
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Macroalgae-derived nitrogen-doped hierarchical porous carbons with high performance for H2 storage and supercapacitors†
Xingxing Wu‡
ab,
Zhongwei Tian‡a,
Langqing Hua,
Sha Huanga and
Jinjun Cai *ab
*ab
aSchool of Chemical Engineering, Xiangtan University, Xiangtan 411105, China. E-mail: caijj@xtu.edu.cn; Fax: +86-731-58298171; Tel: +86-731-58298171
bCAS Key Laboratory of Bio-based Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China
First published on 27th June 2017
Abstract
Using biomass as a precursor for N-doped carbons is critical for future energy storage. Herein, macroalgae pollutants of Enteromorpha prolifera were used to obtain N-doped carbons from carbonization and activation after freeze-drying treatment. The pore structure and surface chemistry of carbons can be altered by adjusting the activation conditions with surface areas up to 3345 m2 g−1 consisting of hierarchical pores, and rich N (1.5–3.7%) groups, making algae a good choice for N-doped carbons as adsorbents and electrode materials. Significantly, H2 uptake is high, up to 7.05 wt% at −196 °C and 20 bar, and an exceptional uptake of 2.71 wt% is also observed at 1 bar, among the largest data relative to state-of-art biomass-based carbons. Electrochemical test results confirmed that the carbon with the largest surface area had a capacitance of 440 F g−1 at 1 A g−1 in 6 M KOH electrolyte while the carbon with the largest microporous surface area had good cycling stability with 87% initial capacitance after 5000 cycles due to its hierarchical pores with high surface areas providing many active sites for better facilitating electrolyte diffusion.
Introduction
The energy crisis and environmental pollution situation has become more and more serious due to an exhaustive usage of fossil fuels. Today, the generation-storage of energy resources in a renewable fashion is probably the greatest challenge. Among these alternative energy sources, H2 is one of the cleanest fuels and is regarded as the most promising candidate because its heating value is about three times larger than petroleum, and its combustion creates only water without pollutants or greenhouse gas emission.1–4 However, effective storage is the greatest bottleneck to realize actual application of H2 energy despite diverse existing methods for H2 production. Porous materials with high surface areas and controlled pore sizes is attractive for H2 storage due to the fact that H2 physisorbed on the adsorbents can be released reversibly.5 Compared with the emerging micro-polymers, zeolites, and MOFs, carbons are particularly of interest for gas storage due to their good stability against moisture and corrosive flue gas. Of late, there are increasing evidence to suggest that carbons with high surface areas are highly effective in gas storage application.6–8 It is thought that carbons with tuned pores might be used to solve climate change especially in relation to high-density H2 storage. Moreover, the fabrication of devices with highly-efficient energy storage has great importance in solving issue of energy crisis. Supercapacitors received great interest considering that they have fast charge capabilities, long-term cycling life and larger energy density.9–11 To meet the requirements for supercapacitors and H2 storage, the key point is the design of suitable adsorbents and electrode materials with simple preparation process to precisely control pore structure of materials.Carbons are of great interest in gas storage and supercapacitors besides from catalysis supports, carbon dots, gas separation and water treatment in view of their hydrophobic nature of surface, high surface area and good chemical stability.10 Significantly, activated carbons still stand out for their low-cost and high surface areas larger than 2000 m2 g−1. As for H2 storage application, carbons with narrow pores in 0.6–0.9 nm range are regarded as the most essential point.1,3 Moreover, high surface area is important in ensuring high uptake at high pressure and can sometimes compensate for the less efficiency of pores larger than 1.0 nm, and KOH activation is the best way for fabricating carbons with such characteristics.6 Sevilla et al.12 reported carbons from the carbonization of polythiophene and KOH activation with H2 uptake of up to 6.64 wt% at 20 bar and −196 °C. The present largest H2 uptake reported on carbons from KOH activation is up to 7.08 wt% at 20 bar and −196 °C.13 As for supercapacitors, carbons are also promising electrode materials especially for the one with large surface area and moderate mesopores.14 The presence of mesopores is very beneficial to facilitate electrolyte diffusion especially at high speed and also can ensure high energy and power densities. Wu et al.15 have ever reported hierarchical carbons from hydrothermal carbonization of bamboo with surface area of 168.6 m2 g−1 showing energy density of 37.8 W h kg−1 in 1 M KOH and the presence of B, N heteroatoms can enhance specific capacitance and energy density. Porous carbons continue to consist of primary choice for H2 storage and supercapacitors due to their microstructure and cost-availability. Studies on activation process which can be allowed to better control over pore sizes and surface areas are also important for the future development. In addition, the nature of precursor had a critical role in determining microstructure of carbons and also enabled heteroatoms-doping on the framework.16 How to achieve large-scale production of carbons with high-performance and cost effective is an important factor for practical utilization in gas storage and supercapacitor. Therefore, it is an urgent task to develop an effective route for preparing carbons with high-surface-areas on a large scale. In this context, searching for a series of secure, renewable, and sustainable sources as the carbonaceous precursors is required.
Biomass is highly attractive for the large-scale preparation of carbons considering that it can be ingested from nature, and hemicelluloses and lignin in biomass have been widely explored to obtain carbons.10,17–19 Although macroalgae are not lignocelluloses in structure, they otherwise known as seaweeds involved proteins endowing them to be promising precursors.20,21 Enteromorpha prolifera (EP) is a typical marine macroalgae rapidly growth due to eutrophication, and a large-scale of EP drift gathering will cause an occurrence of green tide off the coast. Moreover, the growth environment of EP made them have lavish pores in structure to suffice respiration, and there are about 1 billion tons of wet EP harvested while only 1% wastes collected after cleanup every year.22 It is said that a reasonable way for overcoming macroalgae pollution is to transform them into high-value-added matters rather than make them naturally decay. We have previously obtained hierarchical carbons from carbonization of EP with surface area of 422 m2 g−1.23 Herein, we applied EP as precursor to obtain carbons through carbonization and KOH activation after freeze-drying with aims to reach the better EP utilization for protecting marine environment. The conjunction of KOH activation and carbonization is the most common route to obtain carbons with surface areas even up to 3900 m2 g−1 using biomass as precursors.9 Although there are already some studies about carbons from EP as electrode materials,22,24–28 they all applied traditional drying and did not evaluate their potential application as the adsorbents for H2 adsorption, to the best of our knowledge. Several studies have demonstrated that freeze-drying can greatly promote to form hierarchical pores.23,29 Depending on the activation conditions, carbons have tuned hierarchical pores and surface-doping chemistry with surface areas up to 3345 m2 g−1. Significantly, the carbons can be obtained on a large-scale using natural algae, reducing ocean pollution and creating economic benefit. Motivated by the attractive properties of carbons including rich heteroatoms, high surface area and hierarchical pores, we performed gas adsorption for H2 storage and electrochemical measurement to exploit their application related to the clean energy storage. The result indicated that hierarchical carbons from EP can be promising adsorbents for H2 storage and electrode materials for supercapacitors, opening an avenue to design new carbons from the full utilization of sustainable resources.
Experimental section
Preparation of EP-based hierarchical carbons
The fresh EP were collected from the coast of Qingdao, China, washed with distilled water, and dried using a freeze-drying route based on our previous reports.23 Dried EP were cut into pieces and pressed up to 6 MPa for compaction (Fig. 1a), and pre-carbonized at 500 °C under N2 flow for 2 h. The obtained char were ground with KOH in an agate mortar at a KOH/char mass ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 4:1 under an illumination of infrared light to prevent moisture in air. Activation performed at 600–800 °C at about 2 °C min−1 for 1 h under N2 flow of 80 mL min−1, and the derived samples were treated with 4 M HCl for 3 h at 60 °C to remove inorganic impurities, washed with distilled water until a neutral pH derived, and then dried in an oven at 120 °C overnight. The ultimate yield of EP algae-derived carbons is about 19 wt% calculated on the basis of per gram of dried algae used. Carbons referred to AEPC-x-T where ‘x’ denotes KOH/char ratio and ‘T’ refers to temperature.
:1 or 4:1 under an illumination of infrared light to prevent moisture in air. Activation performed at 600–800 °C at about 2 °C min−1 for 1 h under N2 flow of 80 mL min−1, and the derived samples were treated with 4 M HCl for 3 h at 60 °C to remove inorganic impurities, washed with distilled water until a neutral pH derived, and then dried in an oven at 120 °C overnight. The ultimate yield of EP algae-derived carbons is about 19 wt% calculated on the basis of per gram of dried algae used. Carbons referred to AEPC-x-T where ‘x’ denotes KOH/char ratio and ‘T’ refers to temperature.
 | ||
| Fig. 1 (a) Photos for fresh EP; SEM images for (b) AEPC-2-600, (c) AEPC-2-700, (d) AEPC-2-800, and (e) AEPC-4-800; representative TEM images for (f) AEPC-4-800. | ||
Materials characterization
Structure features were studied by powder X-ray diffractometer (XRD, Bruker D8Advance) and Raman spectrometer (LabRAM UVNIR, Jobin-Yvon). Morphology was performed on a scanning electron microscopy (SEM, Hitachi S4800) and transmission electron microscopy (TEM, Tecnai G2 F20, 200 kV). Surface-doping chemistry was characterized by the X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi). N2 adsorption isotherm and pore size distributions (PSD) were performed on a static volumetric sorption analyzer (ASAP2020, Micrometrics, USA). Before measurements, all samples were evacuated under vacuum for 15 h at 200 °C.Gas adsorption and electrochemical measurements
H2 adsorption measurements at −196 °C of 20 bar were performed on an Intelligent Gravimetric Analyzer (IGA, Hiden), which incorporates microbalance capable of weight with resolution of ±0.1 μg. Ultrahigh-purity (99.99%) of H2 were especially further purified using alumina, zeolites, and carbons before introduction into system. All results were corrected for buoyancy effect based on previous parameters,3,6 and samples degassed (10−6 bar) at 200 °C for 4 h before measurements. Electrochemical properties measured in two-electrode cell with 6 M KOH electrolyte on CHI 660A workstation. Electrodes were prepared by pressing a mixture of 70 wt% as-prepared carbon, 20 wt% polyvinylidene fluoride (PVDF) and 10 wt% acetylene black onto nickel foams (d, 10 mm) and vacuum dried at 120 °C. A sandwich-type capacitor consisted of two similar symmetrical electrodes separated by nylon separator, and each electrode was quantified to contain ∼7.0 mg of as-obtained carbon materials. Electrochemical impedance spectroscopy (EIS) measurements were recorded by a sweeping frequency from 100 kHz to 10 mHz at voltage amplitude of 5 mV. Cyclic voltammetry (CV) curves were recorded in the 10–80 mV s−1 range and galvanostatic charging–discharging (GCD) performed in the 1–8 A g−1 range. Cycling performance recorded at constant current density of 1 A g−1 on a Neware BTS cell test apparatus. The specific capacitance of supercapacitors was calculated according to the following formulas reported elsewhere:9,30–32where parameter I (A) is the discharge current, m (g) is the total mass of active materials, Δt (s) is the discharge time, and ΔV (V) is the potential range in the discharge process.

Results and discussion
Morphology structures of carbons were studied by SEM and TEM observations shown in Fig. 1 and S1,† which are irregular in morphology with worm-like pores, indicating that KOH activation process is very complicated. The laminar surface of carbons was smooth as shown in Fig. 1b–e while the increase of activation temperatures will cause the decrease of smooth degree and AEPC-2-800 even has some molten state in morphology (Fig. 1d). Typical SEM image for AEPC-2-800 in Fig. 1d has complete bulk morphology while AEPC-4-800 in Fig. 1e obtained from the same temperature with more activator agents has some coarse morphology and evident fractures and big holes. Typical HRTEM image of AEPC-4-800 in Fig. 1f revealed that the skeleton of carbons is homogeneous and consists of highly developed worm-like micropores with amorphous carbon framework. Micropores can result from KOH activation process and pyrolysis of EP at high temperature, and characteristics of carbons can be altered with activation condition. The HRTEM image for AEPC-2-800 has distinct lattice fringe and disordered short graphene layers can be seen in HRTEM images for other carbons (Fig. S1†), forming randomly distributed uniform micropores with highly defective, resulting in high porosity in carbons. Moreover, hierarchical meso–micropores and channels can be clearly observed in HRTEM images (Fig. S1†). The network of carbons consists of closely linked interconnected pores, forming hierarchical pores and providing a favorable path for fast transportation and penetration of electrolyte ions.29,33 Representative XRD pattern and Raman spectra with deconvoluted peaks for AEPC-4-800 were listed in Fig. S2† and the shape of spectra for carbons is very similar to each others just as can be observed in Fig. S3.† As shown in Fig. S2,† XRD pattern is almost a flat line without any peaks for inorganic impurities. The typical two characteristic peaks at around 2θ = 24, 44° attributing to the (002) and (101) plane of carbons were essentially not observed, indicating that they are wholly amorphous structures with low graphitization. Compared to a broad diffraction at around 2θ = 24° for the carbons from direct carbonization,23,34 all the present carbons are absent for all peaks indicating that KOH activation can greatly decrease graphitization due to the breakdown of aligned structure in carbon framework with intercalation of potassium compounds. Moreover, the diffraction intensity in the low angle region of less than 10° in Fig. S2† is extremely strong, probable due to the presence of high quantities micropores.35 Raman spectra for AEPC-4-800 in inset Fig. S2† showed two prominent broad asymmetry peaks at around 1346.3 (D-band) and 1607.6 cm−1 (G-band) with the latter slightly higher than the former one on the first glance, ascribing to the imperfect structure and C–C stretching vibration mode of graphene with sp2 configuration, respectively.23,36 Raman spectra can be splitted into four peaks centered at around 1191, 1346, 1534, and 1607 cm−1, and the former two at around 1191 and 1346 cm−1 indicated defective structures of carbons while a band at 1534 cm−1 indicated an amorphous structure of carbons.16 It can be clearly observed from deconvoluted peaks that the band at 1534 cm−1 is distinguished when compared to the peak of ideal graphitic sp2 carbon at 1607 cm−1, confirming amorphous properties of carbons. Moreover, it is well-known that the intensity ratio for D-band and G-band (ID/IG) can be used to determine graphitic degree of carbons and the calculated ID/IG value from these fitted peaks for AEPC-4-800 in inset Fig. S2† is about 1.29, representing highly disordered/porous nature of carbons with very low graphitization, which is very accordance with the almost no diffraction in XRD pattern.The effect of activation conditions on pore structure of carbons was evaluated by N2 isotherms and the corresponding pore size distributions (PSD) were calculated using density functional theory (DFT) from adsorption branch. The detailed physical properties of carbons were shown in Table 1. As shown in Fig. 2a, AEPC-2-600 and AEPC-2-700 are type I isotherms with a plateau at relative pressure of 0.1, indicating they are microporous materials. AEPC-2-800 and AEPC-4-800 have an increment in N2 amounts with a ‘knee’ turning point up to pressure of 0.4, a plateau with unapparent hysteresis, indicating the presence of some small-sized mesopores.37 Yue et al.26 have ever reported hierarchical carbons from EP using conventional drying before carbonization and activation with an evident hysteresis loop in isotherms. In addition, N2 isotherms for EP-based carbons from the direct carbonization are type IV isotherms with hysteresis loop at pressure range of 0.45–1.0.23 However, all the present carbons had no hysteresis loop and upward tendency even as pressure close to 0.98, indicating the absence of large-sized mesopores or macropores. Interestingly, isotherms of EP-based carbons using naturally drying and KOH activation reported by Wang et al.24 also have no hysteresis loop, yet showing an upward tendency as the pressure close to 0.9. As observed from Fig. 2b, KOH activation has a remarkable effect on the pore structure of carbons, showing hierarchical structure with most pores in micropores range less than 1.2 nm and small-sized mesopores less than 4.0 nm. It is believed that such hierarchical structure is benefit for gas storage especially at high pressure, and also desirable for facilitating ions transportation and contact of electrode materials with electrolyte. Compared with hierarchical pores at 1.5 and 5.0 nm for the carbons from direct carbonization in our previous works,23 KOH activation greatly enlarged the surface areas and pore volumes with pore size gradually shifting towards smaller pores. It can be seen from Fig. 2b that the most probable pore size of AEPC-2-800 is 2.0 nm in contrast to 2.5 nm for AEPC-4-800. Changes in pore sizes are probable due to the partial collapse of pores between existing large-sized mesopores and generation of new micropores on the surface of carbons.12 Using KOH to activate carbons is a traditional route to create pores involved reaction between KOH and carbon: 6KOH + 2C ↔ 2K + 3H2 + 2K2CO3.33,38 The reaction between potassium and carbon during an etching process is responsible for creating micropores, and decomposition of K2CO3 bringing small molecules will also contribute micropores. KOH activation can cause the breakage of aligned structure in carbons, comprising the reason for low graphitization just as confirmed from XRD and Raman spectra in Fig. S2 and S3.†
| Samples | SBETa (m2 g−1) | SDFTb (m2 g−1) | Vtc (cm3 g−1) | Dfd (nm) | XPS composition (at%) | H2 uptakes (wt%) | |||
|---|---|---|---|---|---|---|---|---|---|
| C | N | O | 1 bar | 20 bar | |||||
| a Surface areas calculated from BET method at relative pressure of 0.03–0.25, and data in parentheses are micropore areas from t-plot method.b Surface areas from DFT method.c Total pore volume obtained from N2 adsorption data at relative pressure of 0.96, and data in parentheses are micropore volumes.d Average pore size calculated from 4Vt/SBET. | |||||||||
| AEPC-2-600 | 1758 (1653) | 1716 | 0.74 (0.66) | 1.69 | 85.69 | 3.48 | 10.83 | 2.06 | 4.43 |
| AEPC-2-700 | 2086 (1994) | 2112 | 0.86 (0.79) | 1.65 | 87.07 | 3.71 | 9.22 | 2.48 | 5.24 |
| AEPC-2-800 | 2825 (2373) | 2839 | 1.39 (1.08) | 1.97 | 89.51 | 2.27 | 8.22 | 2.67 | 6.58 |
| AEPC-4-800 | 3345 (2266) | 3315 | 1.73 (1.04) | 2.06 | 91.63 | 1.53 | 6.84 | 2.71 | 7.05 |
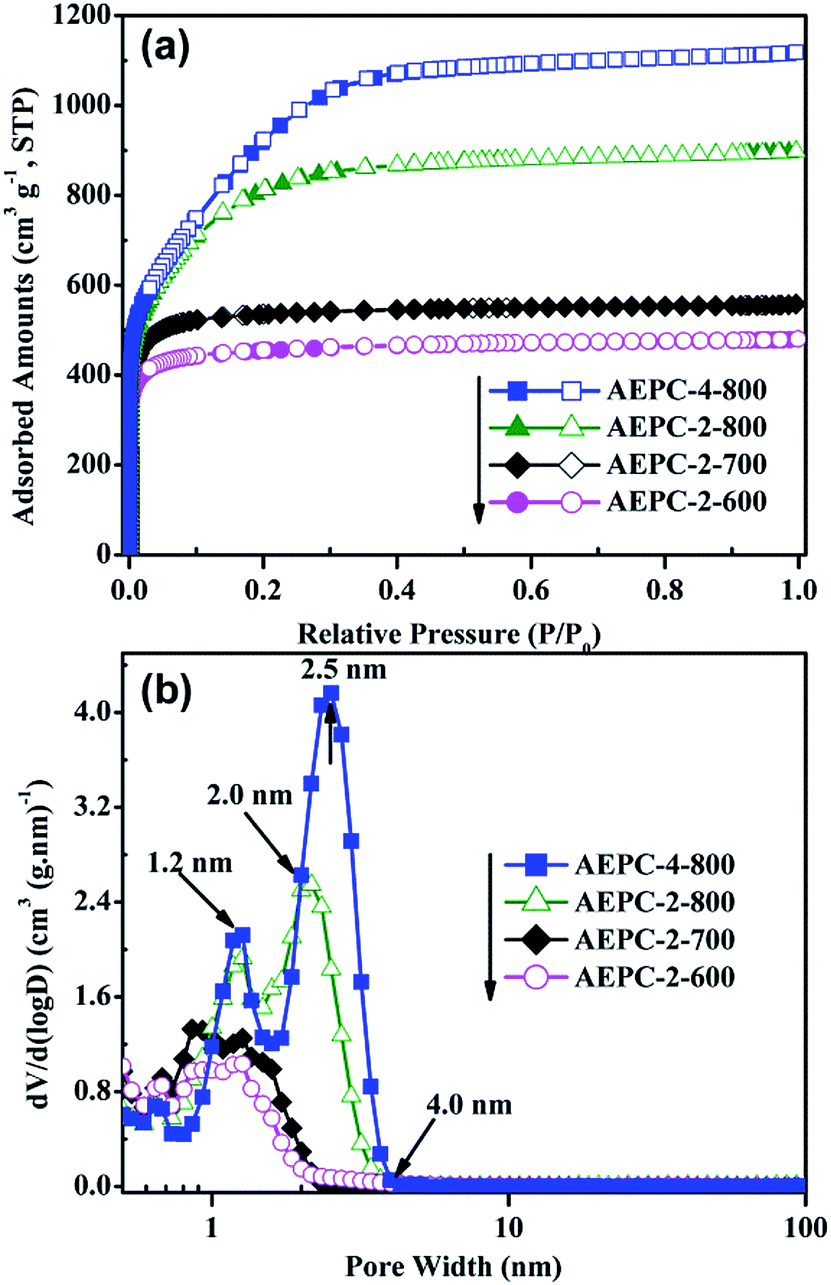 | ||
| Fig. 2 (a) N2 sorption isotherms for carbons at −196 °C, and (b) corresponding pore size distributions. | ||
As can be seen from Table 1, the BET surface area and pore volume of carbons are significantly affected by KOH activation where the corresponding value is in the range of 1758–3345 m2 g−1 and 0.74–1.73 cm3 g−1, respectively. Significantly, AEPC-4-800 has the highest BET surface area of 3345 m2 g−1 with micropore area of 2266 m2 g−1, constituting the largest value for EP-based carbons,22–27 to the best of our knowledge. Moreover, AEPC-2-700 has the BET surface area of 2086 m2 g−1 and pore volume of 0.86 cm3 g−1 while the counterparts from micropores are also highly up to 1994 m2 g−1 and 0.79 cm3 g−1, indicating high quantities of well-developed micropores in structure. As observed from Table 1 that the micropore surface areas and volumes gradually increase as an increase of activation temperature while the two parts decrease as further increase of activator concentration. For example, both BET surface area and pore volume for AEPC-4-800 are larger than AEPC-2-800 while the latter has a little larger micropore surface area and volume with value highly up to 2373 m2 g−1 and 1.08 cm3 g−1, respectively. This high porosity of carbons indicated that both the selection of precursor and synthesis process are critical in determining performance. It should be admitted that multi-points BET method can usually overestimate the surface areas in many cases, and data can be varied with the pressure range.2 An alternative DFT method was also used to calculate surface areas and it can be seen from Table 1 that surface areas from two methods are very close, and the DFT surface area of AEPC-4-800 is also highly up to 3315 m2 g−1. Comparison between BET and DFT area confirmed that using adsorbed N2 amounts at relative pressure range of 0.03–0.25 as multi-points to calculate surface area for high-surface-area carbons is reasonable and acceptable to some extent.3 Moreover, the average pore size for all carbons is in the range of micropores except for AEPC-4-800 with pore size of around 2.06 nm in Table 1. These carbons with high porosity motivated us to examine their potential application for H2 storage and supercapacitors, and the presence of small-sized mesopores can exert benefit on H2 uptake through the decrease of repulsion interaction between adsorbed molecules, and also facilitate electrolyte fast transportation in the charge–discharge process.
XPS measurement was used to evaluate the heteroatom-doping chemistry. As expected, the XPS survey spectra in Fig. 3 indicated the presence of O, N groups in carbons besides the characteristic C elements with energy at around 533, 401 and 285 eV, respectively, and detailed heteroatom contents were listed in Table 1. In general, the presence of some O, N-based functional groups will definitely enhance hydrophilicity and provide extra pseudocapacitance for the applied electrode materials.39 As observed from Fig. 3 and Table 1, these carbons are abundant in surface active O, N-based groups, and increasing activation temperature will largely increase C while decrease O, N contents due to the partially enhancement of carbonization degree and consumption of O, N elements during alkaline activation. The contents of O elements varied in the range of 6.84–10.83 at% and the percentage of N elements varied in the range of 1.53–3.71 at%. Although the existing O, N groups may have some detrimental effects for H2 storage,1,3 they are well-known to enhance hydrophilicity and defective sites, benefiting for CO2 capture and charge storage with introduction of extra pseudocapacitance.7,14 Multiple C chemical states on the surface of carbons were confirmed by high resolution of C1s peak (Fig. 3b), which can be deconvoluted into peaks at around 284.5, 285.1 and 287.4 eV, corresponding to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–O/CN and CO/C–N, respectively.14,40 High resolution O1s XPS spectra in Fig. S4† are found to be combination of three peaks at around 531.7, 533.4, and 536.2 eV, attributing to the –OH/C–O, CO and C–O–OH, respectively.14,24 The results indicated that the surface of carbons has some phenol, lactone, and carboxyl groups, which will partially contribute to the pseudo-capacitance besides from an improvement of wettability of carbons. As observed from Table 1 that N contents decrease as an increase of activation degree in carbons because heteroatoms are chemically unstable during high temperature activation. The contents of N in AEPC-2-800 and AEPC-4-800 are of 2.27 and 1.53 at%, respectively, which are much lower compared to the carbons from carbonization with surface areas of 422 m2 g−1,23 suggesting that it is not easy to obtain carbons with high porosity and high N contents in the meantime. To further evaluate the nature of N elements in carbons, N1s XPS spectra were deconvoluted into three peaks in Fig. 4 with the energy at around 398.9, 400.4, and 402.3 eV, corresponding to pyridinic, pyrrolic, and quaternary N, respectively.14,40 There is no peak associated with N–O bond usually appeared at energy above 403 eV, indicating that oxygen atoms in carbons are wholly bonded to the skeleton carbon. High-resolution N1s XPS peaks in Fig. 4 mainly consisted of pyrrolic N together with small amounts of pyridinic and quaternary N while the former two parts are almost equal in the carbon from direct carbonization.23 Pyrrolic N usually located at the edges of carbons induced more defectives, well-elucidating the broad D band with high intensity in Raman spectra in inset Fig. S3.† The largely reduced pyridinic N in carbons suggested that pyridinic N is less stable than pyrrolic N at high temperature. KOH activation will accelerate the consumption of pyridinic N, and the presence of active pyrrolic N is considered to provide pseudo-capacitance.15,33 KOH activation treatment has greatly changed pore structure and surface chemistry of carbons, including surface area, pore volume, ratio of micro/mesopores, and heteroatoms contents. It is generally believed that the existing O, N-species are highly concerned in modifying electron donor–acceptor and acid–basic characteristics of carbons,41,42 which might have great effects on their gas adsorption and electrochemical capacitive performance.
C, C–O/CN and CO/C–N, respectively.14,40 High resolution O1s XPS spectra in Fig. S4† are found to be combination of three peaks at around 531.7, 533.4, and 536.2 eV, attributing to the –OH/C–O, CO and C–O–OH, respectively.14,24 The results indicated that the surface of carbons has some phenol, lactone, and carboxyl groups, which will partially contribute to the pseudo-capacitance besides from an improvement of wettability of carbons. As observed from Table 1 that N contents decrease as an increase of activation degree in carbons because heteroatoms are chemically unstable during high temperature activation. The contents of N in AEPC-2-800 and AEPC-4-800 are of 2.27 and 1.53 at%, respectively, which are much lower compared to the carbons from carbonization with surface areas of 422 m2 g−1,23 suggesting that it is not easy to obtain carbons with high porosity and high N contents in the meantime. To further evaluate the nature of N elements in carbons, N1s XPS spectra were deconvoluted into three peaks in Fig. 4 with the energy at around 398.9, 400.4, and 402.3 eV, corresponding to pyridinic, pyrrolic, and quaternary N, respectively.14,40 There is no peak associated with N–O bond usually appeared at energy above 403 eV, indicating that oxygen atoms in carbons are wholly bonded to the skeleton carbon. High-resolution N1s XPS peaks in Fig. 4 mainly consisted of pyrrolic N together with small amounts of pyridinic and quaternary N while the former two parts are almost equal in the carbon from direct carbonization.23 Pyrrolic N usually located at the edges of carbons induced more defectives, well-elucidating the broad D band with high intensity in Raman spectra in inset Fig. S3.† The largely reduced pyridinic N in carbons suggested that pyridinic N is less stable than pyrrolic N at high temperature. KOH activation will accelerate the consumption of pyridinic N, and the presence of active pyrrolic N is considered to provide pseudo-capacitance.15,33 KOH activation treatment has greatly changed pore structure and surface chemistry of carbons, including surface area, pore volume, ratio of micro/mesopores, and heteroatoms contents. It is generally believed that the existing O, N-species are highly concerned in modifying electron donor–acceptor and acid–basic characteristics of carbons,41,42 which might have great effects on their gas adsorption and electrochemical capacitive performance.
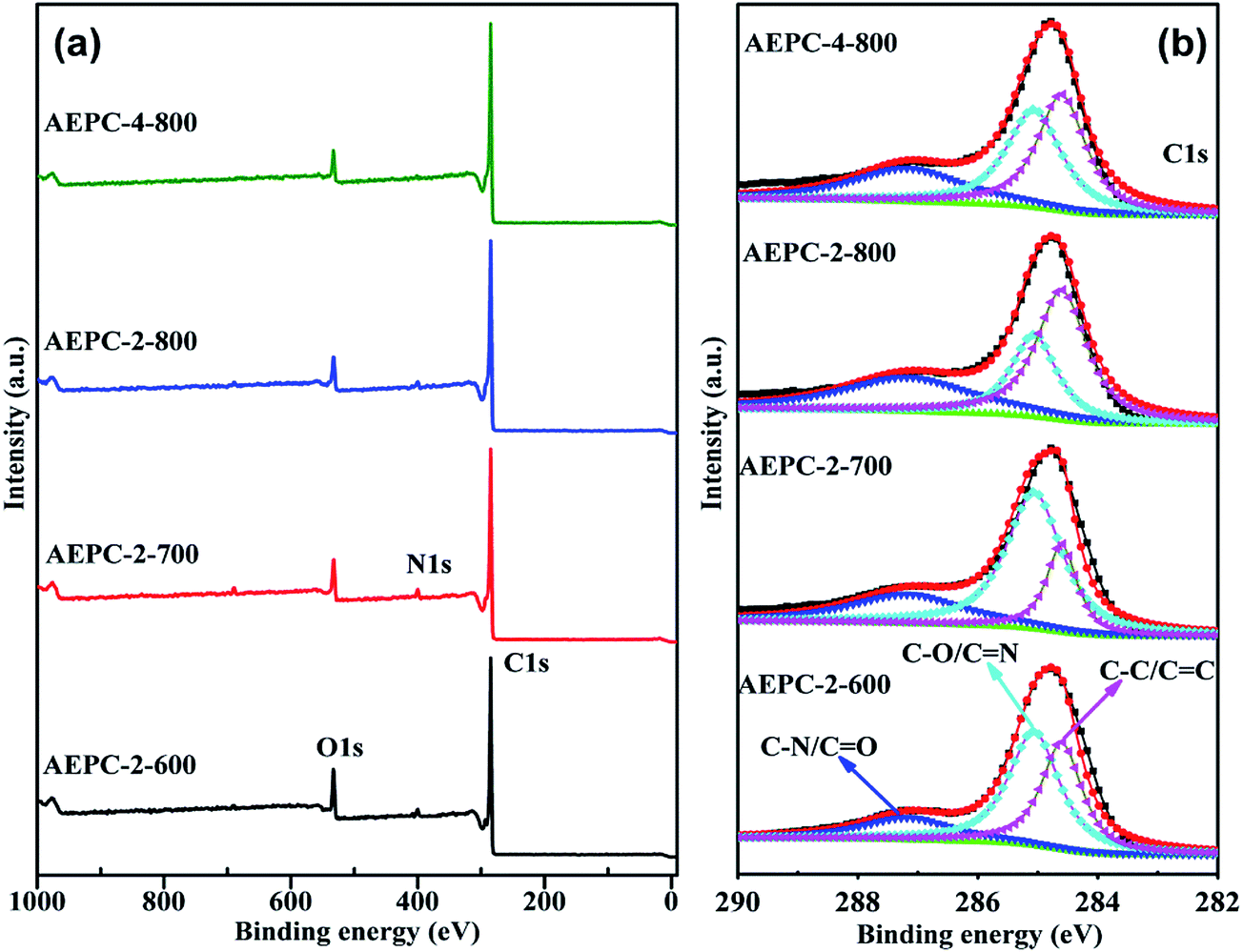 | ||
| Fig. 3 The XPS survey spectra and high-resolution C1s region of the activated carbons. | ||
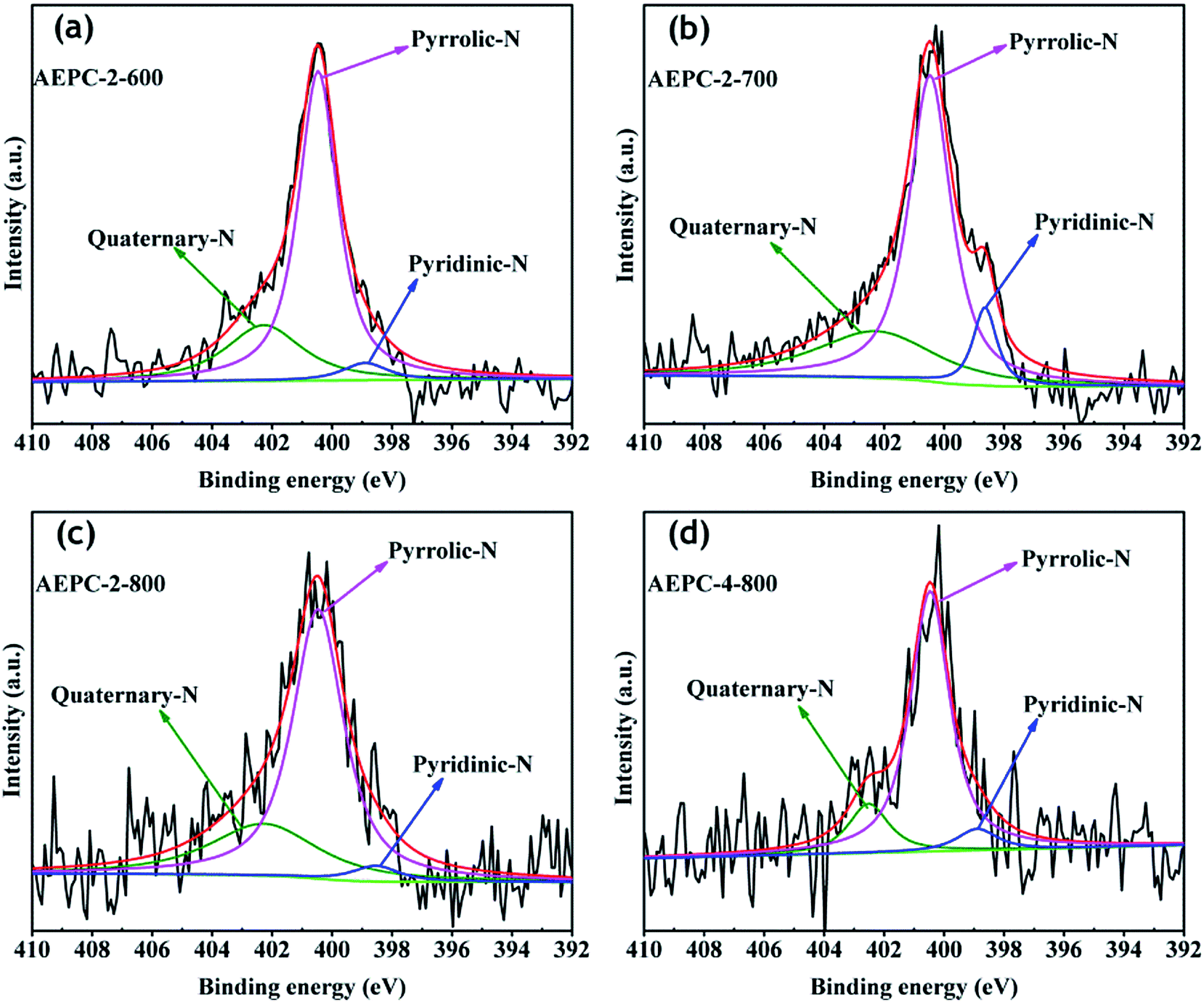 | ||
| Fig. 4 High-resolution N1s XPS spectra (a) AEPC-2-600, (b) APEC-2-700, (c) AEPC-2-800 and (d) AEPC-4-800. | ||
H2 storage performance on carbons at −196 °C were evaluated by the IGA instrument using our previous parameters to correct buoyancy effect,3,6 which has also been verified in others' report.37 H2 isotherms at 1 and 20 bar were shown in Fig. 5 and the detailed uptakes were listed in Table 1. In our previous works,3,6 we analyzed adsorption credibility by comparing the results of D2 isotopes on the system, indicating that mass ratio of adsorbed D2/H2 is close to 2:1 and corresponding molar ratio is at around 1.02. The H2 uptakes are in the range of 2.06–2.71 wt% at 1 bar and 4.43–7.05 wt% at 20 bar, respectively. Significantly, AEPC-4-800 showed H2 uptake of highly up to 7.05 wt% at 20 bar with an exceptional high uptake of 2.71 wt% at 1 bar, and both are among the largest data for biomass-derived carbons, to the best of our knowledge. Generally, H2 uptakes are highly related to the pore structure of carbons, especially with micropore surface area and pore volume inherited from pores in the range of 0.6–0.9 nm. The situation for H2 storage in Fig. 5 is approximately in line with monolayer adsorption mechanism, that is, surface area determined the uptake. As shown in Table 1, AEPC-4-800 in series of carbons has the largest surface area and pore volume, showing the largest H2 uptake both at 1 and 20 bar. The highest H2 uptake of AEPC-4-800 is also partially attributed to low contents of N groups revealed by XPS spectra, considering that the presence of heteroatoms has some detrimental effect on interaction between pore wall and adsorbed H2 molecules.1,3 Importantly, H2 uptake on AEPC-2-700 with relative lower surface area is slightly larger than APEC-2-800 and AEPC-4-800 as pressure less than 0.4 bar (Fig. 5a). Combined with PSDs in Fig. 2b, AEPC-2-700 showed higher porosity with more pores in the 0.6–0.9 nm range, contributing significant effect to H2 storage at low pressure. Comparatively, H2 uptakes at 20 bar are proportional to the surface area and total pore volume (Fig. 5b), illustrating the important role of surface areas and pore volumes in H2 storage at high pressure.6 Moreover, the growth trend for H2 adsorption on the AEPC-2-600 and AEPC-2-700 is moderate at high pressure based on observation isotherms in Fig. 5b as the pressure increase, while the trend is still very clear for AEPC-2-800 and AEPC-4-800 contained hierarchical pores in 1.0–4.0 nm range. No saturation for H2 isotherms is observed for all carbons as pressure up to 20 bar, indicating that H2 uptakes can be further enhanced if pressure continues to increase. We dare to speculate that these hierarchical pores are very beneficial for H2 storage on AEPC-2-800 and AEPC-4-800 at high pressure due to the following reasons: (1) high surface areas and pore volumes of hierarchical carbons can partially compensate low adsorption efficiency in small-sized mesopores; (2) the repulsion interactions between adsorbed H2 molecules in pores are also increased as pressure increase, and these small-sized mesopores may provide fast transportation, and then ease interactions between H2 molecules to some extent.
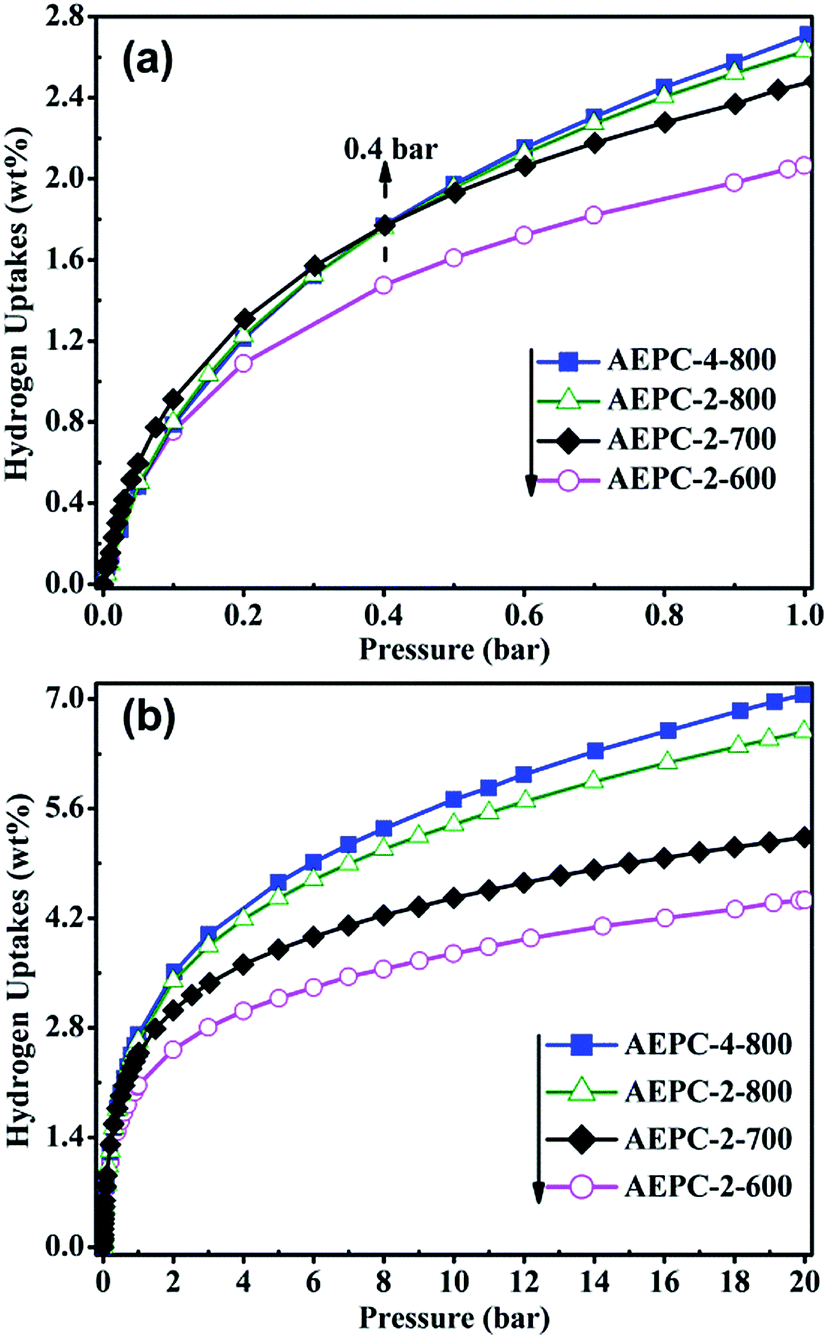 | ||
| Fig. 5 H2 adsorption isotherms at −196 °C for the carbons at (a) 1 bar, and (b) 20 bar. | ||
The electrochemical performance of the EP-based carbons was evaluated in 6 M KOH aqueous electrolyte, after fabricating into symmetric supercapacitor. Typical CV curves of four capacitors at a scan rate of 80 mV s−1 were shown in Fig. 6a, and all curves except for AEPC-2-600 capacitor show quasi-rectangular shapes with moderate distortion, indicating the coexistence of ideal electric double layer capacitance and pseudocapacitance.15,26 CV curves at various scan rates from 10 to 80 mV s−1 for AEPC-2-600, AEPC-2-700 and AEPC-4-800 capacitors were shown in Fig. S5.† Considering the fact that O-based groups in series of carbons are highly up to 10.8 at% as confirmed from XPS spectra in Fig. 3a and Table 1, it can be inferred that pseudo-capacitance is mainly caused by redox reaction between O-based group and electrolyte ions on the surface of carbons.39 Generally, the capacitance is in direct proportion to the area of CV curves, under the same scan rate. The area of CV curve for AEPC-4-800 capacitor at 80 mV s−1 is obviously larger than that of other carbon-based capacitors, indicating that AEPC-4-800 has better capacitive behavior which is largely related to the largest surface area.30,34,41,42 In addition, the existing O, N-based groups is not the decisive factor for the induced capacitance, considering that AEPC-2-600 with the highest N, O content has much inferior capacitance. It should be noted that the severe distortion of CV curve for AEPC-2-600 in Fig. 6a and S5† is probable due to the presence of large amounts of pores less than 1.0 nm with high volume of micropores which is not easy to activate for electrolyte ions to diffuse and transport and then difficult for rapid formation of an electric double layer. This result implied that the presence of some mesopores is needed for high energy storage characteristics of supercapacitors. CV curves in Fig. 6b for AEPC-2-800 at various scan rate (10–80 mV s−1) have a similar quasi-rectangular shape without much distortion and the plateau current increases as increase of scan rate, indicating excellent ions transportation behaviors even at high speed. This is due to the reason that hierarchical pores in AEPC-2-800 can reduce the transportation distance of electrolyte ions and the resistance of diffusion, and the existing O, N-species in the carbon are highly concerned in modifying the electron donor–acceptor and acid–basic characteristics of carbons, improving the capacitance by reversible faradic redox reactions in aqueous electrolyte.30,41,42 The typical GCD curves at 1 A g−1 in Fig. 6c have similar isosceles triangular shapes without severe voltage decrease except for AEPC-2-600, showing good reversibility and high charge–discharge efficiency. GCD curve for AEPC-2-600 capacitor in Fig. 6c has severe nonlinear discharge profile, attributing to the presence of pseudocapacitive behavior from the highest content of O, N-based groups in the sample (Table 1). Comparatively, GCD curve for AEPC-4-800 capacitor has much longer charge–discharge time than that of capacitors consist of other carbons, indicating the higher electrochemical activity and accessible capacitance. However, the discharge profile of AEPC-4-800 has some distortion from linear shape, implying high equivalent series resistance due to its low graphitization and conductivity as confirmed from XRD and Raman spectra in Fig. S2.† The specific capacitance of AEPC-4-800 at 1 A g−1 is as high as 440 F g−1, which is higher than value for AEPC-2-800 (315.6 F g−1), AEPC-2-700 (262 F g−1) and AEPC-2-600 (312 F g−1). The distortion of AEPC-2-800 in series of capacitors is the least indicating good cyclability due to optimum hierarchical pores (Fig. 2b) where micropores less than 0.5 nm in AEPC-2-600 are too narrow for rapid formation of double-layer and hierarchical pores larger than 2 nm in AEPC-4-800 are too wide.33 GCD curves for AEPC-2-800 at various current densities from 1 to 8 A g−1 were shown in Fig. 6d and others were shown in Fig. S5.† It can be found that all curves in Fig. 6d still maintain symmetric and linear at increased current density, which is a typical characteristic of an ideal capacitor. As current density increased to 2, 4, and 8 A g−1, the capacitance of AEPC-2-800 slightly decreased to 268, 248, and 231 F g−1, respectively, due to sterical limitation of materials over which ions can only partially penetrate into micropores. The specific capacitance of AEPC-2-800 at 8 A g−1 maintain 73% capacitance compared the value at 1 A g−1, attributing to the well-developed hierarchical pores of carbons that facilitate rapid electrolyte transfer and relative high content of heteroatoms-doping that imparts good wettability, which has been widely demonstrated in other's works.34,43 Although AEPC-4-800 capacitor has the largest initial capacitance than AEPC-2-800 due to the higher fraction of mesoporous surface area and larger mesopore size, the too low capacitance retention coming from low conductivity limits its significance. Therefore, AEPC-2-800 is a more preferential electrode materials for supercapacitors because AEPC-2-800 also has some mesopores with slight smaller pore sizes besides from the higher contents of N, O-based groups.
 | ||
| Fig. 6 (a) CV curves at 80 mV s−1, (b) CV curves of AEPC-2-800 capacitor at various scan rates, (c) GCD curves at 1 A g−1, (d) GCD curves of AEPC-2-800 capacitor at various current densities. | ||
To further investigate the electrochemical behaviors of the synthesized carbons, electrochemical impedance spectroscopy (EIS) plots of the series EP-derived carbons-based capacitors in 6 M KOH electrolyte were measured and provided in Fig. 7a. As observed from inset Fig. 7a, all curves show two main traits: a depressed semicircle in high frequency range and a sloped line in low frequency range. It is well-known that the semicircle corresponds to charge-transfer resistance (Rct), resulting from the resistance of electrode itself and contact resistance between electrode and current collector, while the sloped line in low frequency corresponds to diffusion-controlled process.31,39 All lines are sloped yet not vertical in the low frequency suggesting collaborative nature of electric double layer capacitor and pseudo-capacitor,34 in accordance with the nature of existing O, N-based groups on the surface of carbons (Fig. 3). It can be seen from Fig. 7a that intercept with real axis in high-frequency for AEPC-2-600, AEPC-2-700, AEPC-2-800 and AEPC-4-800 capacitor, representing the equivalent series resistance (ESR), is separately of 0.60, 2.54, 1.43 and 6.31 Ω. The relative smaller ESR values for AEPC-2-600 and AEPC-2-800 capacitor are related to the highest contents of N, O-based groups in the former and optimum hierarchical pores in the latter, which will decrease diffusion resistance. In addition, the Rct values calculated from the semicircle diameter in Fig. 7a varied to be 14.31, 6.98, 5.32, and 35.59 Ω for AEPC-2-600, AEPC-2-700, AEPC-2-700, and AEPC-4-800, respectively. The lowest Rct value in series of capacitors for AEPC-2-800 implied the faster charge transfer with better electrolyte accessibility into pores of electrode materials,34,39 due to the reason that AEPC-2-800 has the highest microporous surface area combined with abundant hierarchical mesopores. Moreover, the moderate contents of N, O-based groups in the carbon can also improve pore affinity to aqueous electrolyte and decrease ion-diffusive resistance.43 Comparatively, the largest ESR value of 6.31 Ω and Rct of 35.59 Ω for AEPC-4-800 capacitor is probable due to the most inferior conductivity, considering that AEPC-4-800 is derived from the most severe activation with the lowest graphitization as confirmed from XRD and Raman spectra (Fig. S3†) and the lowest content of heteroatoms-doping from XPS spectra (Fig. 3 and Table 1). As can be observed from inset Fig. 7a, the intercept value with real axis in low-frequency for AEPC-2-800 capacitor is about 6.75 Ω, which is the smallest in series capacitors, illustrating that AEPC-2-800 can do charge–discharge process more rapidly with high power density.9,31 The cycling performance of the series carbon-based capacitors was evaluated by GCD measurement at the current density of 2 A g−1 for 5000 cycles, as shown in Fig. 7b and S5.† As can be seen from Fig. 7b, the capacitance retention for AEPC-2-800 is highly up to 96% after charge–discharge process over 2000 cycles, and 87% of its initial capacitance is still retained even charge–discharge process after 5000 cycles, confirming that AEPC-2-800 has a long-term stability as supercapacitor electrode. However, cycling performance of other devices listed in Fig. S6† had much inferior stability especially for AEPC-2-600 and AEPC-4-800 with capacitance retention decrease to 50% only after 100 and 1000 cycles, respectively. Furthermore, the capacitance retention for AEPC-2-700 decrease to 50% after 5000 cycles. The excellent stability for AEPC-2-800 was due to high microporous surface areas with presence of hierarchical pores in critical pore sizes, allowing electrolyte to fast penetrate and provided plenty of favorable active sites for diffusion and transportation of electrolyte. The existence of N, O-based groups on the surface of carbons can also improve conductivity and wettability, which are favorable to overcome kinetic diffusion resistance especially at high-current density, leading to enhancement of capacitive performance. Therefore, EP-based carbons with hierarchical pores could be applied as high performance electrode materials in high-energy-density EDLC applications.
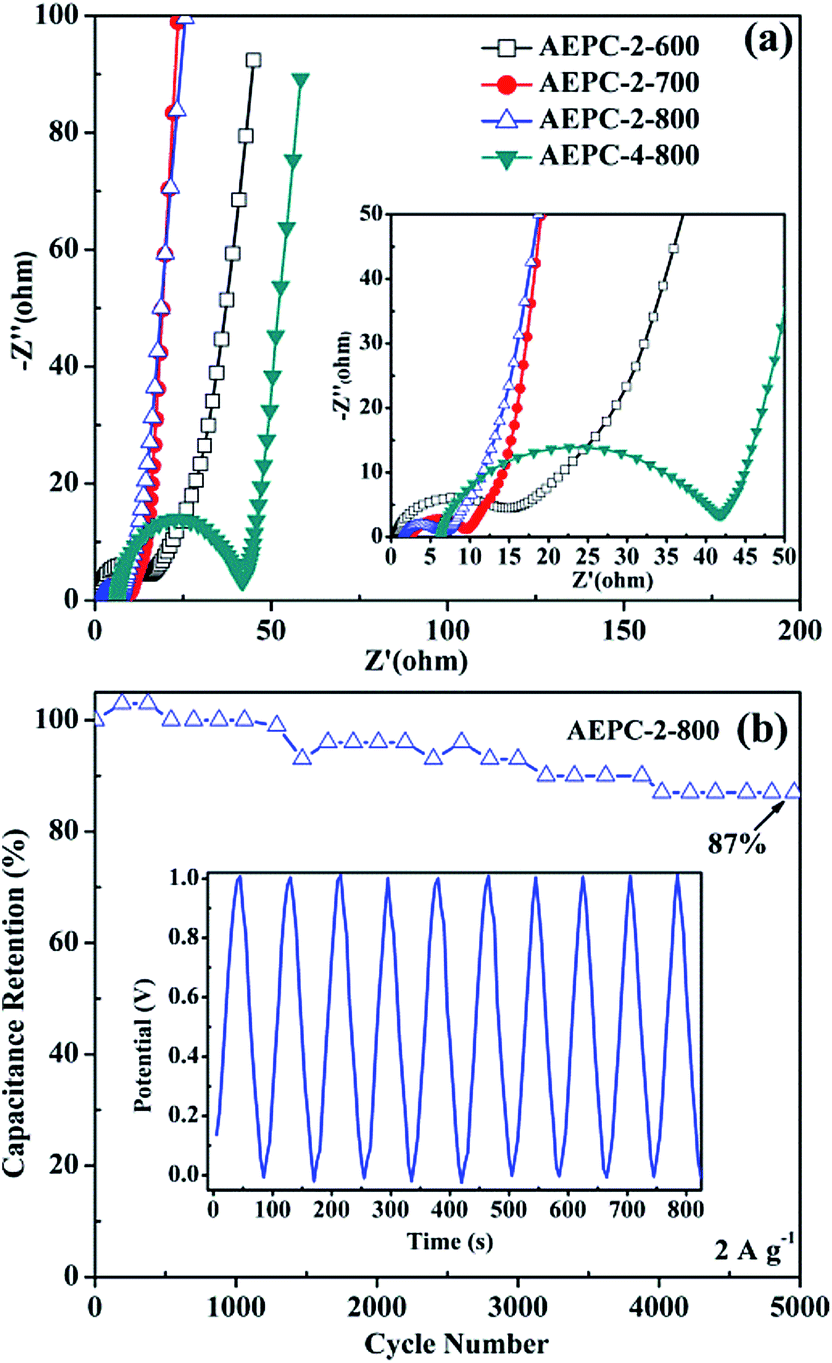 | ||
| Fig. 7 (a) Nyquist plots of four carbon capacitors and inset is the enlarged view of high-frequency region, (b) the cycling performance of AEPC-2-800 capacitor at 2 A g−1 and inset shows the first 10 cycles. | ||
Conclusion
N, O-Doped hierarchical carbons were obtained by carbonization and KOH activation using EP algae as precursors with surface area and pore volume separately highly up to 3345 m2 g−1 and 1.94 cm3 g−1. AEPC-4-800 with the largest surface area shows the highest H2 uptake of 7.05 wt% at 20 bar and also has an exceptional uptake of 2.71 wt% at 1 bar and −196 °C, which is among the largest data reported to date. The remarkable uptakes to adsorb H2 are principally due to the synergistic effects between the existing hierarchical pores, large pore volume and surface area. Moreover, AEPC-4-800 capacitor had the highest capacitance of 440 F g−1 at 1 A g−1 in 6 M KOH electrolyte and AEPC-2-800 capacitor had good cycling stability with 87% of capacitance after 5000 cycles due to its unique hierarchical pores with high-surface-area provided much active sites for the better facilitating electrolyte diffusion.Acknowledgements
We greatly appreciate supports from National Natural Science Foundation of China (21506184), CAS Key Laboratory of Biobased Material (KLBM2016010), PhD Startup of XTU (15QDZ13), Postdoctoral Science Foundation (2015M572258), Project of Postgraduate Research Innovation (CX2017B335), and Education Department of Hunan Province (16C1536). We also gratitude the financial support from Hunan 2011 Collaborative Innovation Center of Chemical Engineering with Environmental Benignity and Effective Resource Utilization.References
- Y. Xia, G. S. Walker, D. M. Grant and R. Mokaya, J. Am. Chem. Soc., 2009, 131, 16493–16499 CrossRef CAS PubMed.
- Z. Yang, Y. Xia and R. Mokaya, J. Am. Chem. Soc., 2007, 129, 1673–1679 CrossRef CAS PubMed.
- J. Cai, L. Li, X. Lv, C. Yang and X. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 167–175 CAS.
- S. J. Yang, T. Kim, J. H. Im, Y. S. Kim, K. Lee, H. Jung and C. R. Park, Chem. Mater., 2012, 24, 464–470 CrossRef CAS.
- Y. J. Heo and S. J. Park, J. Ind. Eng. Chem., 2015, 31, 330–334 CrossRef CAS.
- J. Cai, J. Qi, C. Yang and X. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 3703–3711 CAS.
- D. J. Babu, M. Bruns, R. Schneider, D. Gerthsen and J. J. Schneider, J. Phys. Chem. C, 2017, 121, 616–626 CAS.
- F. Sun, X. Liu, J. Gao, X. Pi, L. Wang, Z. Qu and Y. Qin, J. Mater. Chem. A, 2016, 4, 18248–18252 CAS.
- K. Karthikeyan, S. Amaresh, S. N. Lee, X. Sun, V. Aravindan, Y. G. Lee and Y. S. Lee, ChemSusChem, 2014, 7, 1435–1442 CrossRef CAS PubMed.
- L. Wei and G. Yushin, Nano Energy, 2012, 1, 552–565 CrossRef CAS.
- Y. Zhang, M. Jia, H. Gao, J. Yu, L. Wang, Y. Zou, F. Qin and Y. Zhao, Electrochim. Acta, 2015, 184, 32–39 CrossRef CAS.
- M. Sevilla, A. B. Fuertes and R. Mokaya, Int. J. Hydrogen Energy, 2011, 36, 15658–15663 CrossRef CAS.
- H. Wang, Q. Gao and J. Hu, J. Am. Chem. Soc., 2009, 131, 7016–7022 CrossRef CAS PubMed.
- B. Wang, J. Qiu, H. Feng, E. Sakai and T. Komiyama, Electrochim. Acta, 2016, 190, 229–239 CrossRef CAS.
- H. Chen, D. Liu, Z. Shen, B. Bao, S. Zhao and L. Wu, Electrochim. Acta, 2015, 180, 241–251 CrossRef CAS.
- J. W. Jeon, R. Sharma, P. Meduri, B. W. Arey, H. T. Schaef, J. L. Lutkenhaus, J. P. Lemmon, P. K. Thallapally, M. I. Nandasiri, B. P. McGrail and S. K. Nune, ACS Appl. Mater. Interfaces, 2014, 6, 7214–7222 CAS.
- J. Wang, P. Nie, B. Ding, S. Dong, X. Hao, H. Dou and X. Zhang, J. Mater. Chem. A, 2017, 5, 2411–2428 CAS.
- S. Ravi and S. Vadukumpully, J. Environ. Chem. Eng., 2016, 4, 835–856 CrossRef CAS.
- Z. Gao, Y. Zhang, N. Song and X. Li, Mater. Res. Lett., 2017, 5, 69–88 CrossRef CAS.
- M. Sevilla, C. Falco, M. M. Titirici and A. B. Fuertes, RSC Adv., 2012, 2, 12792–12797 RSC.
- E. Raymundo-Piñero, M. Cadek and F. Béguin, Adv. Funct. Mater., 2009, 19, 1032–1039 CrossRef.
- Z. Zhang, K. Wang, J. D. Atkinson, X. Yan, X. Li, M. J. Rood and Z. Yan, J. Hazard. Mater., 2012, 229–230, 183–191 CrossRef CAS PubMed.
- Z. Tian, Y. Qiu, J. Zhou, X. Zhao and J. Cai, Mater. Lett., 2016, 180, 162–165 CrossRef CAS.
- W. Yu, H. Wang, S. Liu, N. Mao, X. Liu, J. Shi, W. Liu, S. Chen and X. Wang, J. Mater. Chem. A, 2016, 4, 5973–5983 CAS.
- X. Gao, W. Xing, J. Zhou, G. Wang, S. Zhuo, Z. Liu, Q. Xue and Z. Yan, Electrochim. Acta, 2014, 133, 459–466 CrossRef CAS.
- Y. Gao, W. Zhang, Q. Yue, B. Gao, Y. Sun, J. Kong and P. Zhao, J. Power Sources, 2014, 270, 403–410 CrossRef CAS.
- M. Wu, P. Li, Y. Li, J. Liu and Y. Wang, RSC Adv., 2015, 5, 16575–16581 RSC.
- Y. Gao, Q. Yue, B. Gao, Y. Sun, W. Wang, Q. Li and Y. Wang, Chem. Eng. J., 2013, 232, 582–590 CrossRef CAS.
- S. Zhu, J. Li, C. He, N. Zhao, E. Liu, C. Shi and M. Zhang, J. Mater. Chem. A, 2015, 3, 22266–22273 CAS.
- T. Liu, E. Liu, R. Ding, Z. Luo, T. Hu and Z. Li, ChemElectroChem, 2015, 2, 852–858 CrossRef CAS.
- M. Wu, P. Ai, M. Tan, B. Jiang, Y. Li, J. Zheng, W. Wu, Z. Li, Q. Zhang and X. He, Chem. Eng. J., 2014, 245, 166–172 CrossRef CAS.
- J. Liu, Y. Deng, X. Li and L. Wang, ACS Sustainable Chem. Eng., 2016, 4, 177–187 CrossRef CAS.
- K. Wang, N. Zhao, S. Lei, R. Yan, X. Tian, J. Wang, Y. Song, D. Xu, Q. Guo and L. Liu, Electrochim. Acta, 2015, 166, 1–11 CrossRef CAS.
- Z. Tian, M. Xiang, J. Zhou, L. Hu and J. Cai, Electrochim. Acta, 2016, 211, 225–233 CrossRef CAS.
- S. Bai, G. Tan, X. Li, Q. Zhao, Y. Meng, Y. Wang, Y. Zhang and D. Xiao, Chem.–Asian J., 2016, 11, 1828–1836 CrossRef CAS PubMed.
- M. Karnan, K. Subramani, N. Sudhan, N. Ilayaraja and M. Sathish, ACS Appl. Mater. Interfaces, 2016, 8, 35191–35202 CAS.
- Y. Li, D. Li, Y. Rao, X. Zhao and M. Wu, Carbon, 2016, 105, 454–462 CrossRef CAS.
- R. Wang, P. Wang, X. Yan, J. Lang, C. Peng and Q. Xue, ACS Appl. Mater. Interfaces, 2012, 4, 5800–5806 CAS.
- J. Chang, Z. Gao, X. Wang, D. Wu, F. Xu, X. Wang, Y. Guo and K. Jiang, Electrochim. Acta, 2015, 157, 290–298 CrossRef CAS.
- G. Wang, J. Zhang, S. Kuang, J. Zhou, W. Xing and S. Zhuo, Electrochim. Acta, 2015, 153, 273–279 CrossRef CAS.
- C. M. Chen, Q. Zhang, M. G. Yang, C. H. Huang, Y. G. Yang and M. Z. Wang, Carbon, 2012, 50, 3572–3584 CrossRef CAS.
- C. M. Chen, Q. Zhang, X. C. Zhao, B. Zhang, Q. Q. Kong, M. G. Yang, Q. H. Yang, M. Z. Wang, Y. G. Yang, R. Schlogl and D. S. Su, J. Mater. Chem., 2012, 22, 14076–14084 RSC.
- D. W. Wang, F. Li, L. C. Yin, X. Lu, Z. G. Chen, T. R. Gentle, G. Q. Lu and H. M. Cheng, Chem.–Eur. J., 2012, 18, 5345–5351 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05355j |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |