
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ structural modification of graphitic carbon nitride by alkali halides and influence on photocatalytic activity
Yan Xu,
Yinyan Gong *,
Hui Ren,
Wenbo Liu,
Lengyuan Niu,
Can Li and
Xinjuan Liu
*,
Hui Ren,
Wenbo Liu,
Lengyuan Niu,
Can Li and
Xinjuan Liu
Center for Coordination Bond Metrology and Engineering, College of Materials Science and Engineering, China Jiliang University, Hangzhou, Zhejiang 310018, China. E-mail: ygong2007@gmail.com
First published on 26th June 2017
Abstract
A series of graphitic carbon nitride photocatalysts were prepared by thermally polymerizing the mixtures of melamine and alkali halides (MCl, M = Na, K, Rb, Cs) in air. XRD, FTIR, Raman, SEM and TEM were used to investigate the effect of alkali halides on the structure and morphology of g-C3N4. It is found that such prepared samples exhibit reduced in-plane long-range periodic ordering while maintaining the framework of melon strands. However, no significant increase in surface area was observed by BET measurements. Moreover, PL and EIS measurements show that alkali halide modification results in improved separation and transfer of photogenerated electron and holes. Furthermore, the results of UV-vis absorption spectra and ultraviolet photoelectron spectra (UPS) indicate that the presence of alkali halides during the thermal polymerization process can decrease band gap energy and shift the conduction/valence bands of g-C3N4 upward. The variation of structural, electronic, and optical properties depends on the type of cationic ions. Potassium chloride modification gives rise to the best photocatalytic performance among all the samples, which is attributed to the reduced charge carrier recombination, narrower band gap, and higher CB edge. These findings may help in the design of visible-light-driven photocatalysts with enhanced photocatalytic activity.
Introduction
Semiconductor photocatalysts appear to be promising candidates for alleviating the energy shortage and environmental pollution because of their ability to photodecompose toxic pollutants and convert solar energy to light.1–5 Since Fujishima and Honda's pioneering work on water splitting using titanium dioxide (TiO2) electrodes, TiO2-based photocatalysts have drawn great attention due to the abundance and high stability of TiO2.2,6 However, pristine TiO2 has limited photocatalytic efficiency mainly due to its large intrinsic band gap (Eg = 3.2 eV) and fast recombination of photogenerated electron and hole pairs.2 An ideal photocatalyst is expected to have high absorption in the long wavelength region, sufficiently negative/positive conduction band (CB)/valence band (VB) energy levels, fast charge transfer and separation, as well as abundant surface adsorption and reaction sites. In addition to TiO2, various metal oxides, sulfides, nitrides, as well as their composites have been studied.7–9 Although much success has been achieved, the commercial applications of photocatalysts are still hindered by problems involving complicated synthesis process, high fabrication cost, photocorrosion, and poor visible-light response. Therefore, developing stable, inexpensive, and visible-light active photocatalysts is of great importance.Graphitic carbon nitride, a polymeric semiconductor material, appears as an intriguing visible-light-driven photocatalyst due to the moderate band gap (Eg = 2.7 eV), and suitable band edge positions.1 g-C3N4 is commonly prepared by thermal condensation of organic precursors containing both carbon and nitrogen and pristine g-C3N4 typically has a multilayered structure with weak interlayer van der Waals interaction and strong intralayer covalent bonding.10 However, graphitic carbon nitride usually encounters problems such as high recombination rate of photogenerated charge carriers and a low amount of surface active sites, and thus has limited photocatalytic efficiency. To address this issue, different strategies have been proposed including doping,11–13 manipulating microstructure and morphology,14–19 coupling with other semiconductors and noble metals8,20–24 and isotype g-C3N4/g-C3N4 junctions.17
Considering its unique structure, post-growth thermal treatment is frequently used to modify the structural and photocatalytic properties of g-C3N4.15,16,25 For instance, porous structures26 and nanosheets15,16,25 with different surface functional groups have been prepared by thermal exfoliating bulk g-C3N4 in different atmosphere (e.g. air, NH3, and oxygen gas). In addition to interrupting the periodic stacking of carbon nitride monolayer in the vertical direction, researchers also try to break the in-plane long-range arrangement of melon units. It has been proposed27–29 that incomplete deamination of amine-group-containing precursors can result in the formation of abundant hydrogen bonds in the covalent bonding dominated intralayer framework, i.e. the basal plane can be considered as strands of polymeric melon units connected by hydrogen bonds. Kang et al.27 demonstrated that post-growth annealing of bulk g-C3N4 in inert ambient can selectively break these hydrogen bonds and, consequently, enhance the photocatalytic activity due to faster transfer of charge carriers across plane, suppressed recombination of charge carriers and increased visible light absorption.
Alkali metal halides have been used previous to enhance the photocatalytic activity of g-C3N4.30–34 For example, Hu et al. prepared K-doped g-C3N4 by using dicyanadiamide and potassium hydrate as precursors, and such obtained photocatalysts have tunable band edge positions and enhanced mineralization ability.31 Zhang et al. studied K-doped g-C3N4 synthesized by thermal polymerization of dicyanadiamide and KI, and found that the photocatalytic efficiency for phenol and MB degradation was about 3.3 and 5.8 times that of bulk g-C3N4, respectively.32 Moreover, isotype triazine–heptazine based g-C3N4 heterojunctions were synthesized in the media of KCl/LiCl salts, and the photocatalytic hydrogen generation rate is 23 times that of pristine g-C3N4.34 As neither complicated post-growth treatment process nor hazardous chemicals are involve, this eco-friendly method of employing alkali metal halides to improve the photocatalytic performance of g-C3N4 appears intriguing. However, little attention has been given to investigate the influence of different alkali ions on the structural, electronic, optical, and photocatalytic properties of g-C3N4, which will be helpful to select appropriate alkali halides and develop photocatalysts with enhanced photocatalytic performance.
Herein, we synthesized a series of samples using melamine and alkali halides (MCl, M = Na, K, Rb, and Cs) as precursors. XRD, FTIR, Raman, SEM and TEM measurements were carried out to investigate the effect of various cationic ions on the structural and morphological properties of g-C3N4. Moreover, the chemical state of alkali ions and the salt-induced variations of electronic and optical properties of g-C3N4 photocatalysts were studied in detail by X-ray photoemission (XPS), UV-visible diffuse reflectance (DRS), ultraviolet photoelectron (UPS), photoluminescence (PL), and electrochemical impedance (EIS) spectra. Finally, the photocatalytic performance was evaluated by degradation of RhB under visible light irradiation, and the effects of different cationic ions were discussed.
Experimental
Materials
Melamine (CP, Sinopharm Chemical Reagent Co. Ltd), NaCl (AR, Sinopharm Chemical Reagent Co. Ltd), KCl (AR, Aladdin Industrial Corporation), RbCl (AR, Aladdin Industrial Corporation), CsCl (AR, Aladdin Industrial Corporation), and Rhodamine B (RhB, AR, Aladdin Industrial Corporation). All the chemicals were used as received without further purification, and deionized water was used during all the experiments.Synthesis
A series of g-C3N4 samples were prepared using melamine and alkali halides as precursors.35 Typically, 1.5 g of melamine and 3.75 g of alkali halides (MCl, M = Na, K, Rb, Cs) were fully mixed by grinding in an agate mortar for 15 min. The mixture was then transferred into a ceramic crucible covered by a lid and calcinated in a muffle furnace at 550 °C for 4 h at a ramping rate of 2 °C min−1. After being naturally cooled down to room temperature, such obtained products were ground into powder, dispersed in 100 mL DI water and stirred for 4 h. And then, the suspension was centrifuged, washed by water for 3 times, and dried in an oven at 80 °C for 12 h. The collected photocatalysts were labeled as GCN-Na, GCN-K, GCN-Rb and GCN-Cs corresponding to NaCl, KCl, RbCl and CsCl, respectively. For comparison, pristine g-C3N4 was synthesized by heating 1.5 g melamine at 550 °C for 4 h (GCN-0).Characterization
The structure and morphology of all the samples were characterized by XRD (Rigaku D/Max-2400 diffractometer, Japan), FTIR (Thermal Fisher Nicolet i50, US), Raman spectra (inVia, Renishaw, UK) under 785 nm excitation, SEM (SU8010 FE-SEM, Hitachi, Japan) and TEM (JEM-2100 F, JEOL, Japan). The Brunauer–Emmett–Teller (BET) surface area was determined by nitrogen adsorption–desorption isotherms measured on a Micromeritics ASAP2460 system at 77 K. PL spectra were also recorded on inVia spectrometer using the luminescence module under the excitation of 325 nm UV laser. XPS measurements were performed on ESCALAB 250 Xi (Thermal Scientific, UK) using Al-Kα radiation and binding energies were referenced to C 1s line at 284.8 eV from adventitious carbon. The DRS spectra were measured on a UV-Vis-NIR spectrometer equipped with integrated sphere (Lamada 750S, Perkin Elmer, US). The UPS measurements were carried out on a home-built system with HeII (40.8 eV) emission line employed for excitation. A sample bias of −5 V was applied to observe the secondary electron cutoff (SEC). EIS measurements were carried out on an electrochemical working station (CHI660E, Shanghai, China) in 1 × 10−5 M RhB aqueous solution using a three-electrode configuration under daylight. A Pt foil and a standard calomel electrode were served as the counter electrode and the reference electrode, respectively. The working electrodes were prepared by mixing 25 mg photocatalysts in 0.2 mL 2.5% polyvinyl alcohol binder, and the obtained slurry was then coated on a 1 cm × 2.5 cm ITO slice with an effective working area of 1 cm2 by a screen printing method. After that, the electrodes were dried in a vacuum oven at 80 °C for 12 h. For the EIS measurements, the applied bias voltage and ac amplitude were set at open-circuit voltage and 10 mV with the frequency varying in the range of 0.1 Hz–1 MHz.Photocatalytic degradation of RhB
50 mg photocatalysts were dispersed in 50 mL of RhB (2 × 10−5 M) in a quartz tube and magnetically stirred for 60 min in dark to reach an adsorption–desorption equilibrium. A halogen lamp, power regulated at 500 W and equipped with a 400 nm cut-off filter, was used as light source. The lamp was placed vertically in a double-wall jacketed quartz tube with cooling water circulated to maintain a nearly constant temperature during photocatalytic reaction. 4 mL of suspension was withdrawn every 10 min, and the absorbance of RhB solution was measured by Lamada 750S spectrometer at 554 nm after centrifugation. A recycled photocatalytic activity test was carried out according to the above-mentioned procedure. After each run of photocatalytic reaction, the photocatalyst was collected, washed thoroughly by deionized water and dried. To investigate the photocatalytic mechanism, trapping experiments were performed to investigate the main active species during the photocatalytic process. Certain amounts of benzoquinone (BQ), isopropanol (IPA), and ammonium oxalate (AO) were introduced in the above suspension of photocatalysts as the scavengers to detect superoxide radicals (˙O2−), hydroxyl radicals (˙OH), and holes (h+), respectively. The photocatalytic degradation of RhB was treated the same as the photodegradation process.Results and discussion
As presented in Fig. 1a, the XRD pattern of pristine g-C3N4 (GCN-0) exhibited two characteristic diffraction peaks at 2θ ≈ 13.2° and 2θ ≈ 27.6°. The former can be assigned to the (100) crystal plane, representing the in-plane long-range periodic packing and the latter is associated with the (002) crystal plane, representing the stacking of carbon nitride monolayers along the c-axis.10,14,27,36 The absence of 13.2° peak for GCN-Na, GCN-K, GCN-Rb, and GCN-Cs suggests that g-C3N4 synthesized in the media of alkali halides has reduced in-plane ordering. Similar results have been obtained for g-C3N4 prepared by incorporating salts and bases such as KI,32 KCl33 and KOH36 or subjected to post-growth thermal annealing in inert ambient.27 It has been proposed that the intralayer framework of carbon nitride is composed of covalent-bond-dominated polymeric melon strands interconnected by hydrogen bonds.10,27 As the strength of hydrogen bonds is relatively weaker than that of covalent bonds between carbon and nitrogen atoms, it is feasible to selectively break the hydrogen bonds and disturbs in-plane stacking27,28 For our samples, it is plausible that the presence of alkali halides during thermal polymerization of melamine suppresses the formation of hydrogen bonding between melon units, and thus reduces the intra-layer periodic ordering. Regarding to the 27.6° peak, the usage of alkali halides shifts the diffraction angle toward the higher angle side and the extent of variation depends on the type of cationic ions. 2θ of GCN-Na is only slightly increased compared to GCN-0 while a more pronounced shifting of 2θ from 27.8° to 28.4° and 28.2° are observed for GCN-K and GCN-Rb, respectively, suggesting that alkali halides result in decreased inter-layer separation. This is different from the results of alkali ion doping,32,37 and chloride doping,38 in which interstitial Na (or K) and intercalation Cl cause enlarged inter-layer distance. This implies that in our samples neither alkali ions nor chloride ions are doped into the carbon nitride lattice. In addition, the overall diffraction intensity of GCN-Na, GCN-K, GCN-Rb and GCN-Cs are lower than that of pristine g-C3N4, especially for GCN-Cs which has the lowest diffraction intensity. The weakening of overall diffraction intensity implies that the addition of alkali halides deteriorates the crystallinity of g-C3N4.33,36,37 Therefore, the incorporation of MCl (M = Na, K, Rb, Cs) during the thermal polymerization of melamine can reduce the in-plane long-range ordering, decrease inter-layer spacing and lower the crystallinity of g-C3N4, and the extent of structural modulation is related to the type of cationic ions.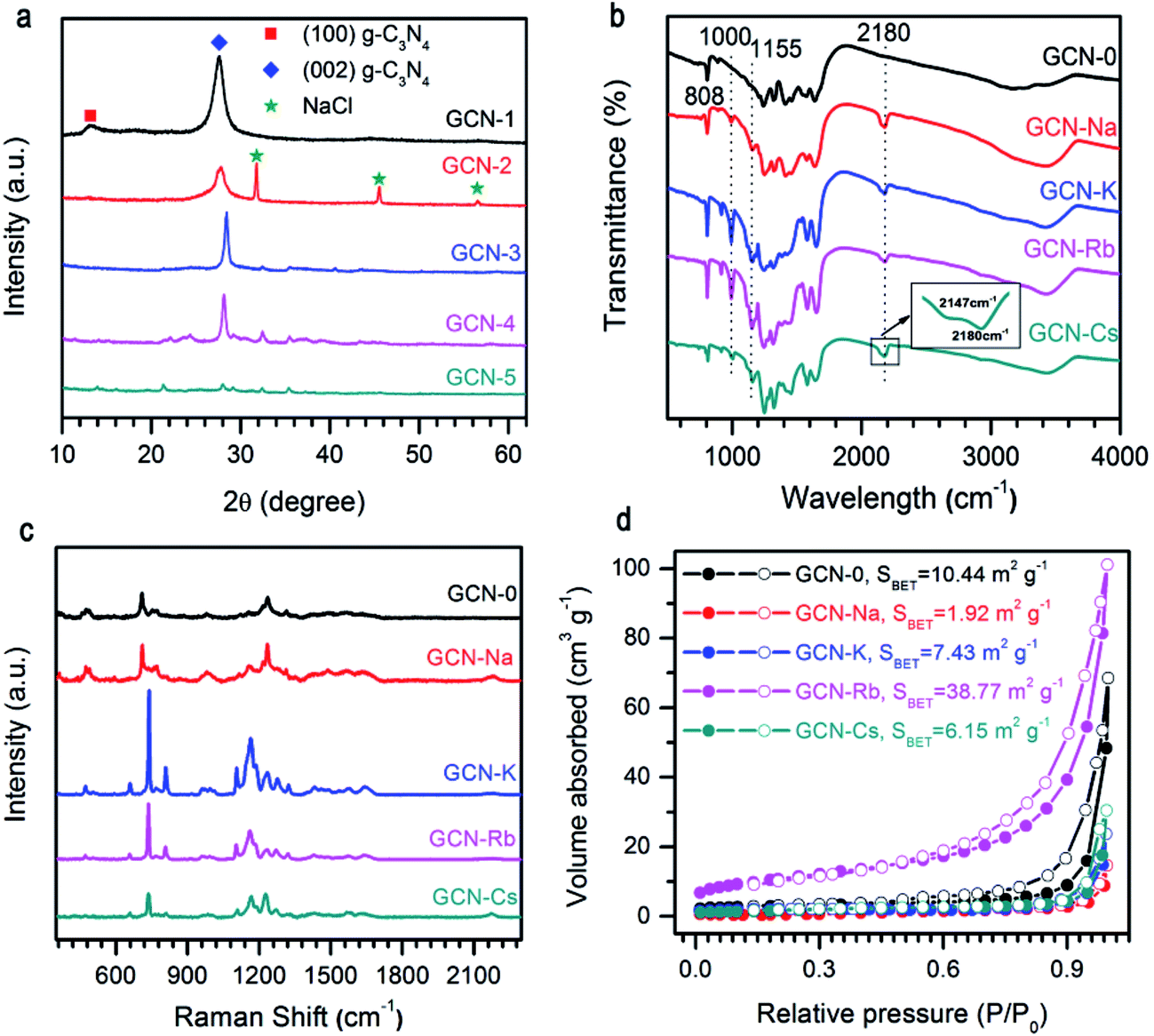 | ||
| Fig. 1 (a) XRD patterns, (b) FTIR spectra, (c) Raman spectra, and (d) N2 adsorption–desorption isotherms of g-C3N4 via MCl (M = Na, K, Rb, Cs) modification. Results of pristine g-C3N4 are plotted together for comparison. The extra diffraction peaks for GCN-Na in (a) is from NaCl residual. | ||
The influence of alkali halides on the structural properties was further investigated by FTIR and Raman spectra due to their high sensitivity to local structure. As shown in Fig. 1b, FTIR spectra of all the samples show comparable characteristic bands at 808 cm−1 and 1200–1900 cm−1 region, which are ascribed to breathing mode of triazine units and stretching vibration modes of aromatic CN heterocycles, respectively,39,40 this confirms that alkali halides only interrupt the intralayer long-range periodic arrangement without modifying the basic atomic building blocks. Compared to pristine g-C3N4, extra peaks at 1000 cm−1, 1155 cm−1, 2147 cm−1 and 2180 cm−1 are observed for GCN-Na, GCN-K, GCN-Rb and GCN-Cs, which can be attributed to hydroxyl groups grafted on the surface of g-C3N4 (ref. 41) and cyano groups (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N).33,36 It is plausible that the presence of alkali ions (Na+, K+, Rb+, and Cs+) could prevent the formation of hydrogen bonds, and then the –OH groups, dissociated from the H2O at high temperatures, will bond to the amino groups presented in g-C3N4. However, a thorough understanding of the underlying mechanism requires a systematic study.10,33,41 The broad bands between 3000 cm−1 and 3600 cm−1 are originated from the terminal amino groups.17 Vibrations of alkali-related groups are not observed, suggesting that the framework of carbon nitride is not changed after salt treatment. Fig. 1c compares NIR Raman spectra of all the samples. It is found that all the samples have similar Raman spectra, suggesting good retention of microstructure. Two Raman bands with complicated structures at 670–800 cm−1 and 1090–1390 cm−1 might be related to breathing modes of s-triazine.42 However, a detailed interpretation of the Raman spectra requires the development of Raman scattering theory of g-C3N4.
N).33,36 It is plausible that the presence of alkali ions (Na+, K+, Rb+, and Cs+) could prevent the formation of hydrogen bonds, and then the –OH groups, dissociated from the H2O at high temperatures, will bond to the amino groups presented in g-C3N4. However, a thorough understanding of the underlying mechanism requires a systematic study.10,33,41 The broad bands between 3000 cm−1 and 3600 cm−1 are originated from the terminal amino groups.17 Vibrations of alkali-related groups are not observed, suggesting that the framework of carbon nitride is not changed after salt treatment. Fig. 1c compares NIR Raman spectra of all the samples. It is found that all the samples have similar Raman spectra, suggesting good retention of microstructure. Two Raman bands with complicated structures at 670–800 cm−1 and 1090–1390 cm−1 might be related to breathing modes of s-triazine.42 However, a detailed interpretation of the Raman spectra requires the development of Raman scattering theory of g-C3N4.
Fig. 1d illustrates the N2 adsorption–desorption isotherms of g-C3N4 synthesized from the mixture of melamine and different alkali halides. All the isotherms are type IV, and the specific surface areas calculated by the BET method (SBET) are 10.44, 1.92, 7.43, 38.77, and 6.15 m2 g−1 for GCN-0, GCN-Na, GCN-K, GCN-Rb and GCN-Cs, respectively. Except GCN-Rb, all the samples have smaller SBET than pristine g-C3N4. Results of XRD, FTIR, Raman and BET measurements indicate that the presence of alkali halides serve as template during thermal condensation of melamine and disturb the long-range periodic arrangement of melon strands through hydrogen bonding while maintains the basic atomic units of C and N atoms in the basal plane. Cationic ions with larger radius cause more affect the structural variation.
The morphologies of the representative samples were investigated scanning electron microscope (SEM) and transmission electron microscope (TEM) analysis, as shown in Fig. 2. As shown in Fig. 2, the pristine g-C3N4 sample is composed of thick sheets while GCN-K has an irregular porous structure. This phenomenon might be ascribed to the reduced in-plane long-range periodic ordering due to the presence of KCl during thermal condensation of melamine. TEM images of both samples are shown in Fig. 3, and both samples display the typical lamellar stacking structure.
 | ||
| Fig. 2 SEM images of GCN-0 (a) and (c), and GCN-K (b) and (d). | ||
 | ||
| Fig. 3 TEM images of GCN-0 (a) and GCN-K (b). | ||
The specific bonding and surface chemical states of representative samples were investigated by XPS spectra. Fig. 4a illustrates the survey scans of GCN-0 and GCN-K, and the observed C, N and O signals are originate from g-C3N4 and adsorbed O-containing species, respectively. In addition, K and Cl are detected in GCN-K, which indicates the existence of KCl residue. The high-resolution XPS spectra of N 1s are shown in Fig. 4b, which can be deconvoluted into three peaks centered at 399.0, 400.4 and 401.6 eV, corresponding to the C–N–C, N–(C)3, and C–N–H groups of melon, respectively.34,43,44 The 401.6 eV peak shifts to 401.3 eV for GCN-K, and the decreasing of binding energy might be related to charge redistribution in the framework of g-C3N4 due to microstructure variation by adding KCl. Fig. 4c plots the high-resolution XPS spectra of C 1s, of which there are two peaks at 284.9 eV and 288.5 eV, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in adventitious carbon and C–N–C coordination of melon, respectively.27,33,44 An extra peak at 287.2 eV is observed for GCN-K, which is tentatively attributed to C–OH. Consistent with FTIR result, the presence of alkali halides during thermal condensation facilitate grafting of hydroxyl groups on the surface of g-C3N4.33,34 As shown in Fig. 4d, the O 1s XPS spectrum of GCN-0 has a single peak at ∼532.5 eV, which is due to the adsorbed water molecules on the surface of g-C3N4.33,34,44–46 The spectrum of GCN-K can be decomposed into two peaks at 532.5 eV and 531.2 eV, corresponding to adsorbed water and N–C–O, respectively.34,46 There are two peaks in the K 2p region of GCN-K located at 293.3 and 296.1 eV, which is consistent with binding energy of KCl.47 The K 2p peaks are higher than the reported values when potassium ions are doped into the interstitial site and form covalent bond with C3N4.32 This is consistent with the XRD result.
C in adventitious carbon and C–N–C coordination of melon, respectively.27,33,44 An extra peak at 287.2 eV is observed for GCN-K, which is tentatively attributed to C–OH. Consistent with FTIR result, the presence of alkali halides during thermal condensation facilitate grafting of hydroxyl groups on the surface of g-C3N4.33,34 As shown in Fig. 4d, the O 1s XPS spectrum of GCN-0 has a single peak at ∼532.5 eV, which is due to the adsorbed water molecules on the surface of g-C3N4.33,34,44–46 The spectrum of GCN-K can be decomposed into two peaks at 532.5 eV and 531.2 eV, corresponding to adsorbed water and N–C–O, respectively.34,46 There are two peaks in the K 2p region of GCN-K located at 293.3 and 296.1 eV, which is consistent with binding energy of KCl.47 The K 2p peaks are higher than the reported values when potassium ions are doped into the interstitial site and form covalent bond with C3N4.32 This is consistent with the XRD result.
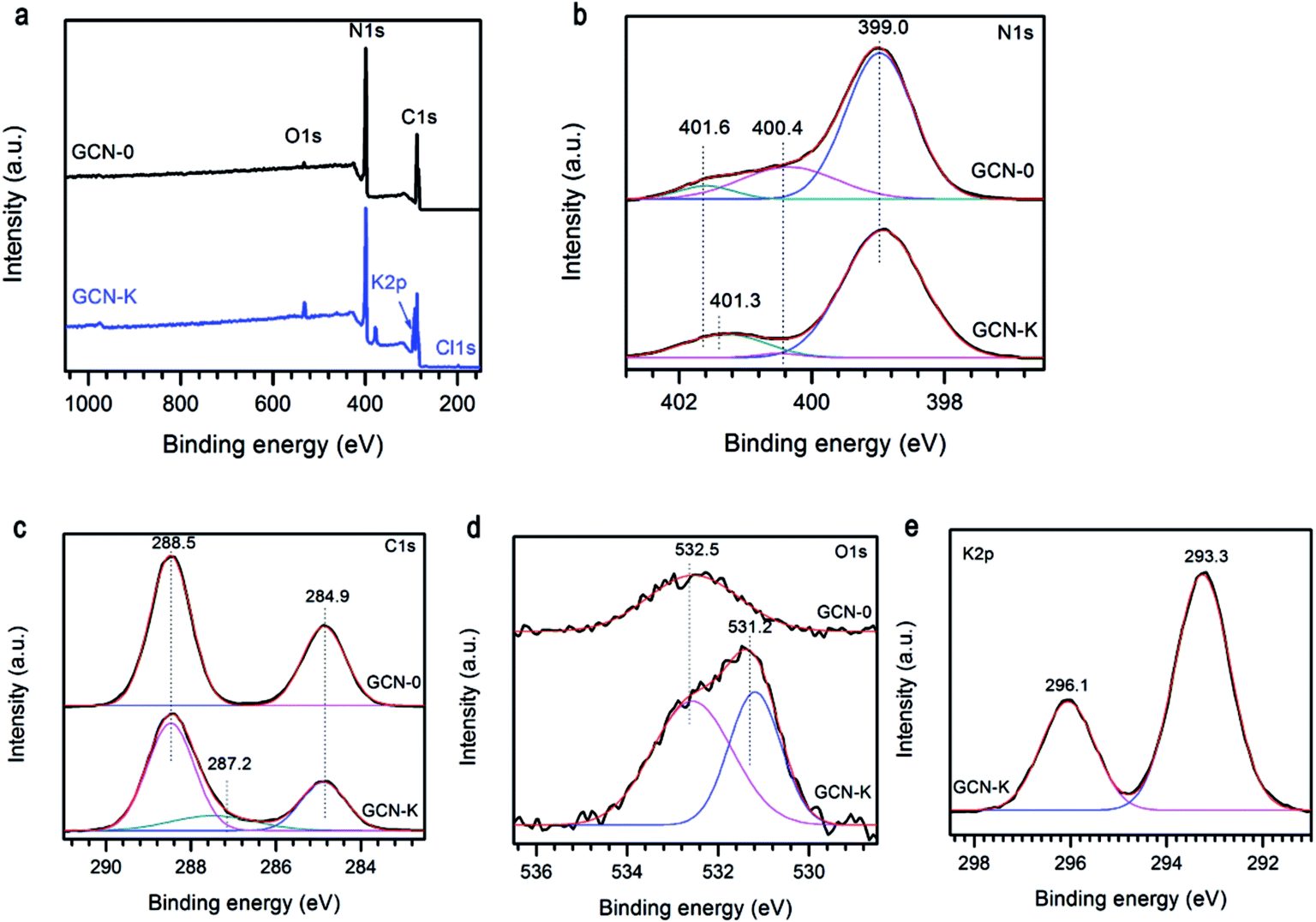 | ||
| Fig. 4 (a) XPS survey spectra, (b) N 1s, (c) C 1s, (d) O 1s, and (e) K 2p high resolution spectra of g-C3N4 prepared from melamine (GCN-0) and mixture of KCl and melamine (GCN-K). | ||
Fig. 5a plots the UV-visible absorption spectra of all the graphitic carbon nitride samples converted from the measured DRS. Compared to GCN-0, the absorption edges of GCN-Na, GCN-K, GCN-Rb and GCN-Cs were red shifted and the variation follows the sequence Rb+ > K+ > Na+ < except for Cs+, i.e. larger cationic ions have more pronounced effect. Steeper absorption edges of GCN-K, GCN-Rb and GCN-Cs than GCN-0 and GCN-Na might be due to the higher degree of structure modification. Band gap energies of semiconductors can be estimated using the Kubelka–Munk function.3,48
| (αhυ)n = A(hυ − Eg) |
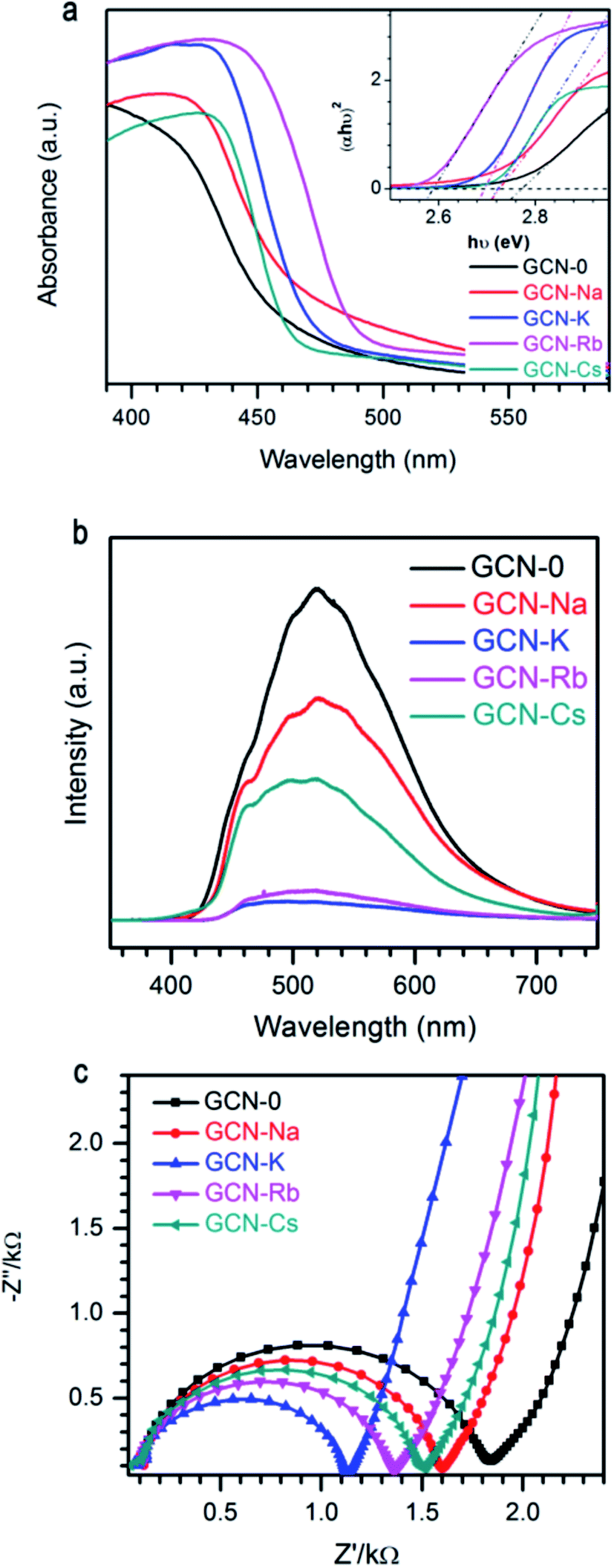 | ||
| Fig. 5 (a) UV-visible spectra, (b) photoluminescence spectra, and (c) EIS Nyquist plot of g-C3N4 via MCl (M = Na, K, Rb, Cs) modification together with pristine g-C3N4. | ||
It is known that electrons will be excited from VB of g-C3N4 to CB under light irradiation, and then the photo-generate charge carriers can migrate to the surface to participate in photocatalytic reactions. Alternatively, the photo-induced electron–hole pairs can recombine and emit light. Thus, photoluminescence (PL) spectroscopy is frequently used to probe the charge separation and recombination in photocatalysts. As shown in Fig. 5b, GCN-0 exhibits a strong and broad PL band centered at ∼515 nm. It is found that PL intensity drops drastically in the sequence of GCN-K > GCN-Rb > GCN-Cs > GCN > Na, where potassium chloride has the most pronounced effect on PL quenching. The much lower PL intensity of GCN-Na, GCN-K, GCN-Rb and GCN-Cs than GCN-0 indicates that the radiative recombination pathways associated with the long-range order of aromatic CN heterocycles were significantly inhibited by alkali halides, especially KCl. The effect of alkali halides on the charge carrier transfer and recombination behavior was further analyzed by electrochemical impedance spectra (EIS). Fig. 5c shows the Nyquist plots of GCN-0, GCN-Na, GCN-K, GCN-Rb and GCN-Cs. In general, the semicircle in the EIS spectra is ascribed to the contribution from charge transfer resistance (Rct) and constant phase element (CPE) at the photocatalyst/electrolyte interface, and the inclined line, resulting from the Warburg impedance, represents the ion-diffusion process in the electrolyte.1,15,17,49–51 From Fig. 5c, it can be seen that the radius of semicircle decreases in the order of GCN-K > GCN-Rb > GCN-Cs > GCN-Na > GCN-0. This indicates that the recombination of electron and hole pairs in GCN-K is more effectively inhibited than that of GCN-0. Therefore, alkali halides result in red shifting of absorption edges and suppressed recombination of photogenerated electrons and holes in graphitic carbon nitride.
Another important factor affecting the performance of a photocatalyst is its CB and VB positions. Fig. 6a and b show the two ends of measured UPS spectra of GCN-0, GCN-Na, GCN-K, GCN-Rb and GCN-Cs. Generally, the ionization energy (equivalent to VB edge (Ev)) of a semiconductor material can be estimated by subtracting the width of the HeII UPS spectra from the excitation energy (40.8 eV) and the CB edge (Ec) can be estimated by Ev − Eg. Fig. 6c plots the extracted Eg and CB/VB positions from DRS and UPS spectra. It is found that alkali halides can shift the CB and VB in upward directions, which is believed to be favorable for producing electrons with higher reducing-ability and strong-oxidation superoxide radicals under light irradiation.
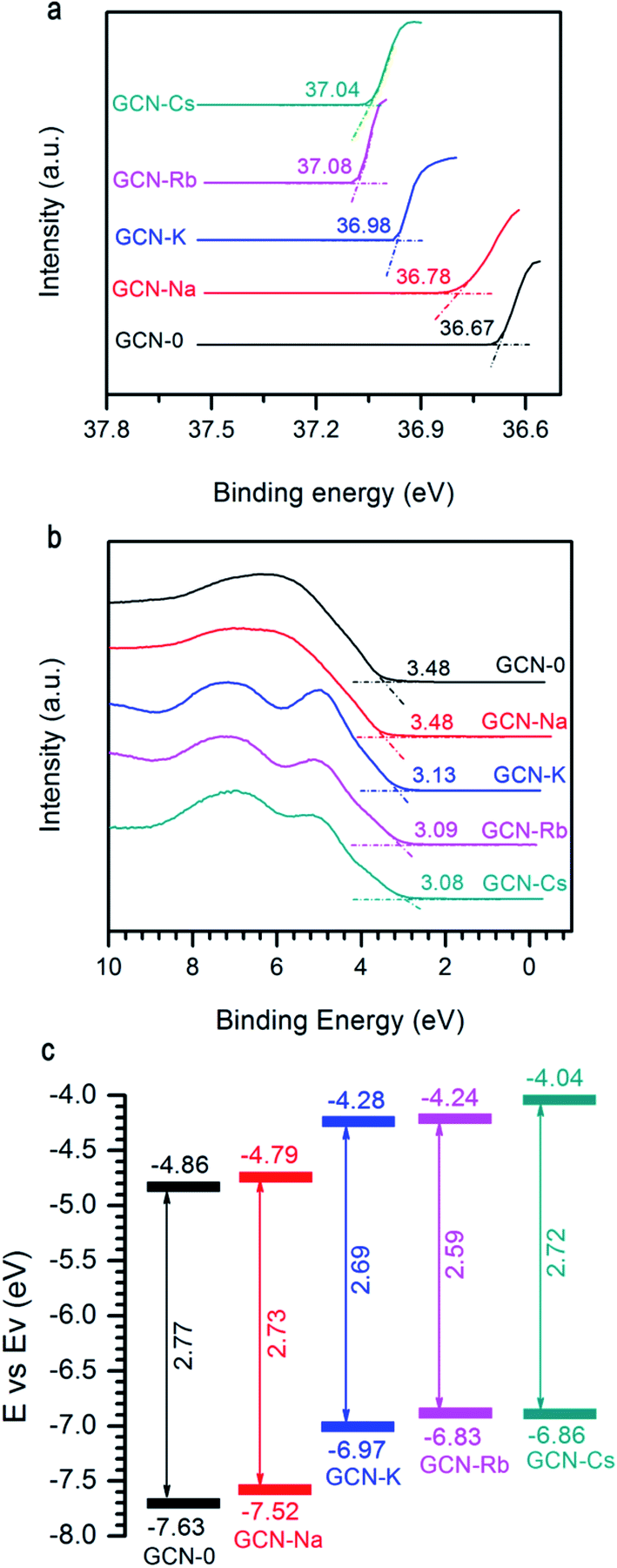 | ||
| Fig. 6 (a) and (b) UPS of GCN-0, GCN-Na, GCN-K, GCN-Rb, and GCN-Cs. (c) Schematic of band gap and band offsets with respect to vacuum level. | ||
The photocatalytic performance of all the samples was evaluated by degradation of RhB solutions under visible light irradiation, see Fig. 7a. When pristine g-C3N4 is used, ∼45% RhB could be decomposed subjected to 40 min light irradiation. Under the same conditions, the degradation efficiency of GCN-K, GCN-Rb and GCN-Cs is 83%, 79% and 52%, respectively. However, GCN-Na has lower photocatalytic degradation efficiency ∼16%. As shown in Fig. 7b and c, the extracted reaction rate constants k of RhB photodegradation are 15 × 10−3, 5 × 10−3, 44 × 10−3, 39 × 10−3, and 17 × 10−3 min−1 for GCN-0, GCN-Na, GCN-K, GCN-Rb, GCN-Cs, respectively. Therefore, alkali halides can improve the photocatalytic performance of graphitic carbon nitride except NaCl. Among all the samples, KCl has the most pronounced effect. To understand the mechanism of this photocatalytic process, trapping experiment has been carried out on GCN-K. As shown in Fig. 7d, the degradation efficiency of RhB is significantly suppressed when benzoquinone (BQ, 2 mM, as a superoxide radicals ˙O2− scavenger) was added. This means that the ˙O2− acts as the main active specie in the photocatalytic degradation. It is plausible that alkali halides modify the microstructure of the obtained graphitic carbon nitride, which in turn affects the charge carrier transfer behavior and reduces their radiative recombination. Moreover, the structure modification also results in enhanced visible-light absorption capability and improved reduction ability of electrons. Therefore, alkali modified samples can have higher photodegradation efficiency than pristine g-C3N4. Since the structure modification depends on the type of cationic ions, the changes of photocatalytic activity is also related to cationic ions. We note that NaCl has the least effect on the structural, electronic, and optical modification of g-C3N4 but much smaller SBET (10.44 vs. 1.92 m2 g−1), which might be responsible for the observed decreasing of photocatalytic activity.
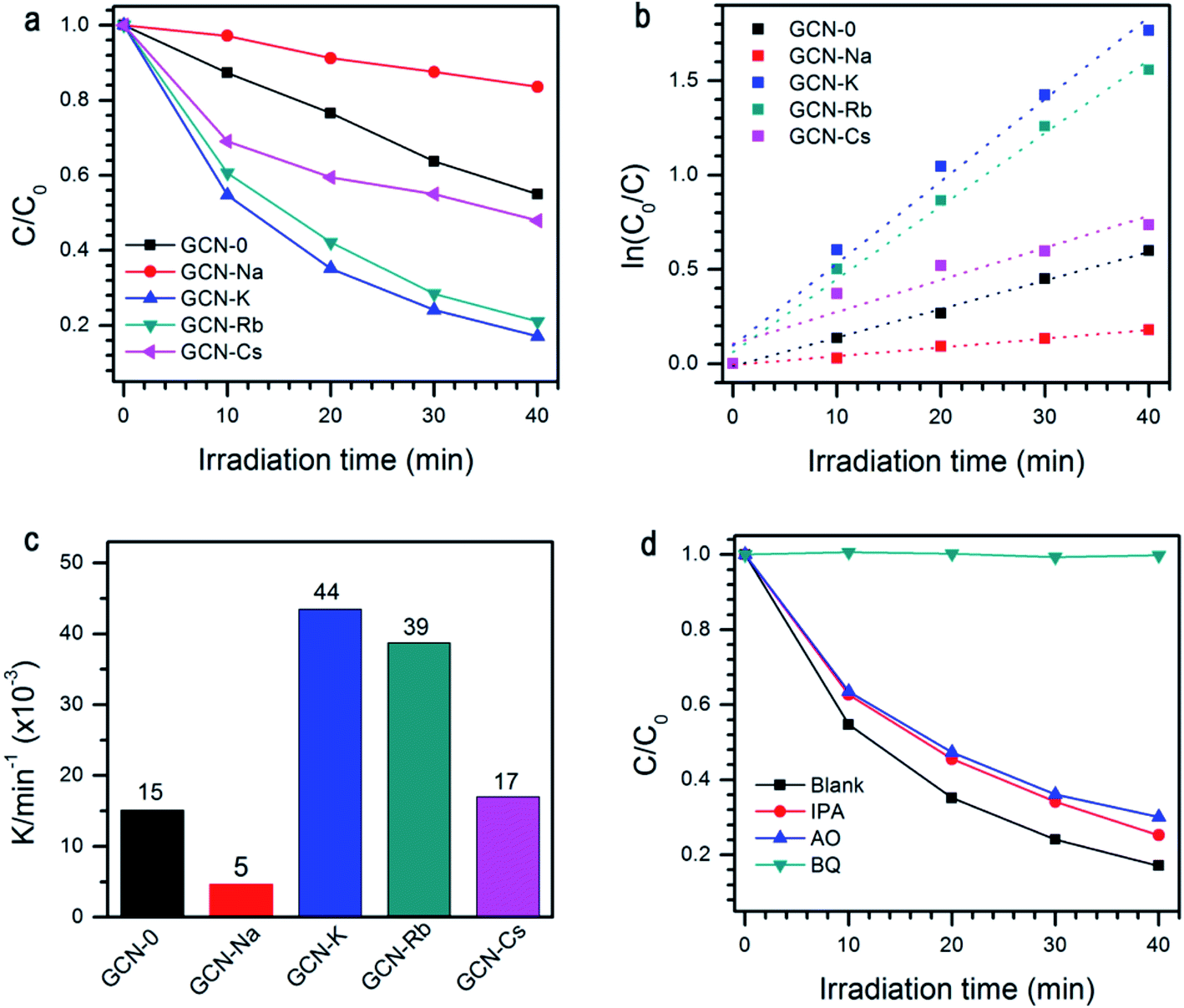 | ||
| Fig. 7 (a) Photocatalytic degradation of RhB by GCN-0, GCN-Na, GCN-K, GCN-Rb and GCN-Cs under visible-light irradiation; (b) first order kinetic fitting data and (c) rate constant k for the photodegradation of RhB by GCN-0, GCN-Na, GCN-K, GCN-Rb and GCN-Cs; (d) GCN-K with adding scavengers of BQ, IPA, and AO. | ||
Stability is very important for the practical applications of photocatalysts. For this purpose, we tested the cycle stability of GCN-K and plotted the results in Fig. 8. Apparently, there is no clear deterioration of the photocatalytic capability after 5 recycles, indicating that GCN-K has good stability.
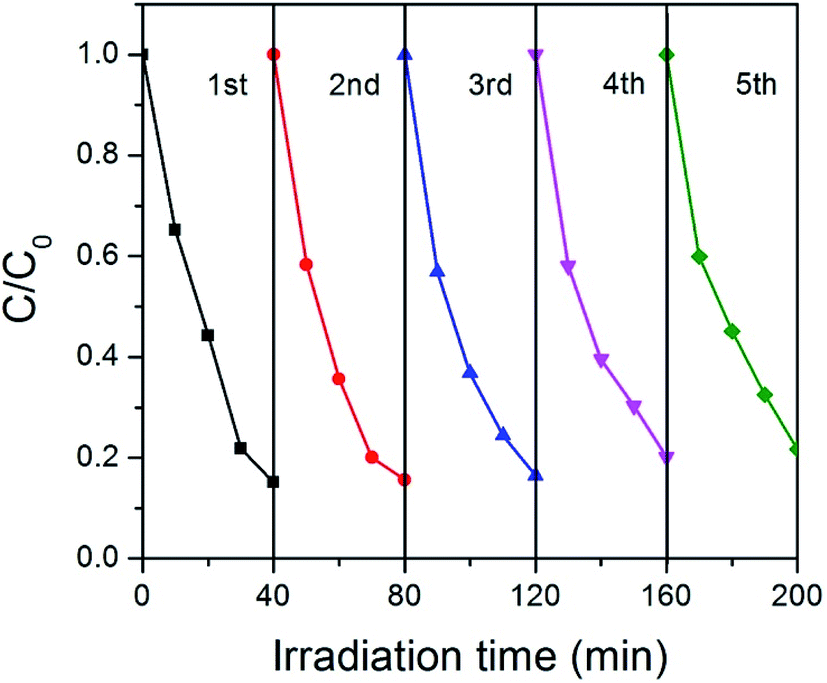 | ||
| Fig. 8 Photocatalytic degradation of RhB on GCN-K for five cycles. | ||
Conclusions
In summary, we investigated the effect of alkali halides on the structural, electronic, optical, and photocatalytic properties of graphitic carbon nitride and compare the effects of different cationic ions. It is found that g-C3N4 photocatalysts synthesized from the mixture of melamine and alkali halides have reduced in-plane long-range periodic ordering, and larger cationic ions have more pronounced effect. This alkali halide salt-induced structural modification facilitates separation and transfer of photogenerated electrons and holes, reduces band gap energies and shifts CB and VB edges in upward direction. The cation-dependent changes of structural, electronic and optical properties result in different photocatalytic performance of GCN-0, GCN-Na, GCN-K, GCN-Rb, and GCN-Cs.Acknowledgements
This work was supported by the National Natural Science Foundation of China [Grant No. 51402277, 51402274, and 21401180].References
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- M. Kapilashrami, Y. Zhang, Y. S. Liu, A. Hagfeldt and J. Guo, Chem. Rev., 2014, 114, 9662–9707 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- D. J. Martin, K. P. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. X. Guo and J. W. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Iwase, S. Yoshino, T. Takayama, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2016, 138, 10260–10264 CrossRef CAS PubMed.
- D. Ma, J. Wu, M. Gao, Y. Xin and C. Chai, Chem. Eng. J., 2017, 316, 461–470 CrossRef CAS.
- Y. Yuan, H. Lu, Z. Ji, J. Zhong, M. Ding, D. Chen, Y. Li, W. Tu, D. Cao, Z. Yu and Z. Zou, Chem. Eng. J., 2015, 275, 8–16 CrossRef CAS.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- Z.-F. Huang, J. J. Song, L. Pan, Z. M. Wang, X. Q. Zhang, J.-J. Zou, W. B. Mi, X. W. Zhang and L. Wang, Nano Energy, 2015, 12, 646–656 CrossRef CAS.
- Z. Z. Lin and X. C. Wang, Angew. Chem., Int. Ed., 2013, 52, 1735–1738 CrossRef CAS PubMed.
- J. Gao, J. Wang, X. Qian, Y. Dong, H. Xu, R. Song, C. Yan, H. Zhu, Q. Zhong, G. Qian and J. Yao, J. Solid State Chem., 2013, 228, 60–64 CrossRef.
- P. Niu, L. L. Zhang, G. Liu and H.-M. Chen, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- P. Xia, B. Zhu, J. Yu, S. Cao and M. Jaroniec, J. Mater. Chem. A, 2017, 5, 3230–3238 CAS.
- Y. Yang, J. Chen, Z. Mao, N. An, D. Wang and B. D. Fahlman, RSC Adv., 2017, 7, 2333–2341 RSC.
- L.-H. Lin, H. H. Ou, Y. F. Zhang and X. C. Wang, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS.
- X. Liu, K. Xu, P. Chen, K. Jia, S. Liu and C. Wu, J. Mater. Chem. A, 2014, 2, 18924–18928 Search PubMed.
- J. Yan, C. Zhou, P. Li, B. Chen, S. Zhang, X. Dong, F. Xi and J. Liu, Colloids Surf., A, 2016, 508, 257–264 CrossRef CAS.
- K. Li, S. Gao, Q. Y. Wang, H. Xu, Z. Y. Wang, B. B. Huang, Y. Dai and J. Lu, ACS Appl. Mater. Interfaces, 2015, 7, 9023–9030 CAS.
- Z. P. Yang, Z. J. Sun, X. Liu, H. X. Jia and P. W. Du, Nanoscale, 2016, 8, 4748–4756 RSC.
- Q. Zhang, H. Wang, S. Chen, Y. Su and X. Quan, RSC Adv., 2017, 7, 13223–13227 RSC.
- Z. Zhang, M. Wang, W. Cui and H. Sui, RSC Adv., 2017, 7, 8167–8177 RSC.
- J. Wang, H. Xu, X. Qian, Y. Dong, J. Gao, G. Qian and J. Yao, Chem.–Asian J., 2015, 10, 1276–1280 CrossRef CAS PubMed.
- L. Yang, J. Huang, L. SHi, L. Cao, Q. Yu, Y. Jie, J. Fei, H. Ouyang and J. Ye, Appl. Catal., B, 2017, 204, 335–345 CrossRef CAS.
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- Y. Kang, Y. Yang, L.-C. Yin, X. Kang, L. Wang, G. Liu and H.-I. Cheng, Adv. Mater., 2016, 28, 6471–6477 CrossRef CAS PubMed.
- Y. Kang, Y. Yang, L.-C. Yin, X. Kang, G. Liu and H.-M. Cheng, Adv. Mater., 2015, 27, 4572–4577 CrossRef CAS PubMed.
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS.
- W. Fang, J. Liu, L. Yu, Z. Jiang and W. Shangguan, Appl. Catal., B, 2017, 209, 631–636 CrossRef CAS.
- S. Hu, F. Li, Z. Fan, F. Wang, Y. Zhao and Z. Lv, Dalton Trans., 2015, 44, 1084–1092 RSC.
- M. Zhang, X. Bai, D. Liu, J. Wang and Y. Zhu, Appl. Catal., B, 2015, 164, 77–81 CrossRef CAS.
- Y. Li, H. Xu, S. Ouyang, D. Lu, X. Wang, D. Wang and J. Ye, J. Mater. Chem. A, 2016, 4, 2943–2950 CAS.
- H. H. Liu, D. C. Chen, Z. Q. Wang, H. J. Jing and R. Zhang, Appl. Catal., B, 2017, 203, 300–313 CrossRef CAS.
- Y. X. Li, S. Ouyang, H. Xu, X. Wang, Y. P. Bi, Y. F. Zhang and J. H. Ye, J. Am. Chem. Soc., 2016, 138, 13289–13297 CrossRef CAS PubMed.
- H. Yu, S. Run, Y. Zhao, T. Bian, Y. Zhao, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2017, 29, 1605148 CrossRef PubMed.
- J. Zhang, S. Hu and Y. Wang, RSC Adv., 2014, 4, 62912–62919 RSC.
- C. Y. Liu, Y. H. Zhang, F. Dong, A. H. Reshak, L. Q. Ye, N. Pinna, C. Zeng, T. R. Zhang and H. W. Huang, Appl. Catal., B, 2017, 203, 465–474 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CAS.
- Y. Shang, X. Chen, W. Liu, P. Tan, H. Chen, L. Wu and C. Ma, Appl. Catal., B, 2017, 204, 78–88 CrossRef CAS.
- X. L. Wang, W. Q. Fang, H. F. Wang, H. Zhang, H. Zhao, Y. Yao and H. G. Yang, J. Mater. Chem. A, 2013, 1, 14089–14096 CAS.
- P. V. Zinin, L.-C. Ming, S. K. Sharma, V. N. Khabashesku, X. Liu, S. Hong, C. Endo and T. Acosta, Chem. Phys. Lett., 2009, 472, 69–73 CrossRef CAS.
- H. Lan, L. Li, X. An, F. Liu, C. Chen, H. Liu and J. Qu, Appl. Catal., B, 2017, 204, 49–57 CrossRef CAS.
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, Chem. Commun., 2012, 48, 12017–12019 RSC.
- H. Ma, Y. Li, S. Li and N. Liu, Appl. Surf. Sci., 2015, 357, 131–138 CrossRef CAS.
- Y. Bu and Z. Chen, Electrochim. Acta, 2014, 144, 42–49 CrossRef CAS.
- H. H. Park, B. H. Kim, S. H. Song, J. Kwon, B. S. Kong, K. Kang and S. Jeon, Nano Lett., 2012, 12, 2871–2876 CrossRef PubMed.
- J. Navas, A. Sanchez-Coronilla, T. Aguilar, N. C. Hernandez, D. M. de los Santos, J. Z. Sanchez-Marquez, D. C. Fernandez-Lorenzo, R. Alcantaraa and J. Martin-Calleja, Phys. Chem. Chem. Phys., 2014, 16, 3835–3845 RSC.
- Z. Zhao, Y. Sun, Y. Zhang and H. ZHao, RSC Adv., 2015, 5, 39549–39556 RSC.
- X. J. She, L. Liu, H. Y. Ji, Z. Mo, Y. P. Li, L. Y. Huang, D. L. Du and H. Xu, Appl. Catal., B, 2016, 187, 144–153 CrossRef CAS.
- J. Xu, L. Zhang, R. Shi and Y. ZHu, J. Mater. Chem. A, 2013, 1, 14766–14772 CAS.
| This journal is © The Royal Society of Chemistry 2017 |