
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly efficient heterogeneous copper-catalyzed decarboxylative cross-coupling of potassium polyfluorobenzoates with aryl halides leading to polyfluorobiaryls†
Yang Lina,
Mingzhong Cai *b,
Zhiqiang Fanga and
Hong Zhao*a
*b,
Zhiqiang Fanga and
Hong Zhao*a
aSchool of Chemistry & Chemical Engineering, Guangdong Cosmetics Engineering & Technology Research Center, Guangdong Pharmaceutical University, Guangzhou 510006, P. R. China. E-mail: zhaohong1001@sina.com
bCollege of Chemistry & Chemical Engineering, Jiangxi Normal University, Nanchang 330022, P. R. China. E-mail: mzcai@jxnu.edu.cn
First published on 11th July 2017
Abstract
The heterogeneous decarboxylative cross-coupling reaction of potassium polyfluorobenzoates with aryl iodides and bromides was achieved in diglyme or DMAc at 130 or 160 °C in the presence of 10–20 mol% of a 1,10-phenanthroline-functionalized MCM-41-immobilized copper(I) complex, [MCM-41-Phen-CuI], yielding a variety of polyfluorobiaryls in good to excellent yields. This heterogeneous copper(I) complex could easily be prepared via a simple procedure from commercially readily available and inexpensive reagents, exhibited the same catalytic activity as the homogeneous CuI/Phen system, and was recovered by filtration of the reaction solution and recycled at least 8 times without significant loss of catalytic activity.
1. Introduction
Polyfluorobiaryls are important molecules in the pharmaceutical chemistry1 and materials science2 and are often used as starting materials in organic synthesis.3 Subsequently, a variety of methods have been developed to form polyfluorobiaryls. The traditional methods involve the palladium- or copper-catalyzed cross-coupling reactions of pre-functionalized polyfluoroarenes with aryl halides, which require the use of expensive organometallic starting materials such as boronic acids,4 borate salts,5 stannanes,6 and copper species.7 Besides these methods, the palladium- or copper-catalyzed C–H arylation of polyfluoroarenes with aryl halides,8 arylboronic acids,9 organosilicon reagents,10 arenediazonium tetrafluoroborates,11 and aryliodine(III) diacetates,12 has provided an alternative approach to the synthesis of polyfluorobiaryls. Transition-metal-catalyzed decarboxylative cross-couplings using carboxylic acids as aryl sources are an attractive strategy for the construction of biaryl bonds because this method does not use expensive and sensitive organometallic reagents, and generates CO2 instead of organometallic byproducts that are formed in Suzuki–Miyaura, Stille, Kumada, and Negishi reactions.13 Furthermore, structurally diverse carboxylic acids are often inexpensive and commercially readily available. Recently, palladium-, nickel- and copper-catalyzed decarboxylative cross-couplings of polyfluorobenzoic acids or polyfluorobenzoate salts with aryl halides14 and palladium-catalyzed decarboxylative cross-coupling of aromatic carboxylic acids with polyfluoroarenes15 have been reported to be highly efficient methods for the synthesis of polyfluorobiaryls.Although significant progress has been achieved in construction of polyfluorobiaryls by homogeneous Pd-, Ni- and Cu-catalyzed decarboxylative cross-coupling reactions, the use of expensive palladium catalysts and about 10–20 mol% of nickel or copper catalysts has limited their applications in large-scale syntheses and industrial processes because homogeneous catalysis suffers from the problematic separation of the catalyst from the product for re-use.16 Moreover, homogeneous catalysis might result in heavy metal contamination of the desired isolated product, which restricts the application of such systems in electronics and biomedicine. The heterogenization of the existing homogeneous transition-metal catalysts appears to be a logical solution to these problems.17 There has been considerable interest in the development of heterogeneous catalytic systems that can be easily recycled whilst maintaining the inherent activity of the catalytic center. However, to the best of our knowledge, no examples of heterogeneous palladium-, nickel- and copper-catalyzed synthesis of polyfluorobiaryls have been reported until now.
Mesoporous MCM-41 materials have recently emerged as promising supports for immobilization because of their large and uniform pore sizes, ultrahigh surface areas, big pore volumes and rich silanol groups in the inner walls.18 To date, some functionalized MCM-41-immobilized palladium,19 rhodium,20 molybdenum,21 gold22 and copper23 complexes have been successfully used as potentially green and sustainable catalysts in organic reactions. Recently, we reported the first synthesis of MCM-41-immobilized 1,10-phenanthroline-copper(I) complex [MCM-41-Phen-CuI] and found that it is a highly efficient and recyclable heterogeneous catalyst for the C–O coupling reaction between aryl iodides and aliphatic alcohols.24 In continuing our efforts to develop greener synthetic pathways for organic transformations,19d–f,23,24 herein we wish to report a highly efficient, heterogeneous copper(I)-catalyzed decarboxylative cross-coupling of potassium polyfluorobenzoates with aryl halides leading to polyfluorobiaryls in good to excellent yields (Scheme 1).
 | ||
| Scheme 1 Synthesis of polyfluorobiaryls via heterogeneous copper(I)-catalyzed decarboxylative cross-coupling of potassium polyfluorobenzoates with aryl halides. | ||
2. Results and discussion
A series of MCM-41-immobilized 1,10-phenanthroline-copper(I) or (II) complexes [MCM-41-Phen-CuXn] were easily prepared according to the procedure summarized in Scheme 2.24 Firstly, the mesoporous MCM-41 (ref. 25) was condensed with 1-(1,10-phenanthrolin-5-yl)-3-(3-(triethoxysilyl)propyl)urea26 in toluene under reflux for 24 h, followed by the silylation with Me3SiCl in toluene at room temperature for 24 h to afford the 1,10-phenanthroline-functionalized MCM-41 [MCM-41-Phen]. The latter was then reacted with copper salts in acetone under reflux for 24 h to generate a series of MCM-41-immobilized 1,10-phenanthroline copper(I) or (II) complexes [MCM-41-Phen-CuXn] as light green powders.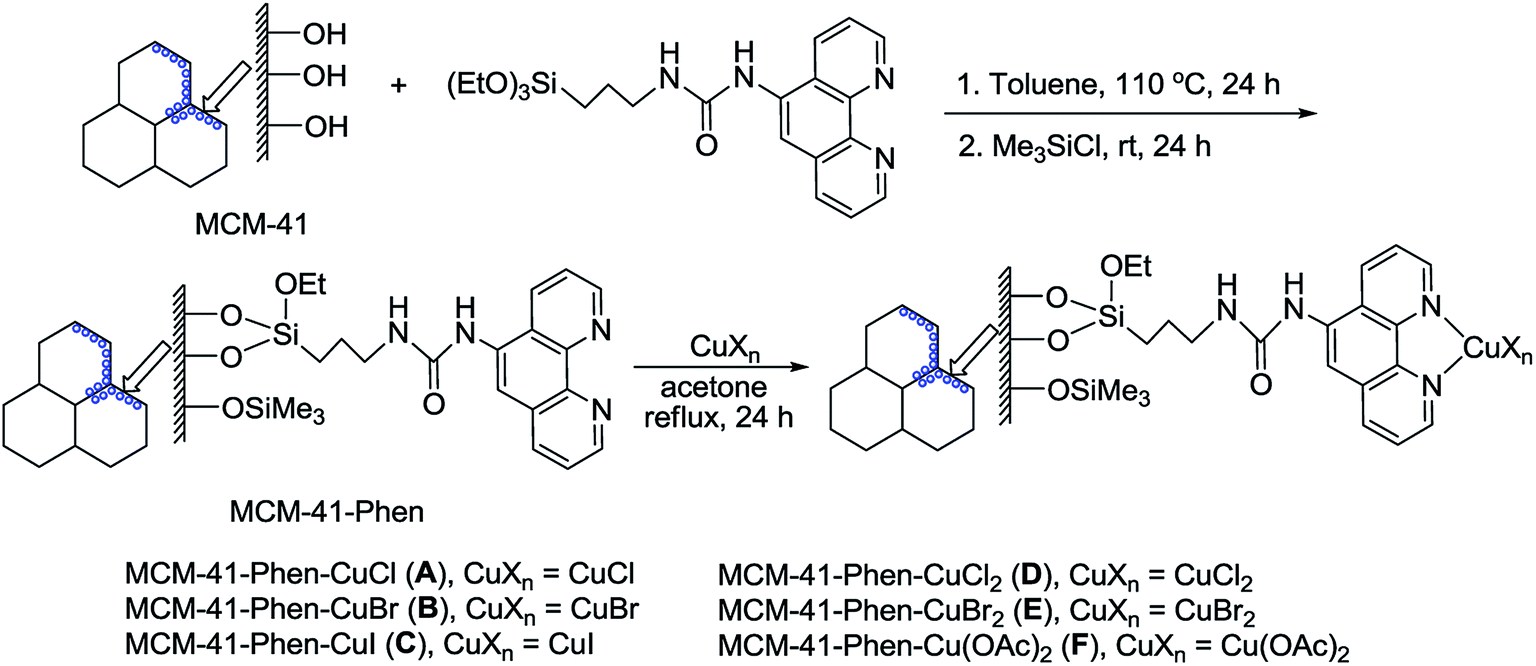 | ||
| Scheme 2 Preparation of MCM-41-immobilized 1,10-phenanthroline copper(I) or (II) complexes. | ||
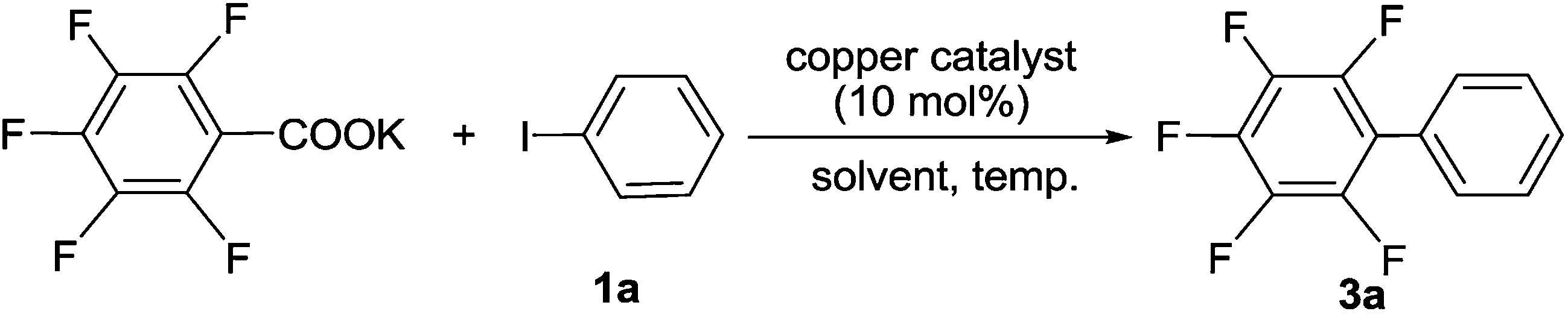
In our initial screening experiments, the reaction of C6F5COOK with PhI was investigated to optimize the reaction conditions, and the results are summarized in Table 1. First, the effect of various immobilized copper complexes on the model reaction was examined by using DMAc as solvent at 130 °C (Table 1, entries 1–6). It is evident that lower yields were obtained when MCM-41-Phen-CuCl [A], MCM-41-Phen-CuBr [B], MCM-41-Phen-CuCl2 [D], and MCM-41-Phen-CuBr2 [E] were used as catalysts, and MCM-41-Phen-CuI [C] could give a moderate yield (entry 3), whilst MCM-41-Phen-Cu(OAc)2 [F] afforded a low yield, so MCM-41-Phen-CuI [C] was finally selected as the catalyst for the reaction. Our next studies focused on the effect of solvent on the model reaction and a significant solvent effect was observed (Table 1, entries 3 and 7–10). When NMP, DMF and DMSO were used as solvents, low yields were obtained (entries 7, 9 and 10). To our delight, an excellent yield of 97% was achieved when diglyme was used as the solvent (entry 8). A possible explanation for this result is that diglyme can coordinate to potassium cation, which facilitates the reaction between the MCM-41-Phen-CuI complex and C6F5COO−. The effect of temperature on the model reaction was also examined (Table 1, entries 8 and 11–13). For the temperatures tested [120, 130, 140 and 150 °C], 130 °C was the best choice (entry 8), and other temperatures were substantially less effective (entries 11–13). Finally, the amount of the immobilized copper catalyst was screened, and 10 mol% loading of copper was found to be optimal, a lower yield was observed and a longer reaction time was required when the amount of the catalyst was decreased (entry 14). Increasing the amount of the copper catalyst could shorten the reaction time, but did not improve the yield of 3a obviously (entry 15). When a homogeneous CuI/Phen was used as the catalyst, the desired product 3a was also isolated in 98% yield (entry 16), which indicating that catalytic activity of MCM-41-Phen-CuI was comparable to that of CuI/Phen combination. When 1.0 equiv. of C6F5COOK was used, the yield of 3a was decreased to 82% (entry 17). Therefore, the optimal catalytic system involved the use of MCM-41-Phen-CuI [C] (10 mol%) and 1.5 equiv. of C6F5COOK in diglyme at 130 °C under Ar for 24 h (Table 1, entry 8).
|
|||||
|---|---|---|---|---|---|
| Entry | Copper catalyst | Solvent | Temp. (°C) | Time (h) | Yieldb (%) |
| a Reaction was performed with C6F5CO2K (0.75 mmol), 1a (0.5 mmol) and copper catalyst (0.05 mmol) in solvent (0.5 mL) under Ar.b Isolated yield based on 1a.c 5 mol% MCM-41-Phen-CuI was used.d 20 mol% MCM-41-Phen-CuI was used.e 1.0 equiv. of C6F5CO2K was used. | |||||
| 1 | A | DMAc | 130 | 24 | 39 |
| 2 | B | DMAc | 130 | 24 | 44 |
| 3 | C | DMAc | 130 | 24 | 49 |
| 4 | D | DMAc | 130 | 24 | 38 |
| 5 | E | DMAc | 130 | 24 | 40 |
| 6 | F | DMAc | 130 | 24 | 18 |
| 7 | C | NMP | 130 | 24 | 41 |
| 8 | C | Diglyme | 130 | 24 | 97 |
| 9 | C | DMF | 130 | 24 | 37 |
| 10 | C | DMSO | 130 | 24 | 24 |
| 11 | C | Diglyme | 140 | 20 | 93 |
| 12 | C | Diglyme | 150 | 12 | 88 |
| 13 | C | Diglyme | 120 | 30 | 79 |
| 14c | C | Diglyme | 130 | 48 | 82 |
| 15d | C | Diglyme | 130 | 12 | 98 |
| 16 | CuI/Phen | Diglyme | 130 | 24 | 98 |
| 17e | C | Diglyme | 130 | 30 | 82 |
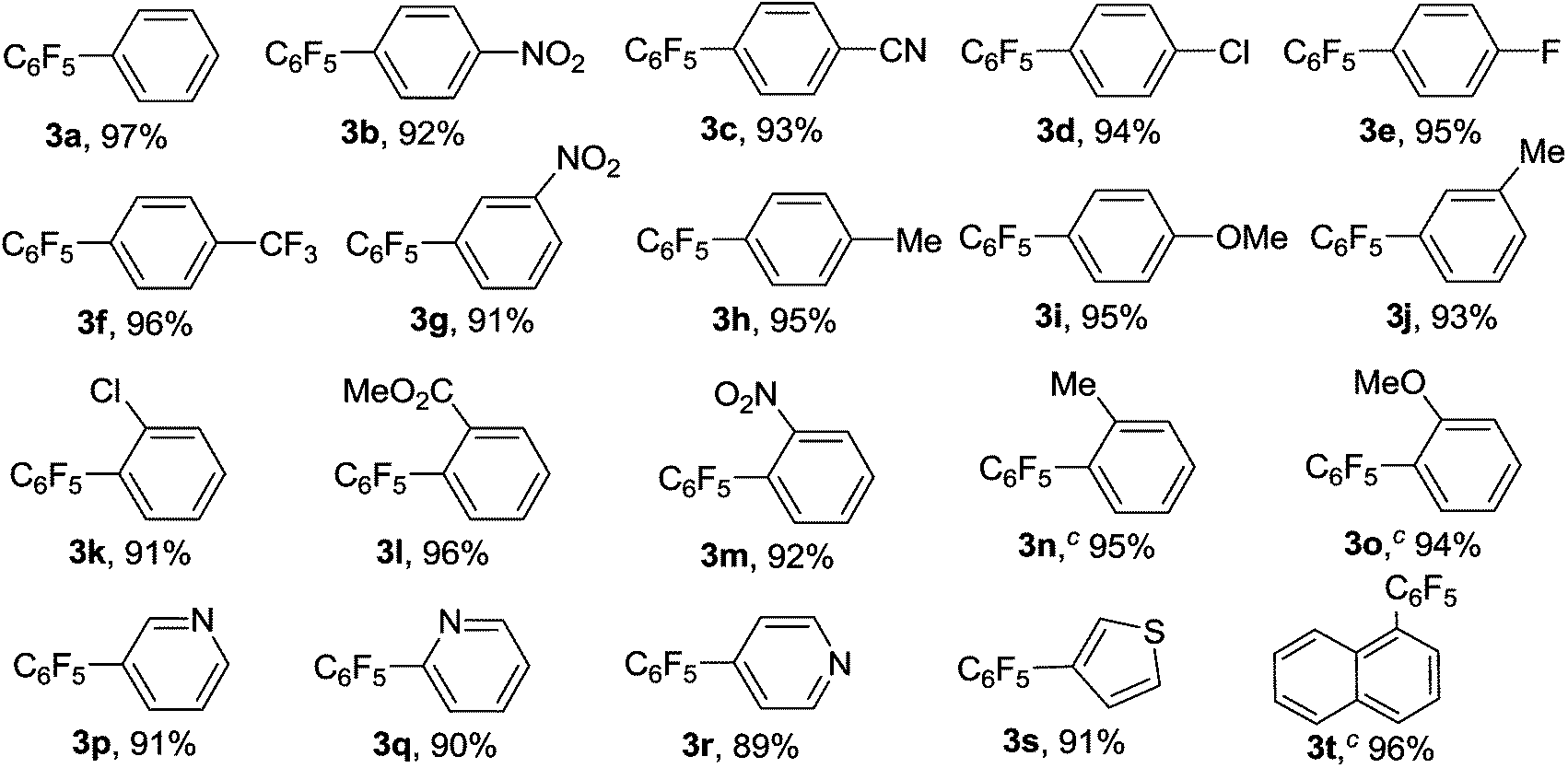
With the optimal reaction conditions established, we tried to investigate the scope and limitations of this heterogeneous copper-catalyzed decarboxylative cross-coupling reaction and the results are summarized in Table 2. As shown in Table 2, both electron-deficient and electron-rich aryl iodides 1b–1j underwent the decarboxylative cross-coupling reaction with C6F5COOK smoothly to afford the corresponding polyfluorobiaryls 3b–3j in 91–96% yields. The results indicated that the electronic natures of the substituents on benzene ring have limited influence on this heterogeneous copper-catalyzed decarboxylative cross-coupling reaction. The sterically congested electron-deficient aryl iodides such as 2-chloroiodobenzene 1k, methyl 2-iodobenzoate 1l and 2-nitroiodobenzene 1m could also be successfully converted into the corresponding polyfluorobiaryls 3k–3m in excellent yields. The electron-rich 2-iodotoluene 1n and 2-iodoanisole 1o showed slightly lower reactivity than the electron-deficient ortho-substituted iodobenzenes, however, the desired products 3n and 3o were also obtained in excellent yields by using 20 mol% of MCM-41-Phen-CuI as catalyst. A range of functional groups such as methyl, methoxy, chloro, fluoro, trifluoromethyl, nitro, cyano and ester were well tolerated. In addition to phenyl iodides, heteroaryl iodides such as 3-, 2-, and 4-iodopyridines 1p–1r and 3-iodothiophene 1s also proved to be good coupling partners and gave the desired products 3p–3s in high yields. Notably, bulky 1-iodonaphthalene 1t could react with C6F5COOK effectively in the presence of 20 mol% of MCM-41-Phen-CuI to furnish the desired product 3t in 96% yield.

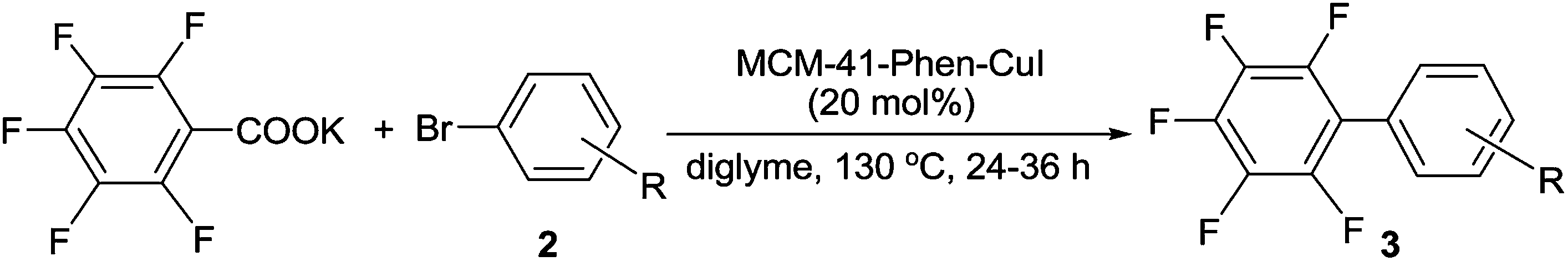
Encouraged by the above results, we further investigated the scope and the generality of the method by varying aryl iodides to aryl bromides and the results are listed in Table 3. Generally, the reactivity of aryl bromides was lower than that of aryl iodides. Under the conditions optimized for aryl iodides, the reaction of bromobenzene with C6F5COOK gave the desired product 3a in only 74% yield after 36 h. However, the yield of 3a could be improved to 91% by increasing the amount of the catalyst to 20 mol%. As shown in Table 3, in the presence of 20 mol% of MCM-41-Phen-CuI, a variety of aryl bromides bearing either electron-withdrawing or electron-donating groups underwent decarboxylative cross-coupling reactions with C6F5COOK smoothly to give the corresponding polyfluorobiaryls 3b–3d, 3u, 3f–3i and 3v in good to excellent yields. The electron-rich aryl bromides exhibited slightly lower reactivity than the electron-deficient ones and longer reaction times were required to complete the reactions. The ortho-substituted bromobenzenes such as 2-chlorobromobenzene and 2-bromotoluene also afforded the desired products 3k and 3n in excellent yields after 36 h. In addition to aryl bromides, heteroaryl bromides proved to be also suitable substrates and could undergo the decarboxylative cross-coupling reaction effectively to give the corresponding polyfluorobiaryls 3p, 3q and 3s in high yields.

The scope of the reaction with respect to fluoroarene was also examined and the results are presented in Table 4. Although diglyme plays an important role in the reactions with potassium pentafluorobenzoate, N,N-dimethyl acetamide (DMAc) can afford better results for fluoroarenes bearing fewer fluorine atoms. In addition, the use of 20 mol% of MCM-41-Phen-CuI and a high temperature of 160 °C were required due to their lower reactivity. Under the optimized reaction conditions, potassium monofluorobenzoate was found to be not suitable substrate and the reactions with aryl iodides generated traces of the desired products. This may be due to the fact that an ortho-F atom in potassium monofluorobenzoate can not activate the carboxylic group effectively. The decarboxylative cross-coupling reactions of potassium 2,6-difluorobenzoate 4a with both electron-poor and electron-rich aryl iodides proceeded smoothly to afford the corresponding 2,6-difluorobiaryls 5a–5e in good to high yields. The coupling reactions could tolerate strong electron-withdrawing or electron-donating groups and could also tolerate ortho substitution. In addition, tri- and tetrafluorobenzoates having two ortho-F atoms 4b, 4c and 4d proved to be also good coupling partners and could undergo the decarboxylative cross-coupling reactions with a range of aryl iodides effectively to furnish the desired coupling products 5f–5p in good to excellent yields. The method provides a quite general and practical route for the synthesis of polyfluorobiaryls having various functionalities under green conditions.


To verify whether the observed catalysis was due to the heterogeneous catalyst MCM-41-1,10-Phen-CuI or to a leached copper species in solution, the reaction of C6F5COOK (1.5 mmol) with PhI (1.0 mmol) in diglyme (1.5 mL) was carried out until an approximately 50% conversion of PhI was reached. Then the MCM-41-1,10-Phen-CuI catalyst was removed from the reaction mixture by hot filtration27 and the filtrate was allowed to react further by addition of C6F5COOK (0.8 mmol) at 130 °C under Ar atmosphere for 20 h. In this case, no significant increase in conversion of PhI was observed, indicating that leached copper species from the supported catalyst (if any) are not responsible for the observed activity. It was confirmed by ICP-AES analysis that no copper species could be detected in the filtrate (below the detection limit). These results rule out any contribution to the observed catalysis from a homogeneous copper species demonstrating that the catalyst was stable during the reaction and the observed catalysis was intrinsically heterogeneous.
A plausible mechanism for the heterogeneous copper(I)-catalyzed decarboxylative cross-coupling of potassium polyfluorobenzoates with aryl halides is illustrated in Scheme 3. Firstly, the reaction of MCM-41-Phen-CuI with C6F5CO2K affords an MCM-41-immobilized 1,10-phenanthroline-Cu(I)–OCOC6F5 intermediate A, which then undergoes decarboxylation via a four-membered-ring transition state to form an MCM-41-immobilized 1,10-phenanthroline-Cu(I)–C6F5 intermediate B. The oxidative addition of Ar–I 1 to intermediate B gives an MCM-41-immobilized 1,10-phenanthroline-ArCu(III)I–C6F5 intermediate C. Finally, reductive elimination of intermediate C produces the desired product 3 and regenerates the MCM-41-Phen-CuI complex to complete the catalytic cycle.
 | ||
| Scheme 3 Plausible mechanism for the heterogeneous Cu-catalyzed decarboxylative cross-coupling reaction. | ||
For the practical application of a heterogeneous catalyst system its stability and recyclability are important factors. We next examined the reusability and stability of the MCM-41-Phen-CuI complex by using coupling reaction between iodobenzene (0.5 mmol) and C6F5CO2K (0.75 mmol) in diglyme (0.5 mL) in the presence of 10 mol% of MCM-41-Phen-CuI at 130 °C under argon atmosphere for 24 h. After carrying out the reaction, the catalyst was separated by simple filtration and washed with distilled water and EtOH. After being air-dried, it can be reused directly without further purification. The recovered catalyst was used in the next run, and almost the same yield of 3a was obtained for 8 consecutive cycles (Fig. 1). In addition, copper leaching in the heterogeneous catalyst was also determined by ICP-AES analysis on the recovered catalyst after eight consecutive runs, which revealing almost the same copper content as the fresh one. In our opinion, the excellent catalytic activity and reusability of the copper catalyst relates to the efficient site isolation, to the optimal dispersion of the active sites on the inner channel walls and to the relatively strong interaction between the bidentate 1,10-phenanthroline ligand and the copper centre supported on the MCM-41. The result is important from the standpoint of green and sustainable chemistry.
 | ||
| Fig. 1 Recycle of MCM-41-Phen-CuI complex. | ||
3. Conclusions
In summary, we have successfully developed a novel, practical and environmentally friendly method for the synthesis of polyfluorobiaryls through the decarboxylative cross-coupling of readily accessible and nonvolatile potassium polyfluorobenzoates with aryl iodides or bromides by using an MCM-41-immobilized 1,10-phenanthroline-copper(I) complex [MCM-41-Phen-CuI] as catalyst. This heterogeneous copper catalyst could be easily prepared by a simple procedure from commercially available and inexpensive reagents and exhibited the same catalytic activity as the homogeneous CuI/Phen system. The reactions generated a variety of polyfluorobiaryls in good to excellent yields and were applicable to various polyfluorobenzoate salts and a wide range of aryl iodides or bromides. In addition, this methodology offers the competitiveness of recyclability of the copper catalyst without any decreases in activity, and the copper catalyst can be readily recovered by a simple filtration and reused at least 8 cycles, thus making this procedure economically and environmentally more acceptable.4. Experimental
All chemicals were reagent grade and used as purchased. All solvents were dried and distilled before use. The MCM-41-Phen-CuCl [A], MCM-41-Phen-CuBr [B], MCM-41-Phen-CuI [C], MCM-41-Phen-CuCl2 [D], MCM-41-Phen-CuBr2 [E] and MCM-41-Phen-Cu(OAc)2 [F] were prepared according to our previous procedure,24 the copper contents were determined to be 0.73 mmol g−1, 0.71 mmol g−1, 0.74 mmol g−1, 0.68 mmol g−1, 0.70 mmol g−1 and 0.67 mmol g−1, respectively. The products were purified by flash chromatography on silica gel. Mixture of EtOAc and hexane was generally used as eluent. 1H NMR spectra were recoded on a Bruker Avance 400 MHz spectrometer with TMS as an internal standard in CDCl3 as solvent. 13C NMR spectra (100 MHz) were recorded on a Bruker Avance 400 MHz spectrometer in CDCl3 as solvent. 19F NMR spectra (376 MHz) were recorded on a Bruker Avance 400 MHz spectrometer in CDCl3 as solvent with trifluorotoluene (TFT) (δ = −67.73 ppm) as an external standard. Melting points are uncorrected. Copper content was determined with inductively coupled plasma atom emission Atomscan16 (ICP-AES, TJA Corporation). HRMS spectra were recorded on a Q-Tof spectrometer with micromass MS software using electrospray ionization (ESI).4.1 General procedure for the decarboxylative cross-coupling of potassium pentafluorobenzoate with various aryl iodides
MCM-41-Phen-CuI (67 mg, 0.05 mmol), potassium pentafluorobenzoate (188 mg, 0.75 mmol) and aryl iodide 1 (0.50 mmol) (if solid) were placed in an oven-dried 10 mL Schlenk tube, the reaction vessel was evacuated and filled with argon for three times. Then aryl iodide (0.50 mmol) (if liquid) and diglyme (0.5 mL) were added with a syringe under a counter flow of argon. The vessel was sealed with a screw cap, stirred at room temperature for 10 min, and then connected to the Schlenk line filled with argon. The reaction mixture was stirred at 130 °C for 24 h. Upon completion of the reaction, the mixture was cooled to room temperature and diluted with ethyl acetate (20 mL) and filtered. The MCM-41-Phen-CuI complex was washed with distilled water (2 × 5 mL) and EtOH (2 × 5 mL), and reused in the next run. The organic layers were washed with water (2 × 20 mL), and then with brine, dried over MgSO4, and filtered. The solvents were removed via rotary vapor and the residue was purified by flash column chromatography on silica gel to provide the product 3.4.2 General procedure for the decarboxylative cross-coupling of potassium pentafluorobenzoate with various aryl bromides
MCM-41-Phen-CuI (135 mg, 0.1 mmol), potassium pentafluorobenzoate (188 mg, 0.75 mmol) and aryl bromide 2 (0.50 mmol) (if solid) were placed in an oven-dried 10 mL Schlenk tube, the reaction vessel was evacuated and filled with argon for three times. Then aryl bromide (0.50 mmol) (if liquid) and diglyme (0.8 mL) were added with a syringe under a counter flow of argon. The vessel was sealed with a screw cap, stirred at room temperature for 10 min, and then connected to the Schlenk line filled with argon. The reaction mixture was stirred at 130 °C for 24–36 h. Upon completion of the reaction, the mixture was cooled to room temperature and diluted with ethyl acetate (20 mL) and filtered. The MCM-41-Phen-CuI complex was washed with distilled water (2 × 5 mL) and EtOH (2 × 5 mL), and reused in the next run. The organic layers were washed with water (2 × 20 mL), and then with brine, dried over MgSO4, and filtered. The solvents were removed via rotary vapor and the residue was purified by flash column chromatography on silica gel to provide the product 3.4.3 General procedure for the decarboxylative cross-coupling of potassium perfluorobenzoates with various aryl iodides
MCM-41-Phen-CuI (135 mg, 0.1 mmol), potassium perfluorobenzoate 4 (0.75 mmol) and aryl iodide 1 (0.50 mmol) (if solid) were placed in an oven-dried 10 mL Schlenk tube, the reaction vessel was evacuated and filled with argon for three times. Then aryl iodide (0.50 mmol) (if liquid) and DMAc (0.8 mL) were added with a syringe under a counter flow of argon. The vessel was sealed with a screw cap, stirred at room temperature for 10 min, and then connected to the Schlenk line filled with argon. The reaction mixture was stirred at 160 °C for 24 h. Upon completion of the reaction, the mixture was cooled to room temperature and diluted with ethyl acetate (20 mL) and filtered. The MCM-41-Phen-CuI complex was washed with distilled water (2 × 5 mL) and EtOH (2 × 5 mL), and reused in the next run. The organic layers were washed with water (2 × 20 mL), and then with brine, dried over MgSO4, and filtered. The solvents were removed via rotary vapor and the residue was purified by flash column chromatography on silica gel to provide the desired product 5.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the National Natural Science Foundation of China (Project No. 21272044) and Project of innovation for enhancing Guangdong Pharmaceutical University, provincial experimental teaching demonstration center of chemistry & chemical engineering for financial support.References
- (a) A. Zahn, C. Brotschi and C. J. Leumann, Chem.–Eur. J., 2005, 11, 2125–2129 CrossRef CAS PubMed; (b) R. E. Mewshaw, R. J. Edsall Jr, C. Yang, E. S. Manas, Z. B. Xu, R. A. Henderson, J. C. Keith Jr and H. A. Harris, J. Med. Chem., 2005, 48, 3953–3979 CrossRef CAS PubMed; (c) S. Purser, P. R. Moor, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (d) M. de Candia, F. Liantonio, A. Carotti, R. De Cristofaro and C. Altomare, J. Med. Chem., 2009, 52, 1018–1028 CrossRef CAS PubMed.
- (a) J. R. Nitschke and T. D. Tilley, J. Am. Chem. Soc., 2001, 123, 10183–10190 CrossRef CAS PubMed; (b) Y. Sakamoto, T. Suzuki, A. Miura, H. Fujikawa, S. Tokito and Y. Taga, J. Am. Chem. Soc., 2000, 122, 1832–1833 CrossRef CAS; (c) V. A. Montes, G. Li, R. Pohl, J. Shinar and P. Anzenbacher, Adv. Mater., 2004, 16, 2001–2003 CrossRef CAS; (d) P. Zacharias, M. C. Gather, M. Rojahn, O. Nuyken and K. Meerholz, Angew. Chem., Int. Ed., 2007, 46, 4388–4392 CrossRef CAS PubMed.
- (a) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed; (b) S. M. Senaweera, A. Singh and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 3002–3005 CrossRef CAS PubMed.
- (a) T. Korenaga, T. Kosaki, R. Fukumura, T. Ema and T. Sakai, Org. Lett., 2005, 7, 4915–4917 CrossRef CAS PubMed; (b) T. Kinzel, Y. Zhang and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14073–14075 CrossRef CAS PubMed.
- (a) H. J. Frohn, N. Y. Adonin, V. V. Bardin and V. F. Starichenko, Tetrahedron Lett., 2002, 43, 8111–8114 CrossRef CAS; (b) H. J. Frohn, N. Y. Adonin, V. V. Bardin and V. F. Starichenko, J. Fluorine Chem., 2002, 117, 115–120 CrossRef CAS.
- P. L. Coe and G. M. Pearl, J. Organomet. Chem., 1971, 31, 55–57 CrossRef CAS.
- R. J. DePasquake and C. Tamborski, J. Org. Chem., 1969, 34, 1736–1740 CrossRef.
- (a) M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754–8756 CrossRef CAS PubMed; (b) M. Lafrance, D. Shore and K. Fagnou, Org. Lett., 2006, 8, 5097–5100 CrossRef CAS PubMed; (c) H.-Q. Do and O. Daugulis, J. Am. Chem. Soc., 2008, 130, 1128–1129 CrossRef CAS PubMed; (d) H.-Q. Do, R. M. Kashif and O. Daugulis, J. Am. Chem. Soc., 2008, 130, 15185–15192 CrossRef CAS PubMed.
- Y. Wei, J. Kan, M. Wang, W. Su and M. Hong, Org. Lett., 2009, 11, 3346–3349 CrossRef CAS PubMed.
- H. Fan, Y. Shang and W. Su, Eur. J. Org. Chem., 2014, 3323–3327 CrossRef CAS.
- X. Zhu, F. Li and W. Su, Tetrahedron Lett., 2013, 54, 1285–1289 CrossRef CAS.
- Z. Fu, Q. Xiong, W. Zhang, Z. Li and H. Cai, Tetrahedron Lett., 2015, 56, 123–126 CrossRef CAS.
- (a) O. Baudoin, Angew. Chem., Int. Ed., 2007, 46, 1373–1375 CrossRef CAS PubMed; (b) L. J. Goossen, N. Rodriguez and K. Goossen, Angew. Chem., Int. Ed., 2008, 47, 3100–3120 CrossRef CAS PubMed; (c) L. J. Goossen, G. Deng and L. M. Levy, Science, 2006, 313, 662–664 CrossRef CAS PubMed; (d) D. Tanaka, S. P. Romeril and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 10323–10333 CrossRef CAS PubMed; (e) C. Wang, I. Piel and F. Glorius, J. Am. Chem. Soc., 2009, 131, 4194–4195 CrossRef CAS PubMed; (f) J. D. Weaver, A. Recio, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846–1913 CrossRef CAS PubMed; (g) W. L. Dzik, P. P. Lange and L. J. Goossen, Chem. Sci., 2012, 3, 2671–2678 RSC; (h) L. J. Goossen and K. Goossen, Top. Organomet. Chem., 2013, 44, 121–141 CrossRef CAS.
- (a) J.-M. Becht, C. Catala, C. Le Drian and A. Wagner, Org. Lett., 2007, 9, 1781–1784 CrossRef CAS PubMed; (b) A. Voutchkova, A. Coplin, N. E. Leadbeater and R. H. Crabtree, Chem. Commun., 2008, 6312–6314 RSC; (c) R. Shang, Y. Fu, Y. Wang, Q. Xu, H.-Z. Yu and L. Liu, Angew. Chem., Int. Ed., 2009, 48, 9350–9354 CrossRef CAS PubMed; (d) R. Shang, Q. Xu, Y.-Y. Jiang, Y. Wang and L. Liu, Org. Lett., 2010, 12, 1000–1003 CrossRef CAS PubMed; (e) L. W. Sardzinski, W. C. Wertjes, A. M. Schnaith and D. Kalyani, Org. Lett., 2015, 17, 1256–1259 CrossRef CAS PubMed.
- H. Zhao, Y. Wei, J. Xu, J. Kan, W. Su and M. Hong, J. Org. Chem., 2011, 76, 882–893 CrossRef CAS PubMed.
- (a) C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889–900 CrossRef CAS; (b) C. J. Welch, J. Albaneze-Walker, W. R. Leonard, M. Biba, J. DaSilva, D. Henderson, B. Laing, D. J. Mathre, S. Spencer, X. Bu and T. Wang, Org. Process Res. Dev., 2005, 9, 198–205 CrossRef CAS; (c) S. Benyahya, F. Monnier, M. Wong Chi Man, C. Bied, F. Ouazzani and M. Taillefer, Adv. Synth. Catal., 2008, 350, 2205–2208 CrossRef CAS; (d) S. Benyahya, F. Monnier, M. Wong Chi Man, C. Bied, F. Ouazzani and M. Taillefer, Green Chem., 2009, 11, 1121–1123 RSC; (e) P.-H. Li and L. Wang, Adv. Synth. Catal., 2006, 348, 681–685 CrossRef CAS; (f) J. Yang, P. Li and L. Wang, Tetrahedron, 2011, 67, 5543–5549 CrossRef CAS; (g) P. Li, L. Wang, L. Zhang and G.-W. Wang, Adv. Synth. Catal., 2012, 354, 1307–1318 CrossRef CAS; (h) L. Zhang, P. Li, C. Liu, J. Yang, M. Wang and L. Wang, Catal. Sci. Technol., 2014, 4, 1979–1988 RSC.
- For selected reviews, see: (a) N. T. S. Phan, M. V. D. Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS; (b) L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS PubMed; (c) R. Akiyama and S. Kobayashi, Chem. Rev., 2009, 109, 594–642 CrossRef CAS PubMed; (d) M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072–1133 CrossRef CAS PubMed; (e) A. Molnar, Chem. Rev., 2011, 111, 2251–2320 CrossRef CAS PubMed; (f) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- (a) C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS; (b) J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T.-W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS; (c) W. Zhou, J. M. Thomas, D. S. Shephard, B. F. G. Johnson, D. Ozkaya, T. Maschmeyer, R. G. Bell and Q. Ge, Science, 1998, 280, 705–708 CrossRef CAS PubMed.
- For selected examples, see: (a) P. C. Mehnert, D. W. Weaver and J. Y. Ying, J. Am. Chem. Soc., 1998, 120, 12289–12296 CrossRef; (b) K. Mukhopadhyay, B. R. Sarkar and R. V. Chaudhari, J. Am. Chem. Soc., 2002, 124, 9692–9693 CrossRef CAS PubMed; (c) J. Y. Ying, C. P. Mehnert and M. S. Wong, Angew. Chem., Int. Ed., 1999, 38, 56–77 CrossRef CAS; (d) M. Cai, G. Zheng and G. Ding, Green Chem., 2009, 11, 1687–1693 RSC; (e) M. Cai, J. Peng, W. Hao and G. Ding, Green Chem., 2011, 13, 190–196 RSC; (f) W. Hao, H. Liu, L. Yin and M. Cai, J. Org. Chem., 2016, 81, 4244–4251 CrossRef CAS PubMed; (g) F. Havasi, A. Ghorbani-Choghamarani and F. Nikpour, New J. Chem., 2015, 39, 6504–6512 RSC.
- (a) S.-G. Shyu, S.-W. Cheng and D.-L. Tzou, Chem. Commun., 1999, 2337–2338 RSC; (b) Y. Yang and R. M. Rioux, Chem. Commun., 2011, 47, 6557–6559 RSC.
- (a) C. D. Nunes, A. A. Valente, M. Pillinger, A. C. Fernandes, C. C. Romao, J. Rocha and I. S. Goncalves, J. Mater. Chem., 2002, 12, 1735–1742 RSC; (b) M. Jia, A. Seifert and W. R. Thiel, Chem. Mater., 2003, 15, 2174–2180 CrossRef CAS.
- (a) A. Corma, E. Gutierrez-Puebla, M. Iglesias, A. Monge, S. Perez-Ferreras and F. Sanchez, Adv. Synth. Catal., 2006, 348, 1899–1907 CrossRef CAS; (b) A. Corma, C. Gonzalez-Arellano, M. Iglesias and F. Sanchez, Angew. Chem., Int. Ed., 2007, 46, 7820–7822 CrossRef CAS PubMed; (c) G. Villaverde, A. Corma, M. Iglesias and F. Sanchez, ACS Catal., 2012, 2, 399–406 CrossRef CAS.
- (a) R. Xiao, H. Zhao and M. Cai, Tetrahedron, 2013, 69, 5444–5545 CrossRef CAS; (b) H. Zhao, W. He, R. Yao and M. Cai, Adv. Synth. Catal., 2014, 356, 3092–3098 CrossRef CAS; (c) M. Cai, R. Yao, L. Chen and H. Zhao, J. Mol. Catal. A: Chem., 2014, 395, 349–354 CrossRef CAS; (d) H. Zhao, Y. Jiang, Q. Chen and M. Cai, New J. Chem., 2015, 39, 2106–2111 RSC; (e) H. Zhao, W. He, L. Wei and M. Cai, Catal. Sci. Technol., 2016, 6, 1488–1495 RSC.
- Y. Lin, M. Cai, Z. Fang and H. Zhao, RSC Adv., 2016, 6, 85186–85193 RSC.
- M. H. Lim and A. Stein, Chem. Mater., 1999, 11, 3285–3295 CrossRef CAS.
- M.-J. Li, Z. Chen, V. Wing-Wah Yam and Y. Zu, ACS Nano, 2008, 2, 905–912 CrossRef CAS PubMed.
- H. E. B. Lempers and R. A. Sheldon, J. Catal., 1998, 175, 62–69 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05711c |
| This journal is © The Royal Society of Chemistry 2017 |