
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intra- and intermolecular-interaction-controlled reversible core–shell structures and photoluminescent properties of lanthanide ion-doped diblock copolymers†
Feifei Xueab,
Hongfei Li*a and
Shichun Jiang *ab
*ab
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: hfli@ciac.ac.cn
bSchool of Materials Science and Engineering, Tianjin University, Tianjin 300072, P. R. China. E-mail: scjiang@tju.edu.cn
First published on 3rd July 2017
Abstract
Lanthanide-based nanotechniques continue to attract considerable attention due to their current range of applications and broad potential in optical devices and biomedicine. Lanthanide ion-loaded block copolymers (BCPs) have gained interest, with questions remaining regarding their luminescence properties and structures. Solutions and films containing polystyrene-block-poly(2-vinylpyridine) (S2VP) and polystyrene-block-poly(4-vinylpyridine) (S4VP) loaded with trivalent lanthanide ions (Ln3+ = Eu3+, Tb3+) were prepared using a solvent displacement method and spin coating approach. Atomic force microscopy and transmission electron microscopy observations confirmed the Ln3+ distribution in the core of S2VP [Ln(x)] micelles and the corona of S4VP[Ln(x)] micelles. The coordination of Ln3+ and the S2VP core segment led to micelle shrinkage and film dewetting with increasing Ln3+ concentration, while the coordination of Ln3+ and the S4VP corona chain swelled the micelle. Photoluminescence investigations of the complex solutions showed typical reddish (Eu3+) and greenish (Tb3+) luminescence, and the emission intensity was enhanced with increasing Ln3+ concentration without typical fluorescence concentration quenching behavior. The luminescence intensity was distinctly enhanced when Ln3+ was distributed in the micellar core. Moreover, the emission spectra of mixed Eu–Tb ions complexes gave addition spectra of the two separated complexes. An orange color emission was observed because Eu3+ was more emissive. These results would improve understanding of ion coordination and lay the foundation for lanthanide ion/BCPs complex applications.
Introduction
Block copolymers (BCPs) are of broad interest in current research across macromolecular chemistry and physics, ranging from the development of new synthetic strategies and molecular architectures to applications of advanced theoretical and computational methods.1 Almost 50 years after the preparation of the first laboratory samples, scientific interest in BCPs continues to grow, not only regarding the properties of their components, but also the particular doping, spatial extent, connectivity, and orientation of their nanodomains. To further explore the novel properties of BCPs and develop their performance, attention has recently been focused on ion/BCP complexes. The added ions strongly affect the thermodynamic, structural, and other properties of ion/BCP complexes, and advanced material with prominent performance can be obtained by selectively choosing ions.2–5Trivalent lanthanide ions (Ln3+) show excellent luminescent characteristics, including high color purity, high fluorescence intensity, and long fluorescence lifetime.6–8 In particular, europium (Eu3+) and terbium (Tb3+) ions, which can emit red and green primary light colors, respectively, have drawn much attention due to their potential application in full color displays. Unfortunately, the direct absorption of lanthanide ions is very weak due to the parity forbidden nature of the 4f transitions. In past years, efforts have been made to overcome these disadvantages by incorporating Ln3+ ions into small molecules, sol–gel precursors, and polymers.9–13 Compared with conventional luminescent materials, lanthanide complexes show excellent mechanical and electronic properties, thermal and chemical stability, biocompatibility, and hydrophobic–hydrophilic balance, while the luminescence features of lanthanide (Ln3+) complexes show high photoluminescence efficiency with a narrow emission spectrum, a large Stokes shifts, and a long fluorescent lifetime. These features offer excellent prospects for the design of new luminescent materials with enhanced desired characteristics and high added value for specific targeted applications. This opens exciting new directions in materials science and related technologies, with noteworthy results for the ecofriendly integration, miniaturization, and multifunctionalization of devices, among others.
In addition to traditional polymers, recent efforts have been made towards lanthanide ion/BCP complexes, especially lanthanide-containing block copolymer micelles.14–17 Amphiphilic BCPs readily undergo microphase separation in selective solvents to form micelles with coronas of the soluble block and cores of the insoluble block, affording the opportunity to achieve discrete nanostructures through a spontaneous process. When such micelles are coordinated by lanthanide ions and transferred on to substrates by spin coating or dip coating, they arrange themselves in 2D arrays spontaneously. Meanwhile, the micellar structure not only provides a confined environment for the lanthanides, to prevent aggregation and reduce vibrational radiationless deactivation, but can also potentially shield the quenching effect. By flexibly controlling the type of lanthanide ions, ion concentration, micelle size, and the inter-micelle spacing, micellar thin films allow potential applications in biological fluorescent labeling, film technology, and more. Therefore, both the influence of lanthanide ions on the thermodynamic and structural properties of the BCP micellar thin films and the influence of BCPs on the luminescent characteristics of lanthanide ions are worth investigating.
Herein, a typical pair of BCPs, polystyrene-block-poly(2-vinylpyridine) (S2VP) and polystyrene-block-poly(4-vinylpyridine) (S4VP), were chosen. The pyridine rings in S2VP and S4VP can form strong coordination bonds with lanthanide ions. Although the only difference between these BCPs is the position of nitrogen in the pyridine ring (ortho vs. para positions), their dipole polarization and Flory–Huggins interaction parameter with polystyrene (PS) are totally different.18–20 Using careful solvent selection, S2VP and S4VP might form opposite micelle structures in the same solvent, meaning that the lanthanide ions are associated with different micelle positions. In this article, by mixing lanthanide ions (Eu3+ or Tb3+) with S2VP and S4VP, the influence of ion distribution (ion loading position) and ion concentration on luminescence features, and the short- and long-range structures of the complex films were investigated and discussed.
Experimental section
Materials
Symmetric S4VP (Mn,PS = 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Mn,P4VP = 22000 g mol−1, polydispersity index = 1.09) and S2VP (Mn,PS = 40500 g mol−1, Mn,P2VP = 40000 g mol−1, polydispersity index = 1.10) diblock copolymers were purchased from Polymer Source and used as received. Europium oxide (99.99%), terbium oxide (99.9%), and 1,10-phenanthroline (Phen) were obtained from Aldrich and stored in a desiccator. All solvents were analytical grade and used without further purification.
000 g mol−1, Mn,P4VP = 22000 g mol−1, polydispersity index = 1.09) and S2VP (Mn,PS = 40500 g mol−1, Mn,P2VP = 40000 g mol−1, polydispersity index = 1.10) diblock copolymers were purchased from Polymer Source and used as received. Europium oxide (99.99%), terbium oxide (99.9%), and 1,10-phenanthroline (Phen) were obtained from Aldrich and stored in a desiccator. All solvents were analytical grade and used without further purification.
Preparation of lanthanide ion/BCP complexes
S2VP and S4VP were dissolved in a mixed solvent of anhydrous ethanol and N,N-dimethylformamide (DMF) (2:1, w/w), and stirred for 5 h to yield a 3 mg mL−1 solution. LnCl3 (EuCl3 or TbCl3) solutions were prepared using the following steps: lanthanide oxides (Eu2O3 or Tb4O7) and excess hydrochloric acid aqueous solution (6 mol L−1) were mixed with stirring. After the solid was completely dissolved, the solution was distilled at 50 °C until the formation of LnCl3·6H2O as white crystals. The crystals were dried under vacuum at 40 °C for 24 h, and then dissolved with anhydrous ethanol to give a 0.1 mol L−1 solution. Phen was dissolved in anhydrous ethanol (20 mg mL−1).
Lanthanide ion/BCP complexes were prepared by mixing the LnCl3, block copolymer, and Phen solutions. LnCl3 solution was added dropwise to block copolymer solution, which was stirred for over 24 h at room temperature to obtain homogeneity. After 24 h, Phen solution was added dropwise to the mixed solution in a Phen/VP molar ratio of 3:2, and stirring was continued for 6 h. The resultant samples were denoted as S4VP[Eu(x)], S4VP[Tb(x)], S2VP[Eu(x)], and S4VP[Tb(x)], where x represents the loading molar ratios of lanthanide ions to pyridine units (x = nLn3+/nVP = 0, 0.3, 0.5, 1.0).
Characterization
The surface topography of thin films on silicon wafers was imaged using atomic force microscopy (AFM) (SPI3800N, Seiko Instruments Inc., Japan) in tapping mode. Cantilevers for AFM measurements with a spring constant of around 2 N m−1 were purchased from Olympus (Tokyo, Japan). Silicon wafers were diced into ∼1 cm2 pieces and cleaned successively in an ultrasonic bath (acetone), “piranha” bath (concentrated H2SO4 and 30% H2O2 (70:30, v/v) at 90 °C), and deionized water, and then blown dry with nitrogen. Films on the cleaned wafer were fabricated by spin-coating.
Transmission electron microscopy (TEM; JEOL-2000) was used to study the nanostructures of the film samples. Samples were obtained by dropping the product solution onto a copper grid for TEM.
Dynamic light-scattering (DLS) measurements were performed on aqueous solutions with a Malvern Zetasizer Nano S (Malvern Instruments, Ltd.) equipped with a 4 mW He–Ne laser light operating at λ = 633 nm. All samples were kept at 25 °C with a scattering angle of 173°.
Fourier-transform infrared (FTIR) spectra were recorded on a Bruker Vertex 70 spectrometer.
Ultraviolet-visible (UV-vis) absorption spectra of the block copolymers and their lanthanide complexes were recorded on a TU-1901 spectrophotometer between 200 and 600 nm at room temperature.
Photoluminescence (PL) measurements were performed at room temperature using an F-280 spectrophotometer with a scanning rate of 1200 nm min−1 at room temperature. The slit widths for both excitation and emission were set at 5 nm.
Emission images of the rare earth complex samples, including powder, solution, and film samples, were taken under a 365 nm UV lamp at room temperature.
Results and discussion
Lanthanide ion distribution in BCP micelles
For diblock copolymers in specific solvent systems, the solubility difference between two blocks makes one block expand and the other collapse, yielding micelles. Fig. 1(a) and (b) show atomic force microscopy (AFM) images of thin films prepared from neat S4VP and S2VP solutions, in which the samples present typical dimple-type patterns, revealing the dried and partially collapsed spherical micellar structure. The microstructures of these micelles were further analyzed by transmission electron microscopy (TEM) (shown in the insets of Fig. 1(a) and (b)). The TEM images presented characteristic core–corona structures with P4VP or P2VP blocks appearing as dark regions due to selective staining with iodine. It could be concluded that, in the binary solvent used here, S4VP and S2VP tended to form opposite micellar structures, with S4VP forming micelles with corona P4VP and core PS, and S2VP forming micelles with corona PS and core P2VP. These opposite micelle structures can be explained by the preferential affinities for the solvent. For S2VP, the affinity of the block for the solvent can be estimated from the solubility parameters of S2VP and solvent due to the weak polarity of the P2VP block. The solubility parameters (δ) of PS, P2VP, and the mixed solvent (ethanol/DMF) were 18.5, 20.4, and 13.0 (δethanol = 13.4, δDMF = 12.1), respectively.21,22 Obviously, the mixed solvent had a preferential affinity for the PS block, inducing a corona PS/core P2VP structure. In contrast, although the only difference between P4VP and P2VP was the nitrogen position in the pyridine ring (ortho vs. para positions), the dipole polarization of P4VP was much greater than that of P2VP. Therefore, the effect of polar interactions between S4VP and solvent on the resultant structure was large. As a result, the P4VP block tended to form the corona due to its strong interactions with ethanol in the solution.23 | ||
| Fig. 1 AFM topography images (1 μm × 1 μm) of (a) S4VP, (b) S2VP, (c) S4VP[Eu(0.5)], (d) S2VP[Eu(0.5)], (e) S4VP[Tb(0.5)], and (f) S2VP[Tb(0.5)]. Insets are corresponding TEM photographs. Scale bar is 100 nm. | ||
When Ln3+ ions were added to the micelle solutions, Ln3+ synchronously coordinated with the corresponding P4VP or P2VP segments due to interactions between Ln3+ and the pyridine ring. Fig. 1(c–f) show the morphologies of thin films prepared from S4VP[Eu(0.5)], S4VP[Tb(0.5)], S2VP[Eu(0.5)], and S2VP[Tb(0.5)] solutions. All samples present micellar structures, with no significant aggregation of Ln3+ was found in these hybrids, indicating a uniform dispersion of Ln3+ within the PVP microdomains. Meanwhile, the opposite micelle structure displayed the same results but with a different distribution of Ln3+ in the micelles, with Ln3+ loaded in the core of S2VP[Ln(x)] micelle and in the corona of S4VP[Ln(x)] micelle, respectively. Scheme 1 describes the Ln3+ distribution in BCP micelles through preferential coordination of the PVP chains and lanthanide ions.
 | ||
| Scheme 1 Schematic illustration of Ln3+ distribution in BCP micelles through preferential coordination of PVP chains to lanthanide ions. | ||
FTIR spectroscopy provided insight into the coordination interactions between S4VP or S2VP and lanthanide ions. Fig. 2 shows the FTIR spectra of S2VP, S2VP–Eu, and S2VP–Tb. The characteristic pyridine ring peaks of S2VP at 1588 cm−1 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C+CN) and 1559 cm−1 (νCN) were shifted to 1590 cm−1 and 1569 cm−1 in S2VP–Eu, and 1592 cm−1 and 1570 cm−1 in S2VP–Tb, respectively. Furthermore, the vibration peaks for –CH2– in the S2VP copolymer at 2920 cm−1 (νasC–H) and 2851 cm−1 (νsC–H) also appeared in the spectra of the complexes. This demonstrated that coordination bonds had formed between Ln3+ and N in the 2VP segments of S2VP and that the backbone structure of the copolymer remained in the complex. P2VP blocks showed a rich basic Lewis base character, with the nitrogen atom incorporated within the aromatic ring tending to share a free-electron pair with the empty f orbitals of Ln3+, causing changes in the electron distributions of those peaks related to the pyridine ring stretching modes. Similar spectral shifts were also observed in S4VP and its complexes (Fig. SI-1, ESI†).
C+CN) and 1559 cm−1 (νCN) were shifted to 1590 cm−1 and 1569 cm−1 in S2VP–Eu, and 1592 cm−1 and 1570 cm−1 in S2VP–Tb, respectively. Furthermore, the vibration peaks for –CH2– in the S2VP copolymer at 2920 cm−1 (νasC–H) and 2851 cm−1 (νsC–H) also appeared in the spectra of the complexes. This demonstrated that coordination bonds had formed between Ln3+ and N in the 2VP segments of S2VP and that the backbone structure of the copolymer remained in the complex. P2VP blocks showed a rich basic Lewis base character, with the nitrogen atom incorporated within the aromatic ring tending to share a free-electron pair with the empty f orbitals of Ln3+, causing changes in the electron distributions of those peaks related to the pyridine ring stretching modes. Similar spectral shifts were also observed in S4VP and its complexes (Fig. SI-1, ESI†).
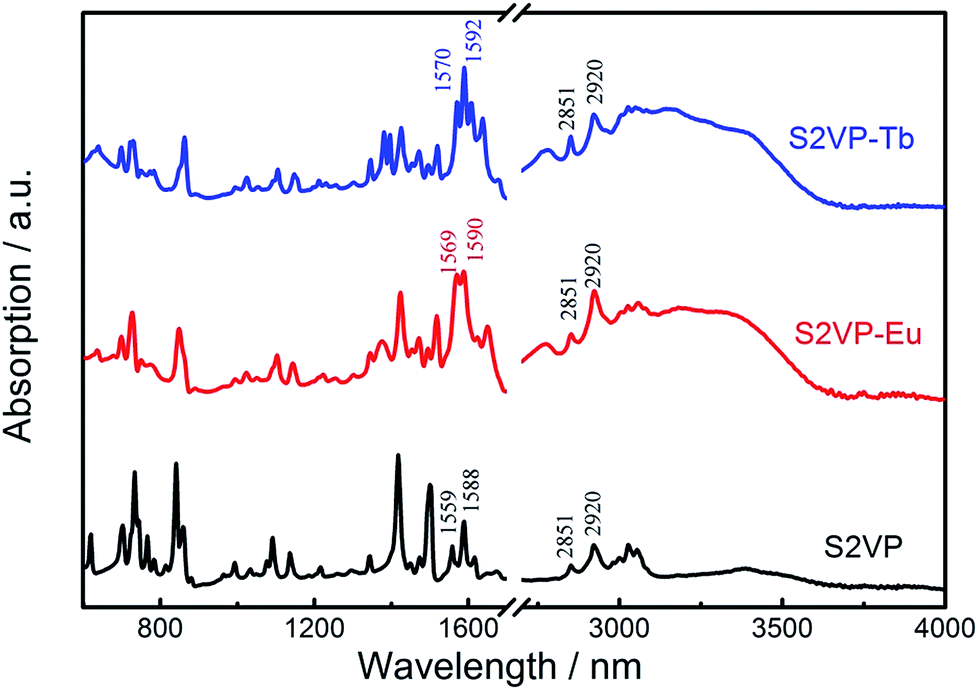 | ||
| Fig. 2 FTIR spectra of S2VP, S2VP–Eu, and S2VP–Tb. | ||
Dependence of micelle size on lanthanide concentration
Different Ln3+ distributions might result in distinct thermodynamic behaviors in short- and long-range structures. For instance, the sizes of S4VP[Ln(x)] and S2VP[Ln(x)] micelles showed the opposite trend with increasing Ln3+ loading ratio (x), with S4VP[Ln(x)] micelle size increasing monotonically with increasing x, while S2VP[Ln(x)] micelle size decreased (Fig. 3).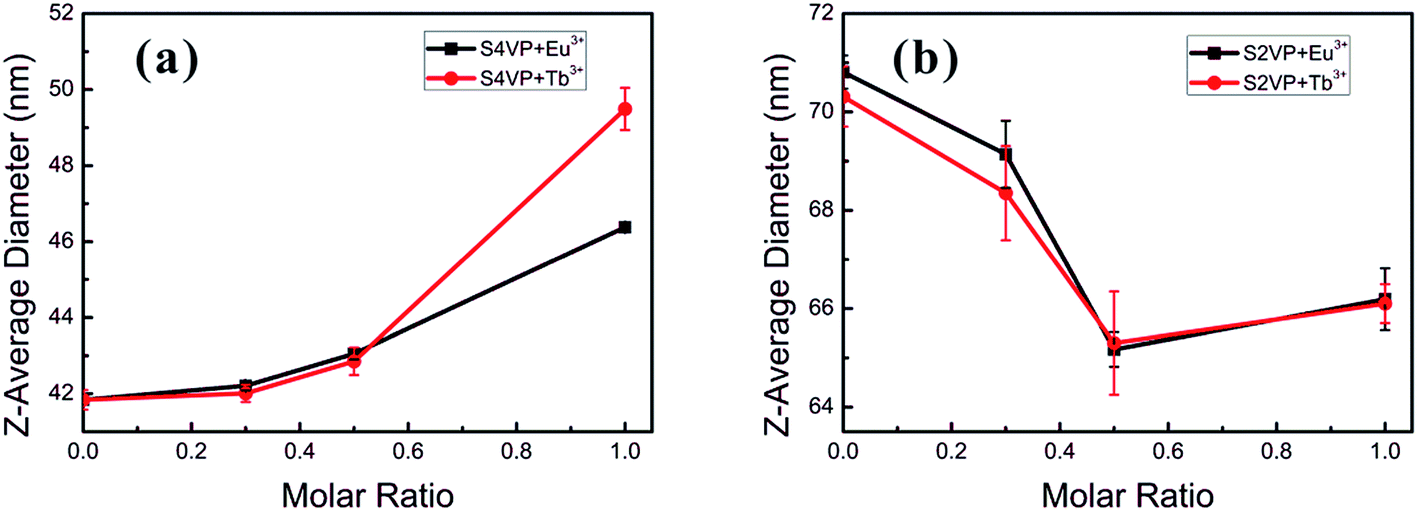 | ||
| Fig. 3 Micelle size of (a) S4VP[Ln(x)] and (b) S2VP[Ln(x)] with increasing the Ln3+ concentration (molar ratio of Ln3+ ranging from 0 to 1.0). | ||
In general, the morphology and size of the micelles are mainly controlled by three factors, namely the extension of the core blocks, surface tension between the core and solvent, and repulsion among the corona chains.24,25 Concerning the S4VP[Ln(x)] micelles, the corona chains were charged with Ln3+, resulting in repulsive interactions among the charged corona, which generally involve a steric contribution, a solvation contribution, and an electrostatic repulsive contribution.22,23 With increasing Ln3+ concentration, the overall charge better compensated the charge of unoccupied pyridine groups and reduced the electrostatic repulsion. Therefore, the interchain repulsive interactions between corona chains became weaker, causing an increase in micelle size to decrease the interfacial energy between the core and solvent. In the S2VP[Ln(x)] micelles, Ln3+ formed interactions with P2VP segments within the micelle core. As only steric–solvation interactions existed in nonionic corona chains, the introduction of Ln3+ mainly affected the extension of the core segments. With increasing Ln3+ concentration, the S2VP[Ln(x)] micelle diameter tended to decrease. We speculated that this was driven by multi-coordinate sites of Ln3+ and the P2VP block (each P2VP block might associate with many Ln3+ ions, and vice versa). This attraction between Ln3+ and the 2VP groups pulled them together, shrinking the micelle and increasing the micelle density. Therefore, we concluded that the Ln3+ distribution was crucial to determining micelle size evolution.
Dewetting phenomena
The stability of the as-spun Ln3+ associated S2VP films was affected by a form of dewetting, which we examined in detail AFM. All Ln3+ doped S2VP-films were dewetted to different extents during the spin coating process (Fig. 4), while films obtained from neat S2VP solution were homogeneous (Fig. SI-2, ESI†). Examining the featureless regions of the dewetted holes using AFM cross-section profiles gave an average thickness of about 5 nm (Fig. SI-3†). This suggested the existence of an adsorbed brush layer, with P2VP/Ln3+ acting as a wetting layer on the polar substrate and a PS layer of lower interfacial energy on top.26–28 In contrast, the films of S4VP[Ln(x)] were homogeneous throughout the Ln3+ concentration range (Fig. SI-4†). Based on the above results, we speculated that these distinct behaviors in film stability originated from the different Ln3+ distributions. | ||
| Fig. 4 AFM topography images (20 μm × 20 μm) of (a) S2VP[Eu(0.3)], (b) S2VP[Eu(0.5)], (c) S2VP[Eu(1.0)], (d) S2VP[Tb(0.3)], (e) S2VP[Tb(0.5)], and (f) S2VP[Tb(1.0)]. | ||
The wetting of a solid surface by a polymer film appears to be result from the combination of short-range intermolecular interactions between polymer and substrate, and long-range dispersive van der Waals forces. According to the literature, two major forms of polymer dewetting have been highlighted.27 In this scenario, as stated above, a layer of non-anchored S2VP[Ln(x)] micelles only partially wetted a monolayer of chemically identical molecules (a monolayer of S2VP[Ln(x)] complexes anchored on substrate), indicating that the relaxation of the thin films followed an autophobic dewetting mechanism.29–35 At the non-wettable interface, the PS corona of micelles autophobically dewetted from a brush of PS blocks, which was analogous to dewetting of a PS homopolymer melt on top of a PS homopolymer brush.36,37 Concerning the driving force, two main factors should be accounted for. Firstly, autophobic dewetting is believed to be primarily driven by entropy, which means that the conformation of the polymer brush layer contacting the substrate is different from that of the other polymer layers. With Ln3+ doping, the interactions between the ion doped P2VP and substrate are much stronger, leading to a strong driving force for autophobic dewetting because of the large reduction in conformational entropy in the wetting layer. Secondly, during evaporation of the deposited film, polymer chains underwent a marked change in conformation, from isolated individual coils to an entangled polymer film. Such non-equilibrated polymer chains might generate residual stresses within the film. Residual stresses are an extra driving force for dewetting, and are often at least the same order of magnitude as the acting capillary forces.35 Any internal or external disturbances, such as heating, dust particles, film defects, or local mechanical forces acting on the film surface, provide a route for nucleation and the relaxation of residual stresses.38 Furthermore, residual stresses arise from the strong biaxial constraint of the polymer film to the substrate. Attraction between Ln3+ and the pyridine groups in S2VP[Ln(x)] micelle cores could cause additional stress acting on the non-equilibrated polymer chains and certainly induce a stronger biaxial film constraint. Therefore, resulting from the complementary driving forces of autophobic behavior and residual stresses, S2VP[Ln(x)] film might rupture and dewet during the spin coating process. Furthermore, the driving force increased with increasing Ln3+ loading ratio, explaining the more obvious dewetting behavior at high Ln3+ concentrations (see Fig. 4).
In S4VP[Ln(x)], Ln3+ coordinated in micelle corona P4VP, as stated above. This association weakened the repulsive interactions between corona chains, resulting in stretching of the corona chain, which is not thought to induce significant residual stresses. Furthermore, although S4VP[Ln(x)] formed an anchored layer on substrate, no autophobic behavior appeared due to the different component in the top layer of anchored molecules (PS) and the corona of micelles (P4VP). Consequently, the addition of Ln3+ had no visible effect on S4VP film stability (Fig. SI-4†).
Photoluminescent properties of aggregates
Based on the different micelle structures of S4VP[Ln(x)] and S2VP[Ln(x)] mentioned above, their luminescence spectra were measured to explore the effect of Ln3+ distribution on luminescence efficiency. Fig. 5 depicts the emission spectra of S4VP[Eu(x)] and S2VP[Eu(x)]. Five characteristic emission peaks arose from transitions between the Russell–Saunders multiplets, 5D07FJ (J = 0, 1, 2, 3, 4). The typical red color of the europium emission was mostly attributed to the strongest transition (5D07F2), with a maximum intensity at 619 nm in our samples. The emission spectra of the Tb3+ complexes (Fig. 6) exhibited four main 5D47FJ transitions (J = 6, 5, 4, 3). The most intense peak, centered at 548 nm, was assigned to the 5D47F5 transition and was responsible for the pure green emission color. For all samples, the luminescent peaks did not show noticeable shifts in position. However, as shown in Fig. 7, the relatively intensities were significantly affected by the Ln3+ concentration and Ln3+ distribution (S2VP[Ln(x)] showed a higher emission intensity than S4VP[Ln(x)] at the same Ln3+ concentration).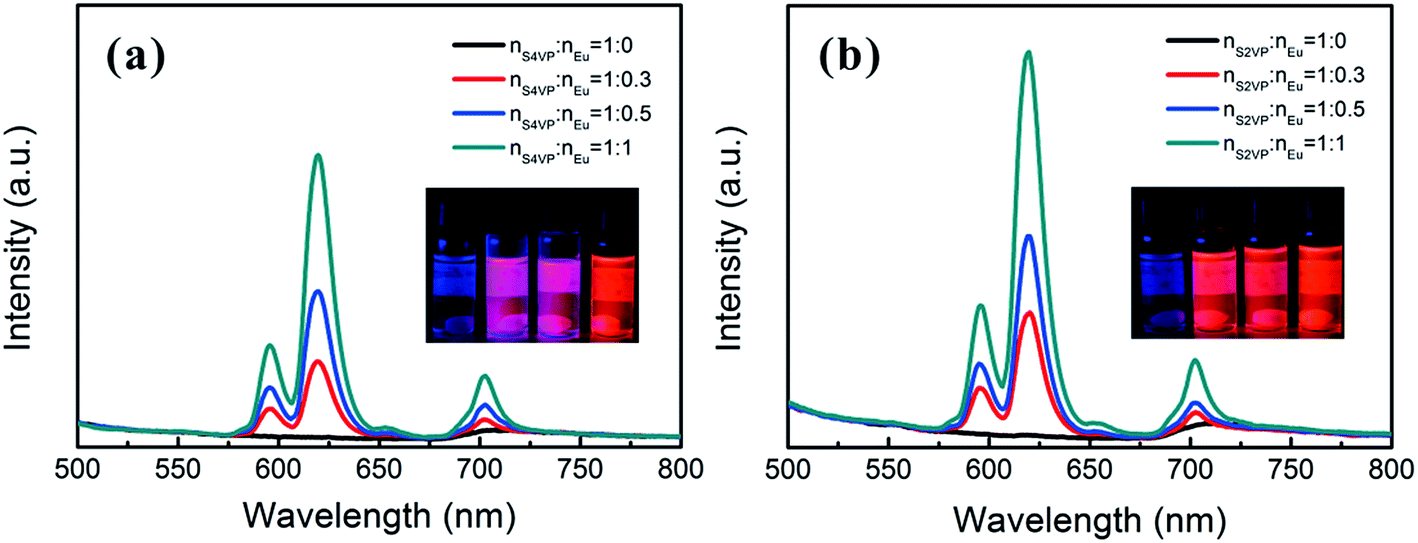 | ||
| Fig. 5 Emission spectra of (a) S4VP[Eu(x)] and (b) S2VP[Eu(x)] with Eu3+ loading ratios ranging from 0 to 1.0. | ||
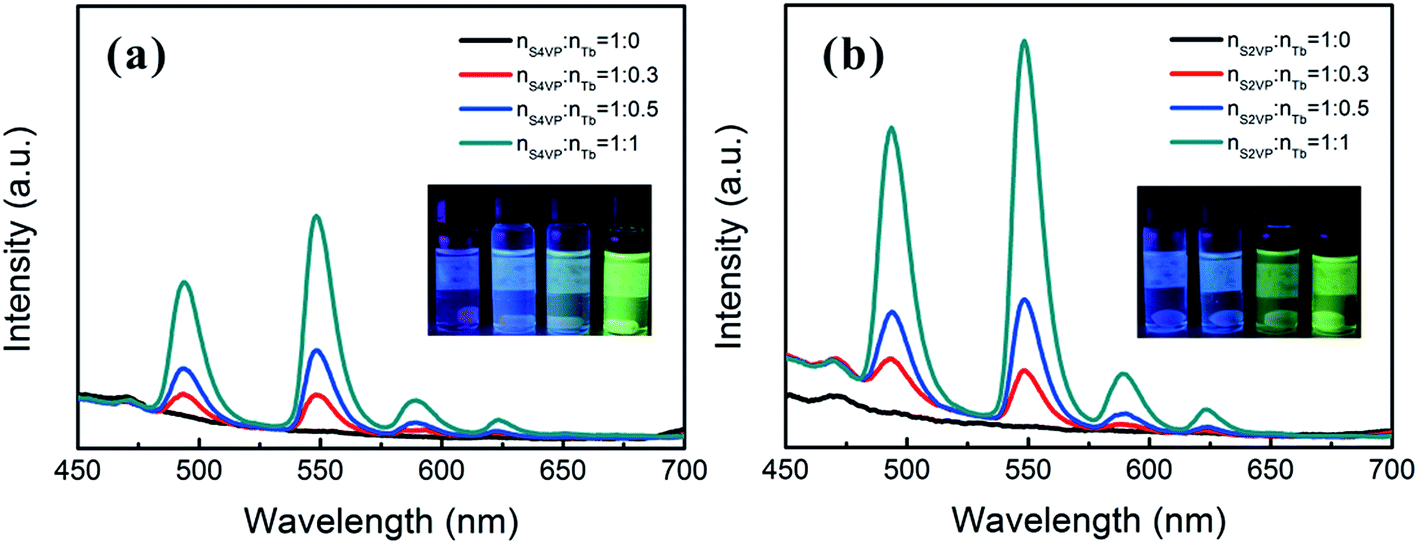 | ||
| Fig. 6 Emission spectra of (a) S4VP[Tb(x)] and (b) S2VP[Tb(x)] with Tb3+ loading ratios ranging from 0 to 1.0. | ||
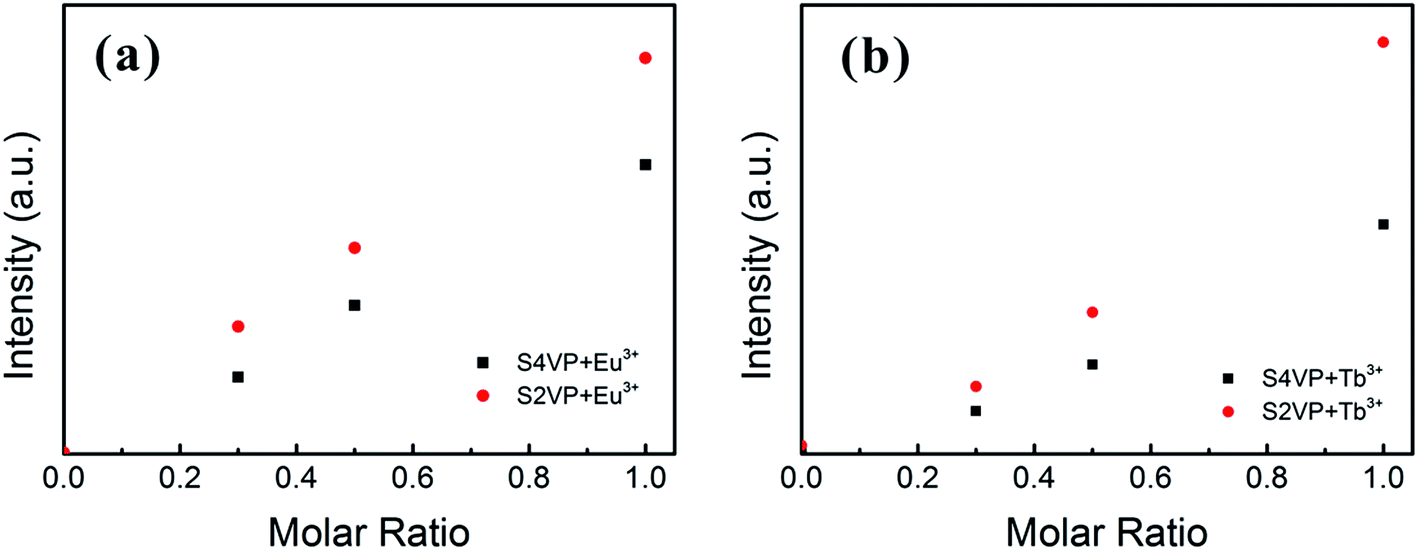 | ||
| Fig. 7 Emission intensity of the (a) 5D0 → 7F2 peak of Eu3+ complexes and (b) 5D4 → 7F5 peak of Tb3+ complexes with increasing the Ln3+ concentration (molar ratio of Ln3+ ranging from 0 to 1.0). | ||
The quantum yield of the luminescence step, ΦLn, expressed how well the radiative processes (characterized by rate constant kr) competed with non-radiative processes (overall rate constant knr).
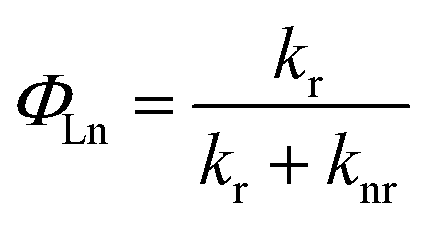
Contributions to knr included electron transfer quenching, back-energy transfer to the sensitizer, and, most importantly, quenching by matrix vibrations.39 Therefore, to explain luminescence changes for various Ln3+ concentrations and distributions, three points should be considered: (i) interactions between Ln3+ and PVP segments; (ii) the isolation effect originating from the aggregate structure; and (iii) the changed Ln3+ coordination sphere.
As mentioned above, the pyridine rings in S2VP and S4VP can form strong coordination bonds with lanthanide ions, which were manifested from FTIR spectra (displayed above) and UV-vis absorption spectra (see Fig. SI-5†). With these coordination bonds, BCP served as an immobilization phase to form a stable rigid structure, which reduced the non-radiative vibronic deactivation (including vibrational and rotational energy transitions) of Ln3+ and resulted in higher energy transfer efficiencies.
The isolation effect of the aggregate structure was revealed by the stronger luminescent intensity of S2VP[Ln(x)] than S4VP[Ln(x)] at the same Ln3+ concentration. Deactivation of luminescence from excited Ln3+ in solution occurred by means of a vibrational energy transfer process involving high-energy vibrations of solvent molecules or of bound ligands. Many investigations have shown that OH oscillators, such as those bound water or solvent alcohol molecules, were the most effective quenchers, both in the solid-state and solution. Therefore, the Ln3+ emission will be enhanced when Ln3+ is located in a relatively hydrophobic environment.40,41 For S2VP[Ln(x)], the hydrophobicity of the exterior PS peripheries shielded quenching of excited Ln3+ and further reduced non-radiative deactivation.17,42 Therefore, when the molar ratio of nLn3+:nVP was fixed, S2VP[Ln(x)] presented a higher emission intensity than S4VP[Ln(x)].
In addition to the structural influences discussed above, the main factor affecting the luminescence property was the change in Ln3+ coordination spheres in complexes. The transitions of Ln3+ consisted mainly of magnetic dipole transitions and electric dipole transitions. Experimental data for the Ln3+ complexes indicated that the magnetic dipole transitions (5D0 → 7F1 for Eu3+ and 5D4 → 7F6 for Tb3+) were largely independent of the chemical surroundings of the ion. The electric dipole transitions (5D0 → 7F2 for Eu3+ and 5D4 → 7F5 for Tb3+), so-called hypersensitive transitions, are sensitive to the symmetry of the coordination sphere.43 Therefore, the intensity ratio (η) of the magnetic dipole transition to the electric dipole transition in the lanthanide complex measured the symmetry of the coordination sphere.43,44 High η value are known to denote a high luminescent monochromaticity for different Ln3+ complexes, with Fig. 8 showing η values for different complexes. As shown in Fig. 8, the monochromaticity of both Eu3+ and Tb3+ increased with increasing Ln3+ concentration, showing that the presence of BCP generally increased the luminescent intensity of the hypersensitive transitions of Ln3+. When Ln3+ was incorporated into the BCP micelles, the complexes exhibited disorder of a certain magnitude. The influences of PVP segments on the coordinative environment resulted in the polarization of Ln3+, which changed the energy-transfer probabilities of the electric dipole transitions, accounting for the increase in luminescent intensity of the peaks at 619 and 548 nm for the Eu3+ and Tb3+ complexes, respectively. Moreover, S4VP[Ln(x)] presented a slightly higher η value than S2VP[Ln(x)], which could be explained by the symmetry of Ln3+ being more easily disrupted when Ln3+ was located in the micellar corona, due to Ln3+ being more likely to contact solvent molecules.
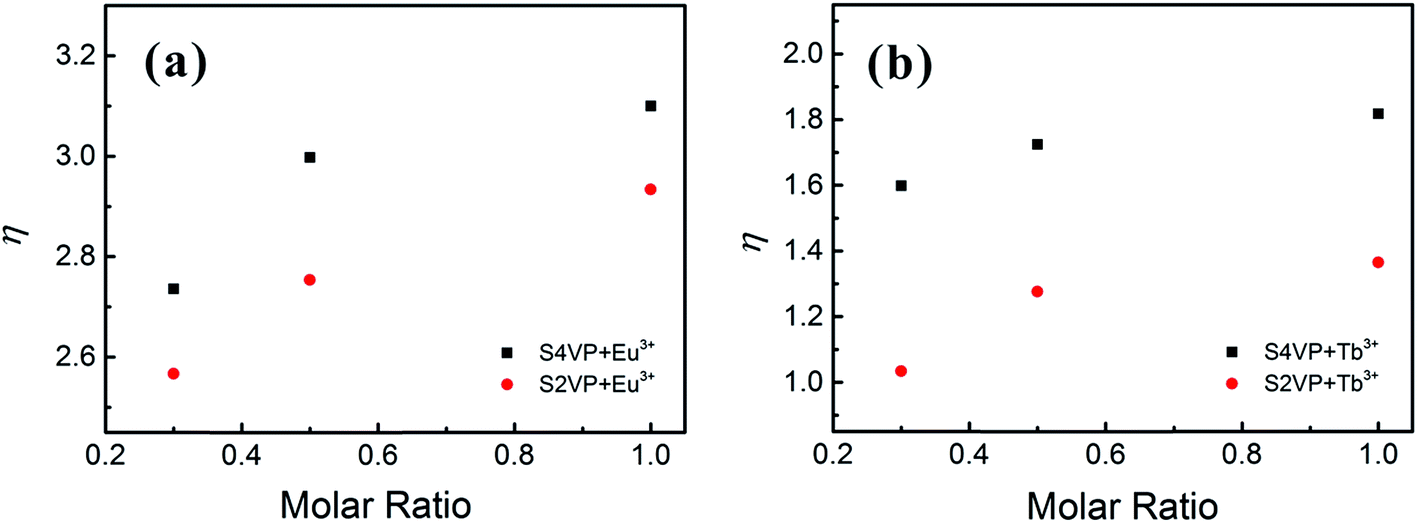 | ||
| Fig. 8 Intensity ratio of the magnetic dipole transition to the electric dipole transition of (a) Eu3+ complexes and (b) Tb3+ complexes with increasing the Ln3+ concentration (molar ratio of Ln3+ ranging from 0.3 to 1.0). | ||
In short, under the cooperative influences of the three factors state above, the coordination of BCP on Ln3+ enhanced the luminescent efficiency of the Ln3+ complexes and protected them from quenching. Especially, when Ln3+ was associated within the micelle core, the protection effect of the hydrophobic shell distinctly enhanced the luminescence intensity.
Using the same procedure, samples were also prepared by the addition of 1:1 (nEu:nTb) lanthanide ions to a solution of ion-free BCP, generating S4VP–Eu0.5Tb0.5 and S2VP–Eu0.5Tb0.5. The Eu–Tb compounds presented interesting luminescent properties. By mixing the appropriate combination of red and green light, orange emission was obtained. The emission spectra of the mixed samples gave addition spectra of the two chromophores (Fig. 9). The orange color was observed because Eu3+ is more emissive. This could be attributed to the possible energy transfer to the low-lying resonance state of Eu3+, 5D0 (17300 cm−1), from the 5D4 of Tb3+ (20400 cm−1).41 However, this possible energy transfer did not induce the total quenching of Tb3+, which could be explained by the existence of three kinds of domains in the mixed complexes: two containing Eu3+ or Tb3+, and the third containing both Eu3+ and Tb3+. Meanwhile, due to protection of the PS shell, S2VP–Eu0.5Tb0.5 presented a higher emission intensity than S4VP–Eu0.5Tb0.5.
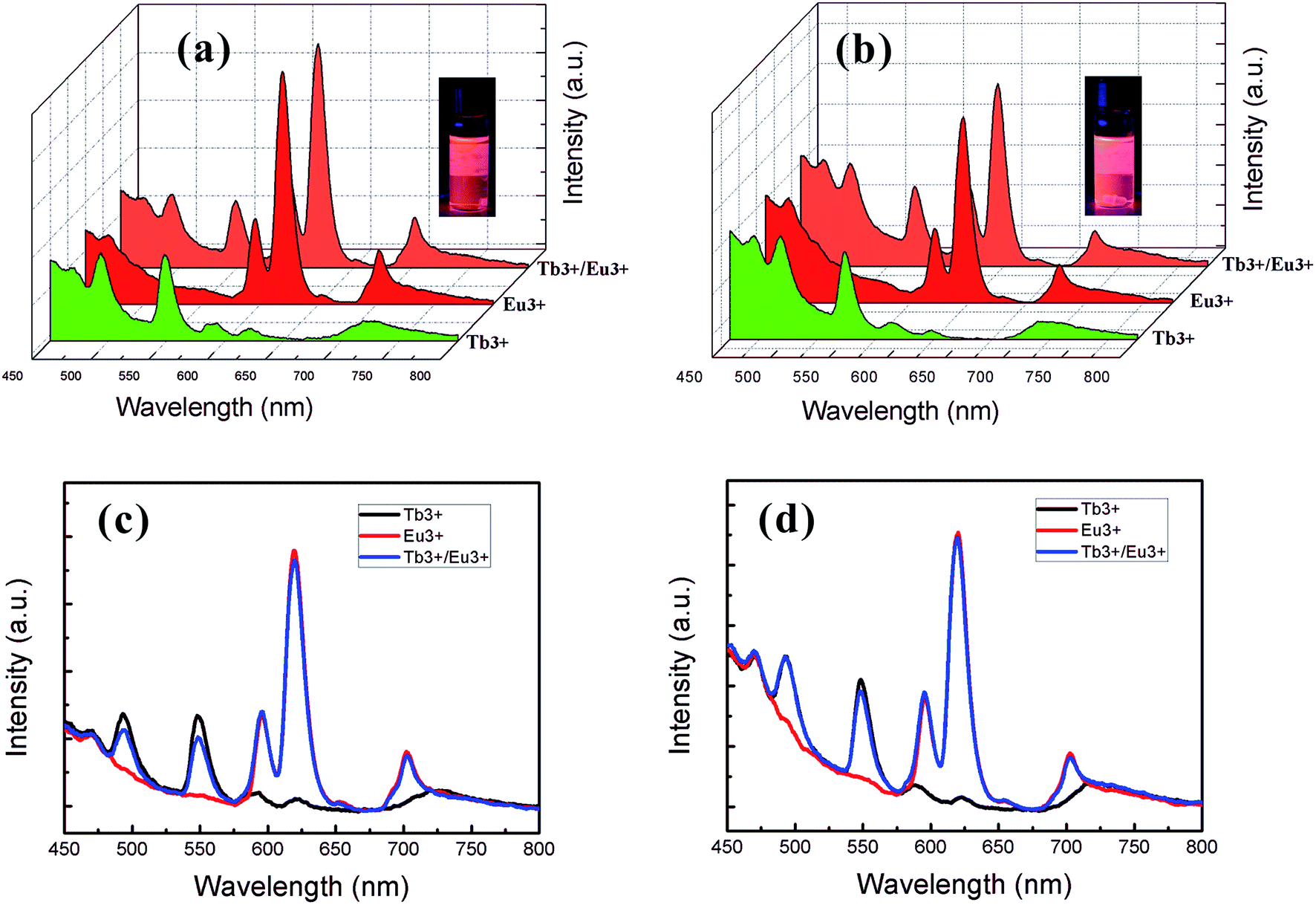 | ||
| Fig. 9 Different views of emission spectra of (a, c) lanthanide ion/S4VP complexes and (b, d) lanthanide ion/S2VP complexes. Green, red and orange profiles correspond to Tb3+, Eu3+, and Tb3+/Eu3+ complexes, respectively. | ||
Conclusions
In summary, the crucial role of Ln3+ ion distribution in micelles, thin films, and the photoluminescent properties of S4VP[Ln(x)] and S2VP[Ln(x)] were investigated in detail. The micelle size, film stability, and luminescent efficiency of these complexes were greatly affected by the Ln3+ ion distribution. With increasing Ln3+ concentration, S2VP[Ln(x)] micelles shrank and films underwent autophobic dewetting due to coordination between Ln3+ and S2VP micelle core segments. In contrast, the S4VP[Ln(x)] micelle size increased and the films remained intact with the coordination of Ln3+ and micelle corona chains. Complexes containing Eu3+ and Tb3+ showed typical reddish and greenish luminescence that are characteristic of f–f transitions in lanthanide ions. The coordination of BCP to Ln3+ enhanced the luminescent efficiency and protected them from quenching. Especially when Ln3+ was located in the micelle cores, the exterior PS shells shielded quenching of excited Ln3+ and markedly enhanced the luminescent intensity of the complexes. Moreover, the emission spectra of mixed Eu–Tb ions complexes give addition spectra of the two separated complexes, resulting in an orange colored emission due to Eu3+ being more emissive. The results reported in this study may improve understanding of ion coordination and lay the foundation for applications of lanthanide ion/BCP complexes.Author contributions
The manuscript was written with the contribution of all authors. All authors have given their approval to the final version of the manuscript.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51573131 and 21374077).References
- T. P. Lodge, Macromol. Chem. Phys., 2003, 204, 265 CrossRef CAS.
- G. S. MacGlashan, Y. G. Andreev and P. G. Bruce, Nature, 1999, 398, 792 CrossRef CAS.
- Z. Gadjourova, Y. G. Andreev, D. P. Tunstall and P. G. Bruce, Nature, 2001, 412, 520 CrossRef CAS PubMed.
- R. P. Pereira, A. M. Rocco and C. Bielschowsky, J. Phys. Chem. B, 2004, 108, 12677 CrossRef CAS.
- A. M. Christie, S. J. Lilley, E. Staunton, Y. G. Andreev and P. G. Bruce, Nature, 2005, 433, 50 CrossRef CAS PubMed.
- N. Sabbatini, M. Guardigli and J. M. Lehn, Coord. Chem. Rev., 1993, 123, 201 CrossRef CAS.
- W. T. Carnall, P. R. Fields and K. J. Rajnak, J. Chem. Phys., 1968, 49, 4424 CrossRef CAS.
- J. C. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC.
- N. E. Wolff and R. J. Pressley, Appl. Phys. Lett., 1963, 2, 152 CrossRef CAS.
- Y. Ueba, E. Banks and Y. Okamoto, J. Appl. Polym. Sci., 1980, 25, 2007 CrossRef CAS.
- Y. Ueba, K. J. Zhu, E. Banks and Y. Okamoto, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 1271 CrossRef CAS.
- J. L. Bender, P. S. Corbin, C. L. Fraser, D. H. Metcalf, F. S. Richardson, E. L. Thomas and A. M. Urbas, J. Am. Chem. Soc., 2002, 124, 8526 CrossRef CAS PubMed.
- X. F. Qiao and B. Yan, Inorg. Chem., 2009, 48, 4714 CrossRef CAS PubMed.
- Y. Cong, J. Fu, Z. Cheng, J. Li, Y. Han and J. Lin, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 2181 CrossRef CAS.
- Z. Li, R. Ma, Y. An and L. Shi, Colloid Polym. Sci., 2010, 288, 1041 CAS.
- Z. Li, R. Ma, A. Li, H. He, Y. An and L. Shi, Colloid Polym. Sci., 2011, 289, 1429 CAS.
- Q. Xu, J. Tang, Y. Wang, J. Liu, X. Wang, Z. Huang, L. Huang, Y. Wang, W. Shen and L. A. Belfiore, J. Colloid Interface Sci., 2013, 394, 630 CrossRef CAS PubMed.
- D. H. Lee, S. H. Han, W. Joo, J. K. Kim and J. Huh, Macromolecules, 2008, 41, 2577 CrossRef CAS.
- D. H. Lee, H. Y. Kim, J. K. Kim, J. Huh and D. Y. Ryu, Macromolecules, 2006, 39, 2027 CrossRef CAS.
- F. Xue, H. Li, J. You, C. Lu, G. Reiter and S. Jiang, Polymer, 2014, 55, 5801 CrossRef CAS.
- D. W. van Krevelen, Properties of polymers: their correlation with chemical structure; their numerical estimation and prediction from additive group contributions, Elsevier, 1990 Search PubMed.
- I. M. Smallwood, Handbook of organic solvent properties: Amold, 1996 Search PubMed.
- W. Zha, C. D. Han, D. H. Lee, S. H. Han, J. K. Kim, J. H. Kang and C. Park, Macromolecules, 2007, 40, 2109 CrossRef CAS.
- H. Shen, L. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1999, 121, 2728 CrossRef CAS.
- L. Zhang and A. Eisenberg, Macromolecules, 1996, 29, 8805 CrossRef CAS.
- J. Meiners, A. Ritzi, M. Rafailovich, J. Sokolov, J. Mlynek and G. Krausch, Appl. Phys. A: Mater. Sci. Process., 1995, 61, 519 CrossRef.
- G. Reiter, Phys. Rev. Lett., 1992, 68, 75 CrossRef CAS PubMed.
- A. Karim, N. Singh, M. Sikka, F. S. Bates, W. D. Dozier and G. P. Felcher, J. Chem. Phys., 1994, 100, 1620 CrossRef CAS.
- J.-U. Sommer and G. Reiter, in Ordered Polymeric Nanostructures at Surfaces, Springer, 2006, p. 1 Search PubMed.
- R. Limary and P. Green, Langmuir, 1999, 15, 5617 CrossRef CAS.
- G. Reiter and R. Khanna, Phys. Rev. Lett., 2000, 85, 2753 CrossRef CAS PubMed.
- T. H. Epps, D. M. DeLongchamp, M. J. Fasolka, D. A. Fischer and E. L. Jablonski, Langmuir, 2007, 23, 3355 CrossRef CAS PubMed.
- D. Yan, H. Huang, T. He and F. Zhang, Langmuir, 2011, 27, 11973 CrossRef CAS PubMed.
- Y. S. Sun, S. W. Chien and J. Y. Liou, Macromolecules, 2010, 43, 7250 CrossRef CAS.
- G. Reiter, M. Hamieh, P. Damman, S. Sclavons, S. Gabriele, T. Vilmin and E. Raphaël, Nat. Mater., 2005, 4, 754 CrossRef CAS PubMed.
- Y. Liu, M. Rafailovich, J. Sokolov, S. Schwarz, X. Zhong, A. Eisenberg, E. Kramer, B. Sauer and S. Satija, Phys. Rev. Lett., 1994, 73, 440 CrossRef CAS PubMed.
- G. Reiter, P. Auroy and L. Auvray, Macromolecules, 1996, 29, 2150 CrossRef CAS.
- X. He, J. Winkel and W. T. S. Huck, Adv. Mater., 2009, 21, 2083 CrossRef CAS.
- M. H. Werts, R. T. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542 RSC.
- W. D. Horrocks Jr and D. R. Sudnick, J. Am. Chem. Soc., 1979, 101, 334 CrossRef.
- X.-P. Yang, C.-Y. Su, B.-S. Kang, X.-L. Feng, W.-L. Xiao and H.-Q. Liu, J. Chem. Soc., Dalton Trans., 2000, 3253 RSC.
- L. Xu, L. Feng, Y. Han, Y. Jing, Z. Xian, Z. Liu, J. Huang and Y. Yan, Soft Matter, 2014, 10, 4686 RSC.
- F. S. Richardson, Chem. Rev., 1982, 82, 541 CrossRef CAS.
- Q. Li, T. Li and J. Wu, J. Phys. Chem. B, 2001, 105, 12293 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional Fig. S1–S5. See DOI: 10.1039/c7ra05705a |
| This journal is © The Royal Society of Chemistry 2017 |