
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational selection of halide ions for synthesizing highly active Au@Pd nanobipyramids†
Fangyan Liua,
Jia Nia,
Hui Haoa,
Wei Wang c,
Wei Chena,
Lijie Zhanga,
Chao Zoua,
Yun Yang*a and
Shaoming Huang*ab
c,
Wei Chena,
Lijie Zhanga,
Chao Zoua,
Yun Yang*a and
Shaoming Huang*ab
aNanomaterials and Chemistry Key Laboratory, Wenzhou University, Wenzhou, Zhejiang 325027, P. R. China. E-mail: bachier@163.com; smhuang@wzu.edu.cn
bSchool of Materials and Energy, Guangdong University of Technology, Guangzhou, Guangdong 510006, P. R. China
cDepartment of Chemistry, University of Bergen, Bergen, Norway
First published on 25th July 2017
Abstract
Pd-based nanostructures with stepped facets possess potential applications in many fields, particularly in catalysis. Generally, crystal growth often only allows the formation of nanostructures with energetically non-stepped facets, so it is desirable to develop methods that can be used to prepare Pd-based nanostructures bounded by stepped facets. Herein, penta-fold twinned (PFT) Au@Pd nanobipyramids (NBs) with stepped {100} facets were synthesized through growing Pd on Au decahedral nanoparticles (NPs) in polyol. During the growth of Au@Pd NBs, Br− was a critical factor, because it has appropriate affinity for the Pd atom and adjusted the growth rate ratio along 〈110〉 and 〈100〉, resulting in the formation of Au@Pd NBs with stepped {100} facets. The product shape and size could be tailored by controlling the reaction conditions. Transmission electron microscopy (TEM), high resolution transmission electron microscopy (HRTEM), energy dispersive spectroscopy (EDS), high angle annular dark field (HAADF) imaging and scanning transmission electron microscopy EDS (STEM-EDS) were used to investigate the structure and growth of the Au@Pd NBs. A growth mechanism involving two stages was elucidated. In the first stage, the growth of Pd clearly occurred along both 〈110〉 and 〈100〉. In the second stage, the growth along 〈110〉 was faster than that along 〈100〉. Furthermore, we also demonstrated that the as-prepared Au@Pd NBs had high catalytic activity, compared with Pd nanocubes and Pd–Au–Pd segmental nanorods.
Introduction
In the past few decades, Pd-based nanostructures have attracted much attention due to their important applications in catalysis.1–18 Their catalytic performance is strongly dependent on the surface atoms, that is, the bounding facets.10–18 Nanostructures with stepped facets exhibit relatively high activity, compared with commonly observed nanostructures.16 Therefore, they could be optimal catalysts. Thus, much effort has been dedicated to developing new technologies for preparing Pd-based nanostructures with stepped facets.19–22 As is well known, growth always prefers to occur at positions with high surface free energy.14 Therefore, the growth rate on a stepped facet is always faster than that on a non-stepped one. This means that a stepped facet often disappears or becomes small after growth. As a result, nanostructures coated with non-stepped facets ({111}, {100}, {110}) often form, such as octahedrons, decahedrons, icosahedrons, tetrahedrons, truncated octahedrons and cubes.23–31 In order to prepare Pd-based nanostructures with stepped facets, the reaction environment should be adjusted to control the growth pattern. Two approaches have been demonstrated to be successful in the synthesis of nanostructures with stepped facets: electrochemical and colloidal chemical routes.19–22 This type of electrochemical technology was mainly developed by Sun's group, and they prepared many noble nanostructures with stepped facets, mainly focusing on Pd and Pt.20,32 They also demonstrated that such Pt and Pd nanostructures with stepped facets have superior catalytic activity. The colloidal chemical route is widely used, due to its excellent tunability over product size, composition and morphology.19,21,22 Various noble nanostructures with stepped facets can be synthesized via colloidal chemical routes. Among the reported nanostructures with stepped facets, penta-fold twinned (PFT) NBs possess important applications in catalysis and optics, due to their unique structure.12,33–46 For example, Au NBs can greatly improve the surface enhanced Raman scattering and allow for the detection of single molecules.41 Wang's group reported that NBs had better optical sensitivity to the environment than Au nanorods.45 So far, most reports on NBs have been focused on Au and rarely on other metals.46 Since Liu and co-workers reported the synthesis of Au NBs,35 numerous synthetic technologies of Au NBs have been developed, so it is not a significant challenge to achieve high-quality Au NBs.33,35,36,45,48,49 For example, Au NBs can be synthesized in low yield through growing Au on small Au decahedral NPs, followed by a shape-selective precipitation, which allows high quality Au NBs to be obtained.35,36,45 Very recently, several groups demonstrated that the optimized synthesis of Au seeds allows for the direct preparation of high-quality Au NBs without the use of shape-selective precipitation.33,48,49 However, for other metals, the syntheses of NBs have been rarely reported. As described above, Pd-based nanostructures are potential catalysts for many organic reactions and fuel cells,10–18 and therefore, it is worth developing methods that can be used to prepare Pd NBs, and explore their catalytic performance.The growth mode often differs depending on the metal, so the synthetic approach for Au NBs is ineffective for other metals. In the synthesis of Au NBs, Ag+ is central to the growth, because Ag(I)-based substances can selectively adsorb or deposit on specific facets of Au nanostructures and modulate their growth.45,50,51 However, Ag(I)-based substances have no significant effect on the growth of Pd nanostructures;52 therefore, this strategy is unable to produce Pd NBs. To synthesize Pd-based NBs, new technologies must be developed. In practice, previous studies are very helpful towards designing a reasonable route for synthesizing Pd NBs. As is well known, NBs are a typical type of PFT nanostructure. Seeded growth is a very popular method for preparing noble nanostructures and in most cases, it allows for the formation of products with the same crystalline features as the seed. Furthermore, PFT nanorods and decahedral NPs are very structurally similar.47,52 Therefore, the utilization of PFT decahedral NPs might be a potential option. Previously, the synthesis of monodispersed Au decahedral NPs was developed in our laboratory.47 Such Au decahedral NPs can often induce the growth of other metals along 〈110〉, allowing for the preparation of one-dimensional nanostructures when appropriate capping agents are present.47,53 Another important goal is to find a substance with similar effects to those of Ag(I)-based substances in the growth of Au NBs. For a decahedral NP, the direction along its five-fold axis is 〈110〉 and the direction perpendicular to 〈110〉 is 〈100〉. NB growth involves the evolution from small sized decahedral NPs to large-sized NBs, implying that growth along both 〈110〉 and 〈100〉 occurs simultaneously.35 Furthermore, the growth rate along 〈110〉 is faster than that of the latter. It is well known that halide ions (Cl−, Br− and I−) have an affinity for Pd and are highly effective for tuning the growth of Pd nanostructures.54–64 For the above three ions, their affinities for noble metal atoms or ions follows this order: I− > Br− > Cl−.61–64 I− can completely block the growth along 〈100〉 even at low levels and facilitate the formation of Pd nanorods.56,65 Cl− it is unable to significantly block the growth along 〈100〉 or promote the selective growth along 〈110〉 even at a very high concentration.65 As a result, non-preferential growth along 〈110〉 and 〈100〉 occurs, always resulting in the formation of spherical NPs. Therefore, Br− might be an appropriate growth-modifier.
Herein, Au decahedral NPs served as seeds and sodium tetrabromopalladate (Na2PdBr4) as an appropriate precursor. Polyol was used as a reducing agent that transforms Pd(II) to Pd. Br− ions released from Na2PdBr4 acted as a growth-modifier and facilitated the growth of Au@Pd NBs. Length-tuned Au@Pd NBs and other shaped Pd–Au nanostructures could be prepared through controlling reaction conditions. Because the prepared Au@Pd NBs are coated with highly active stepped {100} facets, they are more active than other Pd nanostructures (nanocubes, octahedra and Pd–Au–Pd segmental nanorods) in the reduction of p-nitrophenol to p-aminophenol.
Experimental section
Chemicals
Silver nitrate (AgNO3), chloroauric acid (HAuCl4), diethylene glycol (DEG) and poly(diallyldimethylammonium chloride) (PDDA, Mw = 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–500000, 20 wt% in H2O) were purchased from Sigma-Aldrich. Palladium chloride (PdCl2), Na2PdBr4, sodium iodide (NaI), sodium bromide (NaBr) and sodium chloride (NaCl) were bought from Aladdin. No further purifications were needed before use.
000–500000, 20 wt% in H2O) were purchased from Sigma-Aldrich. Palladium chloride (PdCl2), Na2PdBr4, sodium iodide (NaI), sodium bromide (NaBr) and sodium chloride (NaCl) were bought from Aladdin. No further purifications were needed before use.
Preparation of Au decahedral NPs
A slightly modified method to that reported by our group was used to synthesize Au decahedral seeds.47,65 In a typical preparation, 0.5 mL PDDA was added to 20 mL DEG under magnetic stirring (500 rpm), and then 20 μL HAuCl4 (0.48 M) aqueous solution was introduced. After a yellow homogeneous solution had formed, 4 mL DEG solution containing 12 mg AgNO3 was added, and then the mixture was stirred for another 3 min. The resulting solution was placed into an oil bath at 200 °C to promote the reduction of Au(III) to Au. After 30 min, a red colloid formed and then the heating source was removed. The solution was then allowed to slowly cool down to room temperature. In order to remove AgCl, 5 drops of ammonia water (30 vol%) were added to form soluble [Ag(NH3)2]+ and the resulting colloidal solution was subjected to centrifugation (20000 rpm). The precipitate was dispersed in DEG again, collected via centrifugation and then dispersed in 20 mL DEG.
Before the samples were observed using TEM, excess DEG, PDDA and other impurities needed to be removed. Water (9 mL) was mixed with 1 mL of the above colloid precipitate, and then Au decahedral NPs were collected via centrifugation (12000 rpm). The mixture was then dispersed in 9 mL water and precipitated again through centrifugation. After the purification procedure was repeated three times, the precipitate was dissolved in 0.5 mL water.
Preparation of Au@Pd NBs
Briefly, Na2PdBr4 was introduced into 5 mL Au decahedral colloid (the molar ratio of Pd and Au was 8), and then it was stirred for 2 min to form a homogenous solution. N2 was bubbled for at least 30 min to remove O2, and then the solution was heated in an oil bath at 180 °C. During the entire reaction, an atmosphere of N2 was needed. The solution gradually changed from red to gray, indicating that Au@Pd NBs formed. After 5 h, the heating source was removed and the solution was cooled down to room temperature. The purification of Au@Pd NBs was similar to that of Au decahedra, except for the centrifugation speed (6000 rpm).The catalytic reaction of p-nitrophenol to p-aminophenol
An ice-cold NaBH4 solution (2 mL, 60 mM) was mixed with 2 mL p-nitrophenol solution (0.34 mM) and then the mixture was stirred for 15 min. In order to promote the reduction of p-nitrophenol to p-aminophenol, 0.5 mL purified colloid containing NPs (0.002 mg metal) was introduced as a catalyst. Inductively coupled plasma mass spectrometry (OPTIMA8000) was used to precisely determine the used metal catalyst concentration. Before measurement, aqua regia was used to oxidize the metal to soluble ions (be careful: Aqua regia is highly corrosive). A mixture of 3 mL of the above solution containing NaBH4, p-nitrophenol and metal catalyst was quickly transferred to a quartz cuvette and analyzed using a UV-vis spectrometer at regular intervals at room temperature.Characterization
For TEM, HRTEM, energy dispersive spectroscopy (EDS), HAADF and STEM-EDS characterizations, 10 μL of purified aqueous colloid was deposited on a copper grid with a thin carbon film, and was then dried at 50 °C for 20 min. The dried sample was observed using a 300 kV F30 TEM with an attached EDS and STEM detector. All UV-vis spectra were recorded with a Shimadzu 2450 UV-vis spectrophotometer at room temperature.Results and discussion
Structural characterization of Au@Pd NBs
Monodispersed Au decahedral NPs acted as seeds for growing Pd (Fig. S1†). Appropriate conditions allowed for the synthesis of Au@Pd NBs with high yield. The HAADF image at low magnification demonstrates this (Fig. 1A). Compared with Au decahedral NPs, although the length change along 〈100〉 is clear, it is significantly smaller than that along 〈110〉, implying that the growth rate along 〈110〉 was faster than that along 〈100〉. As is well known, the bounding facets of NBs are stepped.42 For a common Au NB, 10 stepped {100} facets are often equivalent and this gives a very regular bipyramid-like shape. However, for the prepared Au@Pd NBs here, the TEM and HAADF images show that the bounding facets are different, and therefore, they have no regular bipyramid-like shape. Their structure can be clearly observed using HAADF. It is known that the contrast of a HAADF image is sensitive to the atomic number of the metal. The atomic number of Pd (46) is smaller than that of Au (79), so the HAADF image can give a clear structure because Au decahedral cores are lighter than Pd shells (Fig. 1A and B). The structure and composition were further confirmed using STEM-EDS technology (Fig. 1C–G). The composition line profile also gave the same structure and elemental distribution (Fig. 1G).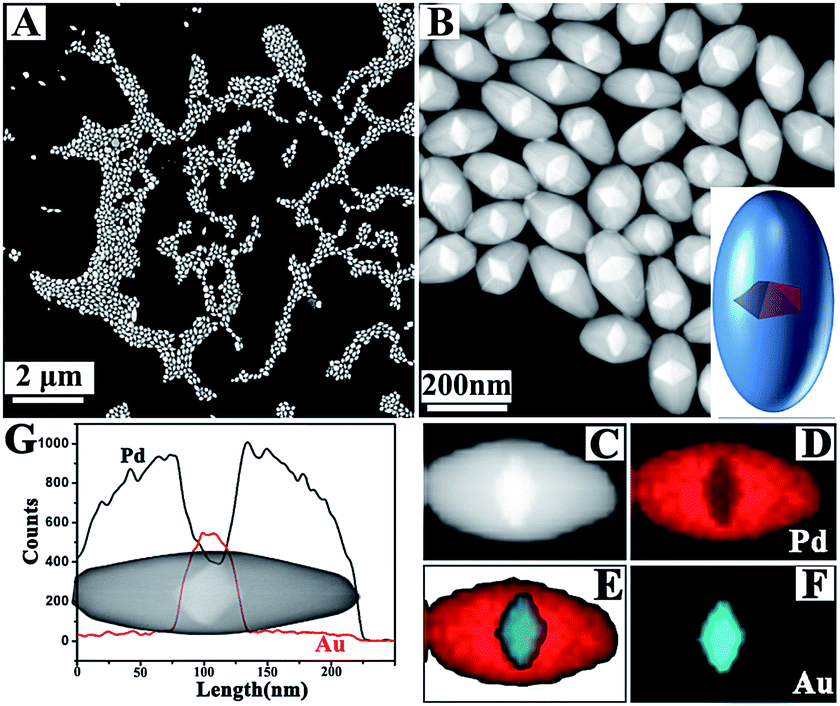 | ||
| Fig. 1 (A, B) Typical HAADF images of Au@Pd NBs; (C–F) elemental mapping results of one Au@Pd NB; (G) compositional line profiles of one Au@Pd NB. | ||
HRTEM was used to further investigate the product structure (Fig. 2). Compared with the HAADF image, the contrast change of a normal TEM image follows an inverse law with atomic number. Metals with a high atomic number are dark because more transmitted electrons are scattered. Therefore, the Au decahedral core is a little darker than the Pd shell in the TEM image (Fig. 2A). The TEM image also shows that the shell thickness of Pd along 〈100〉 is smaller than that along 〈110〉, confirming that the growth occurred preferentially along 〈110〉. The HRTEM images show that the Au@Pd NBs possess typical PFT features and the fast Fourier transform (FFT) pattern also demonstrates that they are PFT structurally (Fig. 2C–G).47 The HRTEM images show a 0.225 nm interplanar spacing, which is attributed to the Pd(111) plane (Fig. 2D–F). Moreover, the HRTEM images also demonstrate that the products have good crystalline features. We also used HRTEM to observe the stepped {100} facet and the results are shown in Fig. S2.† Stepped {100} facets were clearly observed.
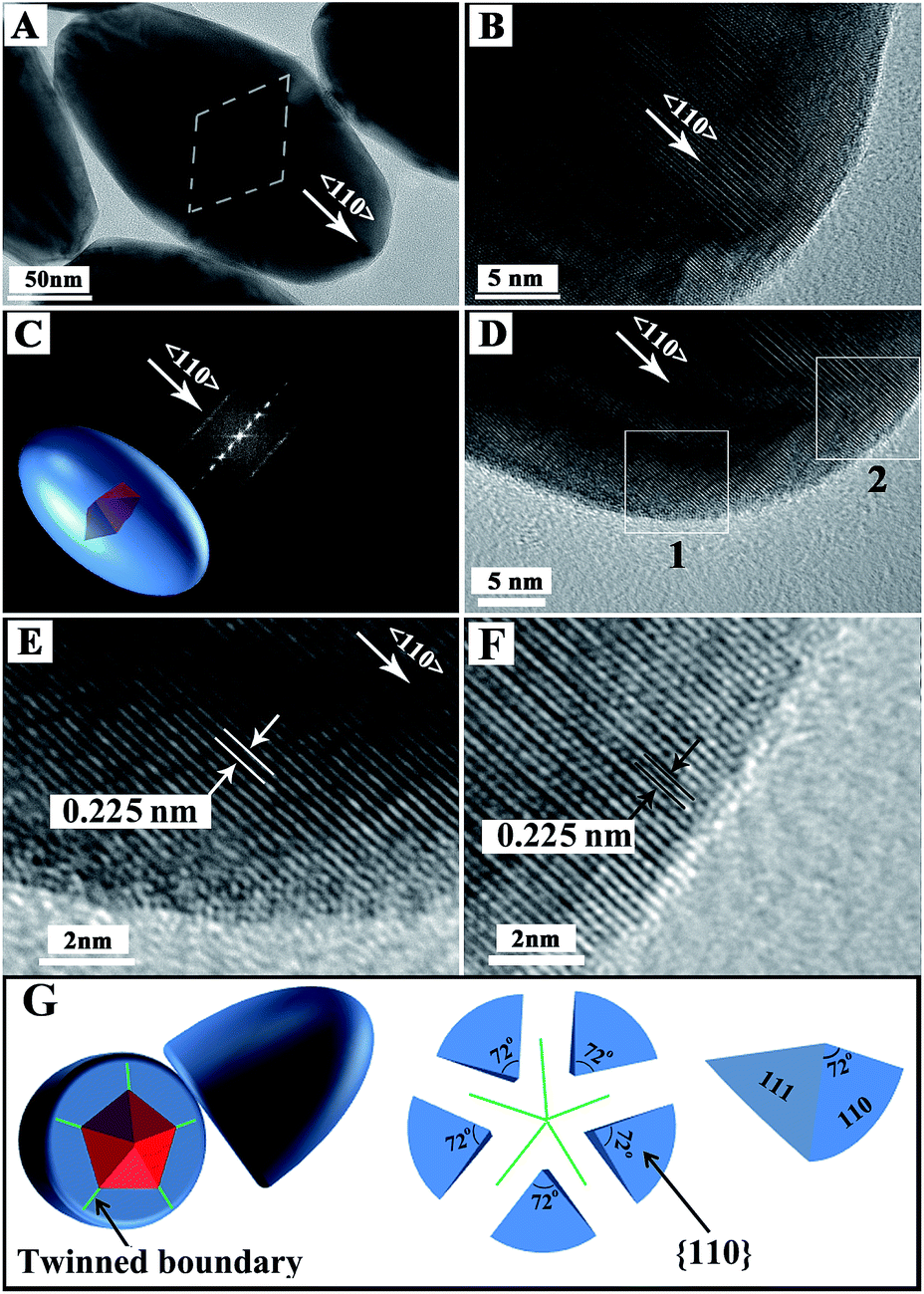 | ||
| Fig. 2 (A–F) Typical TEM and HRTEM images of one Au@Pd NB (E and F are the enlarged images of areas marked by box 1 and box 2 in D, respectively); (G) schematic sketch of Au@Pd NB. | ||
Generally, the pattern of seeded growth is always determined by system energy minimization. When Pd(II) is reduced to Pd, its deposition on Au decahedral seeds can follow two patterns. One is the deposition of Pd atoms onto the seeds and another is the self-nucleation of Pd atoms to form pure Pd NPs. Self-nucleation implies the formation of new particles, which can increase the total metal/solvent interface area, and thus, the system will have higher energy. Therefore, the selective deposition of Pd atoms on the seed surface is favored energetically when growth kinetics is appropriate. In this case, the final products always possess the same crystalline structure as the seeds. Here, the growth of Pd on Au decahedra follows this rule. When PFT Au decahedra were not introduced, the products were mostly single crystalline Pd NPs and very few were PFT Pd NRs (Fig. S3†), confirming that the crystalline structure of the seed was the determinant, that is, Au decahedral seeds induced the growth of Pd atoms.
Effect of reaction time
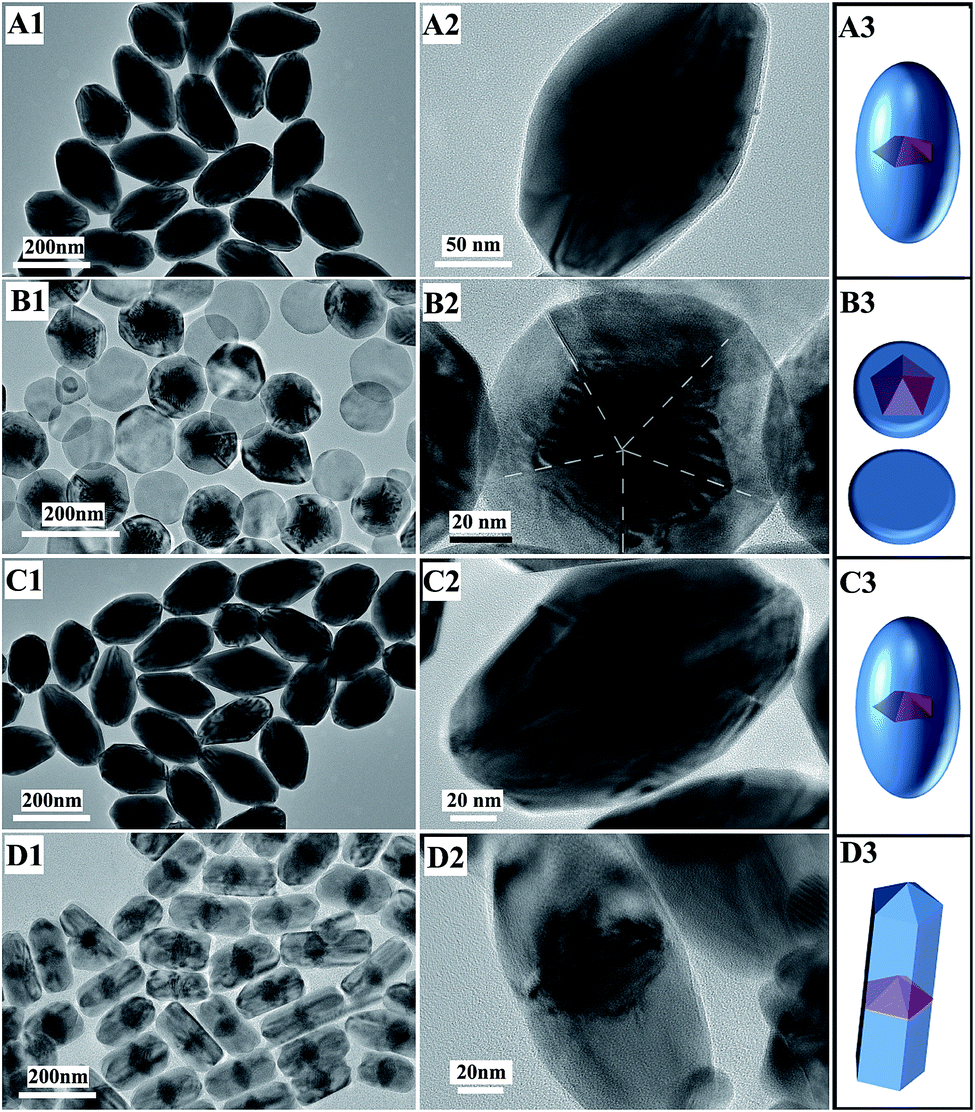
In order to investigate the structure evolution with time, the products at different stages were taken out and observed using TEM. Before growth, the seeds were Au decahedra with uniform shapes (Fig. 3A1–A3) and their edges were close to 45 nm. After a 1.5 h reaction, the growth of Pd on the Au decahedral seeds was observed and Au@Pd NPs were formed (Fig. 3B3). Clearly, compared with Au decahedral seeds, the product has a large transverse length (Fig. 3B2), implying that the growth of Pd along 〈100〉 occurred. More fringes were clearly observed along 〈110〉 because of the large lattice mismatch (4.61%) between the Pd-shell and Au-core.28 This also provides evidence that the growth of Pd along 〈110〉 occurred. Therefore, Pd was deposited on the Au decahedra simultaneously along 〈100〉 and 〈110〉 during this stage. Upon increasing the reaction time to 3 h, some short bipyramid-like products formed, indicating that the growth rate along 〈110〉 was faster than that along 〈100〉 (Fig. 3C1–C3). After the reaction time was further prolonged to 5 h, almost all products were Au@Pd NBs (Fig. 3D1–D3). When the reaction time was more than 5 h, no significant changes in the shape and size were observed. This clearly demonstrates that the growth of Pd was completed within 5 h. | ||
| Fig. 3 TEM images and schematic sketches of products at different times: (A1–A3) 0; (B1–B3) 1.5 h; (C1–C3) 3 h; (D1–D3) 5 h. | ||
Effect of halide ions
In the preparation of Au@Pd NBs, Na2PdBr4 was used as a Pd precursor. As described above, halide ions have an affinity for noble metals, and for this reason, they are always used as growth-modifiers in the preparation of noble nanostructures, especially those containing Au or Pd.55–70 It is commonly believed that they can selectively adsorb on given facets, block growth from occurring there, and promote growth on other facets. This preferential growth causes the formation of nanostructures with specific morphologies. For example, I− ions often adsorb on Pd(100) and facilitate the growth of {100}-faceted Pd nanostructures (nanocube, nanocuboid, PFT nanorod).58,65 In our system, two types of halide ions might play important roles: Cl− ions in PDDA and Br− ions. Although no Br− ions were added, the metal precursor (Na2PdBr4) can release Br− ions in the reduction of Pd(II) to Pd. Previously, it has been demonstrated that Cl− has no decisive effects on the growth of Pd nanostructures.65 Therefore, Br− is possibly crucial to the growth of Au@Pd NBs.In order to clarify this, we carried out a synthesis in which Na2PdCl4 was used as precursor to replace Na2PdBr4. No Au@Pd NBs were observed and decahedral Au-core@Pd-shell NPs were formed (Fig. 4B). Compared to when Au decahedral seeds were used, the product size was clearly increased. This indicates that no preferential growth along 〈110〉 happened when Na2PdCl4 was used, that is, the growth along both 〈100〉 and 〈110〉 occurred simultaneously. Therefore, it is easy to confirm that Br− is critical to promoting growth along 〈110〉 and to blocking growth along 〈100〉. To further confirm this, NaBr was introduced into the above system (the molar ratio of NaBr/Na2PdCl4 was 4). In this case, the products showed almost no distinctive differences to those prepared with Na2PdBr4 (Fig. 4A and C). This further confirms that Br− ions play an important role in the growth of Au@Pd NBs.
 | ||
| Fig. 4 Typical TEM images, HRTEM images and schematic sketches of the products prepared using different precursors and halide ions: (A1–A3) Na2PdBr4; (B1–B3) Na2PdCl4; (C1–C3) Na2PdCl4 and NaBr; (D1–D3) PdCl2 and NaI. | ||
We also carried out growth in the presence of NaI. When NaI was added (the molar ratio of NaI/Na2PdCl4 was 4), the TEM images show that Pd–Au–Pd segmental nanorods formed (Fig. 4D). I− has a very strong affinity for Pd(100). In this case, the growth along 〈100〉 became slow and was blocked almost completely. This is similar to the results reported by our group previously.65 We also carried out a free-halide-ion synthesis in which polyvinylpyrrolidone and Pd(NO3)2 were used as a capping agent and metal precursor, respectively (Fig. S3 and S4†). No preferential growth on Au decahedra was observed and dendritic Au@Pd NPs formed, indicating that halide ions are important to the growth pattern. It is worth noting that solvent also has important impacts on the growth pattern. Previously, it was reported that Br− ions are unable to block the growth of Pd along 〈100〉 in water when I− ions are absent.65 Herein, the results show that Br− ions in DEG can significantly reduce the growth of Pd along 〈110〉 and facilitate the formation of Au@Pd NBs. The difference in the environment possibly changes the affinity between Pd and halide ions.
Aside from the growth pattern on the Au decahedral seeds, halide ions also affected the reduction rate of Pd(II) to Pd(0), that is, the growth kinetics.71 When Na2PdCl4 was used as a precursor, many round Pd nanoplates were observed (Fig. 4B1 and B2), implying that self-nucleation occurred significantly. This self-nucleation is attributed to the oxidation-reduction potential (ORP) differences between various PdX42−/Pd (X = Cl−, Br−, I−) complexes. Compared with the ORP of PdCl42−/Pd, the ORP of PdBr42−/Pd is low because Br− can form more stable complexes with Pd(II) (Table S1†). As a result, the Br− ion-free reduction of Pd(II) to Pd(0) was very fast and consequently, self-nucleation occurred, causing the formation of pure Pd nanoplates (Fig. 4B).54 Once NaBr was introduced, Br− could replace Cl− to form Na2PdBr4. This can be demonstrated by the color and UV-vis spectral changes (Fig. S6†). The Na2PdCl4 solution is light yellow and exhibits an absorption peak at 220 nm (Curve 1 in Fig. S6A and B1†). For the Na2PdBr4 solution, it is brownish red and exhibits a double-peak absorption (Curve 2 in Fig. S6A and B2†). In the case of the solution containing Na2PdCl4 and NaBr (NaBr/Na2PdCl4 = 4), the solution has a similar color and UV-vis spectrum to the Na2PdBr4 solution (Curve 3 in Fig. S6A and B3†). This indicates that Br− replaced Cl− to form Na2PdBr4. Because the ORP of PdBr42−/Pd is lower than that of PdCl42−/Pd, the growth rate decreased and self-nucleation was not observed.
Possible mechanism
We carefully measured the transverse and longitudinal length changes of the products at different stages according to the TEM images (Fig. 5). The longitudinal length of the Au decahedra can be calculated geometrically because they are regular polyhedra. The longitudinal length of the products with a reaction time of 1.5 h is difficult to measure based on the TEM images because their five-fold axis (i.e., longitudinal direction) is always parallel to the electron beam. Therefore, we did not measure the longitudinal length of the products with a reaction time of 1.5 h. However, the evolution of the longitudinal length throughout the growth process can still be observed clearly. The transverse length increased from 65 nm to 110 nm in 1.5 h (Fig. 5A and S7†). This indicates that the growth along 〈100〉 was very fast during the initial 1.5 h. For the products with reaction times longer than 1.5 h (Fig. 5, S7 and S8†), their transverse lengths showed no significant changes (from 110 nm to 119 nm within 3.5 h). However, the longitudinal length increased greatly (from 137 nm to 230 nm within 2 h), indicating that the growth rate along 〈100〉 was very slow, compared with that along 〈110〉. The above results clearly demonstrate that the growth of Pd on Au decahedral seeds can be divided into two stages (Scheme 1). In the first stage, the growth of Pd on Au decahedral seeds occurs along both 〈110〉 and 〈100〉, and thus, the growth preference along 〈110〉 is not significant. It is easy to understand how this growth mode happens through analyzing the change of Br− ions with reaction time. Initially, there are not enough Br− ions in the system because they are bound in Na2PdBr4. For this reason, {100} and {111} are active because no Br− ions adsorb; therefore, growth occurs along both directions. With increasing reaction time (Scheme 1), more Br− ions are released during the second stage because Na2PdBr4 is consumed. There are enough Br− ions that adsorb on {100} densely and decrease the activity there. Therefore, the growth along 〈100〉 becomes slow and the growth rate along 〈110〉 increases relatively.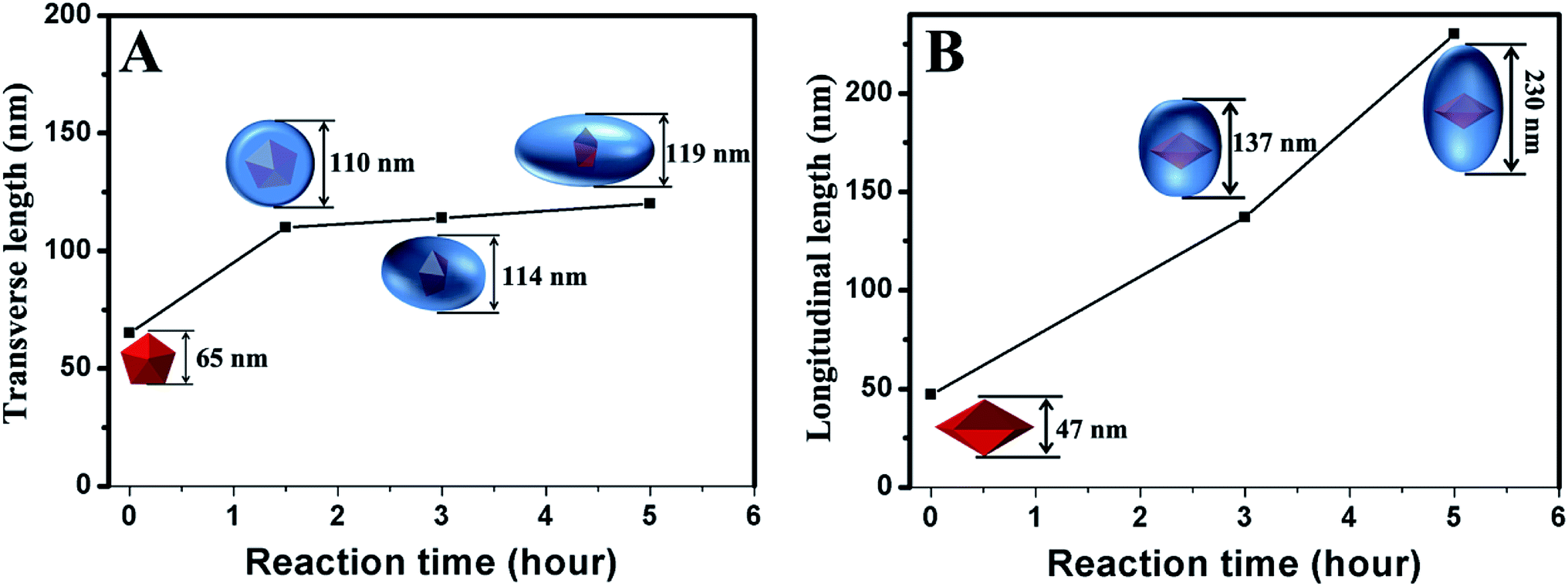 | ||
| Fig. 5 (A) Transverse length and (B) longitudinal length change with reaction time. | ||
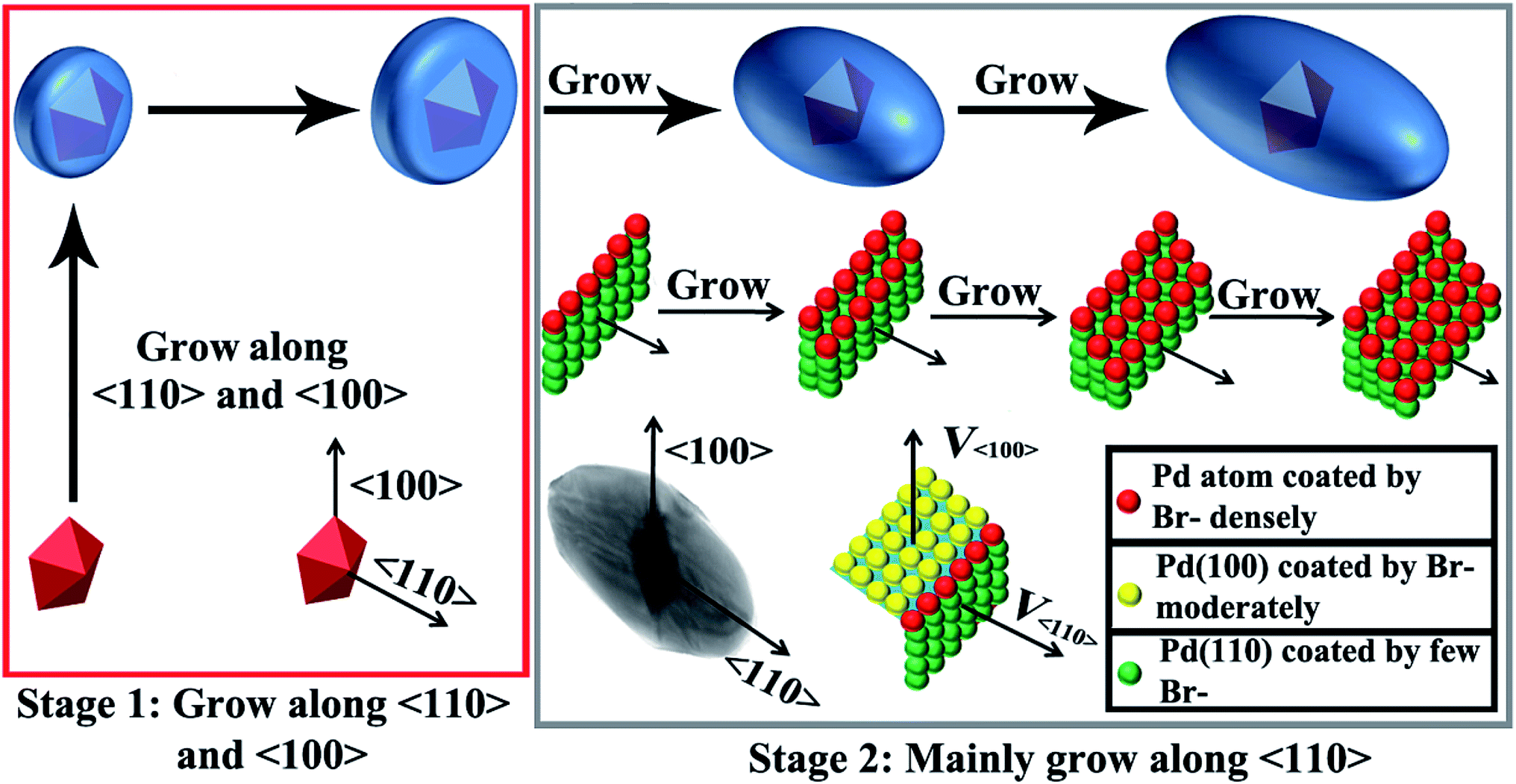 | ||
| Scheme 1 Schematic illustration showing the growth of Au@Pd NBs. | ||
The above results show that the formation of Au@Pd NBs is due to the preferential growth along 〈110〉 during the second stage. It is difficult to figure out exactly how Br− ions cause the preferential growth of Au@Pd NBs during this stage, because there are no appropriate in situ technologies for observing the adsorption of Br−. Here, it is believed that Br− ions during the growth of NBs might have a similar effect to that of Ag(I)-based substances during the growth of Au NBs (Scheme 1).47 During growth, Br− ions selectively adsorb onto Pd{100}, and therefore, the growth rate along 〈100〉 becomes reduced. The growth along 〈110〉 then becomes relatively accelerated. In fact, the atoms on the {110} surface have a different environment. The atoms on the edge marked by the red balls (stage 2 in Scheme 1) have the lowest coordination number, so their surface energy is the highest. Theoretically, the growth rate should be fast in this case. However, they can adsorb more Br− ions because of the high surface free energy. For this reason, Pd{110} is less active, so the growth on this surface is blocked or slower than that on {110} surfaces containing other atoms. This causes the formation of stepped {100} facets and consequently, Au@Pd NBs form. To obtain a clearer mechanism, further studies are underway in our group.
Shape-dependent catalytic performance
As mentioned before, Pd-based nanostructures are good catalysts in many reactions.1–10 Their catalytic performance is highly dependent on composition, structure and shape.1–5 Compared with commonly observed nanostructures, {100}-stepped nanostructures often exhibit good catalytic activity due to the low coordination number of the surface atoms.19–22,72–79 Here, the reduction of p-nitrophenol to p-aminophenol was chosen as a model reaction to investigate the catalytic properties of NBs.71 Furthermore, PFT Pd–Au–Pd segmental nanorods and single crystalline Pd nanocubes and octahedra were also used as catalysts for comparison (Fig. S13, S14 and S16†). Fig. 6A shows the time-dependent UV-vis absorption of the reaction solution when Au@Pd NBs were present as a catalyst. Clearly, the absorption intensity at 397 nm gradually decreased with time, indicating that p-nitrophenol was consumed. However, the absorption at about 297 nm became significant with time because the concentration of p-aminophenol increased. The catalytic performances of various catalysts are shown in Fig. 6B. When no catalyst was used, the reaction rate was very slow. After 30 min, only 8% p-nitrophenol was reduced to p-aminophenol (Curve 1). When {111}-faceted Pd octahedra with edges of 53 nm served as the catalyst (Fig. S16†), p-nitrophenol was still clearly detected (7%) after 30 min (Curve 2). When {100}-faceted Pd nanocubes with edges of 45 nm acted as the catalyst, the reduction of p-nitrophenol to p-aminophenol was completed in 30 min (Curve 3). In the case of Pd–Au–Pd nanorods, 18 min was needed to finish the reduction (Curve 4). When Au@Pd NBs acted as the catalyst, the reduction rate was the fastest and no p-nitrophenol was detected after 9 min (Curve 5). The above results distinctively demonstrate that Au@Pd NBs have the highest catalytic activity, while Pd nanocubes have the lowest. The total surface catalytic sites can be roughly evaluated. When the mass of the metal is constant in four cases, it is easy to know the sequence of their total surface area through analyzing their sizes (Au@Pd-NB < Pd–Au–Pd-nanorod < Pd-octahedron < Pd-cube) (Fig. S17†). However, the catalytic performance follows the reverse order, confirming that both the bounding facet and twinned interface play important roles. It is believed that the high catalytic performance of the Au@Pd NBs is due to the presence of stepped {100} facets on their surfaces. Furthermore, twinned planes are demonstrated to exhibit high activity and possibly make a contribution to the catalytic activity.65 The low activity of the Pd nanocubes and octahedra also supports this conclusion. The Au@Pd NBs and Pd–Au–Pd nanorods used here are PFT structures. However, the Pd nanocubes and octahedral NPs are typical single crystalline nanostructures.56,65 Therefore, the Pd nanocubes and octahedral NPs have low activity because no twinned interfaces are present on their surfaces. The {100}-faceted Pd nanocubes and {111}-faceted octahedra have similar surface areas, but {100} is more active than {111}. Therefore, the Pd nanocubes have a higher activity than the octahedra.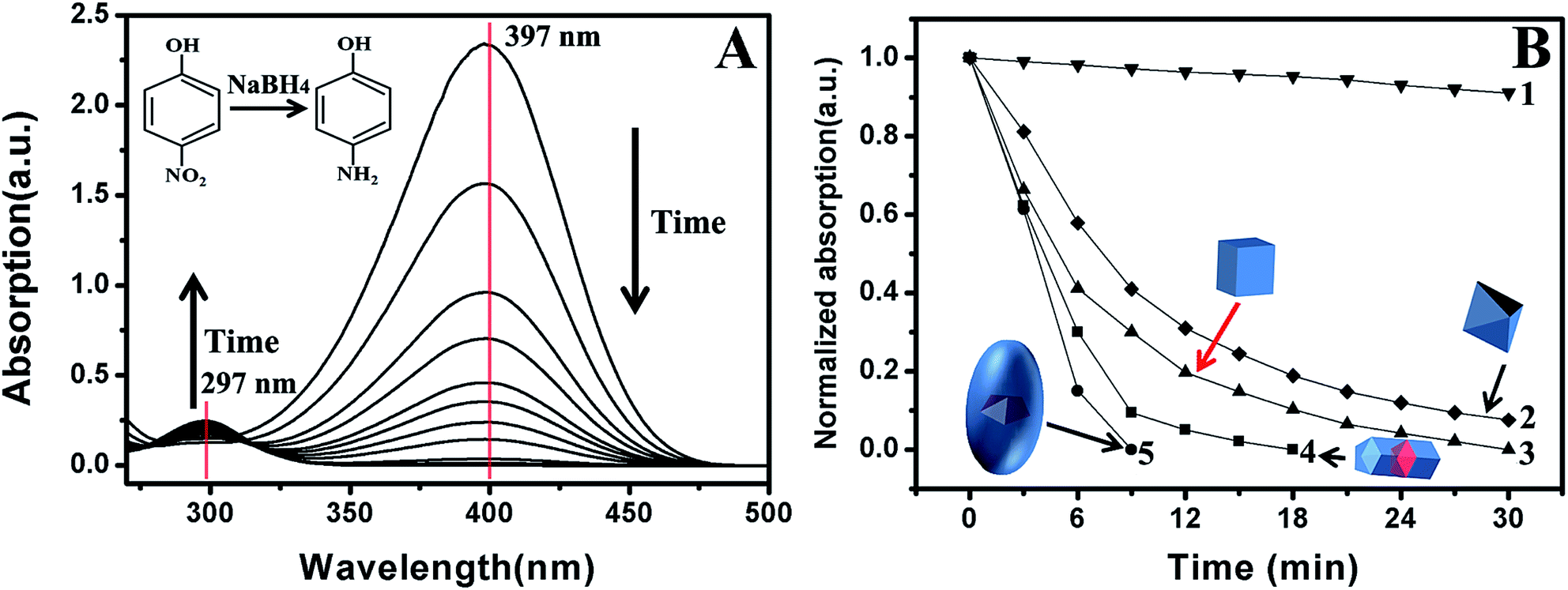 | ||
| Fig. 6 (A) Time-dependent UV-vis absorption of the reaction mixture when Au@Pd NBs were used as catalysts. (B) Normalized absorption at the peak position of p-nitrophenol as a function of time when various Pd-based nanostructures were used as catalysts: (1) without catalyst; (2) Pd octahedra; (3) Pd nanocubes; (4) Pd–Au–Pd segmental nanorods; (5) Au@Pd NBs. | ||
Conclusion
We have demonstrated that PFT Au@Pd NBs can be synthesized through growing Pd on Au decahedral NPs in a polyol system. Br− ions play important roles in the growth of Au@Pd NBs because they have appropriate affinity for Pd(100), which facilitates the formation of stepped {100} facets and preferential growth along 〈110〉. When the affinity is too strong (I−), the growth along 〈100〉 is completely blocked and the formation of Pd–Au–Pd segmental nanorods is preferred. When the affinity is too weak (Cl−), the growth along both 〈100〉 and 〈110〉 is significant and consequently, Au-core@Pd-shell NPs form. It is believed that the growth of Au@Pd NBs mainly involves two stages. In the first stage, Pd atoms grow on Au decahedral NPs along both 〈110〉 and 〈100〉 directions because not enough Br− ions adsorb on Pd(100). In the next stage, more Na2PdBr4 is consumed and, meanwhile, many Br− ions are released. Subsequently, Br− ions adsorb densely onto Pd(100), block the growth along 〈100〉 and, consequently, facilitate the growth of Au@Pd NBs. Compared with single crystalline Pd nanocubes, Pd octahedra and PFT Pd–Au–Pd segmental nanorods, the prepared Au@Pd NBs have high catalytic activity in the reduction of p-nitrophenol to p-aminophenol due to the bounding stepped {100} facets.Acknowledgements
This work was supported by the NSFC (21471117, 21173159 and 51420105002).Notes and references
- X. Zhu, H. Jia, X.-M. Zhu, S. Cheng, X. Zhuo, F. Qin, Z. Yang and J. Wang, Adv. Funct. Mater., 2017, 27, 1700016 CrossRef.
- L. Zhou, Z. K. Liu, H. Zhang, S. Cheng, L.-J. Fan and W. L. Ma, Nanoscale, 2014, 6, 12971–12980 RSC.
- F. Ren, H. Lu, H. Liu, Z. Wang, Y. Wu and Y. Li, J. Mater. Chem. A, 2015, 3, 23660–23663 CAS.
- X. Huang, H. Zhang, C. Guo, Z. Zhou and N. Zheng, Angew. Chem., Int. Ed., 2009, 48, 4808–4906 CrossRef CAS PubMed.
- T. T. Miao, Y. H. Song, C. X. Bi, H. B. Xia, D. Y. Wang and X. T. Tao, J. Phys. Chem. C, 2015, 119, 18434–18443 CAS.
- Y. Li, W. Wang, K. Xia, W. Zhang, Y. Jiang, Y. Zeng, H. Zhang, C. Jin, Z. Zhang and D. Yang, Small, 2015, 11, 4745–4752 CrossRef CAS PubMed.
- L. Sun, Z. C. Zhang, B. Xu and X. Wang, Chem.–Asian J., 2013, 8, 1523–1529 CrossRef CAS PubMed.
- H. You, S. Yang, B. Ding and H. Yang, Chem. Soc. Rev., 2013, 42, 2880–2904 RSC.
- B. Wu and N. Zheng, Nano Today, 2013, 8, 168–197 CrossRef CAS.
- X. Xia, S.-I. Choi, J. A. Herron, N. Lu, J. Scaranto, H.-C. Peng, J. Wang, M. Mavrikakis, M. J. Kim and Y. Xia, J. Am. Chem. Soc., 2013, 135, 15706–15709 CrossRef CAS PubMed.
- D. Kim, Y. W. Lee, S. B. Lee and S. W. Han, Angew. Chem., Int. Ed., 2012, 51, 159–163 CrossRef CAS PubMed.
- Y. Zheng, J. Tao, H. Liu, J. Zeng, T. Yu, Y. Ma, C. Moran, L. Wu, Y. Zhu, J. Liu and Y. Xia, Small, 2011, 7, 2307–2312 CrossRef CAS PubMed.
- L. Zhang, H. Su, M. Sun, Y. Wang, W. Wu, T. Yu and J. Zeng, Nano Res., 2015, 8, 2415–2430 CrossRef CAS.
- M. Jin, H. Zhang, Z. X. Xie and Y. N. Xia, Energy Environ. Sci., 2012, 5, 6352–6357 CAS.
- J. Mao, Y. Liu, Z. Chen, D. Wang and Y. Li, Chem. Commun., 2014, 50, 4588–4591 RSC.
- L. Wei, C. D. Xu, L. Huang, Z. Y. Zhou, S. P. Chen and S. G. Sun, J. Phys. Chem. C, 2016, 120, 15569–15577 CAS.
- D. Chen, J. Li, P. Cui, H. Liu and J. Yang, J. Mater. Chem. A, 2016, 4, 3813–3821 CAS.
- M. Jin, H. Zhang, Z. Xie and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 7850–7854 CrossRef CAS PubMed.
- Z. Quan, Y. Wang and J. Fang, Acc. Chem. Res., 2013, 46, 191–202 CrossRef CAS PubMed.
- S. Liu, N. Tian, A.-Y. Xie, J.-H. Du, J. Xiao, L. Liu, H.-Y. Sun, Z.-Y. Cheng, Z.-Y. Zhou and S.-G. Sun, J. Am. Chem. Soc., 2016, 138, 5753–5756 CrossRef CAS PubMed.
- A.-X. Yin, X.-Q. Min, W. Zhu, W.-C. Liu, Y.-W. Zhang and C.-H. Yan, Chem.–Eur. J., 2012, 18, 777–782 CrossRef CAS PubMed.
- R. G. Weiner, C. J. DeSantis, M. B. T. Cardoso and S. E. Skrabalak, ACS Nano, 2014, 8, 8625–8635 CrossRef CAS PubMed.
- C. J. DeSantis and S. E. Skrabalak, J. Am. Chem. Soc., 2013, 135, 10–13 CrossRef CAS PubMed.
- X. Huang, Y. Li, Y. Li, H. Zhou, X. Duan and Y. Huang, Nano Lett., 2012, 12, 4265–4270 CrossRef CAS PubMed.
- D. Seo, J. H. Park, J. Jung, S. M. Park, S. Ryu, J. Kwak and H. Song, J. Phys. Chem. C, 2009, 113, 3449–3454 CAS.
- W. X. Niu, L. Zhang and G. B. Xu, ACS Nano, 2010, 4, 1987–1996 CrossRef CAS PubMed.
- C. Li, R. Sato, M. Kanehara, H. Zeng, Y. Bando and T. Teranishi, Angew. Chem., Int. Ed., 2009, 48, 6883–6887 CrossRef CAS PubMed.
- F. Fan, F.-R. Fan, D.-Y. Liu, Y.-F. Wu, S. Duan, Z.-X. Xie, Z.-Y. Jiang and Z.-Q. Tian, J. Am. Chem. Soc., 2008, 130, 6949–6951 CrossRef CAS PubMed.
- L. Zhang, Z. Xie and J. Gong, Chem. Soc. Rev., 2016, 45, 3916–3934 RSC.
- H. Zhang, M. Jin, Y. Xiong, B. Lim and Y. Xia, Acc. Chem. Res., 2012, 46, 1783–1794 CrossRef PubMed.
- D. Seo, C. I. Yoo, I. S. Chung, S. M. Park, S. Ryu and H. Song, J. Phys. Chem. C, 2008, 112, 2469–2475 CAS.
- N. Tian, Z. Y. Zhou, N. F. Yu, L. Y. Wang and S. G. Sun, J. Am. Chem. Soc., 2010, 132, 7580–7581 CrossRef CAS PubMed.
- D. Chateau, A. Liotta, F. Vadcard, J. R. G. Navarro, F. Chaput, J. Lermé, F. Lerouge and S. Parola, Nanoscale, 2015, 7, 1934–1943 RSC.
- A. Sánchez-Iglesias, N. Winckelmans, T. Altantzis, S. Bals, M. Grzelczak and L. M. Liz-Marzán, J. Am. Chem. Soc., 2017, 139, 107–110 CrossRef PubMed.
- M. Liu and P. Guyot-Sionnest, J. Phys. Chem. B, 2005, 109, 22192–22200 CrossRef CAS PubMed.
- J.-H. Lee, K. J. Gibson, G. Chen and Y. Weizmann, Nat. Commun., 2015, 6, 7571 CrossRef CAS PubMed.
- Z. C. Canbek, R. Cortes-Huertos, F. Testard, O. Spalla, S. Moldovan, O. Ersen, A. Wisnet, G. Wang, J. Goniakowski, C. Noguera and N. Menguy, Cryst. Growth Des., 2015, 15, 3637–3644 CAS.
- X. L. Zhuo, X. Z. Zhu, Q. Li, Z. Yang and J. F. Wang, ACS Nano, 2015, 9, 7523–7535 CrossRef CAS PubMed.
- W. Rao, Q. Li, Y. Wang, T. Li and L. Wu, ACS Nano, 2015, 9, 2783–2791 CrossRef CAS PubMed.
- W. Liu, D. Liu, Z. Zhu, B. Han, Y. Gao and Z. Tang, Nanoscale, 2014, 6, 4498–4502 RSC.
- E. C. Le Ru, J. Grand, I. Sow, W. R. C. Somerville, P. G. Etchegoin, M. Treguer-Delapierre, G. Charron, N. Félidj, G. Lévi and J. Aubard, Nano Lett., 2011, 11, 5013–5019 CrossRef CAS PubMed.
- J. Burgin, I. Florea, J. Majimel, A. Dobri, O. Ersen and M. Tréguer-Delapierre, Nanoscale, 2012, 4, 1299–1303 RSC.
- X. Zhang, M. Tsuji, S. Lim, N. Miyamae, M. Nishio, S. Hikino and M. Umezu, Langmuir, 2007, 23, 6372–6376 CrossRef CAS PubMed.
- A. Lombardi, M. Loumaigne, A. Crut, P. Maioli, N. Del Fatti, F. Vallée, M. Spuch-Calvar, J. Burgin, J. Majimel and M. Tréguer-Delapierre, Langmuir, 2012, 28, 9027–9033 CrossRef CAS PubMed.
- Q. Li, X. Zhuo, S. Li, Q. Ruan, Q. H. Xu and J. Wang, Adv. Opt. Mater., 2015, 3, 801–812 CrossRef CAS.
- L. Zhang, Q. Chen, X. Wang and Z. Jiang, Nanoscale, 2016, 8, 2819–2825 RSC.
- G. Zhou, Y. Yang, S. Han, W. Chen, Y. Fu, C. Zou, L. Zhang and S. Huang, ACS Appl. Mater. Interfaces, 2013, 5, 13340–13352 CAS.
- S. Gómez-Graña, B. Goris, T. Altantzis, C. Fernández-López, E. Carbó-Argibay, A. Guerrero-Martínez, N. Almora-Barrios, N. López, I. Pastoriza-Santos, J. Pérez-Juste, S. Bals, G. Van Tendeloo and L. M. Liz-Marzán, J. Phys. Chem. Lett., 2013, 4, 2209–2216 CrossRef.
- C. Fang, G. Zhao, Y. Xiao, J. Zhao, Z. Zhang and B. Geng, Sci. Rep., 2016, 6, 36706 CrossRef CAS PubMed.
- S. Hong, K. L. Shuford and S. Park, Chem. Mater., 2011, 23, 5375–5378 CrossRef CAS.
- M. R. Langille, M. L. Personick, J. Zhang and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 14542–14554 CrossRef CAS PubMed.
- D. G. Duff, A. C. Curtis, P. P. Edwards, D. A. Jefferson, B. F. G. Johnson, A. I. Kirkland and D. E. Logan, Angew. Chem., Int. Ed. Engl., 1987, 26, 676–678 CrossRef.
- C. J. Johnson, E. Dujardin, S. A. Davis, C. J. Murphy and S. Mann, J. Mater. Chem., 2002, 12, 1765–1770 RSC.
- B. Jiang, L. Xu, W. Chen, C. Zou, Y. Yang, F. Z. Fu and S. M. Huang, Nano Res., 2017 DOI:10.1007/s12274-017-1562-y.
- H. H. Chen, A. A. Ho and M. H. Huang, J. Am. Chem. Soc., 2009, 131, 9114–9121 CrossRef PubMed.
- Y. Tang, R. E. Edelmann and S. Zou, Nanoscale, 2014, 6, 5630–5633 RSC.
- Y. Yu, Q. B. Zhang, B. Liu and J. Y. Lee, J. Am. Chem. Soc., 2010, 132, 18258–18265 CrossRef CAS PubMed.
- L.-f. Zhang and C.-Y. Zhang, Nanoscale, 2013, 5, 6074–6080 RSC.
- A. Wang, Q. Peng and Y. D. Li, Chem. Mater., 2011, 23, 3217–3222 CrossRef CAS.
- Y. Park, Y. W. Lee, S. W. Kangab and S. W. Han, Nanoscale, 2014, 6, 9798–9805 RSC.
- N. Almora-Barrios, G. Novell-Leruth, P. Whiting, L. M. Liz-Marzań and N. López, Nano Lett., 2014, 14, 871–875 CrossRef CAS PubMed.
- M. L. Personick, M. R. Langille, J. Zhang, N. Harris, G. C. Schatz and C. A. Mirkin, J. Am. Chem. Soc., 2011, 133, 6170–6173 CrossRef CAS PubMed.
- T. H. Ha, H.-J. Koo and B. H. Chung, J. Phys. Chem. C, 2007, 111, 1123–1130 CAS.
- J. S. DuChene, W. Niu, J. M. Abendroth, Q. Sun, W. Zhao, F. Huo and W. D. Wei, Chem. Mater., 2013, 25, 1392–1399 CrossRef CAS.
- L. Xu, K. Wang, B. Jiang, W. Chen, F. Liu, H. Hao, C. Zou, Y. Yang and S. Huang, Chem. Mater., 2016, 28, 7394–7403 CrossRef CAS.
- L. Scarabelli, M. Grzelczak and L. M. Liz-Marzán, Chem. Mater., 2013, 25, 4232–4238 CrossRef CAS.
- T. K. Sau and C. J. Murphy, Langmuir, 2004, 20, 6414–6420 CrossRef CAS PubMed.
- L. Chen, F. Ji, Y. Xu, L. He, Y. Mi, F. Bao, B. Sun, X. Zhang and Q. Zhang, Nano Lett., 2014, 14, 7201–7206 CrossRef CAS PubMed.
- L. Vigderman and E. R. Zubarev, Chem. Mater., 2013, 25, 1450–1457 CrossRef CAS.
- X. Ye, Y. Gao, J. Chen, D. C. Reifsnyder, C. Zheng and C. B. Murray, Nano Lett., 2013, 13, 2163–2171 CrossRef CAS PubMed.
- Y. Holade, R. G. da Silva, K. Servat, T. W. Napporn, C. Canaff, A. Andrade and K. B. Kokoh, J. Mater. Chem. A, 2016, 4, 8337–8349 CAS.
- R. Long, H. Huang, Y. P. Li, L. Song and Y. J. Xiong, Adv. Mater., 2015, 27, 7025–7042 CrossRef CAS PubMed.
- W. Niu, Y. Duan, Z. Qing, H. Huang and X. Lu, J. Am. Chem. Soc., 2017, 139, 5817–5826 CrossRef CAS PubMed.
- L. Wang, Y. Nemoto and Y. Yamauchi, J. Am. Chem. Soc., 2011, 133, 9674–9677 CrossRef CAS PubMed.
- L. Sun, Q. F. Zhang, G. F. Grace Li, E. Villarreal, X. Q. Fu and H. Wang, ACS Nano, 2017, 11, 3213–3228 CrossRef CAS PubMed.
- K. Zhang, D. Bin, B. B. Yang, C. Q. Wang, F. F. Ren and Y. K. Du, Nanoscale, 2015, 7, 12445–12451 RSC.
- R. Zhao, G. Fu, T. Zhou, Y. Chen, X. Zhu, Y. Tang and T. Lu, Nanoscale, 2014, 6, 2776–2781 RSC.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- Y. Ma, Q. Kuang, Z. Jiang, Z. Xie, R. Huang and L. Zheng, Angew. Chem., Int. Ed., 2008, 47, 8901–8904 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05407f |
| This journal is © The Royal Society of Chemistry 2017 |