
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnti-microbial activity of chrysomycin A produced by Streptomyces sp. against Mycobacterium tuberculosis†
Balaji Muralikrishnan‡
 a,
Vipin Mohan Dan‡a,
J. S. Vinodha,
Vellekkatt Jamsheenad,
Ranjit Ramachandrana,
Sabu Thomasb,
Syed G. Dastagere,
K. Santhosh Kumarc,
Ravi Shankar Lankalapallid and
Ramakrishnan Ajay Kumar*a
a,
Vipin Mohan Dan‡a,
J. S. Vinodha,
Vellekkatt Jamsheenad,
Ranjit Ramachandrana,
Sabu Thomasb,
Syed G. Dastagere,
K. Santhosh Kumarc,
Ravi Shankar Lankalapallid and
Ramakrishnan Ajay Kumar*a
aMycobacterium Research Laboratory, Rajiv Gandhi Centre for Biotechnology, Trivandrum, Kerala, India. E-mail: rakumar@rgcb.res.in
bCholera and Biofilm Research Laboratory, Rajiv Gandhi Centre for Biotechnology, Trivandrum, Kerala, India
cChemical and Environmental Biology, Rajiv Gandhi Centre for Biotechnology, Trivandrum, Kerala, India
dChemical Sciences and Technology Division, National Institute for Interdisciplinary Science and Technology, CSIR, Trivandrum, Kerala, India
eNCIM Resource Centre, CSIR-National Chemical Laboratory, Pune, India
First published on 21st July 2017
Abstract
Limited efficacy of the BCG (Bacillus Calmette–Guérin) vaccine against adult tuberculosis and the emergence of resistance to existing anti-tuberculosis drugs compel discovery of novel antibiotics against Mycobacterium tuberculosis. Actinomycetes are still an attractive platform for the discovery of new antimicrobials, especially from untapped natural hotspots, despite the belief that they are an exhausted resource after repeated re-discoveries. Herein we report the isolation and identification of chrysomycin A from an actinomycete isolated from a coastal area in Kerala. We show for the first time that it has antimycobacterial activity. It was found to be bactericidal to planktonic and intracellular M. tuberculosis with an MIC of 3.125 μg mL−1; it is non-hemolytic and has negligible cytotoxicity. The actinomycete that produces chrysomycin A was found to be a Streptomyces sp. through 16S rRNA gene sequencing.
Introduction
Tuberculosis (TB), caused by the airborne pathogen Mycobacterium tuberculosis (MTB) remains a major global health problem. With 1.4 million deaths in 2015, it is still one of the top ten deadly diseases worldwide.1 Antibiotics are the mainstay for the treatment of TB as the only vaccine currently available (the BCG) does not prevent the disease in adults.2 First-line drugs, namely isoniazid, rifampicin, ethambutol and pyrazinamide, are effective in treating TB. However, long duration of treatment (3–6 months) and noncompliance from patients have resulted in the emergence of drug resistant strains that are difficult to kill, and pose a serious threat to global TB control.3,4 Emergence of multi-drug resistant (MDR), extensively drug resistant (XDR) and now totally drug resistant (TDR) strains is on the rise globally.5–7 Therefore, the need of the hour is to discover efficient novel molecules that have novel targets in the bacterium.Actinomycetes have provided us with different classes of antibiotics that play a critical role in treating infectious diseases.8 They are the source of more than 45% of all known antibiotics and 90% of usable antibiotics available in the market.9,10 This is mainly due to their capacity to produce versatile bioactive secondary metabolites through their resourceful biosynthetic gene clusters.11 We hypothesized that ecological biodiversity hotspots such as coastal areas and tropical forests could harbour new strains of actinomycetes from which novel anti-mycobacterials could be isolated.
In this study, we screened culture filtrates of four hundred actinomycetes isolated from coastal areas and Western Ghats of Kerala, India. Ethyl acetate extract of a Streptomyces sp. OA161, was found to inhibit virulent laboratory strain MTB H37Rv. The active principle was purified, subjected to structural characterization and identified as chrysomycin A. This is the first report that chrysomycin A has anti-MTB activity. It was found to be non-cytotoxic to myoblast and epithelial cells and did not lyse red blood cells.
Results and discussion
Screening of culture filtrate extracts for antimicrobial activity
Among four hundred isolates of actinomycetes screened for their antimicrobial activity, ethyl acetate extract of the culture filtrate of the isolate OA161 was found to inhibit the growth of MTB in vitro (ESI Data 1†). The antimicrobial activity assay was extended to a panel of seven more microbes and the results revealed that it inhibited growth of Gram-positive bacteria but not of Gram-negative bacteria (Table 1). Based on the results, M. smegmatis was used as a surrogate organism to identify the active principle in the extract as the assay using MTB at every step of activity-guided separation was laborious and time-consuming. The ethyl acetate extract was fractionated using thin layer chromatography and subsequent bioautography with M. smegmatis revealed an inhibitory zone and the spot was named AMF (antimycobacterial fraction) (ESI Data 2†). Subsequently, the anti-MTB activity of AMF was confirmed by resazurin microtitre assay (REMA).| Bacteria tested | Antimicrobial activity of the ethyl acetate extract | MIC (μg mL−1) of chrysomycin A |
|---|---|---|
| a “+” denotes antimicrobial activity, “−” no activity. | ||
| Mycobacterium tuberculosis H37Rv | + | 3.125 ± 0.5 |
| Mycobacterium smegmatis MC2 155 | + | 1 ± 0.2 |
| Staphylococcus aureus ATCC 25923 | + | 0.5 |
| MRSA (methicillin resistant Staphylococcus aureus) | + | 2 |
| VRE (vancomycin resistant Enterococcus faecalis) | + | 0.5 |
| Escherichia coli ATCC 25922 | − | >25 |
| Pseudomonas aeruginosa ATCC 27853 | − | >50 |
| Vibrio cholerae N16961 | − | >50 |
Identification of the active principle
Separation of AMF using HPLC revealed two prominent peaks with retention times 26.4 minutes (compound 2) and 28.7 minutes (compound 1, ESI Data 3†). While the molecule eluted at 26.4 minutes had negligible activity, the one that eluted at 28.7 minutes had strong anti-MTB activity. The active principle was subjected to structural characterization by extensive NMR analysis which helped in identification of compound 1 as chrysomycin A (Fig. 1) which was further confirmed by comparison of the NMR data with that in the published literature12–15 (ESI Data 4–6†). The AMF, yellow solid (18 mg) after separation through HPLC yielded compound 1 (2.5 mg) and compound 2 (9.5 mg), respectively.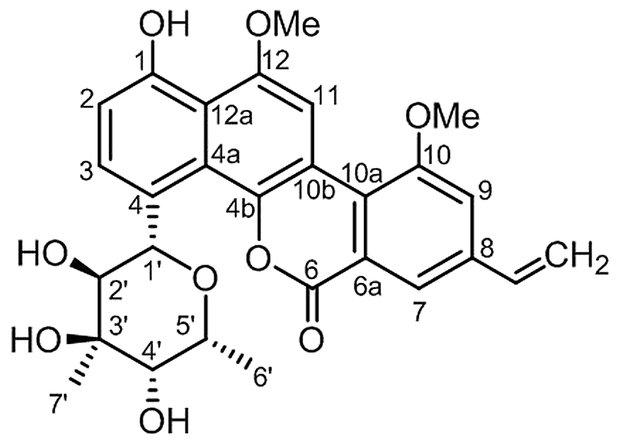 | ||
| Fig. 1 Structure of chrysomycin A. | ||
Antimycobacterial activity of chrysomycin A
Chrysomycin A was tested for antimycobacterial activity against MTB by REMA. Planktonic MTB was inhibited by the molecule at an MIC of 3.125 μg mL−1 and the same MIC was observed with commercially available chrysomycin A (Abcam). The MIC we observed is therefore better than that of two first-line anti-TB drugs, ethambutol (4 μg mL−1) and pyrazinamide (16 μg mL−1).16,17Time-course experiment with free-living MTB revealed that the activity is bactericidal in nature as the CFUs were reduced by three log10 folds at MIC and more at higher concentrations. In fact, no colonies were found when bacteria were treated with chrysomycin A (3.125 μg mL−1) for 24 hours. At this concentration the bactericidal activity was comparable to that of rifampicin (1 μg mL−1). Above 7.8 μg mL−1, which is the IC50 for THP-1-derived macrophages, no colonies were found at any time point checked on planktonic MTB (Fig. 2). Interestingly the compound was also found to be inhibitory to intracellular MTB at the same concentration (Fig. 3). The antimycobacterial activity of chrysomycin A is comparable to that of rifampicin (1 μg mL−1) as the CFU data suggested. This is the first report that chrysomycin A has anti-MTB activity against planktonic as well as intracellular MTB.
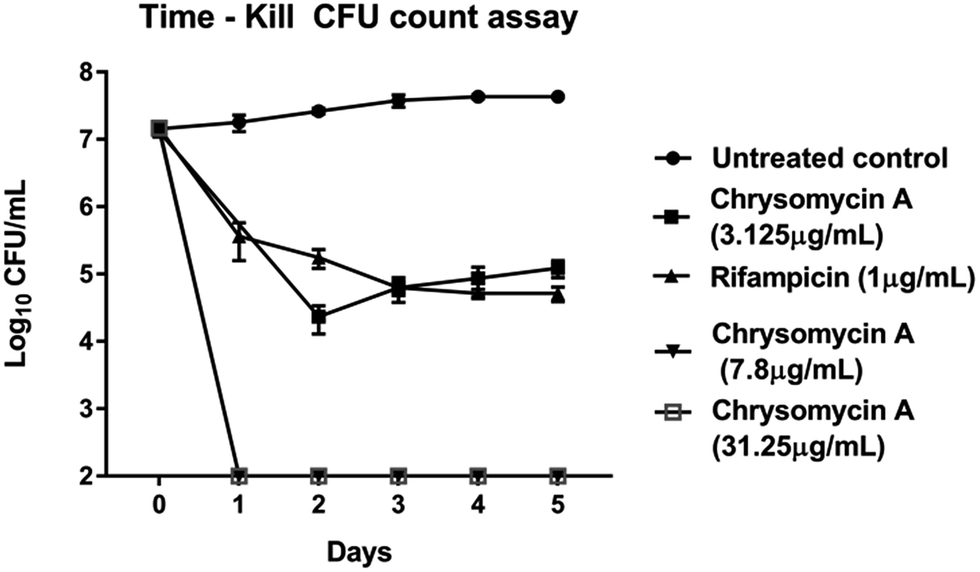 | ||
| Fig. 2 Kinetics of bactericidal activity of chrysomycin A on planktonic MTB. Data presented is mean ± SD of three independent experiments. | ||
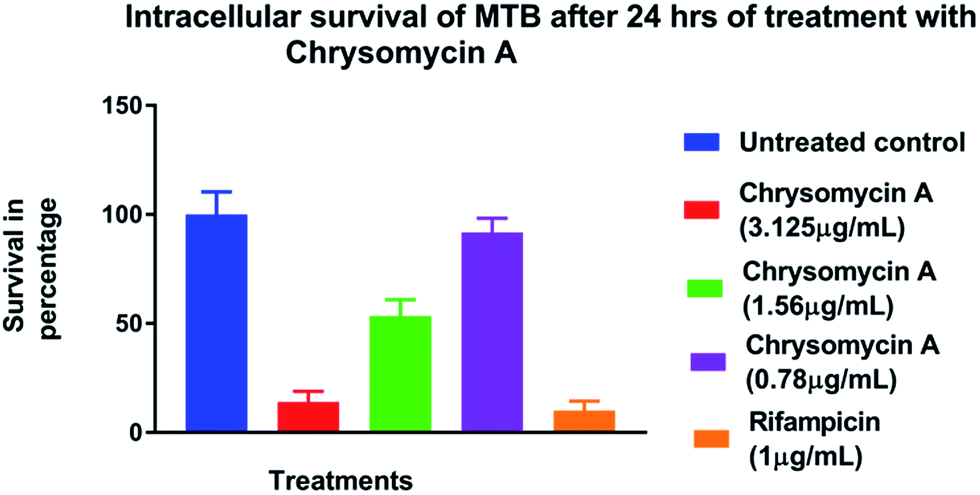 | ||
| Fig. 3 Survival of intracellular MTB in the presence of chrysomycin A. Data represented is mean ± SD of three independent experiments. | ||
Chrysomycin A was found to inhibit M. smegmatis and B. subtilis but not Gram negative bacteria by Strelitz et al.14 Our study also showed that it has the ability to inhibit M. smegmatis at almost the same concentration (0.6 μg mL−1). In addition, it is inhibitory to drug resistant strains of Staphylococcus aureus and Enterococcus faecalis. It was active against MRSA (methicillin resistant Staphylococcus aureus) and VRE (vancomycin resistant Enterococcus faecalis) in vitro with an MIC of 2 μg mL−1 and 0.5 μg mL−1, respectively. It was not inhibitory to Gram negative bacteria tested (E. coli, Pseudomonas aeruginosa and Vibrio cholerae). Interestingly, this molecule has also been reported to cause DNA damage and apoptosis in leukemic cell line HL60.13 Our study therefore highlights the need for re-purposing existing and abandoned drugs as recommended by Boguski et al.18
Chrysomycin A is non-hemolytic
Human erythrocytes were unharmed even at a concentration ten times the MIC against MTB whereas 1% SDS resulted in complete lysis (Fig. 4). This is another attractive characteristic of an ideal antimicrobial lead molecule. MTT assay revealed that cell lines L6 and SW480 were also unaffected even at 31.25 μg mL−1 while THP-1-derived macrophages had an IC50 of 7.8 μg mL−1, but largely remained unharmed at the MIC (3.125 μg mL−1) (Fig. 5).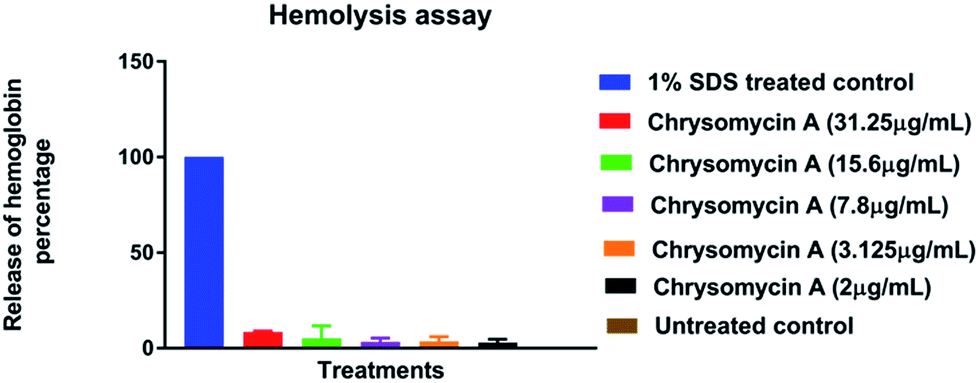 | ||
| Fig. 4 Hemolysis assay: chrysomycin A is non-hemolytic even at ten times the MIC (31.25 μg mL−1). Data presented is mean ± SD of three independent experiments. | ||
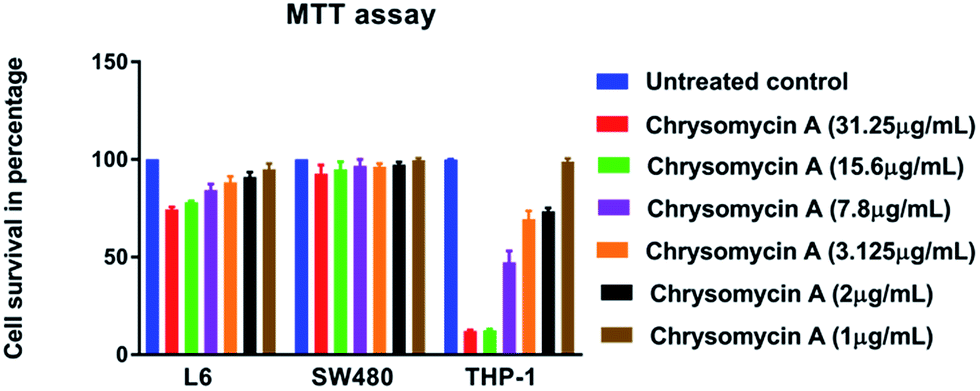 | ||
| Fig. 5 MTT assay: cytotoxicity of chrysomycin A was analyzed on cell lines up to ten times the MIC (31.25 μg mL−1). Little or negligible toxicity was found against L6 (rat myoblast cell line) and SW480 (epithelial cell line from colon). It was relatively more toxic against THP-1-derived macrophages. Data presented is mean ± SD of three independent experiments. | ||
The high cytotoxicity of chrysomycin A on THP-1 cells in comparison with the others was rather puzzling, however we presume that this is perhaps due to the fact that THP-1 cells are derived from human leukemia, and chrysomycin A has been reported to possess anti-leukemic activity.13 Interestingly, when tested on mice, chrysomycin A was not lethal even at 400 mg kg−1 dose and it was, on the other hand, found to increase the life span of the tumour-induced animals.15
Isolate that produces chrysomycin A is a Streptomyces sp. OA161
The partial 16S rRNA gene sequence of our isolate was submitted to GenBank, NCBI (accession number, KX364040). It is 1410 bp in length and 99.07% similar to that of Streptomyces smyrnaeus. A phylogenetic tree was generated using neighbor joining method19 using MEGA 7 software and the isolate forms a separate clade that is characteristic of a separate species (ESI Data 7†).Experimental
General procedures, strains, cell lines and chemicals
Mycobacterium tuberculosis H37Rv was grown in Middlebrook 7H9 medium (Difco) supplemented with 10% OADC (Difco). Other bacteria used in this study are Mycobacterium smegmatis mc2 155, Staphylococcus aureus ATCC 25953, MRSA (methicillin resistant Staphylococcus aureus) clinical strain C10, VRE (vancomycin resistant Enterococcus faecalis) clinical strain E8, E. coli ATCC 25922, Pseudomonas aeruginosa clinical strain R8 and Vibrio cholera N16961 and were grown in Luria Bertani broth (HiMedia, Mumbai). The clinical strains were a kind gift from the Department of Microbiology, Government Medical College, Trivandrum. Nalidixic acid, nystatin, gentamycin, phorbol 12-myristate 13-acetate (PMA) and resazurin were purchased from Sigma; Cell lines L6 (rat myoblast), SW480 (epithelial cell from colon) and THP-1 (Human monocytes) were obtained from National Centre for Cell Science, Pune, India. Cell lines were maintained in RPMI1640 (Sigma) or DMEM (Dulbecco's Modified Eagle's Medium, Gibco) with 10% FBS (Gibco). The extracts and antibiotics were prepared in DMSO and the final concentration of DMSO used in the assays was 2% whenever used for antimicrobial activity.Isolation of actinomycetes
About 10 grams of soil samples were collected in sterile bags from the sub-soil of different ecological niches and were dried under shade. This was suspended in sterile water (1 gm mL−1), serially diluted and plated on selective solid growth media (starch casein agar/actinomycetes isolation agar/ISP-2/ISP-7) with or without nalidixic acid (100 μg mL−1) and nystatin (50 μg mL−1) and incubated at 25 °C for 2 weeks. Once chalky colonies appeared, they were further purified on ISP-2 agar. The pure colonies were stored in the form of stabs or as glycerol stocks at 4 °C and −20 °C, respectively.Purification of anti-mycobacterial principle
Single colony of the actinomycete isolate (OA161) was inoculated in modified ISP-2 medium (50 mL) and grown at 30 °C on a shaker incubator (250 rpm). Two glass marbles were introduced into the medium to prevent clumping of the culture. After 6 days, the culture filtrate was prepared by centrifugation of the culture at 6000 rpm for 15 minutes at room temperature followed by filtration through Whatman 3 filter paper, and passing through 0.2 μm membrane filter. The supernatant was frozen at −80 °C before lyophilization. The lyophilized powder of the culture filtrate was extracted with ethyl acetate, chloroform, acetone and methanol and these extracts were concentrated under vacuum and used in the antimicrobial assay.Resazurin microtitre assay (REMA)
The assay was carried as described by Martin et al.20 Briefly, MTB H37Rv and M. smegmatis were grown in Middlebrook 7H9 medium, turbidity of the culture was adjusted to McFarland standard 1 and further diluted 20 times. For other test strains, Luria Bertani broth (HiMedia) was used for growth. One hundred microlitre of the diluted culture was transferred to the wells of a 96 well microtitre plate (Thermo Fisher). The extracts and purified active principle were dissolved in DMSO and added to a final concentrations of 200 μg mL−1 to 0.78 μg mL−1 in a final volume of 200 μL per well. The plates were incubated for 7 days, with negative (no drug) and positive controls (rifampicin and ciprofloxacin at 1 μg mL−1). MIC was determined after the addition of Resazurin (0.25% solution in water) on the 8th day which either remains blue or changes to pink (after 24 hours) based on the viability of the bacteria. Blue indicates that the bacteria are susceptible (inhibition), and pink indicates the bacteria are resistant to killing (viable) by the extract.Thin layer chromatography and bioautography
Normal phase Silica gel 60 F254 aluminum TLC plates (Merck, India) were used to fractionate the ethyl acetate extract of the culture filtrate of the isolate. Hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Acetone (1:1) was used as the mobile phase. The TLC plate was allowed to develop in a sterile, saturated closed chamber for 35 minutes. The fractionated TLC plate was overlayed with Muller Hinton soft agar containing S. aureus or M. smegmatis. This was allowed to solidify and incubated for 24 hours (S. aureus) or 36 hours (M. smegmatis) at 37 °C. The active fraction was named AMF (antimycobacterial fraction) and this was subjected to HPLC.
:Acetone (1:1) was used as the mobile phase. The TLC plate was allowed to develop in a sterile, saturated closed chamber for 35 minutes. The fractionated TLC plate was overlayed with Muller Hinton soft agar containing S. aureus or M. smegmatis. This was allowed to solidify and incubated for 24 hours (S. aureus) or 36 hours (M. smegmatis) at 37 °C. The active fraction was named AMF (antimycobacterial fraction) and this was subjected to HPLC.
Purification and characterization of the active principle
:1. Four hours after infection, the external bacteria (which were not phagocytosed) were treated with gentamycin (45 μg mL−1), and the cells were subsequently treated with chrysomycin A (3.125 μg mL−1, 1.56 μg mL−1 and 0.78 μg mL−1). Macrophages treated with rifampicin (1 μg mL−1) were taken as positive control and untreated macrophages as negative control. These were incubated at 37 °C for 48 hours and lysed with distilled water. The lysate was plated on 7H10 agar containing 0.1% casitone (Difco) and the colonies were counted after 3–4 weeks of incubation at 37 °C.000 cells were seeded per well in a 96 well plate in triplicates. They were treated with chrysomycin A at different concentrations (1–31.25 μg mL−1) and incubated for 48 hours at 37 °C in the presence of 5% CO2. Twenty microlitre of MTT (3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide) dye (5 mg mL−1) was added to each well and after 4 hours, the reaction was terminated by adding 200 μL of isopropanol and this dissolves the formazan crystals. This was read at 540 nm on an ELISA reader (Bio-Rad).Conclusion
Chrysomycin A, isolated from a new species of Streptomyces, showed bactericidal activity against planktonic as well as intracellular MTB at an MIC of 3.125 μg mL−1. This is the first report of chrysomycin A possessing anti-MTB activity.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
RAK and ST thank financial support from Open Source Drug Discovery (OSDD), Council of Scientific and Industrial Research (CSIR) (OSDD/HCP0001/12FYP/2012-123/Fin/2412), Government of India. BM thanks Council of Scientific and Industrial Research, Government of India, for research fellowship. VMD was supported by Fast Young Scientist Scheme (SERB:SB/FT/LS-322/2012) of Department of Science and Technology, Government of India. VJS thanks OSDD-CSIR for fellowship. SGD thanks financial support from Council of Scientific and Industrial Research, Government of India.References
- W. H. Organization, Global tuberculosis report 2015, World Health Organization, 2015 Search PubMed.
- L. Brandt, J. F. Cunha, A. W. Olsen, B. Chilima, P. Hirsch, R. Appelberg and P. Andersen, Infect. Immun., 2002, 70, 672–678 CrossRef CAS PubMed.
- I. Gelmanova, S. Keshavjee, V. Golubchikova, V. Berezina, A. K. Strelis, G. V. Yanova, S. Atwood and M. Murray, Bull. W. H. O., 2007, 85, 703–711 CrossRef CAS PubMed.
- J. E. de Steenwinkel, T. Marian, G. J. de Knegt, H. A. Verbrugh, R. E. Aarnoutse, M. J. Boeree, M. A. den Bakker, D. van Soolingen and I. A. Bakker-Woudenberg, Antimicrob. Agents Chemother., 2012, 56, 4937–4944 CrossRef CAS PubMed.
- K. Dheda, T. Gumbo, N. R. Gandhi, M. Murray, G. Theron, Z. Udwadia, G. B. Migliori and R. Warren, Lancet Respir. Med., 2014, 2, 321–338 CrossRef PubMed.
- N. R. Gandhi, P. Nunn, K. Dheda, H. S. Schaaf, M. Zignol, D. Van Soolingen, P. Jensen and J. Bayona, Lancet, 2010, 375, 1830–1843 CrossRef.
- M. Klopper, R. M. Warren, C. Hayes, N. C. Gey van Pittius, E. M. Streicher, B. Muller, F. A. Sirgel, M. Chabula-Nxiweni, E. Hoosain and G. Coetzee, Emerging Infect. Dis., 2013, 19(3), 449–455 CrossRef PubMed.
- A. Raja and P. Prabakarana, Am. J. Drug Discovery Dev., 2011, 1, 75–84 CrossRef.
- J. Berdy, J. Antibiot., 2005, 58, 1–26 CrossRef CAS PubMed.
- G. B. Mahajan and L. Balachandran, Front. Biosci., Elite Ed., 2011, 4, 240–253 Search PubMed.
- M. Nett, H. Ikeda and B. S. Moore, Nat. Prod. Rep., 2009, 26, 1362–1384 RSC.
- G. T. Carter, A. A. Fantini, J. C. James, D. B. Borders and R. J. White, J. Antibiot., 1985, 38, 242–248 CrossRef CAS PubMed.
- S. K. Jain, A. S. Pathania, R. Parshad, C. Raina, A. Ali, A. P. Gupta, M. Kushwaha, S. Aravinda, S. Bhushan, S. B. Bharate and R. A. Vishwakarma, RSC Adv., 2013, 3, 21046–21053 RSC.
- F. Strelitz, H. Flon and I. N. Asheshov, J. Bacteriol., 1955, 69, 280–283 CAS.
- U. Weiss, K. Yoshihira, R. Highet, R. J. White and T. T. Wei, J. Antibiot., 1982, 35, 1194–1201 CrossRef CAS PubMed.
- S. G. Franzblau, R. S. Witzig, J. C. McLaughlin, P. Torres, G. Madico, A. Hernandez, M. T. Degnan, M. B. Cook, V. K. Quenzer and R. M. Ferguson, J. Clin. Microbiol., 1998, 36, 362–366 CAS.
- G. Jadaun, C. Agarwal, H. Sharma, Z. Ahmed, P. Upadhyay, J. Faujdar, A. K. Gupta, R. Das, P. Gupta and D. Chauhan, J. Antimicrob. Chemother., 2007, 60, 152–155 CrossRef CAS PubMed.
- M. S. Boguski, K. D. Mandl and V. P. Sukhatme, Science, 2009, 324, 1394–1395 CrossRef CAS PubMed.
- N. Saitou and M. Nei, Mol. Biol. Evol., 1987, 4, 406–425 CAS.
- A. Martin, M. Camacho, F. Portaels and J. C. Palomino, Antimicrob. Agents Chemother., 2003, 47, 3616–3619 CrossRef CAS PubMed.
- J. van Meerloo, G. J. Kaspers and J. Cloos, Cancer cell culture: methods and protocols, 2011, pp. 237–245 Search PubMed.
- B. C. Evans, C. E. Nelson, S. Y. Shann, K. R. Beavers, A. J. Kim, H. Li, H. M. Nelson, T. D. Giorgio and C. L. Duvall, J. Visualized Exp., 2013, e50166 Search PubMed.
- Y. Lee, H. Kim, C. Liu and H. Lee, J. Microbiol. Methods, 2003, 52, 245–250 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectroscopic data and other relevant information for chrysomycin A; extract activity, bioautography and phylogenetic tree supportive data are given here. See DOI: 10.1039/c7ra05576e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |