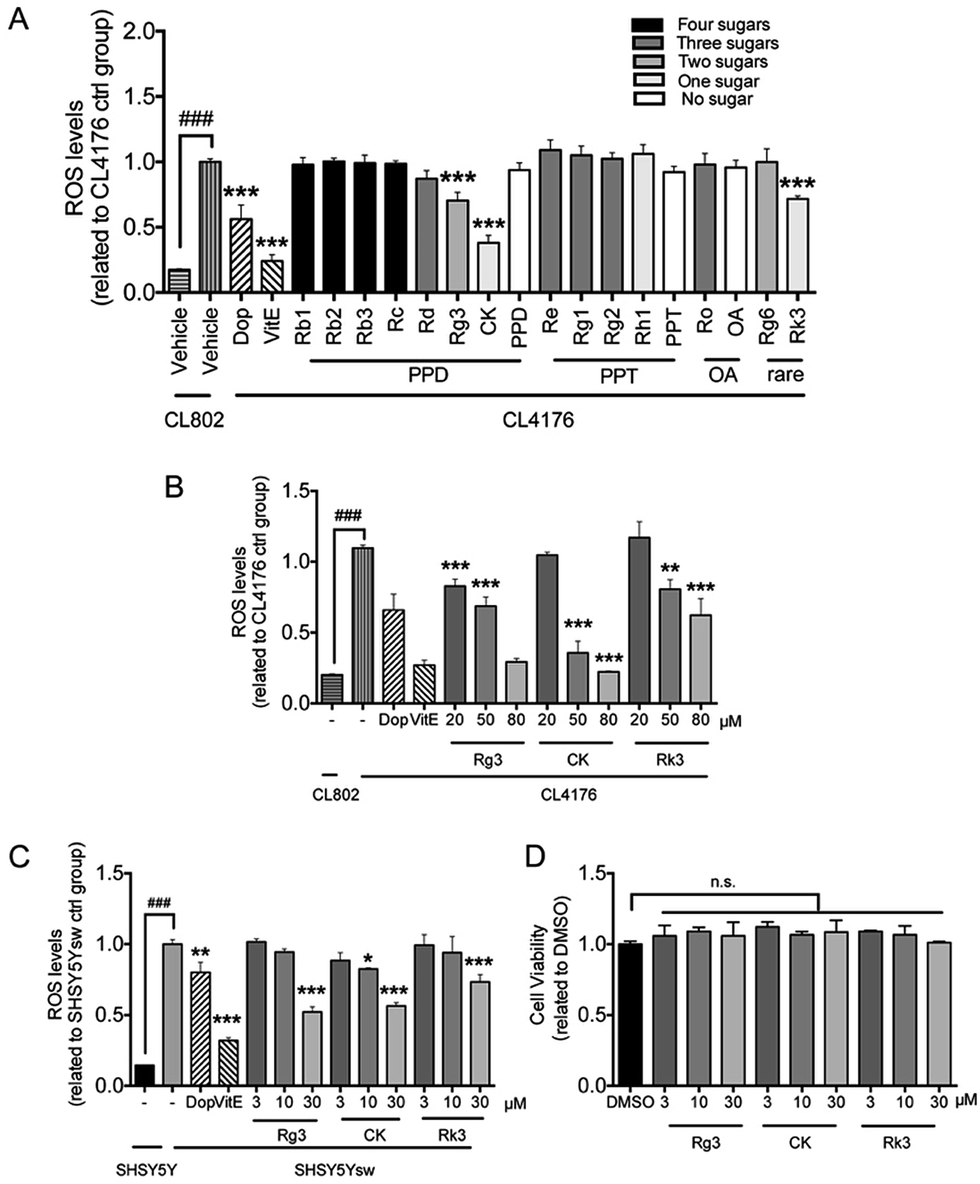 | ||
| Fig. 4 Effect of ginsenosides on the production of ROS Levels of ROS in the C. elegans fed with different ginsenoside (A) or ginsenoside 20(S)-Rg3, CK and Rk3 at different concentrations (B). Age-synchronized groups of transgenic C. elegans strain CL802 or CL4176 at 16 °C for 36 h were temperature upshifted to 25 °C for 36 h followed by the DCF assay for ROS as described in Method section. At least 60 worms from each group were analyzed for the levels of ROS. Results are expressed as fold change of fluorescence in CL4176 control group (treated with vehicle), which is set as 1. Error bars represent the standard error. ###p < 0.001, **p < 0.01, ***p < 0.001 compared with CL4176 control group. (C) Cells were pretreated with vehicle (0.1% DMSO) or ginsenoside Rg3, CK, Rk3 at different concentrations for 48 h followed by administrated with BA (1 μM) for 24 h to stimulate the express of transgene. ROS levels in cells were measured as described in Method section. Results were obtained from 6 independent samples for each compound tested. (D) Primary neurons (DIV 8) were treated with Rg3, CK or Rk3 at different concentration for 24 h followed by measurement of cell viability through MTT assay. Error bars represent the standard error. ###p < 0.001, *p < 0.05, ***p < 0.001 compared with SH-SY5Ysw vehicle treated group. | ||
Next, a transgenic mammal neuron cell line SH-SY5Ysw cells were used to further validate the effect of 20(S)-Rg3, CK and Rk3 on internal expression of Aβ induced ROS production. Dop and VitE were served as positive control. As shown in Fig. 4C, the levels of ROS were significantly higher in Aβ secreting mutant cells SH-SY5Ysw compared to the wild type cells SH-SY5Y. Pretreatment with Rg3, CK and Rk3 for 48 h significantly attenuated the elevation of ROS levels in SH-SY5Ysw cells in a dose dependent manner. Moreover, none of these three saponins showed cytotoxicity in primary neurons (Fig. 4D).
4. Discussion
In the present study, we investigated the in vivo anti-Aβ toxicity effects of different types of ginsenosides using transgenic C. elegans models. We found that (1) Rc, Rd, 20(S)-Rg3, CK that belong to PPD type of ginsenoside, Rg1 that belongs to PPT type, could significantly reduce the formation of Aβ deposits; (2) Rc, Rd, CK, Rk3 and Rg1 inhibit Aβ induced paralysis, (3) 20(S)-Rg3, CK and Rk3 prevented Aβ induced chemotaxis associated learning dysfunction and reduce Aβ induced ROS production both in whole organism and in transgenic mammal cells.Among various in vitro and in vivo models of AD, C. elegans is an ideal model organism for anti-AD drug discovery because of its well-developed genetics, short life cycle, simple structure of nervous system, and low dosage of drug consumption. Moreover, the absence of endogenous Aβ production in C. elegans make researchers could find direct role of Aβ involvement in pathological behaviors.50 Since AD has been associated with aggregation of Aβ and cell death in brain, different Aβ transgenic C. elegans models were used in the present study to validate the potential anti-AD ability of ginsenosides.
In vitro studies have shown that Aβ aggregation start from monomers, then form the oligomer, protofibril and fibril.51 Recent studies showed that the oligomer, rather than the fibril or plaque, play as the main toxic species in the pathogenesis of AD.52,53 We found Rc, Rd, CK and Rg1 reduced Aβ deposits in CL2006 worms through Aβ staining assay (Fig. 1), and significantly delay the Aβ induced paralysis (Fig. 2) in CL4176 worms. Combining the previous observation that paralysis occurs before detectable Aβ deposition formation in C. elegans,54 the paralysis delay effects of Rc, Rd, CK and Rg1 may related to the regulation of Aβ oligomerization processes in worms. On the other hand, it has been found that inducible expression of human Aβ42 in muscle leads to an accumulation of autophagic vesicles in C. elegans which contribute to reducing Aβ aggregation.55 Compounds that promote autophagy–lysosomal process could reduce Aβ deposition and prevented Aβ induced paralysis.56–58 It has been showed that CK,59 Rg3 (ref. 60) could induce autophagy, which may explain the Aβ deposits reducing effect of CK and Rg3 in CL4176 worms (Fig. 1B).
Considering the muscle specific Aβ expressing C. elegans model could not be specific reflect of the pathology in the AD brain. Therefore, we further used the transgenic C. elegans model that has inducible, pan-neuronal expression of Aβ to evaluate in vivo effects of ginsenosides on neuronal Aβ toxicity. The relationship between Aβ expression in neuron cells and defects in chemotaxis associated learning ability in transgenic C. elegans model has been well established.31 Dop, VitE, and four ginsenosides, Rd, Rg3, CK and Rk3 were found to significantly improve Aβ induced defect in associative learning capacity. Numerous studies suggested that the oxidative stress plays a key role in Aβ induced neurotoxic and enhance the progression of AD.61,62 The oxidative stress is found increased in neurons that are exposed to Aβ oligomer63 or neurons surrounded the Aβ plaque in the AD brain.64 Similarly, elevated ROS production was found increased in transgenic C. elegans expressing constitutive35 and inducible Aβ.54 There is a positive correlation between ROS levels and muscle degeneration presenting as paralysis.54 Consistent with the previous report,45 we observed significantly increased ROS production in CL4176 worms in the present study. Recent studies have showed that the inhibitors of acetylcholinesterase altered redox homeostasis and attenuate Aβ induced toxicity.65–67 Our results showed that both Dop and VitE, a known antioxidant, reduced ROS production in CL4176 and SHSY5Ysw transgenic cells (Fig. 4).
Among the ginsenosides that have been studied in the present research, Rg3, CK and Rk3 significantly decreased the in vivo ROS production in a dose dependent manner. The anti-oxidative effects of Rg3, CK and Rk3 were further confirmed in transgenic mammal neuronal cell line SH-SY5Ysw (Fig. 4). It has been reported that both Rg3 (ref. 68) and CK69 could suppress ROS production in bone marrow-derived macrophages and keratinocytes. It was also reported that CK induced autophagy and apoptosis in human colon cancer cells, but not in normal colon cells, through ROS generation.59 These results suggested that the effects of CK on ROS levels are cell-type specific. This might partially explain the effects of CK in SHSY5Ysw transgenic cells, since SHSY5Y is also a bone-marrow derived cell line.70 We found that CK also exhibited cytotoxicity in SHSY5Ysw cells at higher concentrations (Fig. S1†), which indicated that CK inhibits Aβ induced ROS production in a certain concentration range. It is interesting that Rd could delay Aβ induced paralysis, reduce plaque formation and enhance chemotaxis associated learning ability in transgenic C. elegans models, but did not affect ROS production. The p38 MAPK pathway is involved in endoplasmic reticulum homeostasis and immune activation in C. elegans during stress. It has been reported that Rd could promote non-amyloidogenic pathway of amyloid precursor protein processing by regulating MAPK/PI3K pathway.23 Further research is needed to investigate whether Rd ameliorates Aβ induced toxicity through MAPK pathway in C. elegans.
Several in vitro studies suggest that the activity of ginsenosides have relationship with its structure.71–73 Takemoto et al. found that several PPD type saponins, but not PPT or oleanane type saponins, potentiated nerve growth factor (NGF) induced neurite outgrowth.74 Tohda et al. indicated that the PPD type saponins, including Rb1 and Rb3, enhanced axonal and dendritic formation in SK-N-SH cells, while the PPT, ocotillol and oleanane type saponins have no effect on the neurite outgrowth.75 In terms of ginsenosides studied in the present research (Fig. 5), four PPD type saponin, including Rc, Rd, Rg3 and CK, while only one PPT type saponin Rg1, that could significantly reduce Aβ deposits (Fig. 2); three PPD type saponin that Rc, Rd, and CK, while only one PPT type saponin Rg1, that could significantly delay Aβ induced paralysis (Fig. 3). Moreover, only PPD type saponin Rg3 and CK, and rare type Rk3 ameliorated Aβ induced associative learning capacity deficiency and ROS production (Fig. 4 and 5). Our results suggested that different types of aglycone might contribute different anti-Aβ activity. Sugar moiety is another key factor, which influences the activity of ginsenosides.71,76,77 The anti-Aβ effects of tetrasaccharide glycoside, trisaccharide glycoside, disaccharide glycoside, monosaccharide glycoside and sapogenin in different C. elegans models were compared. Firstly, ginsenosides with less sugar moieties showed stronger efficacy in ameliorating Aβ induced toxicity in vivo. This might related to better absorption and cell membrane permeability of the ginsenosides with less sugar moieties and increased hydrophobicity.76 Secondly, the position of sugar linkage seems affect the anti-Aβ activity of ginsenosides. As shown in Tables 2–4, CK has one glucose moiety linked at its C-20 position and showed stronger anti-Aβ activity than PPD. Rc, Rd, Rb1, Rb2 which have the same aglycone type and the same sugar moiety at C-3 position, but different types of sugar moiety linked at C-20 position. Both Rc and Rd showed anti-Aβ activity in different models, while Rb1 and Rb2 have no significant effects. Rg1 and Re along with rare gensinosides Rk3 and Rg6 have different sugar moiety at C-6 position and showed different effects in C. elegans model. Moreover, PPD, PPT and OA showed insignificant effects in the present study. These results indicated that the moiety number and position of ginsenosides are crucial for their anti-Aβ activity.
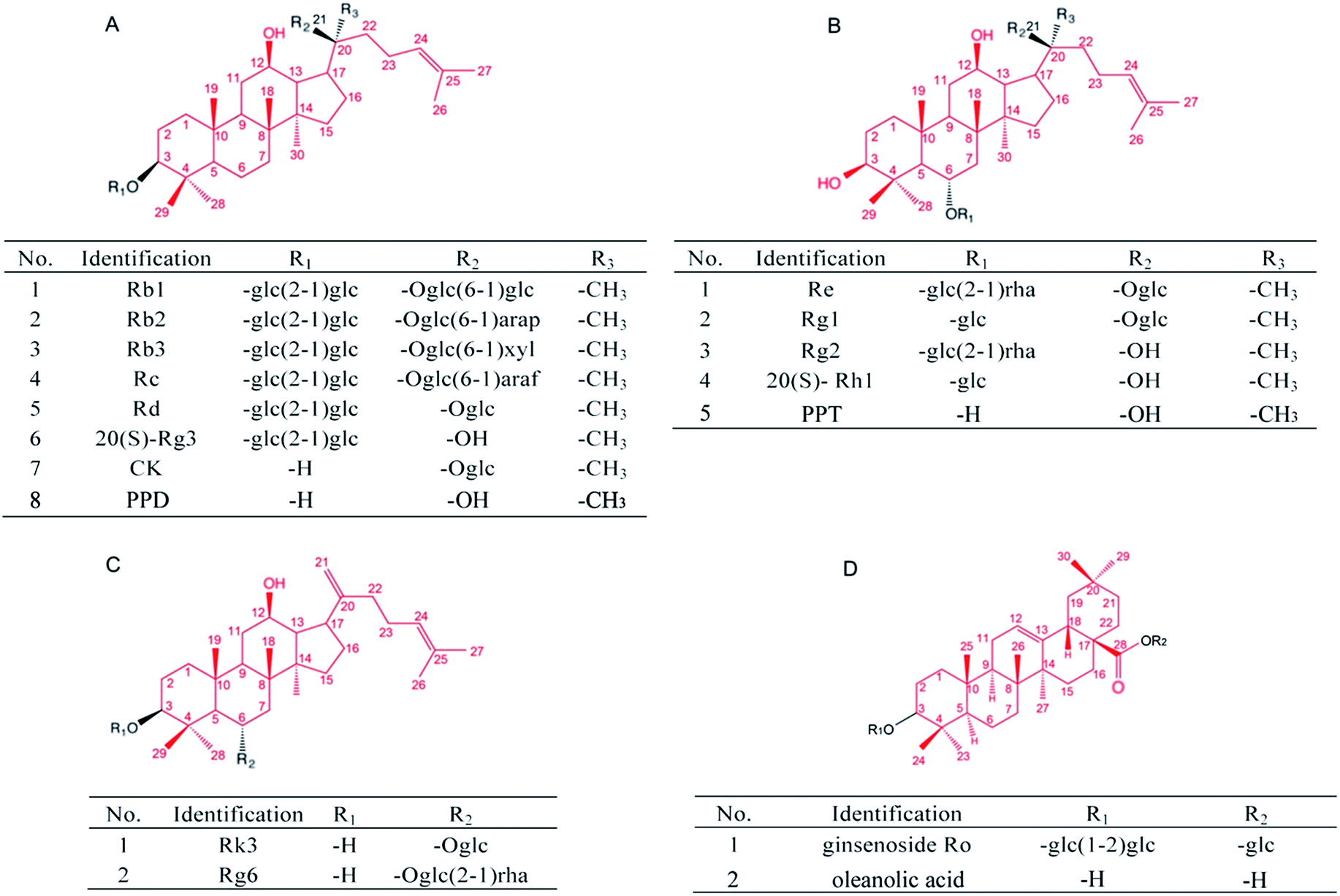 | ||
| Fig. 5 Chemical structures of the studied ginsenosides. (A) Protopanaxadiol (PPD)-type ginsenosides, (B) protopanaxatriol (PPT)-type ginsenosides, (C) rare ginsenosides, (D) oleanane-type ginsenosides. glc, β-D-glucose; rha, α-L-rhamnose; arap, α-L-arabinose (pyranose); araf, α-L-arabinose (furanose); xyl, β-D-xylopyranose. The red line indicated the aglycone structure. | ||
| Identification | Aglycone | No. of sugars | Position of sugars | Anti-Aβ activity | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| C-3 | C-6 | C-12 | C-20 | Paralysis | Stain | CI | ROS | |||
| a *p < 0.05, ***p < 0.001 compared with model control group. | ||||||||||
| Rb1 | PPD | 4 | 2 | 0 | 1-OH | 2 | n.s. | n.s. | n.s. | n.s. |
| Rb2 | 4 | 2 | 0 | 1-OH | 2 | n.s. | n.s. | n.s. | n.s. | |
| Rb3 | 4 | 2 | 0 | 1-OH | 2 | n.s. | n.s. | n.s. | n.s. | |
| Rc | 4 | 2 | 0 | 1-OH | 2 | *** | *** | n.s. | n.s. | |
| Rd | 4 | 2 | 0 | 1-OH | 2 | * | * | * | n.s. | |
| 20(S)-Rg3 | 2 | 2 | 0 | 1-OH | 1-OH | n.s. | * | *** | *** | |
| CK | 1 | 1 | 0 | 1-OH | 0 | *** | *** | *** | *** | |
| PPD | 0 | 1-OH | 0 | 1-OH | 0 | n.s. | n.s. | n.s. | n.s. | |
| Re | PPT | 3 | 1-OH | 2 | 1-OH | 1-OH | n.s. | n.s. | n.s. | n.s. |
| Rg1 | 2 | 1-OH | 1 | 1-OH | 1 | *** | * | n.s. | n.s. | |
| Rg2 | 3 | 1 | 2 | 1-OH | 1-OH | n.s. | n.s. | n.s. | n.s. | |
| 20(S)-Rh1 | 1 | 1-OH | 1 | 1-OH | 1-OH | n.s. | n.s. | n.s. | n.s. | |
| PPT | 0 | 0 | 1-OH | 1-OH | 1-OH | n.s. | n.s. | n.s. | n.s. | |
| Identification | No. of sugars | Position of sugars | Anti-Aβ activity | ||||||
|---|---|---|---|---|---|---|---|---|---|
| C-3 | C-6 | C-12 | C-20 | Paralysis | Stain | CI | ROS | ||
| a ***p < 0.001 compared with model control group. | |||||||||
| Rk3 | 1 | 1-OH | 1 | 1-OH | 0 | *** | n.s. | *** | *** |
| Rg6 | 2 | 1-OH | 2 | 1-OH | 0 | n.s. | n.s. | n.s. | n.s. |
| Identification | Aglycone | No. of sugars | Position of sugars | Anti-Aβ activity | |||||
|---|---|---|---|---|---|---|---|---|---|
| C-3 | C-6 | C-28 | Paralysis | Stain | CI | ROS | |||
| Ro | OA | 3 | 2 | 0 | 1 | n.s. | n.s. | n.s. | n.s. |
| OA | 0 | 1-OH | 0 | 1-OH | n.s. | n.s. | n.s. | n.s. | |
5. Conclusions
Our studies in C. elegans provided a systematic evaluation of ginsenosides on the anti-Aβ effect in vivo. Moreover, based on the uncovered structure–activity relationships, we can select and modify specific ginsenosides to obtain new prodrug with superior anti-AD activity. However, given the differences in mammals and nematode physiology, more study is needed to determine the anti-AD activities of Rc, Rd, Rg3, CK, Rk3 and Rg1 in mammals. Of particular relevance are the questions of whether anti-Aβ species of ginsenosides actually cross the blood brain barrier and whether these ginsenosides activation in the periphery can lead to physiological changes that impact the brain. Nevertheless, our studies add to the growing body of literature indicating that anti-oxidative is neuroprotective.Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81573637, 81421005), “Twelfth-Five Years” Supporting Programs from the Ministry of Science and Technology of China (No. 2015ZX09101043). The authors would like to thank Prof. Christopher D. Link (Institute for Behavioral Genetics, University of Colorado) for generously providing C. elegans strains CL2109 and CL2241. The authors acknowledge Mr David Anderson (Southeast University) for his generous support with language editing of the manuscript, and Dr Bin Wu for his technical support with primary neuron culture.References
- K. Blennow, M. J. de Leon and H. Zetterberg, Lancet, 2006, 368, 387–403 CrossRef CAS.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS.
- D. M. Mann, T. Iwatsubo, N. J. Cairns, P. L. Lantos, D. Nochlin, S. M. Sumi, T. D. Bird, P. Poorkaj, J. Hardy, M. Hutton, G. Prihar, R. Crook, M. N. Rossor and M. Haltia, Ann. Neurol., 1996, 40, 149–156 CrossRef CAS.
- E. M. Snyder, Y. Nong, C. G. Almeida, S. Paul, T. Moran, E. Y. Choi, A. C. Nairn, M. W. Salter, P. J. Lombroso, G. K. Gouras and P. Greengard, Nat. Neurosci., 2005, 8, 1051–1058 CrossRef CAS.
- L. Jia and Y. Zhao, Curr. Med. Chem., 2009, 16, 2475–2484 CrossRef CAS.
- J. Y. Wan, Y. Fan, Q. T. Yu, Y. Z. Ge, C. P. Yan, R. N. Alolga, P. Li, Z. H. Ma and L. W. Qi, J. Pharm. Biomed. Anal., 2015, 107, 89–97 CrossRef CAS.
- H. J. Kim, P. Kim and C. Y. Shin, J. Ginseng Res., 2013, 37, 8–29 CrossRef CAS.
- K. Radad, G. Gille, L. Liu and W. D. Rausch, J. Pharmacol. Sci., 2006, 100, 175–186 CrossRef CAS.
- E. Tachikawa, K. Kudo, K. Harada, T. Kashimoto, Y. Miyate, A. Kakizaki and E. Takahashi, Eur. J. Pharmacol., 1999, 369, 23–32 CrossRef CAS.
- A. S. Attele, J. A. Wu and C. S. Yuan, Biochem. Pharmacol., 1999, 58, 1685–1693 CrossRef CAS.
- L. W. Qi, C. Z. Wang and C. S. Yuan, Nat. Prod. Rep., 2011, 28, 467–495 RSC.
- M. H. Kim, Y. C. Lee, S. Y. Choi, C. W. Cho, J. Rho and K. W. Lee, J. Ginseng Res., 2011, 35, 497–503 CrossRef CAS.
- B.-H. Kim, S.-Y. Lee, H.-J. Cho, S.-N. You, Y.-J. Kim, Y.-M. Park, J.-K. Lee, M.-Y. Baik, C.-S. Park and S.-C. Ahn, Biol. Pharm. Bull., 2006, 29, 2472–2478 CrossRef CAS.
- J.-R. Choi, S.-W. Hong, Y. Kim, S.-E. Jang, N.-J. Kim, M. J. Han and D.-H. Kim, J. Ginseng Res., 2011, 35, 301–307 CrossRef CAS PubMed.
- T. F. Lee, Y. J. Shiao, C. F. Chen and L. C. Wang, Planta Med., 2001, 67, 634–637 CrossRef CAS.
- C. Tohda, N. Matsumoto, K. Zou, M. R. Meselhy and K. Komatsu, Neuropsychopharmacology, 2004, 29, 860–868 CrossRef CAS.
- N. Nishiyama, S. I. Cho, I. Kitagawa and H. Saito, Biol. Pharm. Bull., 1994, 17, 509–513 CrossRef CAS.
- B. Jiang, Z. Xiong, J. Yang, W. Wang, Y. Wang, Z. L. Hu, F. Wang and J. G. Chen, Br. J. Pharmacol., 2012, 166, 1872–1887 CrossRef CAS.
- N. Li, Y. Liu, W. Li, L. Zhou, Q. Li, X. Wang and P. He, J. Ginseng Res., 2016, 40, 9–17 CrossRef CAS PubMed.
- B. Lee, B. Sur, J. Park, S. H. Kim, S. Kwon, M. Yeom, I. Shim, H. Lee and D. H. Hahm, Biomol. Ther., 2013, 21, 381–390 CrossRef CAS PubMed.
- J. Liu, X. Yan, L. Li, Y. Zhu, K. Qin, L. Zhou, D. Sun, X. Zhang, R. Ye and G. Zhao, Neurochem. Res., 2012, 37, 2738–2747 CrossRef CAS.
- X. Zhang, M. Shi, R. Ye, W. Wang, X. Liu, G. Zhang, J. Han, Y. Zhang, B. Wang, J. Zhao, J. Hui, L. Xiong and G. Zhao, Neurochem. Res., 2014, 39, 1363–1373 CrossRef CAS PubMed.
- X. Yan, G. Hu, W. Yan, T. Chen, F. Yang, X. Zhang, G. Zhao and J. Liu, Life Sci., 2017, 168, 16–23 CrossRef CAS.
- Q. Wan, X. Ma, Z. J. Zhang, T. Sun, F. Xia, G. Zhao and Y. M. Wu, Mol. Neurobiol., 2017, 54, 2889–2900 CrossRef CAS.
- C. F. Wu, X. L. Bi, J. Y. Yang, J. Y. Zhan, Y. X. Dong, J. H. Wang, J. M. Wang, R. Zhang and X. Li, Int. Immunopharmacol., 2007, 7, 313–320 CrossRef CAS.
- Y. Z. Wang, J. Chen, S. F. Chu, Y. S. Wang, X. Y. Wang, N. H. Chen and J. T. Zhang, J. Pharmacol. Sci., 2009, 109, 504–510 CrossRef CAS.
- J. Qiu, W. Li, S. H. Feng, M. Wang and Z. Y. He, GMR, Genet. Mol. Res., 2014, 13, 3586–3598 CrossRef CAS.
- J. G. Hou, J. J. Xue, M. R. Lee, M. Q. Sun, X. H. Zhao, Y. N. Zheng and C. K. Sung, Biochem. Biophys. Res. Commun., 2013, 436, 104–109 CrossRef CAS.
- A. L. Lublin and C. D. Link, Drug Discovery Today: Technol., 2013, 10, e115–119 CrossRef CAS.
- C. D. Link, Exp. Gerontol., 2006, 41, 1007–1013 CrossRef CAS.
- L. E. Dosanjh, M. K. Brown, G. Rao, C. D. Link and Y. Luo, J. Alzheimer's Dis., 2010, 19, 681–690 CAS.
- H. F. Epstein and D. C. Shakes, Caenorhibditus Elegans: Modern Biological Analysis of an Organism, Academic Press, 1995 Search PubMed.
- J. Dong, R. Revilla-Sanchez, S. Moss and P. G. Haydon, Neuropharmacology, 2010, 59, 268–275 CrossRef CAS.
- L. Diomede, G. Cassata, F. Fiordaliso, M. Salio, D. Ami, A. Natalello, S. M. Doglia, A. De Luigi and M. Salmona, Neurobiol. Dis., 2010, 40, 424–431 CrossRef CAS.
- J. V. Smith and Y. Luo, J. Alzheimer's Dis., 2003, 5, 287–300 Search PubMed.
- V. Dostal and C. D. Link, J. Visualized Exp., 2010, 44, 2252 Search PubMed.
- S. Alavez, M. C. Vantipalli, D. J. Zucker, I. M. Klang and G. J. Lithgow, Nature, 2011, 472, 226–229 CrossRef CAS.
- W. B. Wood, Developmental Biology, New York, N.Y, 1985, 1988, vol. 5, pp. 57–78 Search PubMed.
- W. M. Nuttley, S. Harbinder and D. van der Kooy, Learning & memory, Cold Spring Harbor, N.Y., 2001, vol. 8, pp. 170–181 Search PubMed.
- J. Wang, Z. Q. Shi, M. Zhang, G. Z. Xin, T. Pang, P. Zhou, J. Chen, L. W. Qi, H. Yang, X. Xu and P. Li, J. Alzheimer's Dis., 2015, 43, 465–477 CAS.
- T. Pang, Y. J. Wang, Y. X. Gao, Y. Xu, Q. Li, Y. B. Zhou, L. Xu, Z. J. Huang, H. Liao, L. Y. Zhang, J. R. Gao, Q. Ye and J. Li, Acta Pharmacol. Sin., 2016, 37, 741–752 CrossRef CAS.
- C. D. Link, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9368–9372 CrossRef CAS.
- C. D. Link, C. J. Johnson, V. Fonte, M. Paupard, D. H. Hall, S. Styren, C. A. Mathis and W. E. Klunk, Neurobiol. Aging, 2001, 22, 217–226 CrossRef CAS.
- C. D. Link, A. Taft, V. Kapulkin, K. Duke, S. Kim, Q. Fei, D. E. Wood and B. G. Sahagan, Neurobiol. Aging, 2003, 24, 397–413 CrossRef CAS.
- Y. Wu, Z. Wu, P. Butko, Y. Christen, M. P. Lambert, W. L. Klein, C. D. Link and Y. Luo, J. Neurosci., 2006, 26, 13102–13113 CrossRef CAS.
- O. Hobert, J. Neurobiol., 2003, 54, 203–223 CrossRef CAS.
- T. Matsuura, T. Sato and R. Shingai, Zool. Sci., 2005, 22, 1095–1103 CrossRef.
- R. Sultana, M. Perluigi and D. A. Butterfield, Acta Neuropathol., 2009, 118, 131–150 CrossRef CAS PubMed.
- M. W. Dysken, P. D. Guarino, J. E. Vertrees, S. Asthana, M. Sano, M. Llorente, M. Pallaki, S. Love, G. D. Schellenberg, J. R. McCarten, J. Malphurs, S. Prieto, P. Chen, D. J. Loreck, S. Carney, G. Trapp, R. S. Bakshi, J. E. Mintzer, J. L. Heidebrink, A. Vidal-Cardona, L. M. Arroyo, A. R. Cruz, N. W. Kowall, M. P. Chopra, S. Craft, S. Thielke, C. L. Turvey, C. Woodman, K. A. Monnell, K. Gordon, J. Tomaska and G. Vatassery, Alzheimer's Dementia, 2014, 10, 36–44 CrossRef PubMed.
- Y. Wu and Y. Luo, Curr. Alzheimer Res., 2005, 2, 37–45 CrossRef CAS PubMed.
- G. Bitan, M. D. Kirkitadze, A. Lomakin, S. S. Vollers, G. B. Benedek and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 330–335 CrossRef CAS.
- A. Salminen, J. Ojala, T. Suuronen, K. Kaarniranta and A. Kauppinen, J. Cell. Mol. Med., 2008, 12, 2255–2262 CrossRef CAS.
- M. E. Larson and S. E. Lesne, J. Neurochem., 2012, 120(suppl. 1), 125–139 CrossRef CAS.
- J. Drake, C. D. Link and D. A. Butterfield, Neurobiol. Aging, 2003, 24, 415–420 CrossRef CAS.
- M. L. Florez-McClure, L. A. Hohsfield, G. Fonte, M. T. Bealor and C. D. Link, Autophagy, 2007, 3, 569–580 CrossRef CAS.
- C. Regitz, L. M. Dussling and U. Wenzel, Mol. Nutr. Food Res., 2014, 58, 1931–1940 CrossRef CAS.
- J. Li, X. Cui, X. Ma and Z. Wang, Exp. Gerontol., 2017, 89, 78–86 CrossRef CAS.
- C. Regitz, E. Fitzenberger, F. L. Mahn, L. M. Dussling and U. Wenzel, Eur. J. Nutr., 2016, 55, 741–747 CrossRef CAS.
- A. D. Kim, K. A. Kang, H. S. Kim, D. H. Kim, Y. H. Choi, S. J. Lee, H. S. Kim and J. W. Hyun, Cell Death Dis., 2013, 4, e750 CrossRef CAS PubMed.
- M. Sun, C. Huang, C. Wang, J. Zheng, P. Zhang, Y. Xu, H. Chen and W. Shen, Biochem. Biophys. Res. Commun., 2013, 441, 169–174 CrossRef CAS.
- M. Pohanka, Curr. Med. Chem., 2014, 21, 356–364 CrossRef CAS.
- G. Perry, R. J. Castellani, K. Hirai and M. A. Smith, J. Alzheimer's Dis., 1998, 1, 45–55 CAS.
- N. Izuo, T. Kume, M. Sato, K. Murakami, K. Irie, Y. Izumi and A. Akaike, ACS Chem. Neurosci., 2012, 3, 674–681 CrossRef CAS.
- H. Xie, S. Hou, J. Jiang, M. Sekutowicz, J. Kelly and B. J. Bacskai, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7904–7909 CrossRef CAS PubMed.
- P. Atukeren, M. Cengiz, H. Yavuzer, R. Gelisgen, E. Altunoglu, S. Oner, F. Erdenen, D. Yuceakin, H. Derici, U. Cakatay and H. Uzun, Biomed. Pharmacother., 2017, 90, 786–795 CrossRef CAS PubMed.
- M. Pohanka, M. Hrabinova, F. Zemek, L. Drtinova, H. Bandouchova and J. Pikula, Neuroendocrinol. Lett., 2011, 32(suppl. 1), 95–100 CAS.
- T. Zhang, F. Tian, J. Wang, S. Zhou, X. Dong, K. Guo, J. Jing, Y. Zhou and Y. Chen, Cell Stress Chaperones, 2015, 20, 787–792 CrossRef CAS PubMed.
- Y. M. Shin, H. J. Jung, W. Y. Choi and C. J. Lim, Mol. Biol. Rep., 2013, 40, 269–279 CrossRef CAS.
- T. T. Cuong, C. S. Yang, J. M. Yuk, H. M. Lee, S. R. Ko, B. G. Cho and E. K. Jo, Life Sci., 2009, 85, 625–633 CrossRef CAS.
- R. A. Ross, B. A. Spengler and J. L. Biedler, J. Natl. Cancer Inst., 1983, 71, 741–747 CAS.
- K. Quan, Q. Liu, J. Y. Wan, Y. J. Zhao, R. Z. Guo, R. N. Alolga, P. Li and L. W. Qi, Sci. Rep., 2015, 5, 8598 CrossRef CAS PubMed.
- G. X. Li and Z. Q. Liu, Food Chem. Toxicol., 2008, 46, 886–892 CrossRef CAS PubMed.
- S. Chae, K. A. Kang, U. Youn, J. S. Park and J. W. Hyun, J. Food Biochem., 2010, 34, 31–43 CrossRef.
- Y. Takemoto, T. Ueyama, H. Saito, S. Horio, S. Sanada, J. Shoji, S. Yahara, O. Tanaka and S. Shibata, Chem. Pharm. Bull., 1984, 32, 3128–3133 CrossRef CAS.
- C. Tohda, N. Matsumoto, K. Zou, M. R. Meselhy and K. Komatsu, Jpn. J. Pharmacol., 2002, 90, 254–262 CrossRef CAS.
- S. A. Nag, J. J. Qin, W. Wang, M. H. Wang, H. Wang and R. Zhang, Front. Pharmacol., 2012, 3, 25 Search PubMed.
- H. Teng, L. Chen, T. Fang, B. Yuan and Q. Lin, J. Funct. Foods, 2017, 28, 306–313 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05717b |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |