
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decoration of TiO2/g-C3N4 Z-scheme by carbon dots as a novel photocatalyst with improved visible-light photocatalytic performance for the degradation of enrofloxacin†
Yuehan Su ,
Ping Chen,
Fengliang Wang,
Qianxin Zhang,
Tiansheng Chen,
Yingfei Wang,
Kun Yao,
Wenying Lv and
Guoguang Liu*
,
Ping Chen,
Fengliang Wang,
Qianxin Zhang,
Tiansheng Chen,
Yingfei Wang,
Kun Yao,
Wenying Lv and
Guoguang Liu*
School of Environmental Science and Engineering, Institute of Environmental Health and Pollution Control, Guangdong University of Technology, Guangzhou, 510006, China. E-mail: liugg615@163.com
First published on 6th July 2017
Abstract
A novel visible-light-driven carbon dot (CDs)/TiO2/g-C3N4 photocatalyst was successfully synthesized by doping CDs in TiO2 nanoparticles and the surface of g-C3N4 nanosheets via a facile hydrothermal process and was confirmed by characterization methods. UV-vis diffuse reflectance spectra (DRS) revealed that CDs/TiO2/g-C3N4 showed obvious additional absorption in the 370–450 nm region. DMPO spin-trapping ESR spectra demonstrated the existence of O2˙− and ·OH. The photocatalytic activity of the CDs (1.0 wt%)/TiO2/g-C3N4 was remarkably enhanced as compared to that of the single components (TiO2 and g-C3N4) and double component (TiO2/g-C3N4) towards the degradation of enrofloxacin (ENX) under visible-light irradiation. About 91.6% of ENX was decomposed by CDs (1.0 wt%)/TiO2/g-C3N4 in 1 h, which is nearly 3.95 times, 4.82 times, and 1.69 times that for TiO2, g-C3N4, and TiO2 (90.0 wt%)/g-C3N4, respectively. Scavenging experiments revealed that O2˙− and ·OH played key roles during the photocatalytic degradation of ENX. This study provides a simple and convenient method to modify materials with enhanced photocatalytic performance, and the CDs/TiO2/g-C3N4 catalyst is efficient, stable, and reusable for environmental practical applications.
Introduction
In the past decade, use of solar-energy-driven semiconductors as photocatalysts to remove aquatic contaminants became one of the most promising approaches.1–3 As is known, semiconductor titanium dioxide (TiO2) plays a significant role in the field of photocatalysis due to its nontoxicity, inexpensiveness, high activity, etc.4 However, TiO2 has a band gap of approximately 3.2 eV, which requires ultraviolet irradiation with wavelengths less than 385 nm for activation. That is, this excitation wavelength can activate electron transfer from the TiO2 valence band (VB) to the conduction band (CB). Consequently, the photocatalyst utilizes less than 5% of the energy available in sunlight, which extremely restricts its application in practice.5–7 More recently, a metal-free semiconductor, graphitic carbon nitride (g-C3N4), has been successfully synthesized through a facile thermal polycondensation method.8,9 It is reported that g-C3N4 exhibits high photocatalytic activity due to its delocalized conjugated π structures, leading to relatively rapid photoinduced charge separation and slow charge recombination.10 Furthermore, the low band gap (2.70 eV) capacitates g-C3N4 to efficiently harvest sunlight within the visible-light region. Although g-C3N4 possesses better photocatalytic activity as compared to TiO2, it is far from enough to meet the demands in practical use. Various attempts including surface sensitization, chemical modification, and coupling with other semiconductor materials have been made to increase the photocatalytic efficiency of TiO2 and g-C3N4.9,11,12 Much interest has recently been concentrated on the fabrication of TiO2/g-C3N4 heterostructures, which extend the absorption to the visible-light region.13–15 However, to the best of our knowledge, h+ from EVB of g-C3N4 is incapable of generating ·OH under visible light; in addition to the abovementioned shortcomings of TiO2, the photocatalytic performance of TiO2/g-C3N4 is still unsatisfactory.Carbon dots (CDs), with an appealing group of zero-dimensional nanostructures, have attracted extensive research interest in the photocatalytic field due to their low-cost, aqueous dispersibility, chemical stability, unique photoinduced electron transfer, and electron reservoir properties.16 Hence, photocatalysts with the incorporation of CDs have achieved excellent effects in the past few years. For example, Guo et al. synthesized a carbon quantum dot/carbon nitride hybrid photocatalyst to improve the photocatalytic degradation of methyl orange.17 Wu et al. used CDs as a solid-state electron mediator for the preparation of BiVO4/CD/CdS Z-scheme photocatalyst, which showed excellent photocatalytic activity and stability for overall water splitting under visible light.18 Recently, Yu et al. reported a novel CD/ZnFe2O4 composite photocatalyst with enhanced photocatalytic performance for the removal of NOx, demonstrating that CDs acted as an electron reservoir and transporter as well as a powerful energy-transfer component.19 Due to the outstanding photocatalytic properties, in this study, CDs were first used for the decoration of the TiO2/g-C3N4 Z-scheme through a facile hydrothermal-polymerized method.
Pharmaceuticals and personal care products (PPCPs), as emerging contaminants in recent years, have received unprecedented concern because of their potential hazard to the ecological environment and human health.20 According to the previous study, PPCPs have been confirmed to be ubiquitous throughout the world,21–24 and even worse, the drinking water treatment plants are also included.25,26 Consequently, it is extremely urgent to develop efficient methods for the elimination of PPCPs in aquatic environments.
In this study, the visible-light-driven CD/TiO2/g-C3N4 ternary composites were first synthesized and used as photocatalysts for the degradation of PPCPs. Herein, enrofloxacin (ENX), largely used as veterinary bactericides, was taken as targeted PPCPs under the effects of the as-prepared materials. The structural and optical properties of the composites were investigated in detail by TEM, XRD, XPS, PL, etc. In addition, the influence of the TiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :g-C3N4 molar ratio and doping amount of CDs on the photocatalytic performance of these composite photocatalysts was studied in an attempt to illuminate the potential mechanisms. The CD/TiO2/g-C3N4 ternary composites were efficient towards the decomposition of refractory pollutants because of the synergic effect of accelerated charge separation and an extended light harvesting region. It was anticipated that this study could be instrumental in studying the removal of PPCPs and fabrication of novel ternary composite photocatalysts.
:g-C3N4 molar ratio and doping amount of CDs on the photocatalytic performance of these composite photocatalysts was studied in an attempt to illuminate the potential mechanisms. The CD/TiO2/g-C3N4 ternary composites were efficient towards the decomposition of refractory pollutants because of the synergic effect of accelerated charge separation and an extended light harvesting region. It was anticipated that this study could be instrumental in studying the removal of PPCPs and fabrication of novel ternary composite photocatalysts.
Experimental
Materials
Enrofloxacin (ENX, 98% purity) was acquired from TCI Reagent Co. Ltd. (China). Degussa P25 (commercial TiO2) was purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). Dicyandiamide was commercially purchased from Aladdin (China). Acetonitrile and methanol were obtained from CNW Technologies GmbH (Germany, HPLC-grade). Other reagents (e.g., citric acid, urea, acetic acid, sulfuric acid, sodium hydroxide, benzoquinone, isopropanol, potassium iodide, and sodium azide) were of analytical grade and used without further purification. Deionized (DI) water from a Milli-Q apparatus (Germany) was used in the whole experiment.Synthetic procedures
000 rpm for 15 min to remove large particles. Finally, the remaining solution was transferred to the vacuum drier for 3 h at 80 °C to obtain a brownish black solid (C-dots).Characterization
Thermogravimetric (TG) analysis was conducted using STA-449C Jupiter (NETZSCH Corporation, Germany). The interlayer structure of the samples was characterized by a transmission electron microscope (TEM, JEM-2100HR). The crystal structure and phase purity of the obtained materials were tested by X-ray diffraction (XRD, BRUKER D8 ADVANCE) equipped with Cu Kα radiation (λ = 0.154178 nm). The UV-vis diffuse reflection spectra (DRS) of the as-prepared samples were analyzed via a UV2450 UV-vis spectrophotometer (Shimadzu) using BaSO4 as a reflectance standard. X-ray photoelectron spectroscopy (XPS) was used to analyze the ionic characteristics, which was conducted using a Thermo VG ESCALAB 250 spectrometer with Al Kα radiation at 1486.6 eV. Fourier transform infrared (FT-IR) spectra were obtained using a Nicolet 6700 spectrophotometer (Thermofisher). Photoluminescence (PL) spectra were obtained using a FluoroMax-4 fluorescence spectrophotometer (HORIBA Jobin Yvon).Photocatalytic experiments
Photocatalytic activity tests of the photocatalysts were carried out in a XPA-7 rotary photochemical reactor (Fig. 1, Nanjing Xujiang machine plant). A 350 W xenon lamp with a 420 nm cut-off filter was used as the visible-light irradiation source. For each experiment, 50 mL of a 4 mg L−1 ENX aqueous solution with 1.0 g L−1 catalysts was introduced into a 50 mL quartz tube, which was adjusted to a neutral pH via 1% NaOH and H2SO4 solution. Before the light irradiation, the reaction solution was stirred in the dark for 30 min to reach adsorption equilibrium for ENX on the photocatalyst. The samples were filtered through 0.22 μm millipore filters to remove the nanophotocatalyst. ENX was analyzed by high-performance liquid chromatography (HPLC), and the chromatographic conditions of ENX are listed as follows: column: Zorbax Eclipse XDB-C18, 4.6 × 150 mm, 5 μm; temperature: 40 °C; mobile phase: methanol/Milli-Q water containing 0.2% formic acid (65:25 v/v); flow rates: 1 mL min−1; injection volume: 20 μL; and detection wavelength: 278 nm.
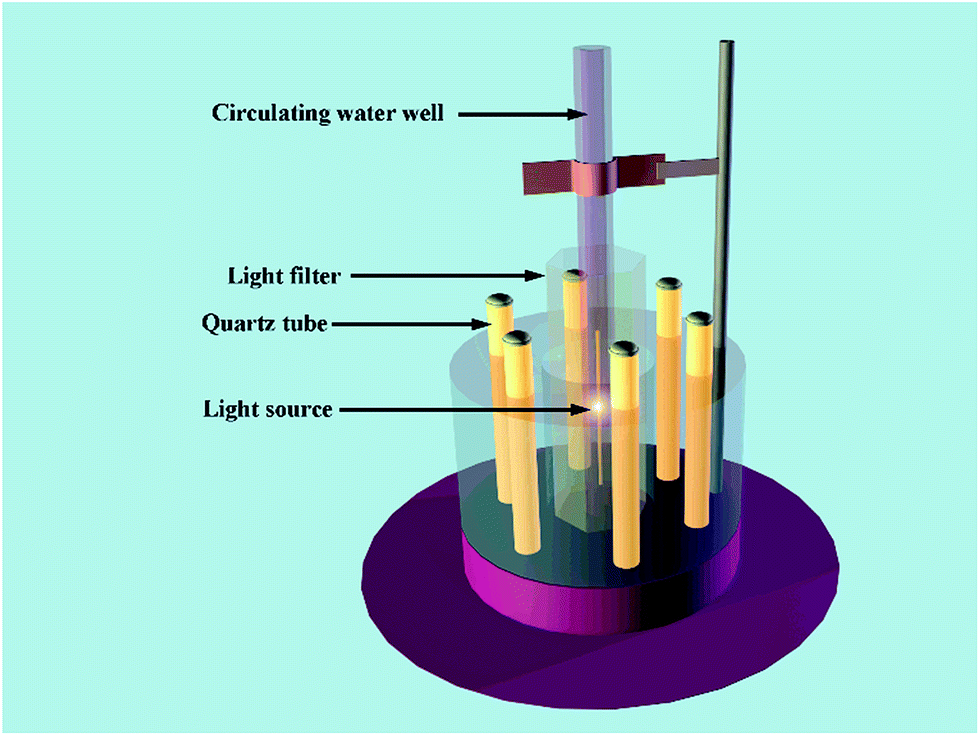 | ||
| Fig. 1 Rotary photochemical reactor with a 350 W xenon lamp and 420 nm cut-off filter. | ||
The quenching experiments were employed to prove the existence of reactive species in the ENX photodegradation process. Herein, 0.0001 mol L−1 benzoquinone (BQ, O2˙− scavenger), 0.01 mol L−1 isopropanol (IPA, ·OH scavenger), 0.01 mol L−1 potassium iodide (KI, ·OH and h+ scavenger), and 0.075 mol L−1 sodium azide (NaN3, 1O2 scavenger) were used as quenchers in the experiments.30–33
Results and discussion
Characterization
To determine the real contents of g-C3N4 in TiO2/g-C3N4, thermogravimetric analysis was performed from 30 °C to 800 °C at a heating rate of 10 °C min−1 under air conditions. It can be observed from Fig. 2 that the burning of g-C3N4 occurs in the temperature range of 500–650 °C, and the pure TiO2 presents almost no weight loss, which is in accordance with the previous report.34 As shown in Fig. 2, the real contents of g-C3N4 can be easily calculated from the weight remained after combustion.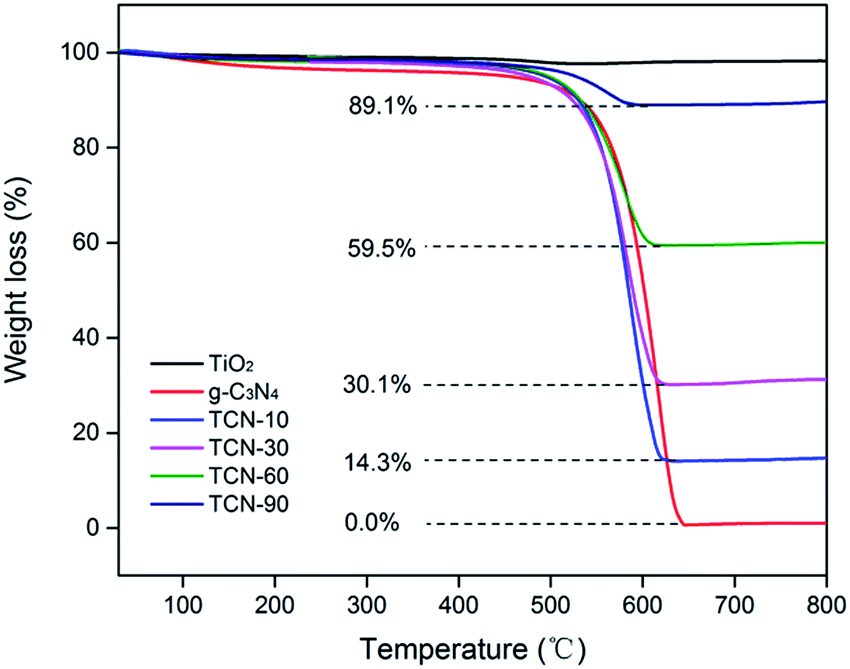 | ||
| Fig. 2 TG analysis of the as-prepared samples. | ||
Fig. 3 shows the TEM and HRTEM images of the obtained materials. It can be clearly observed that the g-C3N4 displays a graphite-like layered structure with the agglomeration of TiO2 on the surface (Fig. 3A and B). CDs, as the decoration of TiO2/g-C3N4 heterojunction, are embedded in TiO2 nanoparticles and deposited onto the surface of g-C3N4 nanosheets (Fig. 3C and D). The HRTEM images of the CDs/TiO2/g-C3N4 samples (Fig. 3E) clearly reveal an interplanar spacing of 0.35 nm, which authenticates the existence of TiO2 (anatase 101).35 HRTEM images of CDs and the inset in Fig. 3F show CDs (average size: ∼5 nm) with a lattice spacing of 0.22 nm corresponding to the (100) facet of graphite.36
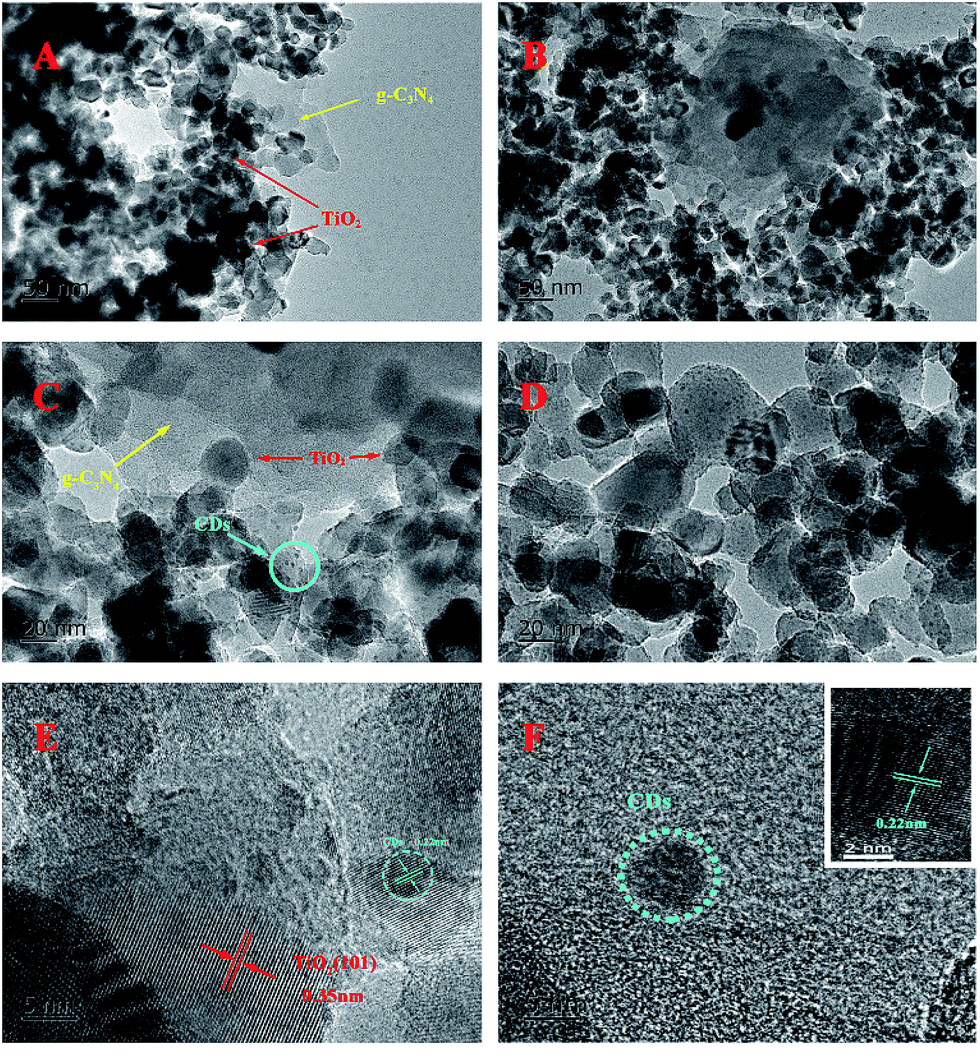 | ||
| Fig. 3 (A and B) TEM images of TCN-90; (C and D) TEM images of CTCN-1; (E) HRTEM image of CTCN-1; and (F) HRTEM image of CDs. Inset: HRTEM image of CDs with obvious crystal lattice. | ||
XRD was carried out to reveal the phase composition of the samples (Fig. 4). As shown in Fig. 4A, no impurity diffraction peaks are observed in the XRD patterns of TiO2 and g-C3N4, which can be indexed to anatase (JCPDS 21-1272), rutile phases (JCPDS 21-1276) of TiO2, and hexagonal phase of g-C3N4 (JCPDS 87-1526). TCN-90 samples show relatively strong peaks at the position of R(100), indicating the introduction of g-C3N4. After decoration of CDs, the characteristic diffraction peaks of TiO2/g-C3N4 remain unchanged with the increasing mass contents of CDs. It can be concluded that CDs have a negligible effect on the crystalline structure of the TiO2/g-C3N4 Z-scheme, decorated via a facile hydrothermal process. For others, no new diffraction peaks appear due to the low doping content of CDs.
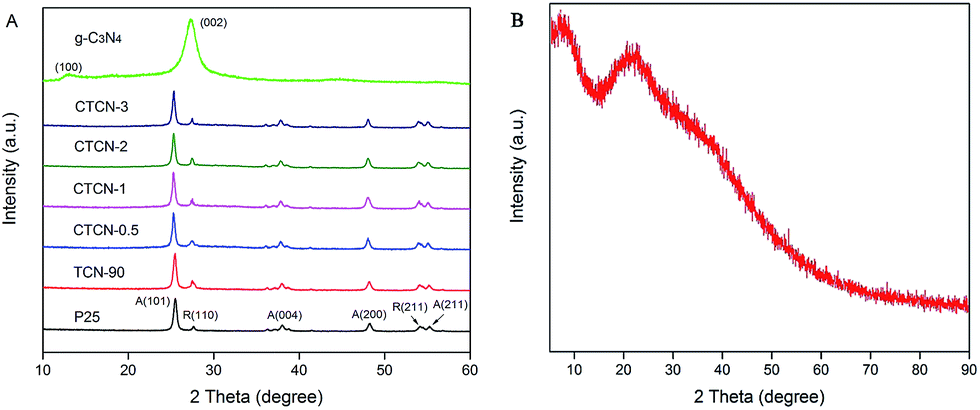 | ||
| Fig. 4 (A) XRD patterns of TiO2, g-C3N4, TCN-90, CTCN-0.5, CTCN-1, CTCN-2, and CTCN-3; (B) XRD patterns of CDs. | ||
To study the surface chemical composition and chemical status of CD/TiO2/g-C3N4, XPS analysis was carried out, and the results are shown in Fig. 5. It can be concluded that the ternary composites primarily consist of C, N, Ti, and O elements (see Fig. 5A). High-resolution spectra of C 1s at 288.88 eV and 284.78 eV are shown in Fig. 5B. The former can be attributed to the N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds, whereas the latter is assigned to the C–C coordination including sp2-hybridized C atoms present in g-C3N4.37 High-resolution spectra of C 1s of TCN-90 are provided as a contrast. It can be observed that C–C at 288.88 eV of CTCN-1 occupies relatively larger percentage than TCN-90 due to the doping of CDs. The N 1s high-resolution spectrum in Fig. 5C shows three individual peaks with the binding energies of 400.98 eV, 399.98 eV, and 398.78 eV, which can correspond to C–N–H, N–(C)3, and CN–C, respectively.38 From Fig. 5D, due to the phenomena of spin–orbit separation, two peaks at the binding energies of 458.88 eV (Ti 2p3/2) and 464.58 eV (Ti 2p1/2) suggest the existence of Ti4+ in the composite.39 The O 1s high-resolution spectrum in Fig. 5E is observed at the binding energies of 532.08 eV and 530.48 eV, which are ascribed to the O–H bond and Ti–O bond, respectively.40,41 The former can be connected to the presence of a hydroxyl group on the surface of the CDs/TiO2/g-C3N4 composite, which is in line with the FT-IR analysis in Fig. 6.
N bonds, whereas the latter is assigned to the C–C coordination including sp2-hybridized C atoms present in g-C3N4.37 High-resolution spectra of C 1s of TCN-90 are provided as a contrast. It can be observed that C–C at 288.88 eV of CTCN-1 occupies relatively larger percentage than TCN-90 due to the doping of CDs. The N 1s high-resolution spectrum in Fig. 5C shows three individual peaks with the binding energies of 400.98 eV, 399.98 eV, and 398.78 eV, which can correspond to C–N–H, N–(C)3, and CN–C, respectively.38 From Fig. 5D, due to the phenomena of spin–orbit separation, two peaks at the binding energies of 458.88 eV (Ti 2p3/2) and 464.58 eV (Ti 2p1/2) suggest the existence of Ti4+ in the composite.39 The O 1s high-resolution spectrum in Fig. 5E is observed at the binding energies of 532.08 eV and 530.48 eV, which are ascribed to the O–H bond and Ti–O bond, respectively.40,41 The former can be connected to the presence of a hydroxyl group on the surface of the CDs/TiO2/g-C3N4 composite, which is in line with the FT-IR analysis in Fig. 6.
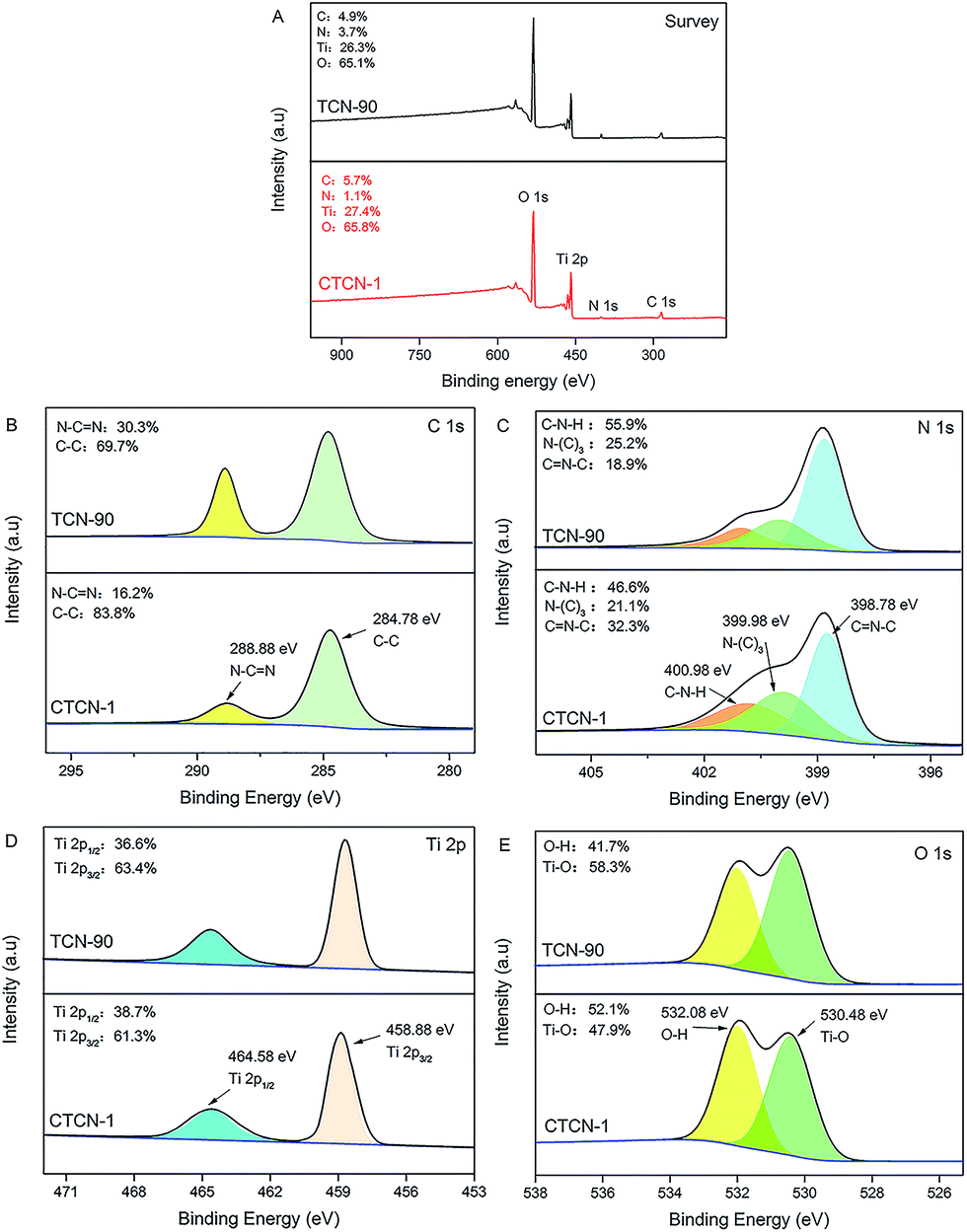 | ||
| Fig. 5 X-ray photoelectron spectra (XPS) survey spectrum of CTCN-1 and TCN-90 (A); high-resolution spectra of the C 1s (B), N 1s (C), Ti 2p (D), and O 1s (E) of CTCN-1 and TCN-90. | ||
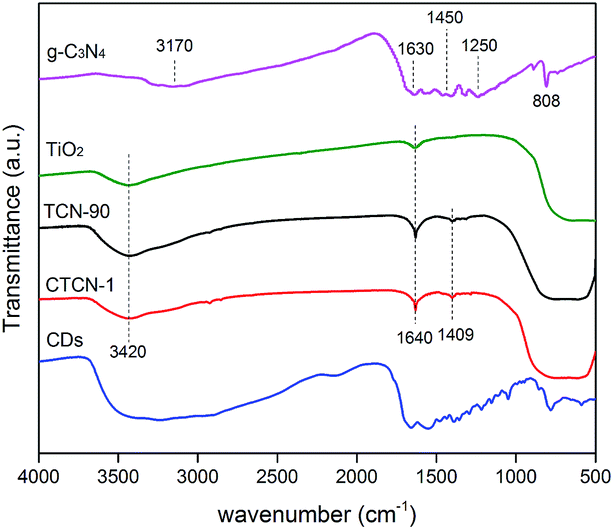 | ||
| Fig. 6 FT-IR spectra of the as-prepared samples. | ||
The FT-IR spectra of the TiO2 microspheres, g-C3N4, TCN-90 composite, and CTCN-1 composite are shown in Fig. 6. For TiO2, a wide absorption band can be observed at 500–700 cm−1, which is attributed to Ti–O stretching.42 The peaks at 1640 cm−1 and 3420 cm−1 are related to the bending vibration of O–H and N–H.43 For g-C3N4, the peak at 1630 cm−1 corresponds to the C–N heterocycle stretching vibration modes, whereas the peaks at 1450 cm−1 and 1250 cm−1 can be assigned to aromatic C–N stretching vibration modes.9 The peaks at 808 cm−1 and 3170 cm−1 are in accordance with the characteristic breathing mode of triazine units and N–H.15 A peak at 1409 cm−1 is observed in the FTIR spectra of both TCN-90 and CTCN-1, which can be interpreted as the introduction of g-C3N4 and CDs.
The optical properties of the as-prepared samples were characterized by the DRS technique, and the results are shown in Fig. 7A. TiO2 and g-C3N4 each revealed a basal absorption band at ∼390 nm and ∼475 nm, respectively, which was consistent with previous reports.31,44,45 g-C3N4/TiO2 possesses the absorption features of single TiO2 and g-C3N4, which indicates the existence of g-C3N4. As expected, CD/TiO2/g-C3N4 shows obvious additional absorption in the 370–450 nm region when doped with different amounts of CDs. The absorption edge of CTCN-1 ternary composite shows a remarkable red shift towards a higher wavelength at ∼450 nm; this suggests that the ternary composite is able to utilize the visible-light region better. Herein, Kubelka–Munk transformation was used to estimate the band gap energies of the semiconductors according to eqn (1).
| (αhv)(1/2) = A(hv − Eg) | (1) |
| EVB = X − Ec + 0.5Eg | (2) |
| ECB = EVB − Eg | (3) |
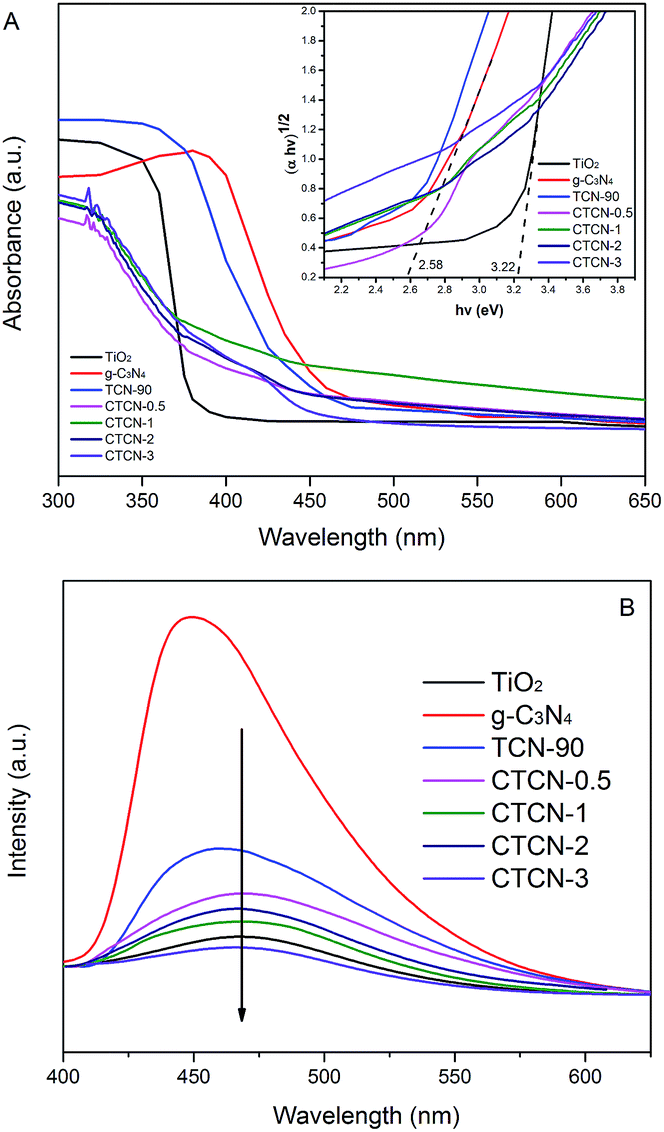 | ||
| Fig. 7 UV-vis diffuse reflectance spectra. Inset: band gap of the as-prepared photocatalyst determined from the (αhv)1/2 versus photon-energy (A) and photoluminescence spectra under 350 nm excitation (B) of the photocatalysts. | ||
PL spectra were obtained (Fig. 7B) to detect the recombination of electron–hole pairs. Generally, lower PL intensity corresponds to lower recombination of electron–hole pairs, contributing to higher photocatalytic activity.47 The g-C3N4 has an intensive emission peak at around 460 nm, which can be attributed to the fast recombination of the photoinduced electron–hole pairs of g-C3N4. The intensities of the peaks for CTCN-0.5, CTCN-1, CTCN-2, and CTCN-3 are much lower than those for TCN-90, revealing that CDs conduce to slow the recombination rate of electron–hole pairs. Further, the emission peak of CDs/TiO2/g-C3N4 shows an obvious red-shift from 455 nm to 470 nm after doping of CDs, which demonstrates the existence of CDs from another perspective. The PL results confirm the importance of CDs/TiO2/g-C3N4 in blocking the recombination of electrons and holes.
PL spectra of CDs are shown in Fig. 8, which demonstrate superior up-converted photoluminescence property of CDs. When CDs were excited by long-wavelength light (from 600 nm to 900 nm), the up-converted emissions were significantly located in the range from 350 nm to 600 nm. These results imply that CDs might enhance the photocatalytic efficiency via the conversion of near-infrared wavelength to visible light, which markedly promotes the light utilization of the photocatalysts.
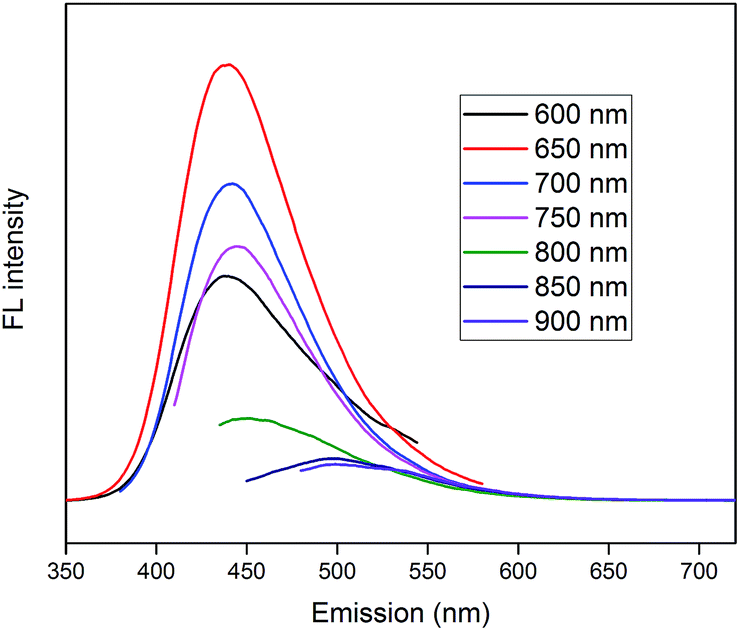 | ||
| Fig. 8 Up-converted PL spectra of CDs. | ||
Photocatalytic performance of composites
Fig. 9 displays the photocatalytic degradation of ENX over various photocatalysts. As can be seen, the self-photolysis of ENX could be neglected in the blank experiment. For TCN-90 (see Fig. 9A), the concentration of ENX is reduced to 45.95% in 1 h. This means TCN-90 performs better than other doping ratios of g-C3N4, which confirms that the small doping ratio of g-C3N4 is in favour of ENX degradation. After doping of CDs (1 wt%), enhanced photocatalytic activity was exhibited by both TiO2 and g-C3N4, which could be attributed to the up-converted PL property. In Fig. 9B, we drew a comparison between TiO2, TiO2/CDs, g-C3N4, g-C3N4/CDs, TCN-90, CTCN-0.5, CTCN-1, CTCN-2, and CTCN-3 in the photocatalytic degradation of ENX. CTCN-1 was found to be the most optimal photocatalyst among all the photocatalysts as it reduced ENX to 8.34% in 1 h, which corresponds to the DRS results in Fig. 7A. Compared with the single-component (TiO2 and g-C3N4) samples and two-component samples (TiO2/CDs, g-C3N4/CDs, TCN-90), CD/TiO2/g-C3N4 exhibits remarkably enhanced photocatalytic activity, owing to the introduction of CDs as an electron reservoir and transporter. However, further CD loading (>1.0 wt%) leads to decreased degradation rates, which can be attributed to the inner filter effect of CDs via scrambling for the absorption of photons and reduction of the formation of reactive species.48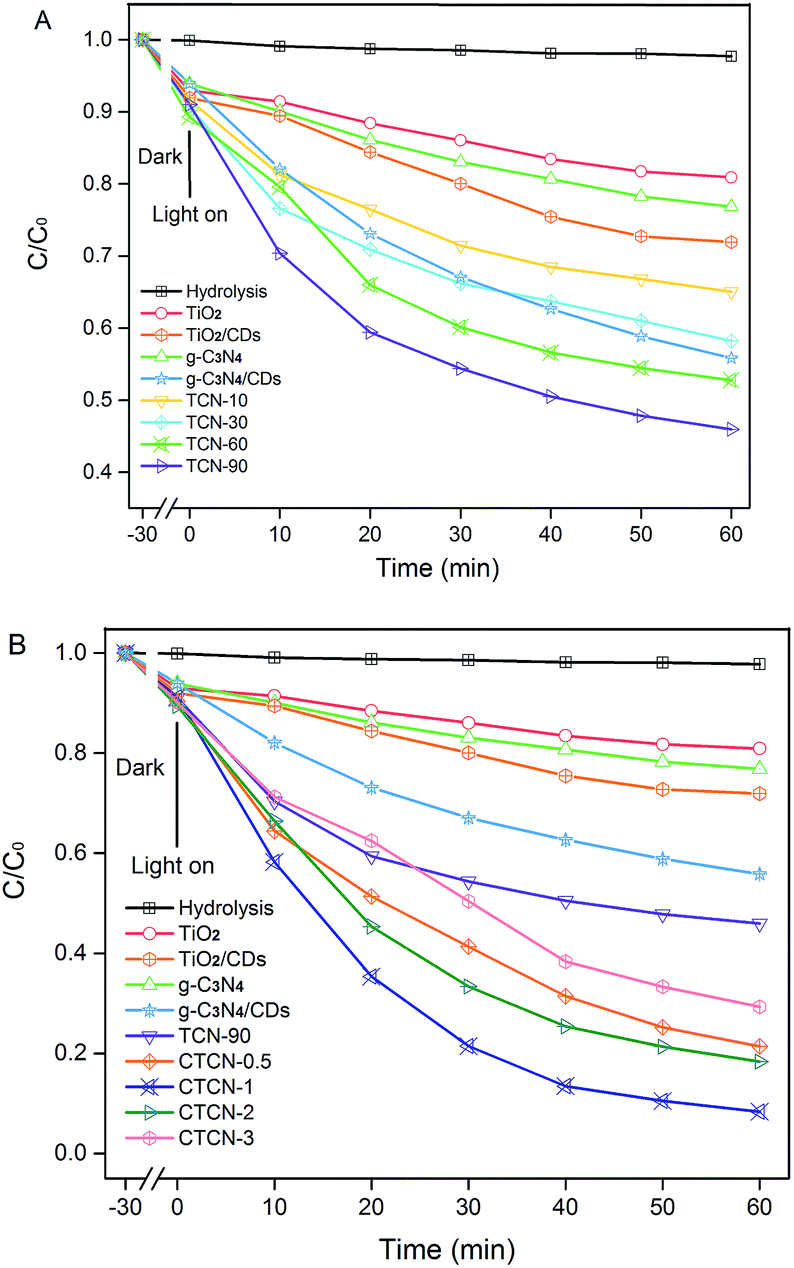 | ||
| Fig. 9 Photocatalytic activity of TiO2/g-C3N4 binarys (A) and CDs/TiO2/g-C3N4 ternarys (B) based on the photocatalytic degradation of ENX under visible-light irradiation (λ > 420 nm). | ||
Reusability is a critical factor for practical utilization. To investigate the stability of CD/TiO2/g-C3N4, five photocatalytic experiment cycle runs were completed, and the results are shown in Fig. 10. As a result, 88.7% of the ENX was decomposed in 60 min in the fifth cycle, which was slightly reduced as compared to that in the first round (91.6%). Thus, the CTCN-1 can be identified as a stable photocatalyst.
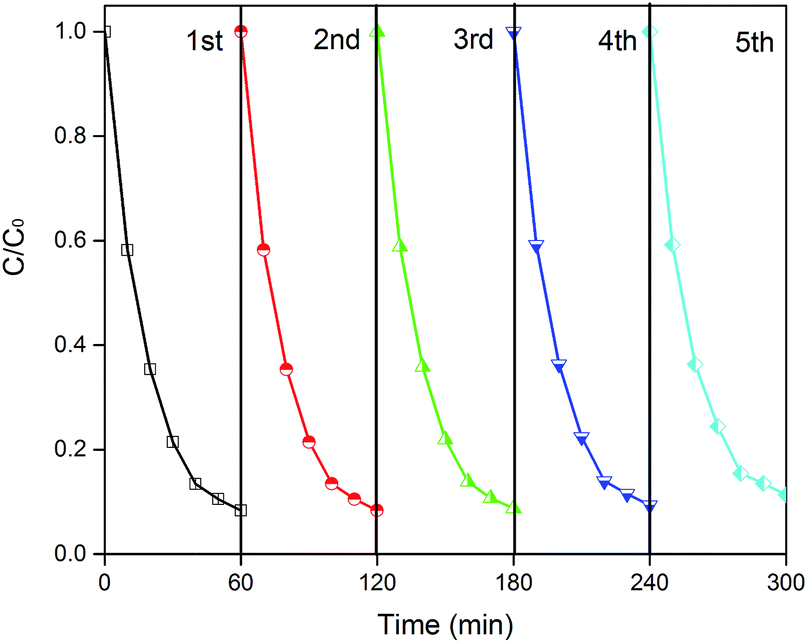 | ||
| Fig. 10 The repeated photocatalytic experiments for CTCN-1. | ||
To study the generation of radicals in the photocatalytic system under visible-light irradiation (λ > 420 nm), the ESR spin-trap with DMPO technique was used. As shown in Fig. 11, the signals of O2˙− and ·OH increase with time elapsing in 10 minutes, whereas no signals appear in the dark. The weak signals of O2˙− and ·OH radicals in TiO2 can be attributed to fast recombination of photogenerated electron–hole pairs.49 Because the VB position of g-C3N4 (+1.36 eV) is more negative than the standard redox potential of ·OH/OH− (+1.99 eV) and ·OH/H2O (+2.73 eV), h+ of g-C3N4 fails to provide ·OH by oxidizing OH− or H2O.50 The signals of ·OH radicals appear in g-C3N4 due to the reaction of O2˙− and h+.51 Compared with TCN-90, the signals of O2˙− radicals are greatly enhanced for CTCN-1, indicating that CDs play a significant role as electron mediator to improve the photocatalytic activity of CD/TiO2/g-C3N4. Simultaneously, the signals of O2˙− radicals are also improved, demonstrating the increased photocatalytic activity of CD-modified TiO2/g-C3N4.
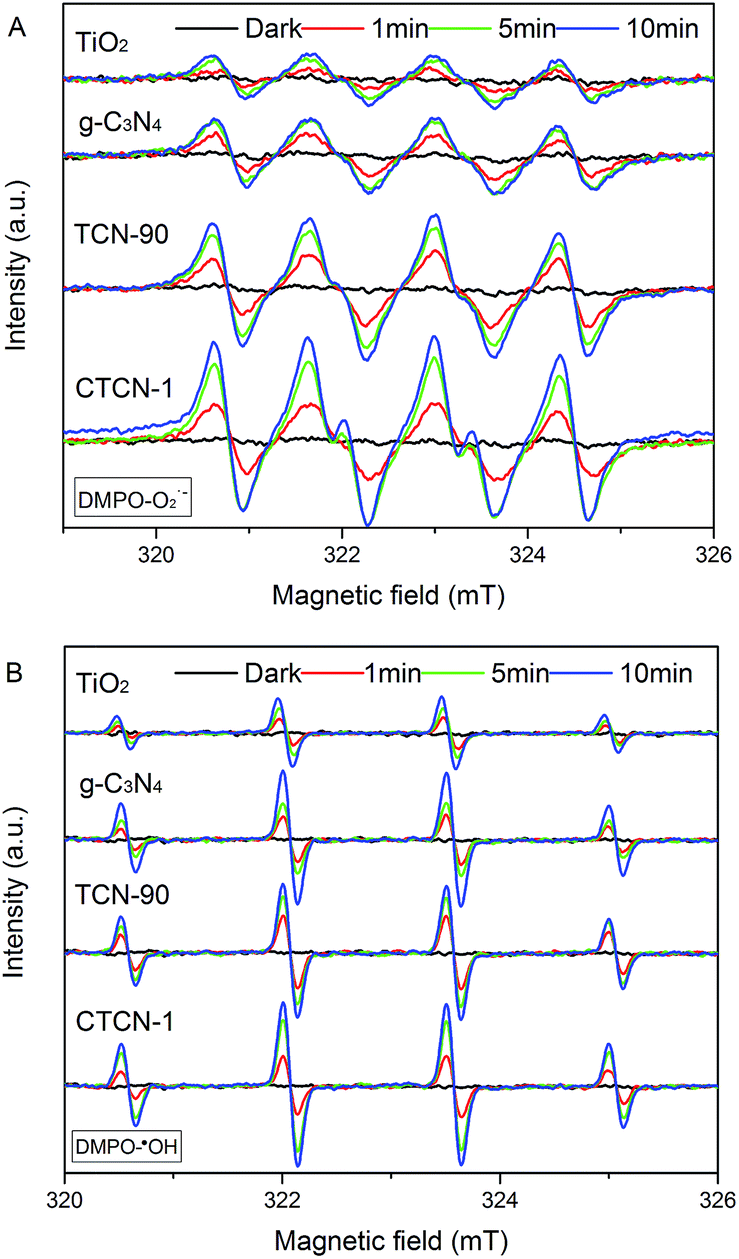 | ||
| Fig. 11 DMPO spin-trapping ESR spectra under visible-light irradiation (λ > 420 nm) in aqueous solutions (A) and ethanol solutions (B). | ||
Fig. 12 shows the kinetic constants of TiO2, g-C3N4, TCN-90, and CTCN-1 in the presence of different quenchers. For the addition of isopropanol (IPA, ·OH scavenger) and KI (·OH and h+ scavenger), a slight decrease can be observed in the degradation of ENX by CTCN-1, indicating the minor role of h+. For the TCN-90 composite, similar results were obtained, whereas a slight decrease of k was observed after the addition of NaN3 (1O2 scavenger) for the TCN-90 and CTCN-1 composites. Although benzoquinone (BQ, O2˙− scavenger) has a slight effect in the presence of TCN-90, it plays a quite important role in CTCN-1 probably because CDs can act as electron mediators and generate more O2˙−.
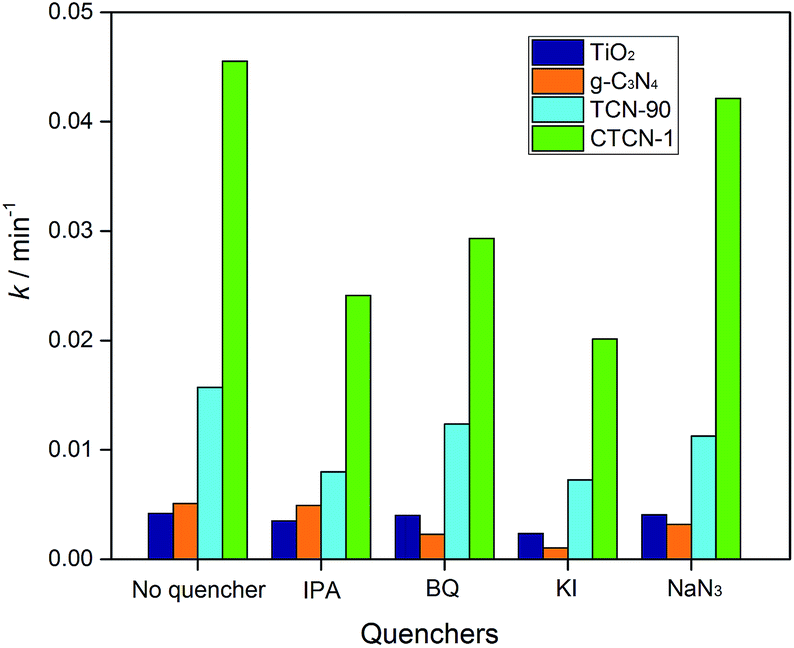 | ||
| Fig. 12 Kinetic constants of the as-prepared photocatalysts with different quenchers. | ||
Based on the abovementioned results and the previous study,43,52–54 the potential photocatalytic mechanisms are proposed in Fig. 13. For the conventional TiO2/g-C3N4 heterojunctions, electron–hole pairs are formed at the ECB and EVB of TiO2 and g-C3N4 under visible light. Subsequently, h+ generates at the EVB of TiO2 and transfers to the EVB of g-C3N4, and the e− from ECB of g-C3N4 can transfer to the ECB of TiO2. However, the h+ from EVB of g-C3N4 cannot oxidize OH− or H2O to generate ·OH because of the negative valence band position of g-C3N4 (+1.51 eV), which is less than the standard redox potential of ·OH/OH− (+1.99 eV) and ·OH/H2O (+2.73 eV). The e− from TiO2 may be captured by molecular oxygen to generate O2˙− radicals,55 which can directly attack pollutants. If TiO2/g-C3N4 follows this mechanism, h+ should play a major role in the photocatalytic system, whereas IPA has minor effects. However, the quenching experiments and ESR spectra results have contradicted this hypothesis. Moreover, the results of the 1O2 quenching experiment are inconsistent with this mechanism. Thus, the TiO2/g-C3N4 photocatalyst follows the direct Z-scheme type mechanism.
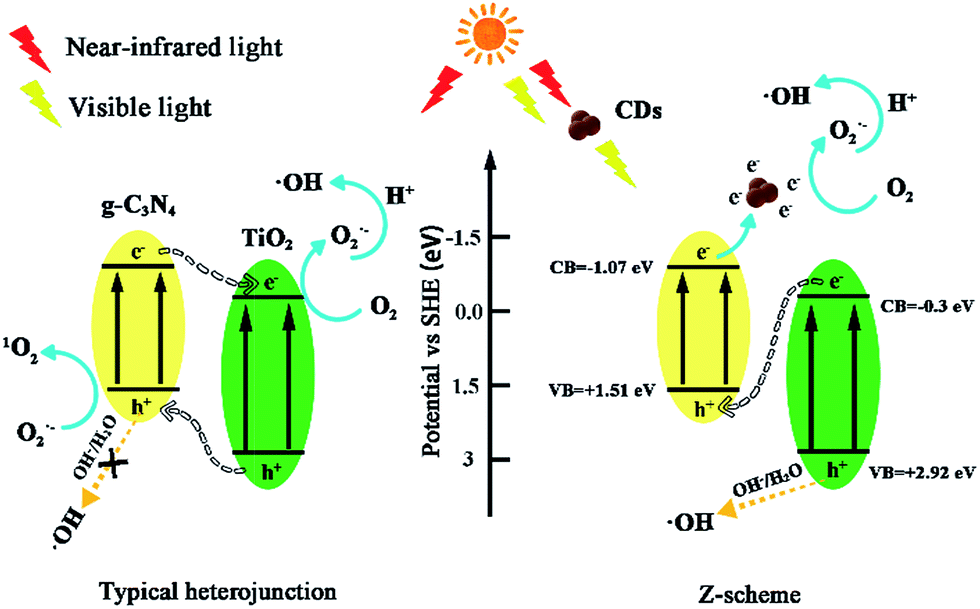 | ||
| Fig. 13 Photocatalytic mechanism scheme in conventional TiO2/g-C3N4 heterojunctions and TiO2/g-C3N4 Z-scheme with the introduction of CDs. | ||
For the TiO2/g-C3N4 Z-scheme with the introduction of CDs, the near-infrared light wavelength could be converted to visible light due to the up-converted PL properties of CDs53 Herein, the presence of CDs can facilitate a Z-scheme transfer mechanism, which differs from the general heterostructured mechanism. The general heterostructured mechanism can hardly interpret the formation of ·OH, and the transfer speed of electrons and holes might be very slow.56 The active species, h+, O2˙−, 1O2, and ·OH can effectively degrade ENX into small molecule compounds or directly into final products such as CO2 and H2O.
Conclusions
In this study, CDs/TiO2/g-C3N4 was synthetized via a facile hydrothermal method, which exhibited high photocatalytic performance towards the degradation of ENX under visible-light irradiation. The results of the characterization verified the formation of CD/TiO2/g-C3N4, in which the CDs were embedded in TiO2 nanoparticles and deposited onto the surface of the g-C3N4 nanosheets. The enhanced photocatalytic performance of CDs/TiO2/g-C3N4 might be attributed to the unique up-converted PL property, efficient charge separation, and efficient electron transportation of CDs. The synergistic effects could be concluded as follows: (1) the extension of visible-light response scope; (2) efficient photogenerated electron–hole pair separation; and (3) retardation of charge recombination. The active species h+, O2˙−, 1O2, and ·OH were produced in the Z-scheme system. The present study confirms that the CD/TiO2/g-C3N4 catalyst is efficient, stable, and reusable for environmental applications.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21377031 and 21677040) and the Scientific and Technical Projects of Guangdong Province (No. 2013B020800009).Notes and references
- S. M. Lam, J. C. Sin and A. R. Mohamed, Mater. Sci. Semicond. Process., 2016, 47, 62–84 CrossRef CAS.
- O. Mehraj, N. A. Mir, B. M. Pirzada, S. Sabir and M. Muneer, J. Mol. Catal. A: Chem., 2014, 395, 16–24 CrossRef CAS.
- M. J. Nalbandian, M. Zhang, J. Sanchez, Y. H. Choa, D. M. Cwiertny and N. V. Myung, J. Mol. Catal. A: Chem., 2015, 404, 18–26 CrossRef.
- G. W. Cui, W. L. Wang, M. Y. Ma, M. Zhang, X. Y. Xia, F. Y. Han, X. F. Shi, Y. Q. Zhao, Y. B. Dong and B. Tang, Chem. Commun., 2013, 49, 6415–6417 RSC.
- T. H. Tan, J. Scott, H. N. Yun, R. A. Taylor, K. F. Agueyzinsou and R. Amal, ACS Catal., 2016, 6(3), 1870–1879 CrossRef CAS.
- L. Pan, S. Wang, J. Xie, L. Wang, X. Zhang and J. J. Zou, Nano Energy, 2016, 28, 296–303 CrossRef CAS.
- L. Pan, S. Wang, J. J. Zou, Z. F. Huang, L. Wang and X. Zhang, Chem. Commun., 2014, 50, 988–990 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- L. Ge, F. Zuo, J. Liu, Q. Ma, C. Wang, D. Sun, L. Bartels and P. Feng, J. Phys. Chem. C, 2012, 116, 13708–13714 CAS.
- B. Chai, J. Yan, C. Wang, Z. Ren and Y. Zhu, Appl. Surf. Sci., 2016, 391, 376–383 CrossRef.
- S. Li, F. Zheng, S. Cai, W. Liang and Y. Li, Sens. Actuators, B, 2013, 188, 280–285 CrossRef CAS.
- D. R. Baker and P. V. Kamat, Adv. Funct. Mater., 2010, 19, 805–811 CrossRef.
- X. Zhong, M. Jin, H. Dong, L. Liu, L. Wang, H. Yu, S. Leng, G. Zhuang, X. Li and J. G. Wang, J. Solid State Chem., 2014, 220, 54–59 CrossRef CAS.
- H. Tang, S. Chang, L. Jiang, G. Tang and W. Liang, Ceram. Int., 2016, 42, 18443–18452 CrossRef CAS.
- M. Reli, P. Huo, M. Šihor, N. Ambrožová, I. Troppová, L. Matějová, J. Lang, L. Svoboda, P. Kuśtrowski and M. Ritz, J. Phys. Chem. A, 2016, 120, 8564–8573 CrossRef CAS PubMed.
- X. Xu, Z. Bao, Z. Gang, H. Zeng and J. Hu, ACS Appl. Mater. Interfaces, 2016, 8, 14118–14124 CAS.
- Y. Guo, P. Yao, D. Zhu and G. Cheng, J. Mater. Chem. A, 2015, 3, 13189–13192 CAS.
- X. Wu, J. Zhao, L. Wang, M. Han, M. Zhang, H. Wang, H. Huang, Y. Liu and Z. Kang, Appl. Catal., B, 2017, 206, 501–509 CrossRef CAS.
- H. Yu, Y. Liang, Y. Rao, D. Zhu, J. Cao, Z. Shen, W. K. Ho and S. C. Lee, Environ. Sci. Technol., 2017, 51, 2924–2933 CrossRef PubMed.
- J. L. Liu and M. H. Wong, Environ. Int., 2013, 59, 208–224 CrossRef CAS PubMed.
- G. R. Boyd, H. Reemtsma, D. A. Grimm and S. Mitra, Sci. Total Environ., 2003, 311, 135 CrossRef CAS PubMed.
- B. J. Richardson, P. K. Lam and M. Martin, Mar. Pollut. Bull., 2005, 50, 913 CrossRef CAS PubMed.
- B. Kasprzykhordern, R. M. Dinsdale and A. J. Guwy, Water Res., 2009, 43, 363–380 CrossRef CAS PubMed.
- R. Shen and S. A. Andrews, Water Res., 2011, 45, 944–952 CrossRef CAS PubMed.
- T. Lin, S. Yu and W. Chen, Chemosphere, 2016, 152, 1 CrossRef CAS PubMed.
- J. Radjenović, M. Petrović, F. Ventura and D. Barceló, Water Res., 2008, 42, 3601–3610 CrossRef PubMed.
- S. Qu, X. Wang, Q. Lu, X. Liu and L. Wang, Angew. Chem., Int. Ed., 2012, 51, 12215–12218 CrossRef CAS PubMed.
- F. Dong, L. Wu, Y. Sun, M. Fu, Z. Wu and S. C. Lee, J. Mater. Chem., 2011, 21, 15171–15174 RSC.
- J. Yu, S. Wang, J. Low and W. Xiao, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC.
- G. Li, X. Nie, J. Chen, Q. Jiang, T. An, P. K. Wong, H. Zhang, H. Zhao and H. Yamashita, Water Res., 2015, 86, 17 CrossRef CAS PubMed.
- H. Xiao, J. Zhu and A. Thomas, RSC Adv., 2015, 5, 105731–105734 RSC.
- G. Li, K. H. Wong, X. Zhang, C. Hu, J. C. Yu, R. C. Chan and P. K. Wong, Chemosphere, 2009, 76, 1185–1191 CrossRef CAS PubMed.
- J. Cao, B. Xu, B. Luo, H. Lin and S. Chen, Appl. Surf. Sci., 2011, 257, 7083–7089 CrossRef CAS.
- Y. He, L. Zhang, X. Wang, Y. Wu, H. Lin, L. Zhao, W. Weng, H. Wan and M. Fan, RSC Adv., 2014, 4, 13610–13619 RSC.
- X. Sun, W. Dai, G. Wu, L. Li, N. Guan and M. Hunger, Chem. Commun., 2015, 51, 13779 RSC.
- X. Zhai, P. Zhang, C. Liu, T. Bai, W. Li, L. Dai and W. Liu, Chem. Commun., 2012, 48, 7955 RSC.
- J. A. Singh, S. H. Overbury, N. J. Dudney, M. Li and G. M. Veith, ACS Catal., 2012, 2, 1138–1146 CrossRef CAS.
- S. J. Yang, J. H. Cho, G. H. Oh, K. S. Nahm and C. R. Park, Carbon, 2009, 47, 1585–1591 CrossRef CAS.
- X. Yang, J. Qin, J. Yan, K. Chen, X. Yan, Z. Du, L. Rong and T. Hua, Appl. Catal., B, 2015, 166–167, 231–240 CrossRef CAS.
- Y. Chen, W. Huang, D. He, S. Yue and H. Hong, ACS Appl. Mater. Interfaces, 2014, 6, 14405 CAS.
- L. A. D. Silva, V. A. Alves, S. C. D. Castro and J. F. C. Boodts, Colloids Surf., A, 2000, 170, 119–126 CrossRef.
- J. Yu, S. Wang, J. Low and W. Xiao, Phys. Chem. Chem. Phys., 2013, 15, 16883 RSC.
- Y. Chen, W. Huang, D. He, Y. Situ and H. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 14405 CAS.
- N. Boonprakob, N. Wetchakun, S. Phanichphant, D. Waxler, P. Sherrell, A. Nattestad, J. Chen and B. Inceesungvorn, J. Colloid Interface Sci., 2014, 417, 402–409 CrossRef CAS PubMed.
- Z. F. Huang, J. Song, L. Pan, Z. Wang, X. Zhang, J. J. Zou, W. Mi, X. Zhang and L. Wang, Nano Energy, 2015, 12, 646–656 CrossRef CAS.
- J. Tauc, Mater. Res. Bull., 1970, 5, 721–729 CrossRef CAS.
- X. Zhou, B. Jin, L. Li, F. Peng, H. Wang, H. Yu and Y. Fang, J. Mater. Chem., 2012, 22, 17900–17905 RSC.
- J. Pan, Y. Sheng, J. Zhang, J. Wei, P. Huang, X. Zhang and B. Feng, J. Mater. Chem. A, 2014, 2, 18082–18086 CAS.
- K. Lv, X. Li, K. Deng, J. Sun, X. Li and M. Li, Appl. Catal., B, 2010, 95, 383–392 CrossRef CAS.
- D. Wu, B. Wang, W. Wang, T. An, G. Li, T. W. Ng, H. Y. Yip, C. Xiong, H. K. Lee and P. K. Wong, J. Mater. Chem. A, 2015, 3, 15148–15155 CAS.
- J. Ng, X. Wang and D. D. Sun, Appl. Catal., B, 2011, 110, 260–272 CrossRef CAS.
- Y. Li, K. Lv, W. Ho, F. Dong, X. Wu and Y. Xia, Appl. Catal., B, 2017, 202, 611–619 CrossRef CAS.
- F. Wang, P. Chen, Y. Feng, Z. Xie, Y. Liu, Y. Su, Q. Zhang, Y. Wang, K. Yao and W. Lv, Appl. Catal., B, 2017, 207, 103–113 CrossRef CAS.
- Y. He, L. Zhang, M. Fan, X. Wang, M. L. Walbridge, Q. Nong, Y. Wu and L. Zhao, Sol. Energy Mater. Sol. Cells, 2015, 137, 175–184 CrossRef CAS.
- J. Liu, Y. Yang, N. Liu, Y. Liu, H. Huang and Z. Kang, Green Chem., 2014, 16, 4559–4565 RSC.
- Y. He, L. Zhang, B. Teng and M. Fan, Environ. Sci. Technol., 2015, 49, 649 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05485h |
| This journal is © The Royal Society of Chemistry 2017 |