
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn optimized 3D carbon matrix for high rate silicon anodes†
Xuejun Bai *a,
Min Houa,
Zhaoyu Yua,
Chan Liua,
Hui Caoab,
Dong Wangab and
Junjie Fuc
*a,
Min Houa,
Zhaoyu Yua,
Chan Liua,
Hui Caoab,
Dong Wangab and
Junjie Fuc
aShanghai Aerospace Power Technology Co., LTD, Shanghai, 201615, P. R. China. E-mail: renee1125@163.com
bShanghai Institute of Space Power-sources, Shanghai, 200245, China
cShanghai Geophysical Branch, Sinopec Offshore Oilfield Services Company, Shanghai, 201208, China
First published on 3rd July 2017
Abstract
A free-standing 3D porous carbon matrix was fabricated by a solution-based self-assembly process, using graphene aerogel (GA) as the matrix and CNTs as the structural enhancer. CNTs, with high electrical conductivity and mechanical strength, formed into a 3D scaffold along with GA. Silicon particles, with an oxidation coating (Si@SiOx), were embedded into this GA matrix, and twined around the CNTs to increase conductivity of the Si@SiOx agglomerations and bonding force with the GA matrix. Electrodes constructed with the Si@SiOx/CNTs/GA containing 44 wt% Si@SiOx exhibit a stable storage capacity of 905 mA h g−1 at 4 A g−1 and 1500 mA h g−1 at 0.1 A g−1 after 150th cycles with 93% capacity retention compared with the 10th cycle capacity. These outstanding rate performances and cycling stability are attributed to the enhanced 3D porous matrix, which provides abundant internal space for volume changes of Si@SiOx, easy penetration of electrolyte, fast electron and ion transfer speed, and high mechanical strength.
1 Introduction
For current commercial lithium-ion batteries (LIBs), graphite is used as an anode material with a 372 mA h g−1 theoretical capacity.1 The low energy density severely limits the applications of LIBs, especially in the automotive field. As a promising alternative, silicon (Si) has been considered as the leading anode material for next generation LIBs because of its high theoretical capacity of ∼4200 mA h g−1, high reserves in the earth, and low charge/discharge potential.2–5 However, the large volumetric changes of Si (>300%), during Li+ insertion/extraction processes, lead to rapid capacity degradation, severe pulverization of the electrode, and destruction of the electrically conductive network.6–9 For better implementation of Si in LIBs, the introduction of a second phase as a conductive matrix is considered to obtain high stability Si based anodes, such as carbon-based matrices: amorphous carbon,10–14 graphite,15 carbon fibers (CFs),16–19 carbon nanotubes (CNTs),20 and graphene.21–23 Si–graphene composites, such as graphene-encapsulated Si particles and Si–graphene nanosheets, show improved electrochemical performances due to their excellent electronic properties, flexibility, large specific surface area, good mechanical properties, and high thermal and chemical stability.24–26 Particularly, the hierarchical porous carbon matrixes, such as mesoporous carbon fiber, ordered hierarchical carbon, and ordered mesoporous carbon, show better loading and fast Li+ storage ability in LIBs.27–31 This hierarchical structure also shows positive influence in EDLC, electrochemical hydrogen storage, catalyst and other application area.32–34 Graphene aerogel (GA) with 3D porous network structure has attracted much more attention,35–38 as it provides more micropores for electrolyte infiltration and free Li+ diffusion. This increases rate capacity and power density of the LIBs in previous reports.39–41 However, it still remains challenging to future increase electron transport kinetics and overall mechanical strength.In this work, CNTs, with ultra-high electrical conductivity, flexibility, and mechanical strength, had been uniformly employed in graphene oxide suspension via ultrasonic dispersion. In our previous work, we reported a technique to form Si@SiOx core–shell nanoparticles by ozone treatment, which can improve dispersibility of Si nanoparticles in graphene oxide suspension.42 In the resulting free-standing 3D Si@SiOx/CNTs/GA composite, CNTs self-assembled into a 3D scaffold along GA and small Si@SiOx aggregation. Based on the enhanced stable-porous 3D matrix, Si@SiOx/CNTs/GA showed excellent storage capacity and cycling stability at high current density.
2 Experimental
2.1 Sample preparation
The Si@SiOx nanoparticles with 4.2 nm oxidization layer were oxidized from Si nanoparticles (∼50 nm, 99.9%, US Research Nanomaterials, Inc) at 200 °C as reported earlier.42Typically, Si@SiOx nanoparticles (10, 5, 2.5 mg respectively) and 10 mg of CNTs (99.9%, US Research Nanomaterials, Inc) were dispersed in 3 mL of ethanol and sonicated for 1 h. 2 mL of GO suspension (5 mg mL−1) was added and sonicated for 1 h to obtain a stable suspension. 80 mg of L-ascorbic acid (99%, Sigma Aldrich) was then added to the suspension and sonicated for 20 min to dissolve the acid. This mixture was then kept at 80 °C for 8 h to initiate pre-reduction and the self-assembly process. The resulting solid was washed repeatedly with distilled deionized (DDI) water and then freeze-dried. Finally, the solid was heated at 800 °C in a flow of Ar for 2 h for further reduction of GO, to obtain the Si@SiOx/CNTs/GA composites (Si@SiOx/CNTs/GA-1, -2, and -3). The process of fabrication the Si@SiOx/CNTs/GA composite was shown in Fig. 1.
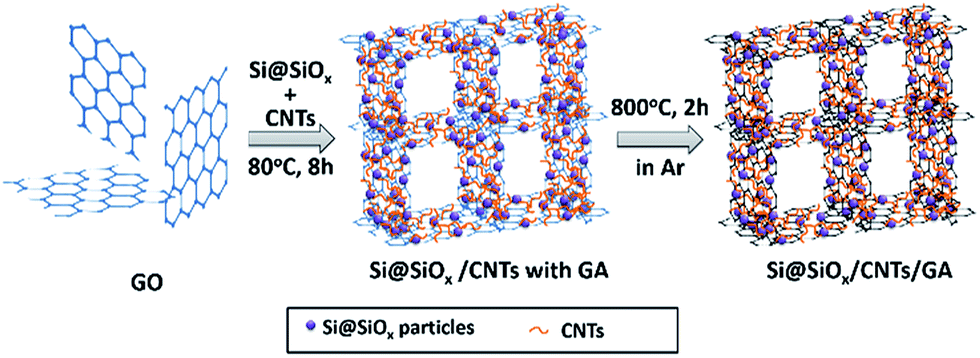 | ||
| Fig. 1 Schematic illustration of procedure to fabricate Si@SiOx/CNTs/GA composites. | ||
2.2 Sample characterization
X-ray diffraction (XRD) was carried out by a Rigaku D/max 2550 V X-ray diffractometer using Cu-Kα irradiation. Raman spectrum was measured on a T64000 triple Raman system with a 514.5 nm Ar-ion laser. X-ray photoelectron spectroscopy (XPS) was conducted with Thermo Scientific ESCALAB 250Xi using Al Kα (1486.6 eV) excitation. Thermogravimetric analysis (TGA) was recorded on a Netzsh TG 209 F1 Analyzer, from room temperature to 800 °C with a ramp rate of 15 °C min−1 in 15 mL min−1 lab air. Microstructure images were collected using high-resolution field emission scanning electron microscopy (FESEM, Hitachi SU8030). Cycled electrodes were removed from dissembled coin cells and rinsed in dimethyl carbonate (DMC) in an argon-filled glovebox before characterization by FESEM.2.3 Electrochemical measurements
Electrochemical measurements were conducted at room temperature using CR2032 coin cells, with Li as the counter electrode and Celgard 2400 film as the separator. The Si@SiOx/CNTs/GA composites, cut into circular discs of approximately 5 mm in diameter, were used as self-standing work electrodes. The electrolyte was 1 M LiPF6 in ethylene carbonate/dimethyl carbonate (EC/DMC 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol. ratio). The cells were assembled in an argon-filled glove box (H2O, O2 < 1 ppm). Galvanostatic cycling measurements were performed with a BT2000 Potentiostat/Galvanostat system (Arbin Instruments) in the voltage range of 0.01–2 V. The current setting for cell tests and the specific capacity calculation were based on the total mass of Si@SiOx/CNTs/GA composites. Cyclic voltammogram (CV), at a scan rate of 0.1 mV s−1 within 2–0.01 V, and EIS, at frequency 106–0.01 Hz, were recorded by electrochemical workstation (Autolab PGSTAT128N).
:1 vol. ratio). The cells were assembled in an argon-filled glove box (H2O, O2 < 1 ppm). Galvanostatic cycling measurements were performed with a BT2000 Potentiostat/Galvanostat system (Arbin Instruments) in the voltage range of 0.01–2 V. The current setting for cell tests and the specific capacity calculation were based on the total mass of Si@SiOx/CNTs/GA composites. Cyclic voltammogram (CV), at a scan rate of 0.1 mV s−1 within 2–0.01 V, and EIS, at frequency 106–0.01 Hz, were recorded by electrochemical workstation (Autolab PGSTAT128N).
3 Results and discussion
X-ray diffraction pattern of CNTs and Si@SiOx/CNTs/GA-1 are compared in Fig. 2a. The marked peaks are corresponded to crystalline Si, suggesting the Si structure is preserved in the whole synthesis process. The peak of GO at 12° isn't observable, and is replaced by a broader peak at ∼25.8°,43,44 which overlaps with the sharp peak of CNTs. The Raman spectrum of Si@SiOx/CNTs/GA-1 (Fig. 2b) shows five peaks clearly. A D-band at 1332 cm−1 indicates presence of disordered carbon, and a G-band at 1594 cm−1 and a 2D-band at ∼2880 cm−1 assign to the strong tangential mode of graphitized structure.45 LA-band and TO-band below 500 cm−1 are related to Si.46 In C 1s XPS spectra (Fig. 2c), there are three peaks: two weak peaks at 286.2 eV and 287.5 eV, representing O–C–O/C–O and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety respectively, and an intense peak at 284.8 eV which represents the C/C moiety.47 All the data above show CNTs/GA matrix is partially graphitized which can facilitate the electron transfer during electrochemical reactions. Three peaks in XPS Si 2p spectra (Fig. 2d) indicate of presence of three valence states of Si: Si4+, Si2+, and Si0.48 The contents are 41%, 11%, and 48%. SiOx shell around Si helps improve dispersibility and structure stability in GA.
O moiety respectively, and an intense peak at 284.8 eV which represents the C/C moiety.47 All the data above show CNTs/GA matrix is partially graphitized which can facilitate the electron transfer during electrochemical reactions. Three peaks in XPS Si 2p spectra (Fig. 2d) indicate of presence of three valence states of Si: Si4+, Si2+, and Si0.48 The contents are 41%, 11%, and 48%. SiOx shell around Si helps improve dispersibility and structure stability in GA.
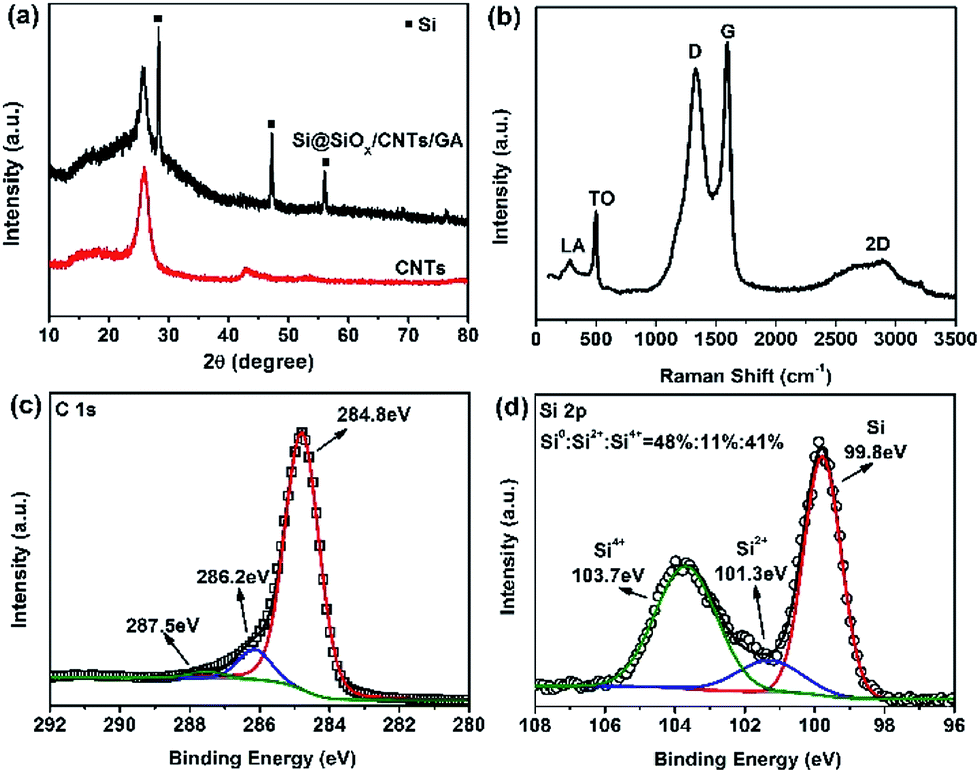 | ||
| Fig. 2 (a) XRD patterns of CNTs and Si@SiOx/CNTs/GA-1; (b, c and d) Raman spectrum, high resolution C 1s XPS spectra, and Si 2p XPS spectra of Si@SiOx/CNTs/GA-1, respectively. | ||
The contents of Si@SiOx in these composites were determined by TGA, shown in Fig. 3. As the crystal structure of Si@SiOx is relative stable during TGA test, as shown in Fig. S1.† From the remaining weight after heating above 700 °C, the Si@SiOx loading in samples Si@SiOx/CNTs/GA-1, -2, and -3 were determined to be 44 wt%, 29 wt%, and 20 wt% respectively. Si@SiOx/CNTs/GA composites with different Si@SiOx contents have similar 3D network structures (Fig. 4). The interconnecting micropores are open cells in nature, bounded by walls of ultrathin layers of stacked graphene sheets. CNTs are uniformly distributed on graphene sheet, forming a conductive flexible framework to support GA as a scaffold. The overall dispersion of Si@SiOx nanoparticles in GA has been great improved by oxidization layer, but small agglomerations also exist. As the SiOx layer form hydrogen bonds (Si–O–H–O–C) with organic groups on GO. GO anchored with Si@SiOx nanoparticles acted as a building block and self-assembled into a 3D network, driven by π–π stacking interactions. Then during reduction, the Si@SiOx nanoparticles can be anchored tightly and uniformly in GA.35,49 Some CNTs twine around Si@SiOx nanoparticles to increase conductivity of Si@SiOx agglomerations and bonding force with GA matrix. The EDS spectra of Si@SiOx/CNTs/GA-1 is shown in Fig. S2.†
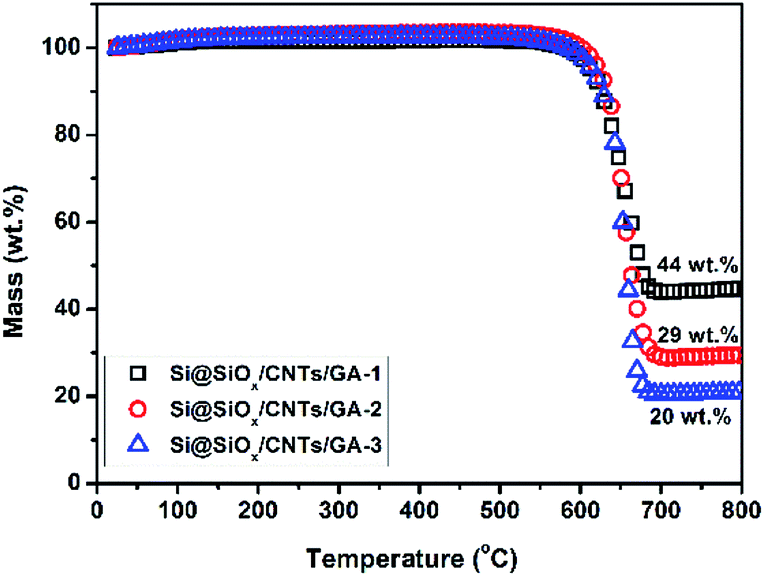 | ||
| Fig. 3 TGA curves of Si@SiOx/CNTs/GA composites. | ||
 | ||
| Fig. 4 SEM images of Si@SiOx/CNTs/GA composites: (a, b) Si@SiOx/CNTs/GA-1, (c, d) Si@SiOx/CNTs/GA-2, (e, f) Si@SiOx/CNTs/GA-3. | ||
The capacities of the Si@SiOx/CNTs/GA composites tested at different current densities (0.1, 0.5, 1, 2, and 4 A g−1) are shown in Fig. 5a. The initial coulombic efficiencies of Si@SiOx/CNTs/GA-1, -2, and -3 are 57%, 60%, and 62%. The large initial capacity loss is mainly due to the formation of SEI film on the surface of porous CNTs/GA matrix and stable products of Li+ with SiOx during lithiation. The increase of Si@SiOx loading doesn't show obvious effects on the cyclic stability, which shows that the hierarchical porous 3D structure of GA could effectively accommodate drastic volumetric variation of Si@SiOx during cycling.50 Compared to the capacity of 1613 mA h g−1 at 10th cycles at 0.1 A g−1, the capacity at 150th cycles reaches 1504 mA h g−1 with 93% capacity retention. The cyclic stability of Si@SiOx particles has been improved a lot (Fig. S3†). As the rate capacity retention shown in Fig. 5b, the capacities of Si@SiOx/CNTs/GA-1 are 1705 mA h g−1 at 0.1 A g−1 and 905 mA h g−1 at 4 A g−1, and the rate capacity retention at 4 A g−1 is 53%. The rate capacity retention of Si@SiOx/CNTs/GA is much better than that of Si@SiOx/GA (with similar Si@SiOx content) shown in previous research.42 CNTs improve flexibility and conductivity of GA matrix, maximizing electron transmission speed. They also increase the bonding force between Si@SiOx and GA, minimizing detachment of Si@SiOx from GA.
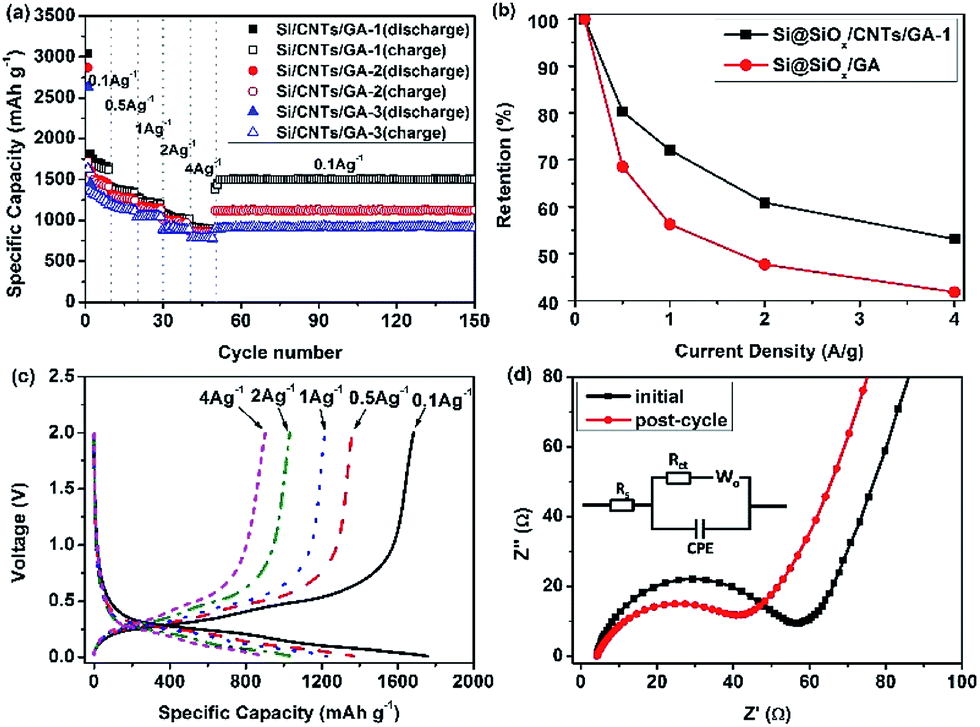 | ||
| Fig. 5 (a) Rate cyclic performance of Si@SiOx/CNTs/GA composites, (b) capacity retention at different current densities of Si@SiOx/CNTs/GA-1 and Si@SiOx/GA composite, (c) galvanostatic charge–discharge profiles of Si@SiOx/GA-1 at various current densities as marked, and (d) the electrochemical impedance spectra of Si@SiOx/CNTs/GA-1 composites at initial state and post-cycle. The insert is the equivalent circuit used for the analysis. | ||
The discharge–charge curves for Si@SiOx/CNTs/GA-1 (Fig. 5c) show the typical behavior of Si. The increase of current density shows little effects on the shape of these curves. The discharge plateaus are stable below 0.5 V, and charge ranges appear between 0.3 and 0.6 V. The electrode polarization doesn't increase obviously with current density. EIS of Si@SiOx/CNTs/GA-1 at initial state and post-cycle are shown in Fig. 5d. The Nyquist plots are fitted through the use of an equivalent circuit (Fig. 5d insert). The semicircle in the high-frequency region is associated with charge transfer impedance (Rct) and constant phase element of the electrode/electrolyte interface (CPE). The line in the low-frequency region is the Warburg impedance (Wo), which is related to the diffusion of Li+ into the bulk of the electrode. The fitting data shows in Table 1. Rs is very low, indicating good electrical conductivity of Si@SiOx/CNTs/GA-1 during rate cycle test. Rct decreases after the 150th rate cycle, demonstrating better electrolyte infiltration and faster Li+ transfer speed.35,51,52 This is consistent with excellent rate cyclic performance of these composites.
| Sample | State | Rs (Ω) | CPE-T | CPE-P | Rct (Ω) |
|---|---|---|---|---|---|
| Si@SiOx/GA-1 | Initial | 4.6 | 0.00015 | 0.7641 | 49 |
| Si@SiOx/GA-1 | Post-cycle | 4.2 | 0.00014 | 0.7868 | 32.3 |
The structure stability of Si@SiOx/CNTs/GA composite is further studied by post-cycling SEM images of Si@SiOx/CNTs/GA-1 electrode (Fig. 6). The electrode retains similar original porous 3-D structure as Fig. 3a. The pore size is still same, but graphene wall is thicker. No crack and damage can be found. Si@SiOx nanoparticles and CNTs show no sign of additional agglomeration post-cycling, marked by red-circles in Fig. 6b. With supports of CNTs, GA shows improved elasticity and stability during lithiation/de-lithiation of Si@SiOx nanoparticles.
 | ||
| Fig. 6 SEM images of Si@SiOx/CNTs/GA-1 anode after rate cycles test. | ||
4 Conclusions
A 3D porous carbon matrix was fabricated successfully via a solution-based self-assembly process and applied in anodes of LIBs. In this matrix, the interconnecting micropores are open cells in nature, bounded by walls of ultrathin layers of stacked graphene sheets. CNTs formed a conductive flexible scaffold supporting GA to strengthen overall mechanical properties. CNTs/GA matrix provided large specific surface areas for efficient loading of Si@SiOx nanoparticles. The abundant micropores effectively enhanced electrolyte infiltrate, Li+ diffusion, and electron transport kinetics. They also provided enough space to accommodate volume changes of Si during Li+ insertion/extraction processes. Electrodes constructed with Si@SiOx/CNTs/GA composites showed excellent outstanding rate capability (1705 mA h g−1 at 0.1 A g−1, and 905 mA h g−1 at 4 A g−1) and good cyclic stability (93% capacity retention between 10th and 150th cycles at 0.1 A g−1). The outstanding results provide an optimized structural design and a promising modification method of carbon matrixes for high rate LIBs.Acknowledgements
This work was supported by Shanghai Pujiang Program 17PJ1430100, Program of Shanghai Technology Research Leader 17XD1420600, Shanghai Science and Technology Innovation Action Plan 15DZ1201001 and 16111106001, Minhang District Leader. Assistance from the research groups of Prof. Harold H. Kung, Prof. Jiaxing Huang and Prof. Samuel Stupp of Northwestern University for parts of the tests was acknowledged.Notes and references
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed
.
- T. Kim, Y. H. Mo, K. S. Nahm and S. M. Oh, J. Power Sources, 2006, 162, 1275–1281 CrossRef CAS
- H. Ma, F. Y. Cheng, J. Chen, J. Z. Zhao, C. S. Li, Z. L. Tao and J. Liang, Adv. Mater., 2007, 19, 4067–4070 CrossRef CAS
- H. Kim and J. Cho, Nano Lett., 2008, 8, 3688–3691 CrossRef CAS PubMed
- H. Tao, L. Fan and X. Qu, Electrochim. Acta, 2012, 71, 194–200 CrossRef CAS
- A. M. Wilson, B. M. Way, J. R. Dahn and T. Vanbuuren, J. Appl. Phys., 1995, 77, 2363–2369 CrossRef CAS
- B. C. Kim, H. Uono, T. Sato, T. Fuse, T. Ishihara and M. Senna, Solid State Ionics, 2004, 172, 33–37 CrossRef CAS
- C. Ma, C. Ma, J. Wang, H. Wang, J. Shi, Y. Song, Q. Guo and L. Liu, Carbon, 2014, 72, 38–46 CrossRef CAS
- X. Zhou, Y. Yin, L. Wan and Y. Guo, Adv. Energy Mater., 2012, 2, 1086–1090 CrossRef CAS
- A. A. Arie, O. M. Vovk and J. K. Lee, J. Electrochem. Soc., 2010, 157, A660–A665 CrossRef CAS
- Q. Si, K. Hanai, T. Ichikawa, A. Hirano, N. Imanishi, Y. Takeda and O. Yamamoto, J. Power Sources, 2010, 195, 1720–1725 CrossRef CAS
- J. Xie, N. Imanishi, T. Zhang, A. Hirano, Y. Takeda and O. Yamamoto, Mater. Chem. Phys., 2010, 120, 421–425 CrossRef CAS
- J. Liu, N. Li, M. D. Goodman, H. Zhang, E. Epstein, B. Huang, Z. Pan, J. Kim, J. Choi, X. Huang, J. Liu, K. J. Hsia, S. J. Dillon and P. V. Braun, ACS Nano, 2015, 9, 1985–1994 CrossRef CAS PubMed
- W. Li, Y. Tang, W. Kang, Z. Zhang, X. Yang, Y. Zhu, W. Zhang and C. S. Lee, Small, 2015, 11, 1345–1351 CrossRef CAS PubMed
- I. Saleh and R. Raj, J. Power Sources, 2016, 310, 18–25 CrossRef CAS
- A. Magasinski, P. Dixon, B. Hertzberg, A. Kvit, J. Ayala and G. Yushin, Nat. Mater., 2010, 9, 353–358 CrossRef CAS PubMed
- S. M. Jang, J. Miyawaki, M. Tsuji, I. Mochida and S. H. Yoon, Carbon, 2009, 47, 3383–3391 CrossRef CAS
- H. Liu, W. Qiao, L. Zhan and L. Ling, New Carbon Mater., 2009, 24, 124–130 CrossRef CAS
- L. Wang, C. X. Ding, L. C. Zhang, H. W. Xu, D. W. Zhang, T. Cheng and C. H. Chen, J. Power Sources, 2010, 195, 5052–5056 CrossRef CAS
- W. Wang and P. N. Kumta, ACS Nano, 2010, 4, 2233–2241 CrossRef CAS PubMed
- S. Chou, J. Wang, M. Choucair, H. Liu, J. A. Stride and S. Dou, Electrochem. Commun., 2010, 12, 303–306 CrossRef CAS
- J. K. Lee, K. B. Smith, C. M. Hayner and H. H. Kung, Chem. Commun., 2010, 46, 2025–2027 RSC
- C. Sun, Y. Deng, L. Wan, X. Qin and G. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 11277–11285 CAS
- J. Luo, X. Zhao, J. Wu, H. Jang, H. H. Kung and J. Huang, J. Phys. Chem. Lett., 2012, 3, 1824–1829 CrossRef CAS PubMed
- X. Zhao, C. M. Hayner, M. C. Kung and H. H. Kung, Adv. Energy Mater., 2011, 1, 1079–1084 CrossRef CAS
- W. Zhang, P. Zuo, C. Chen, Y. Ma, X. Cheng, C. Du, Y. Gao and G. Yin, J. Power Sources, 2016, 312, 216–222 CrossRef CAS
- L. Wang, C. Yang, S. Dou, S. Wang, J. Zhang, X. Gao, J. Ma and Y. Yu, Electrochim. Acta, 2016, 219, 592–603 CrossRef CAS
- Y. Sha, X. Xu, L. Li, R. Cai and Z. Shao, J. Power Sources, 2016, 314, 18–27 CrossRef CAS
- Y. Xing, Y. Wang, C. Zhou, S. Zhang and B. Fang, ACS Appl. Mater. Interfaces, 2014, 6, 2561–2567 CAS
- M. S. Kim, D. Bhattacharjya, B. Fang, D. S. Yang, T. S. Bae and J. S. Yu, Langmuir, 2013, 29, 6754–6761 CrossRef CAS PubMed
- B. Fang, J. H. Kim, M. S. Kim and J. S. Yu, Acc. Chem. Res., 2013, 46, 1397–1406 CrossRef CAS PubMed
- B. Fang, M. Kim, J. H. Kim and J. S. Yu, Langmuir, 2008, 24, 12068–12072 CrossRef CAS PubMed
- B. Fang, J. H. Kim, M. S. Kim, A. Bonakdarpour, A. Lam, D. P. Wilkinson and J. S. Yu, J. Mater. Chem., 2012, 22, 19031–19038 RSC
- B. Fang, A. Bonakdarpour, M. S. Kim, J. H. Kim, D. P. Wilkinson and J. S. Yu, Microporous Mesoporous Mater., 2013, 182, 1–7 CrossRef CAS
- R. Wang, C. Xu, J. Sun, L. Gao and H. Yao, ACS Appl. Mater. Interfaces, 2014, 6, 3427–3436 CAS
- X. Liu, J. S. Cui, J. B. Sun and X. Zhang, RSC Adv., 2014, 4, 22601–22605 RSC
- S. Lu, Y. Chen, X. Wu, Z. Wang and Y. Li, Sci. Rep., 2014, 4, 4629 CrossRef PubMed
- K. Han, J. Shen, C. M. Hayner, H. Ye, M. C. Kung and H. H. Kung, J. Power Sources, 2014, 251, 331–337 CrossRef CAS
- R. Tian, Y. Zhang, Z. Chen, H. Duan, B. Xu, Y. Guo, H. Kang, H. Li and H. Liu, Sci. Rep., 2016, 6, 1–9 CrossRef PubMed
- L. Zhang, Z. Wang, H. Wang, K. Yang, L. Wang, X. Li, Y. Zhang and H. Dong, J. Alloys Compd., 2016, 656, 278–283 CrossRef CAS
- Y. Zhang, H. Liu, Z. Zhu, K. W. Wong, R. Mi, J. Mei and W. M. Lau, Methods Mol. Biol., 2016, 1336, 179–209 Search PubMed
- X. Bai, Y. Yu, H. H. Kung, B. Wang and J. Jiang, J. Power Sources, 2016, 306, 42–48 CrossRef CAS
- H. Xiang, K. Zhang, G. Ji, J. Y. Lee, C. Zou, X. Chen and J. Wu, Carbon, 2011, 49, 1787–1796 CrossRef CAS
- J. Wang, C. Zhong, S. Chou and H. Liu, Electrochem. Commun., 2010, 12, 1467–1470 CrossRef CAS
- B. Wang, X. Li, X. Zhang, B. Luo, M. Jin, M. Liang, S. A. Dayeh, S. T. Picraux and L. Zhi, ACS Nano, 2013, 7, 1437–1445 CrossRef CAS PubMed
- J. Wu, X. Qin, H. Zhang, Y. He, B. Li, L. Ke, W. Lv, H. Du, Q. Yang and F. Kang, Carbon, 2015, 84, 434–443 CrossRef CAS
- Y. Xu, Z. Lin, X. Huang, Y. Wang, Y. Huang and X. Duan, Adv. Mater., 2013, 25, 5779–5784 CrossRef CAS PubMed
- X. Yuan, H. Xin, X. Qin, X. Li, Y. Liu and H. Guo, Electrochim. Acta, 2015, 155, 251–256 CrossRef CAS
- Y. Teng, H. Zhao, Z. Zhang, Z. Li, Q. Xia, Y. Zhang, L. Zhao, X. Du, Z. Du, P. Lv and K. Swierczek, ACS Nano, 2016, 10, 8526–8535 CrossRef CAS PubMed
- B. Fang, M. S. Kim, J. H. Kim, S. Lim and J. S. Yu, J. Mater. Chem., 2010, 20, 10253–10259 RSC
- D. Wang, J. Yang, X. Li, D. Geng, R. Li, M. Cai, T. K. Sham and X. Sun, Energy Environ. Sci., 2013, 6, 2900–2906 CAS
- C. Zhong, J. Wang, Z. Chen and H. Liu, J. Phys. Chem. C, 2011, 115, 25115–25120 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05647h |
| This journal is © The Royal Society of Chemistry 2017 |