
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of fluorophthalocyanines bearing four 2-(2-thienyl)ethoxy moieties: from the optimization of the fluorine substitution to chemosensing†
Amélie Wannebroucq,
Rita Meunier-Prest,
Jean-Claude Chambron,
Claire-Hélène Brachais,
Jean-Moïse Suisse and
Marcel Bouvet *
*
Institut de Chimie Moléculaire de l'Université de Bourgogne, UMR CNRS 6302, Univ. Bourgogne Franche-Comté, 9 avenue Alain Savary, 21078 Dijon Cedex, France. E-mail: marcel.bouvet@u-bourgogne.fr; Fax: +33-380-396-098; Tel: +33-380-396-086
First published on 23rd August 2017
Abstract
The energy levels of the HOMO/LUMO Frontier orbitals and the electronic properties of phthalocyanine macrocycles can be tuned by the introduction of substituents. Starting from tetrafluorophthalonitrile, we studied the substitution of fluorine atoms by (2-thienyl)ethoxy moieties. An optimization of the experimental conditions (nature and stoichiometry of the alcohol and base, temperature) allowed us to obtain the monoalkoxy derivative with a very good yield. It was fully characterized using 19F and 1H NMR spectroscopies, thermal analysis and X-ray diffraction on single crystals. Then, the corresponding zinc phthalocyanine was synthesized, characterized by means of 19F and 1H NMR spectroscopies, thermal analysis, and also by electronic spectroscopy and electrospray mass spectrometry. The unsymmetrical zinc phthalocyanine bearing also four (2-thienyl)ethoxy moieties was prepared by the mixed condensation of the tetraalkoxyphthalonitrile with the tetrafluorophthalonitrile. The phthalocyanines were used to build an electronic device, a p-type Molecular Semiconductor – Doped Insulator heterojunction (MSDI), in combination with the lutetium bisphthalocyanine as a molecular semiconductor, and their chemosensing behavior towards ammonia was studied.
Introduction
The energy levels of highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO) are responsible for the electronic and optical properties of molecular materials which are at the origin of their use in devices, such as organic light emitting diodes (OLED), organic field-effect transistors (OFET) or organic photovoltaic cells. Thus, it is of the utmost importance to be able to tune the energy of molecular orbitals.The nature of majority charge carriers in the channel of OFETs1,2 and their charge transport properties3,4 can be modified by the introduction of substituents. Tuning molecular orbitals energy is especially important in heterojunctions, in which the alignment of HOMO and LUMO determines the energy barrier at the interface between the materials, and by extension the current–voltage characteristics of the device.5,6 More generally, the molecular orbitals energies affect the interfaces between molecular materials, and between molecular materials and electrodes.7 Different substituents can be introduced to stabilize the orbitals, such as fluorine or carboxylate moieties, whereas alkoxy or amine groups shift up the energy levels of orbitals. In addition, the engineering of side chains allows for solution processing,8–10 generally at room temperature, which is compatible with plastic substrates used in flexible electronics.11,12 It is also required to develop printable and flexible sensors.13
Research and innovation in the field of chemical sensors should deal at first with new sensing materials. Most of the sensing materials are metal oxides, which require high temperatures for operation (200–400 °C) and even higher ones for their synthesis. On the contrary, molecular materials like polymers or macrocyclic compounds, such as porphyrin and phthalocyanine derivatives, can be deposited by solution processing, at room temperature.
All the general aforementioned considerations on side chains engineering and on the tuning of orbital energy are especially relevant for these macrocyclic molecules. Thus, the introduction of alkyl or alkoxy chains on lanthanide phthalocyanines, such as the lutetium bisphthalocyanine LuPc2, and lanthanide porphyrins allowed for solution processing of OFETs14 and chemical sensors.15–17 Photophysical properties of phthalocyanines or phthalocyanine analogs also depend on the presence of electron-accepting or donating moieties. Previously, the introduction of fluorine atoms on the phthalocyanine ring led to n-type semiconductors useful to get n-type OFETs18 and p–n heterojunctions,19 which can be used as gas sensors.20 A recent review on phthalocyanine-based OFETs shows the interests of phthalocyanine complexes in organic electronics.21 Among other examples of chemical sensors, we can cite conductometric sensors based on thioalkyl-substituted LuPc2, deposited by a jet spray technique, to detect volatile organic compounds,22 amphiphilic europium triple decker complexes deposited by quasi-Langmuir–Schaefer technique, for the detection of ozone,16 or alkoxy lanthanide complexes deposited as Langmuir–Blodgett and Langmuir–Schaefer films used in voltammetric sensors.23
In the present paper, we report on the synthesis of two new phthalocyanines bearing both electron-donating and electron-withdrawing substituents. While most molecular materials exhibit p-type conductivity, molecules bearing withdrawing substituents such as perfluorophthalocyanines can form n-type materials. However, it is of interest to have ambipolar materials, capable to exhibit both p-type and n-type conductivities, depending on the operating conditions.24,25 This is the reason why we chose to introduce electron-donating substituents on a fluorinated phthalocyanine. In addition, we wanted to introduce some electrochemically-active moiety to open the door to other applications in electrochemistry. All these considerations led us to choose a thienyl-containing ethoxy group. This moiety was used by T. Nyokong on metallophthalocyanines,26 for their electrocatalytic activity to determine L-cysteine concentrations in solution. The thienyl-containing ethoxy group was introduced by starting from 4-nitrophthalonitrile. The synthesis of fluorinated phthalocyanines bearing electron-donating moieties has been reported, but always by starting from a mixture of two phthalonitriles, one bearing the electron-donating substituent and the other being the tetrafluorophthalonitrile.27,28 Such a mixed condensation reaction leads to a mixture of molecules with five different chemical compositions, AnB4−n, with n = 0 to 4, if we call A and B the two substituted phthalonitrile molecules, the formula A2B2 corresponding to two regioisomers.29 Herein we aim at introducing four electro-donating groups on a fluorinated phthalocyanine. Two possibilities are available. We could use the mixed condensation method, which starts from tetrafluorophthalonitrile (F4PN) and a phthalonitrile bearing the four electro-donating groups. Another approach would be to start from a monoalkoxy-trifluorophthalonitrile synthesized from F4PN. Using this method, the phthalocyanine formation is expected to lead to a mixture of four regioisomers of C4h, D2h, C2v and CS symmetry, close to the statistical ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2:4, respectively, if their reactivity is identical to each other.30 So, we need to synthesize the 4-{2-(2-thienyl)ethoxy}-3,5,6-trifluorophthalo-nitrile (TF3PN, 1a) and the tetra-3,4,5,6-{2-(2-thienyl)ethoxy}-phthalonitrile (T4PN, 4).
:1:2:4, respectively, if their reactivity is identical to each other.30 So, we need to synthesize the 4-{2-(2-thienyl)ethoxy}-3,5,6-trifluorophthalo-nitrile (TF3PN, 1a) and the tetra-3,4,5,6-{2-(2-thienyl)ethoxy}-phthalonitrile (T4PN, 4).
In this work, we will discuss at first the selective synthesis of these two precursors 1a and 4, and their full characterization by means of 19F and 1H NMR spectroscopies and thermal analysis, and, in the case of 1a, X-ray crystal structure analysis. The synthesis of the zinc phthalocyanine 5, obtained from 1a (Schemes 1 and 2), and of its analogue 6, obtained by reaction of 4 with F4PN (Scheme 3) will be reported next.
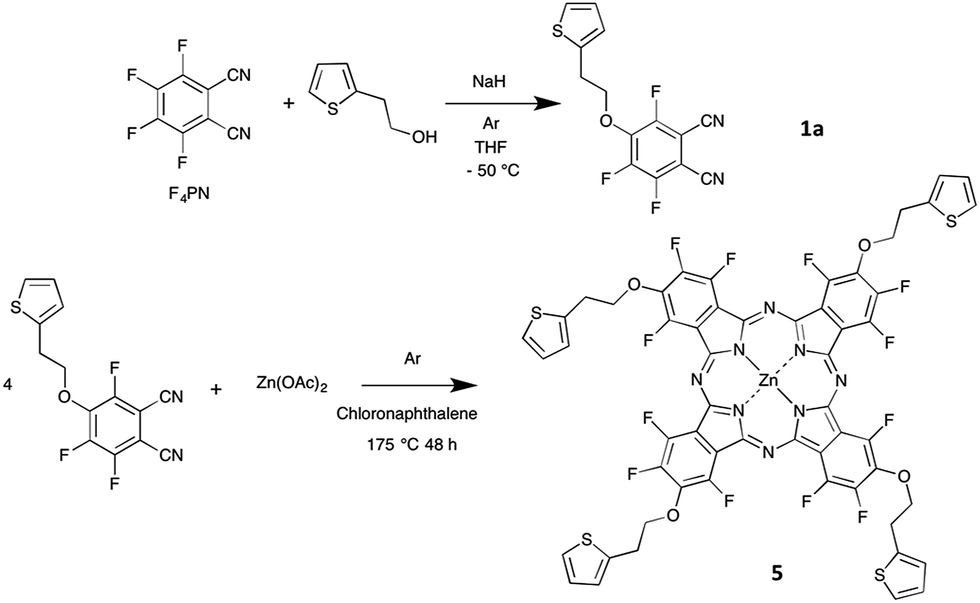 | ||
| Scheme 1 Synthesis of the phthalocyanine 5 (the isomer C4h is shown) and its precursor 1a. | ||
 | ||
| Scheme 2 List of all the phthalonitrile derivatives identified during the reactions of 2-(2-thienyl)ethanol with tetrafluorophthalonitrile. | ||
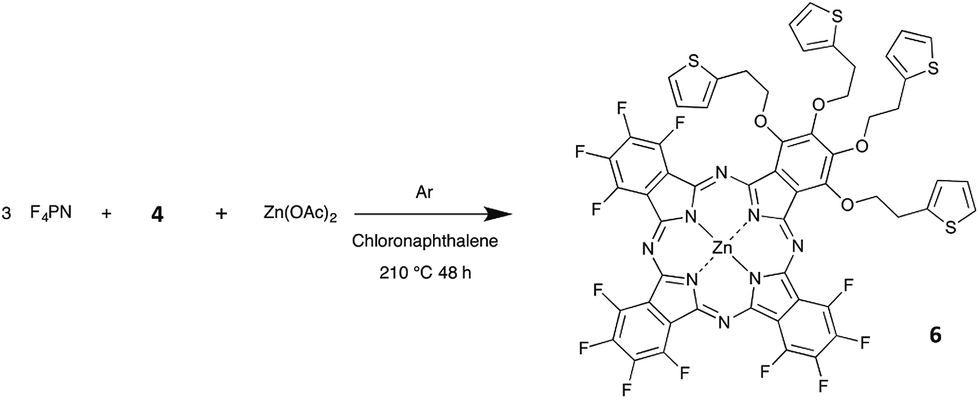 | ||
| Scheme 3 Synthesis of the phthalocyanine 6 from F4PN and 4 as precursors. | ||
Finally, the new phthalocyanines will be incorporated into a MSDI conductometric transducer, namely a molecular semi-conductor – doped insulator heterojunction, together with the lutetium bisphthalocyanine LuPc2, and the first data on ammonia chemosensing will be presented.16,31,32
Results and discussion
Syntheses
The 4-{2-(2-thienyl)ethoxy}-3,5,6-trifluorophthalonitrile (1a) was synthesized by reacting 2-(2-thienyl)ethanol with tetrafluorophthalonitrile (F4PN) in the presence of sodium hydride NaH in dry tetrahydrofuran (THF). These conditions were chosen because the usual method,33 that is to say the reaction of the alcohol in dimethylformamide (DMF), in the presence of potassium carbonate as a base, at 100 °C, over 24 h, was not efficient enough for the monosubstitution to occur. We only observed a partial conversion into 1a no matter the base we used, namely potassium carbonate33 or even metallic sodium,34 either at room temperature or at −10 °C. Having a selective reaction is crucial for this nucleophilic aromatic substitution since up to four substitutions can be formed. Using 19F NMR spectrum analysis of the crude reaction mixture we determined the relative amount of the products obtained in the course of the synthesis of 1a run at room temperature to be as follows: unreacted F4PN (56%), 1a (26%), the by-products 1b (6%), 2a (4%), 2b (5%), 2d (1%), 3a (1%) and 3b (1%). It is noteworthy to mention that the by-product 2c is not visible on the spectrum, being masked by the signal of 1b (Fig. S1†).The optimized conditions include the preparation of an alcoholate solution, with NaH as a base, at 0 °C and its very slow transfer by a cannula to a solution of F4PN cooled at −50 °C. In these conditions, the formation of the alcoholate is not quantitative and an excess of alcohol (1.3 eq.) and NaH (2 eq.) is necessary to get the target molecule in a very good yield, 93% after purification by chromatography. It is worth mentioning that raising the temperature and/or the rate of the addition of the alcoholate leads to a poorer selectivity despite the stoichiometry being the same. In such cases, the main by-product is the phthalonitrile monosubstituted by the alkoxy chain in ortho position with regard to a cyano group (1b). Even with the excess of alcohol and NaH used in the optimized conditions, the di-substitution reaction was limited to 5%, and the corresponding products (2a–d, mainly 2a and 2b, Scheme 2) could be separated by chromatography. Also, after optimization, F4PN was fully converted, mainly into 1a (93%), with 2% of 1b and 5% of 2a–d, as identified by 19F NMR of the crude reaction mixture.
The synthesis of 4 can be achieved by using classical conditions (K2CO3/DMF) for the nucleophilic substitution.33,35 However, in the present study, traces of non-fully substituted intermediates complicated the isolation of the target compound. That was not the case when the conditions worked out for 1a (NaH/THF) were used. However, the experimental conditions are simpler than those of 1a, since the addition does not require the same level of control as for the selective mono-substitution, in particular regarding the addition rate. The yield after purification was 46%.
Phthalocyanine 5 was obtained by heating 1a with zinc acetate in chloronaphthalene (Scheme 1), at 175 °C. This temperature was chosen after thermogravimetric analysis of 1a. After a few hours, the solution turned deep green. The mixture was held at this temperature for two more days. Compound 5 was precipitated by addition of cyclohexane and purified by washing with ethanol (yield = 77%). Compound 6 was obtained by reaction of 4 with F4PN in a molar ratio 1 to 3, with zinc acetate in chloronaphthalene, at 210 °C (Scheme 3). The mixture was purified at first by silica gel column chromatography to separate the perfluorinated phthalocyanine (blue), then further purified on preparative silica gel plates. Compound 6 was obtained as a green product (yield = 0.6%). Both phthalocyanines were characterized by mass spectrometry.
NMR analyses
The syntheses carried out to optimize the reaction conditions allowed us to isolate the trifluorophthalonitrile monosubstituted in ortho position with regard to a cyano group (1b), and also the di- and tri-substituted species, existing respectively as four (2a–d) and two (3a–b) regioisomers (Scheme 2). All compounds were characterized by 19F NMR spectroscopy, the chemical shifts and coupling constants allowing for a complete attribution of the signals. 19F NMR revealed to be a particularly powerful analytical method to identify the different isomers. The data are displayed in Table 1. JF–F couplings on a benzene ring are well known.36 JF–F ortho is very near 20 Hz whereas JF–F varies in a broad range for meta (−20 Hz to +20 Hz) and for para (+5 Hz to +20 Hz) positions. However, in the case of methoxy-fluorophthalonitrile derivatives34 coupling constants were found to be ca. 20, 14 and 9 Hz for JF–F ortho, meta and para, respectively. These coupling constants are very close to those we observed for 1a. Three doublets of doublets (dd) were observed for both mono-substituted molecules. In compound 1a, they appear at −120.1, −127.6 and −137.8 ppm (Fig. 1), and for compound 1b at −129.1, −136.7 and −143.1 ppm. For disubstituted molecules, among the four isomers, two are symmetrical, 2a and 2d, each showing one singlet. The unsymmetrical disubstituted isomers, 2b and 2c, are identified by two doublets with the same coupling constants that match with meta (2b, JF–F 12.3 Hz) and ortho (2c, JF–F 20.1 Hz) coupling. Finally, each trisubstituted derivative is identified by one singlet, at −122.6 and −133.5 ppm for 3a and 3b, respectively. For all these molecules, the chemical shifts appear between −120.1 and −129.1 ppm for fluorine atoms in ortho of cyano groups, and between −131.9 and 143.1 ppm for fluorine atoms in para positions. A more accurate way to attribute these chemical shift ranges is to identify the two nearest neighboring moieties. Thus, the signals of fluorine atoms between one cyano group and one alkoxy group lie between −120.1 and −122.8 ppm, those between two alkoxy groups are located between −131.9 and −133.5 ppm and those between one alkoxy group and another fluorine atom between −136.7 and −143.1 ppm.| 1a | 1b | 2a | 2b | 2c | 2d | 3a | 3b | |
|---|---|---|---|---|---|---|---|---|
| R1 | −120.1 | −121.7 | −122.8 | −122.6 | ||||
| R2 | −136.7 | −139.1 | ||||||
| R3 | −137.8 | −143.1 | −131.9 | −139.8 | −139.1 | −133.5 | ||
| R4 | −127.6 | −129.1 | −121.7 | −129.0 |
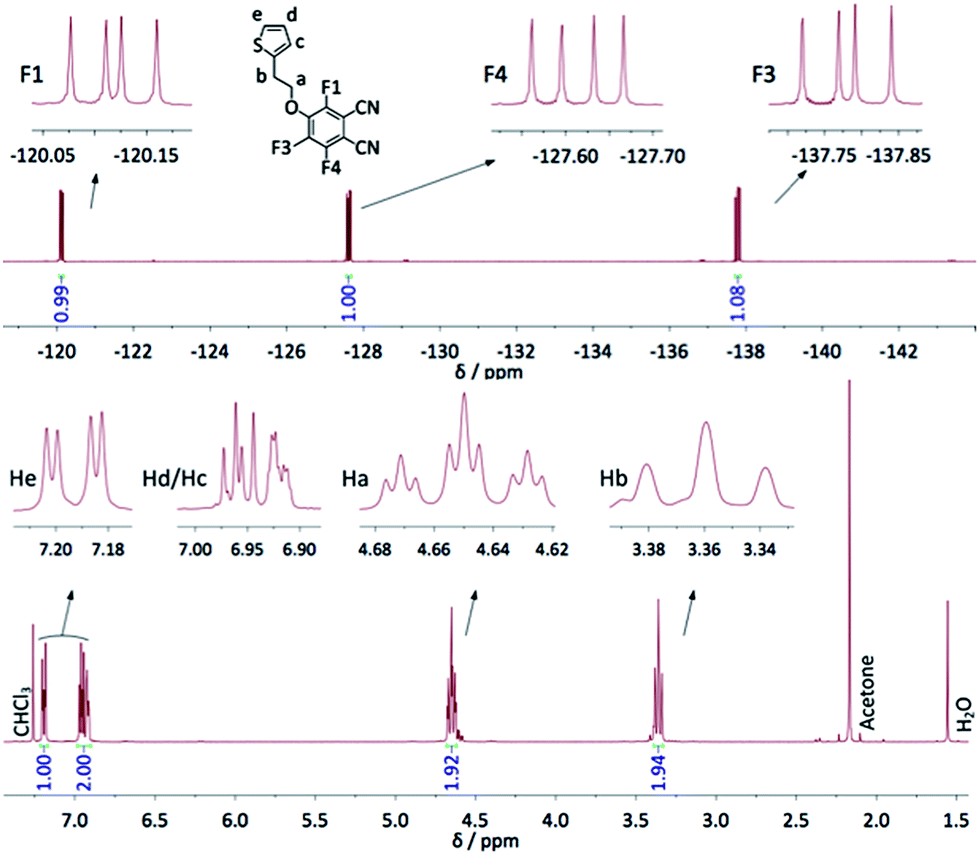 | ||
| Fig. 1 1H (bottom) and 19F NMR (top) spectra of 1a in CDCl3, with the integration values; the identification of all H and F atoms is given. | ||
In the 1H NMR spectrum of 1a, the ethylene bridge appears as two triplets. One of them is a triplet of triplets that results from a coupling (1.6 Hz) with the two fluorine atoms in ortho position of the alkoxy chain. This coupling is not visible in the 19F NMR spectrum by a lack of resolution. For the thienyl group, the H in ortho of the sulfur atom appears as a doublet of doublets, the H in ortho of the chain as a multiplet and the third H as a pseudo doublet of doublets. The 1H NMR spectrum of 4 exhibits 4 triplets in the region of the ethylene bridges (3–4.5 ppm), indicating that the four substituents are not equivalent: those in ortho position relatively to the cyano groups are different from those in meta position. The spectrum is provided in the ESI (Fig. S2†). The two triplets attributed to the CH2 connected to the O atoms appear at 4.15 and 4.26 ppm, i.e. at lower chemical shift compared to 1a, due to the absence of F atoms. This effect is smaller for the CH2 connected to the thienyl group, with two triplets at 3.08 and 3.28 ppm, vs. 3.36 in 1a. The signals assigned to the protons of thienyl moieties appear as three signals. The first one, centered at 7.16 ppm, assigned to He, appears as two pseudo triplets that actually result from the superimposition of two doublets of doublets slightly shifted from each other because of the lack of symmetry of the molecule. The coupling constants of He with Hd and Hc are 5.4 and 1.5 Hz, respectively. The multiplet at 6.84 ppm, assigned to Hc, matches to only two protons, the signal of the two other Hc protons being coincident with the multiplet assigned to Hd, centered at 6.93 ppm (Fig. S2†).
In the 19F and 1H NMR spectra of phthalocyanine 5 (Fig. 2), broad signals result from the existence of different isomers and from a possible aggregation in DMSO, despite the polarity of this solvent. The 19F NMR spectrum shows mainly three signals, at −133.2, −142.5 and −146.3 ppm that can be attributed to F1, F4, and F3, respectively. They are shifted by ca. 13 and 15 ppm for the fluorine atoms in ortho position with regard to the macrocycle, compared to the chemical shifts in the starting phthalonitrile 1a, and by only 9 ppm for the F atom farther from the cycle, as a result of the ring current effect. The broadest signals are attributed to F atoms in ortho positions of the pyrrole subunit (F1 and F4) since they are more influenced by the effect of the neighboring groups. On the contrary, the third signal (F3) is sharper than the others because it lies farther from the neighboring cycles, no matter the symmetry of the phthalocyanine isomer. The presence of other smaller and thinner signals, near the main signals attributed to F1 and F4, is due to the existence of different isomers of the phthalocyanine, as mentioned in the introduction when we start from nonsymmetrical phthalonitriles. The 1H NMR shows one multiplet for the thienyl moieties, between 7 and 7.5 ppm that integrates for 12 protons, whereas for the CH2 groups three broad signals are visible, at 3.72, 4.53 and 5.04 ppm, which integrate for 12 protons. The signals at 3.72 and 5.04 ppm that have the same shape and height, can be attributed to Hb and Ha, respectively. This corresponds to shifts of +0.39 and +0.36 ppm, respectively, compared to 1a. The third signal, at 4.53 ppm, can also be attributed to Ha, but of other isomers, with a shift of −0.15 ppm. Thus, from such a small negative shift of the signal of Hb, it is possible to deduce that a fourth signal lies in the water peak that would correspond to the 4 missing protons. All these phenomena observed in 1H NMR spectrum result from the presence of several isomers, of the ring current effect and of a possible aggregation of the phthalocyanine 5. Recording the 1H NMR spectrum of 6 was not possible because of its high tendency to aggregate.
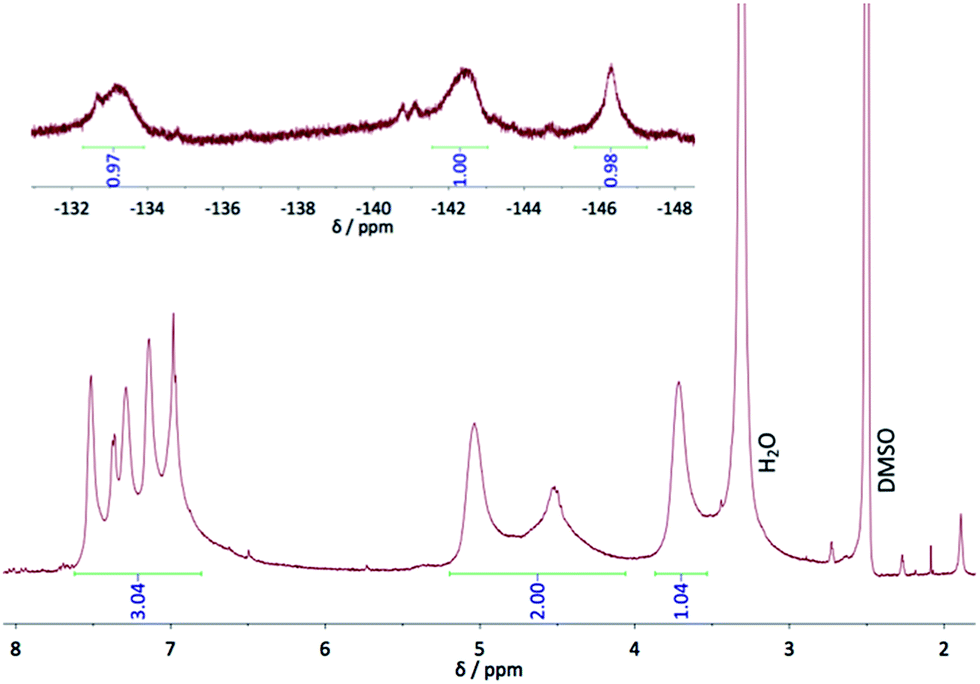 | ||
| Fig. 2 1H (bottom) and 19F (top) NMR spectra of 5 in DMSO-d6. | ||
Mass and infra-red analyses
High resolution mass spectrometry of compound 1a was carried out by electrospray, in positive ionization mode. It displays a parent ion at m/z = 309.0297 that corresponds to [M + H]+. The mass spectrum of 4 obtained by MALDI-ToF exhibits a signal at 671.0654, assigned to [M + K]+.Compound 5 was also identified by electrospray mass spectrometry, in the same conditions, with a parent ion at m/z = 1297.0381 that corresponds to the monoprotonated phthalocyanine [M + H]+ (Fig. S3†). The mass spectrum of 6 obtained by MALDI-ToF exhibits also a signal at 1297.0806, assigned to [M + H]+ (Fig. S4†).
The IR spectrum of 1a shows additional C–H aromatic (3108 and 3086 cm−1) and aliphatic vibrations (2978 and 2964 cm−1) compared to the starting material. Moreover, the CN vibration peak, at 2239 cm−1 and 2232 cm−1 for 1a and 4, respectively, was shifted by −7 and −14 cm−1 compared to the starting material, in accordance with the replacement of withdrawing substituents by electron-donating moieties on the aromatic ring.
Cyclic voltammetry
The cyclic voltammogram (CV) of complex 5 was registered in DMF containing 0.1 M Bu4NPF6 at various scan rates (Fig. 3). Two reduction steps can be identified at −0.84 V (Ep,red1, irreversible system) and −1.05 V (reversible system,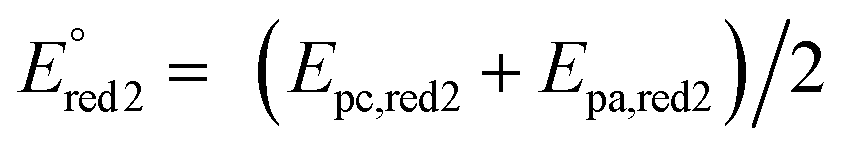 ) vs. SCE, which can be attributed to the successive reductions of the phthalocyanine ring, corresponding to the redox couples Pc2−/Pc3− and Pc3−/Pc4−, respectively. One oxidation peak attributed to the first oxidation of the phthalocyanine ring appears at +0.97 V (Ep,ox1).
) vs. SCE, which can be attributed to the successive reductions of the phthalocyanine ring, corresponding to the redox couples Pc2−/Pc3− and Pc3−/Pc4−, respectively. One oxidation peak attributed to the first oxidation of the phthalocyanine ring appears at +0.97 V (Ep,ox1).
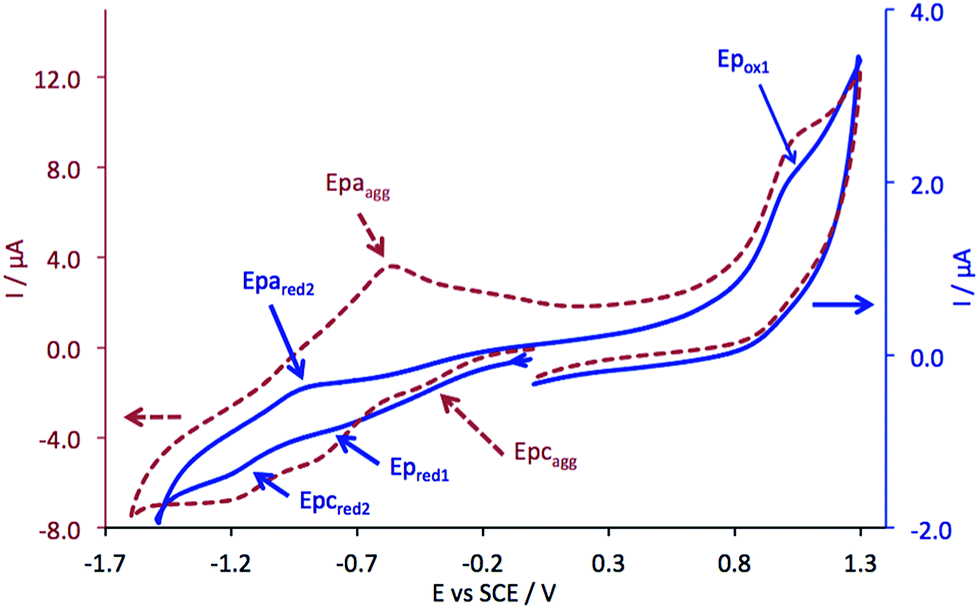 | ||
| Fig. 3 CV of 5, on Pt disc electrode, in DMF containing 0.1 M Bu4NPF6, at a scan rate of 0.1 V s−1, at a concentration of 10−3 mol L−1 (solid blue line) and at 4 × 10−3 mol L−1 (aggregation, dashed red line). | ||
These data were obtained using a freshly polished working electrode. After few CVs, an additional cathodic peak appears at ca. −0.4 V vs. SCE.
Besides, when we started from a solution at a higher concentration (4 × 10−3 mol L−1), this cathodic peak also appeared at −0.4 V (Epc,agg), whereas a new anodic peak, at −0.56 V (Epa,agg), increased cycle after cycle. This is due to the adsorption of aggregates at the surface of the working electrode.
Even though electrochemical potentials are determined for molecules in electrolytic solutions, they can be used to estimate the energy values of the HOMOs and LUMOs Frontier orbitals. These are known to be responsible for the electronic properties of the corresponding materials, in the solid state. A consensus exists to calculate them from the onset potential, which is defined as the potential at which the initial electron transfer from the HOMO, or to the LUMO, arises on the cyclic voltammogram, by the increase of the anodic or cathodic current, respectively37–40 They are calculated from the redox potentials determined in solution versus SCE as reference electrode:
| EHOMO = −(Eox1/SCE + 4.4) |
| ELUMO = −(Ered1/SCE + 4.4) |
The energies of the HOMO and LUMO of 5 are −5.37 and −3.46 eV, respectively, and −5.44 and −3.97 eV respectively for Zn(F16Pc).41 This explains that the energy barrier observed in MSDI heterojunctions are lower when using 5 rather than the perfluorinated phthalocyanine as sublayer.
Electronic absorption spectroscopy
The electronic absorption spectrum of 5 in chloronaphthalene (Fig. 4) exhibits two main absorption bands in the UV-visible region: The Soret band at 363 nm (logε = 4.37) and the Q band at 695 nm (logε = 4.85), as expected for a phthalocyanine derivative. Two smaller bands associated to vibrations can be found at 625 nm (logε = 4.14) and as a shoulder at 666 nm. The Q band of 5 undergoes a bathochromic shift (10 nm) compared to the perfluorinated zinc phthalocyanine.42 In this solvent, the Beer–Lambert law is valid, at least up to 10−5 mol L−1. This is not the case in dimethylformamide (λmax = 687 nm) where aggregation occurs above 2 × 10−6 mol L−1. As a result there is an increase of the band at 651 nm, which is attributed to H aggregates.43,44
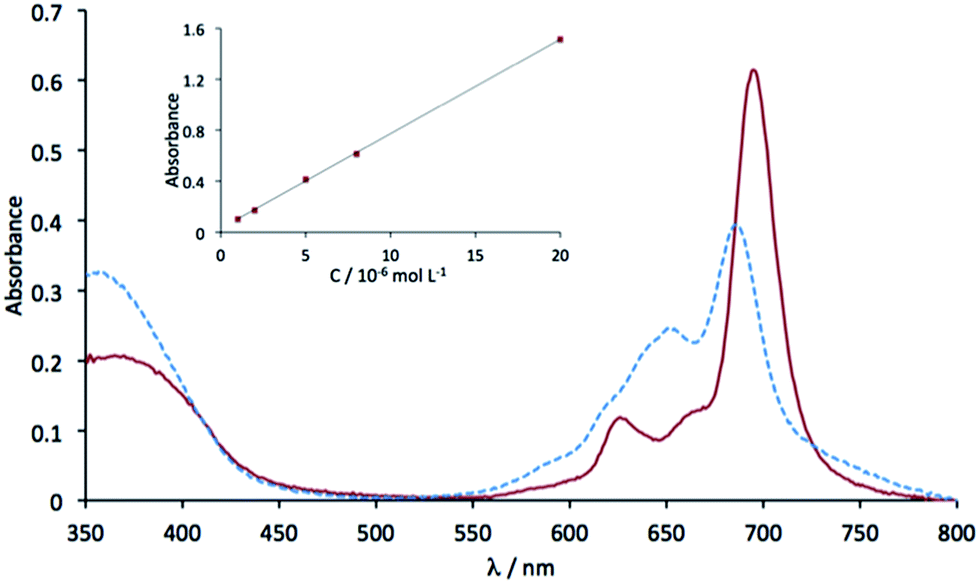 | ||
| Fig. 4 UV-visible absorption spectra of 5 in chloronaphthalene (solid red line) and in dimethylformamide (dotted blue line), both at 8 × 10−6 mol L−1. The inset shows the variation of the absorbance at the maximum wavelength as a function of the concentration, in chloronaphthalene. | ||
The phthalocyanine 6 exhibits the same kind of spectrum (Fig. S5†) as 5. In chloronaphthalene the Q band can be found at 697 nm and the Soret band at 378 nm. The only difference is the value of the full width at half maximum of the Q band that is larger in 6 than in 5, namely 37 nm against 27 nm, at a concentration of 8 × 10−6 mol L−1. It indicates that for 6 aggregation already started at this concentration. In CH2Cl2 the Q band appears at 688 nm and the Soret band at 351 nm (Fig. S5†), but the spectrum shows the presence of aggregates.
Thermal analysis
1a exhibits a fusion peak at 59 °C accompanied by a huge supercooling phenomenon. The DSC analysis shows that crystallization occurs at +22 °C during the cooling stage from the liquid phase, at 1 °C min−1 (Fig. S6†). 4 exhibits a DSC thermogram with fusion (+23.2 °C) and crystallization (52.6 °C) temperatures rather close to those of 1a, with an important supercooling phenomenon as well. However, a major difference is that the crystallization phenomenon of 4 occurs during the heating stage, as shown in the second thermal cycle of Fig. S7.† This means that the four thienylethoxy substituents prevent 4 from crystallizing easily. Both 1a and 4 have been analyzed also by TGA, at a heating rate of 5 °C min−1, under nitrogen (Fig. S8†). 1a shows only one weight loss step, higher than 99%, starting from 180 °C and finishing at 250 °C, whereas 4 exhibits a weight loss of ca. 50% between 260 °C and 320 °C. For the synthesis of phthalocyanines, due to the presence of fluorine atoms on the starting phthalonitrile, we cannot use the classical method that takes place in basic conditions in an alcohol as a solvent, usually n-hexanol (boiling point 160 °C). High temperature conditions are needed. TGA allows us to know the highest reaction temperature compatible with the stability of the starting phthalonitrile.The TGA of 5 carried out at a heating rate of 2 °C min−1 showed two weight loss steps (Fig. S9†). The first one (23%), from 200 to 330 °C, can be attributed to the loss of thienyl groups (calc. 26%), and the second one (68%), between 330 and 635 °C, can be attributed to the loss of all the macrocycle (calc. {–CH2CH2O}4F12Pc 69%). The residue (7%) mainly corresponds to the zinc atom (calc. 5%).
X-ray analysis of 1a
1a crystallizes in the monoclinic system (space group P21/c). The thienyl substituent was found to be disordered over two positions (75%/25%) (Fig. 5). Within a molecule, the two rings make an angle of 59.45(5)° relatively to each other. The thienyl ring of one molecule is almost parallel to the benzene ring of a neighboring molecule, with an angle of 10.15(6)° between their mean planes. The intermolecular distances between neighboring molecules are more interesting. The distance between the centroid of the benzene ring, defined from atoms C7 to C12, and the centroid of the thienyl ring is Ct–Ct = 3.8148(2) Å. The distance between the centroid of the benzene ring and the mean plane of the thienyl ring is 3.408 Å, and the one between the centroid of the thienyl ring, defined from S1 and C1 to C4, and the mean plane of the benzene ring is 2.795 Å. The shortest intermolecular distance can be found between one C atom of the thienyl ring and one C atom of the benzene ring, C(4)–C(8) = 3.275 Å. It is significantly shorter than the sum of van der Waals radii (3.40 Å). Thus, the crystal packing is controlled by some π–π interactions between the thienyl (πD) and phthalonitrile (πA) rings (Fig. 5). The carbon–carbon distances are 3.275–3.758 Å between the aromatic rings. The projection on the (b, c) plane showing these π–π interactions makes clearly visible the existence of chains parallel to the c-axis. Crystallographically speaking, these molecules correspond to each other by a glide plane perpendicular to [0, 1, 0] with glide component [0, 0, ½]. Eventhough the withdrawing effect of cyano groups disappears in the case of the phthalocyanine molecule, π–π interactions could be preserved between thienyl rings and the electron poor benzene rings due to the presence of the fluorine atoms. Even though we have not structural information on the thin films of phthalocyanines, these intermolecular π–π interactions should also play a role in the packing of molecules in the solid state.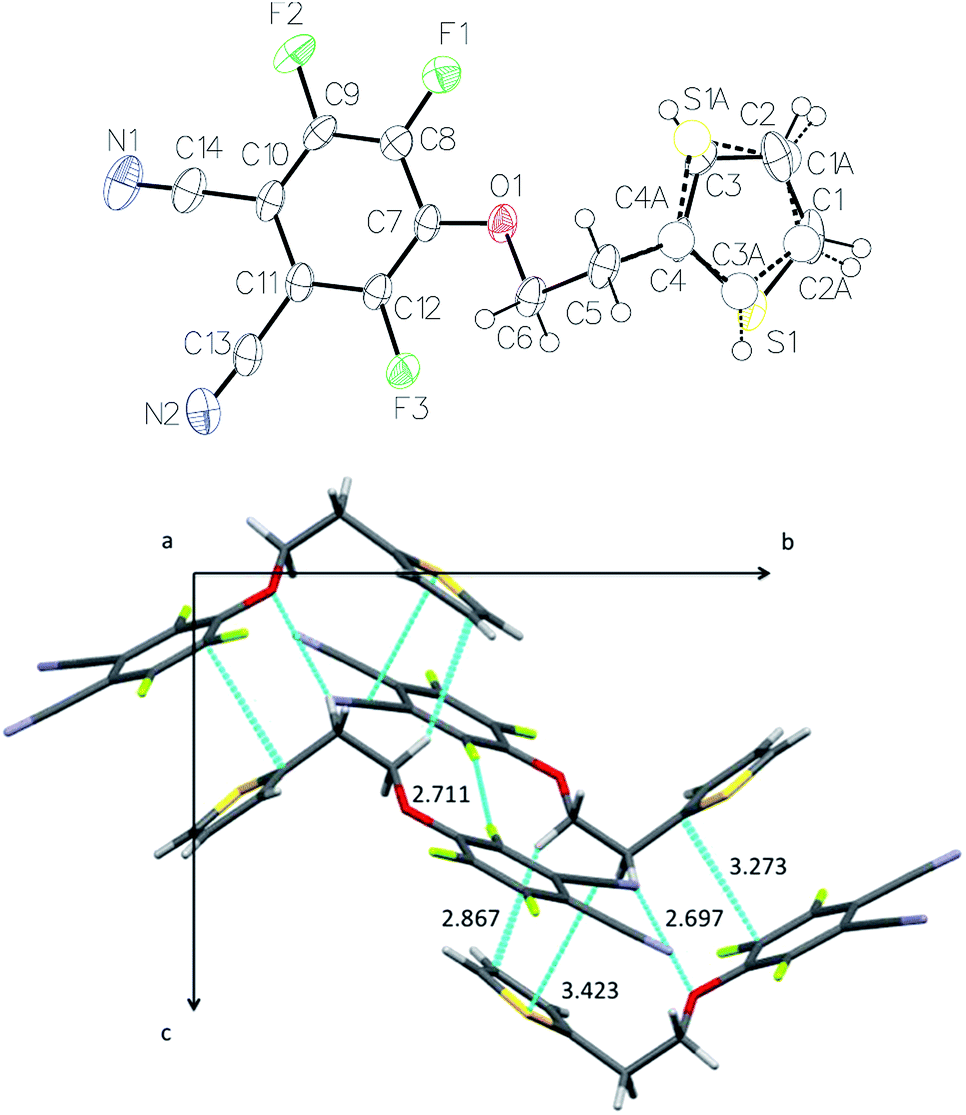 | ||
| Fig. 5 X-ray crystal structure of 1a: ORTEP representation (30% probability level) (top) and projection on the (b, c) plane (bottom). The shortest intermolecular distances are indicated (in Å). | ||
Electrical and sensing properties
Films of 5 and 6 were obtained by solvent cast, from 85 μL of DMF solutions, at concentrations of 3.3 × 10−3 and 2.77 × 10−3 mol L−1, respectively, deposited dropwise on interdigitated electrodes (IDE). The substrates were heated at 60 °C in a closed desiccator in order to fully evaporate the solvent and increase the homogeneity of the films. Electrical measurements in ambient atmosphere showed that these films were too resistive to register I–V characteristics, at least up to +10 V, with the present IDEs. Only, an upper limit value for the conductivity can be guessed at. Their conductivity is lower than 5 × 10−10 Ω−1 cm−1, i.e. lower than for nonsubstituted phthalocyanine films in the same conditions.45 However, it was possible to build MSDI heterojunctions, by vacuum evaporation of lutetium bisphthalocyanine, LuPc2, 50 nm in thickness, on films of 5 and 6. MSDIs are devices recently designed and patented by one of the authors.31,46 The I–V characteristics obtained with the 5/LuPc2 and 6/LuPc2 MSDIs are symmetrical but not linear (Fig. S10 and S11†), contrarily to what is observed for a LuPc2 resistor in the same potential range.31 The non-linearity is greater for the 6/LuPc2 MSDI, with a smaller current, ca. 0.6 μA at 10 V, against 2.60 μA at the same bias voltage for the 5/LuPc2 MSDI. This shows that the energy barrier between the molecular materials or the one between the sublayer and the electrodes is higher with 6 than with 5. This could be the result of a segregation between the fluorinated and nonfluorinated parts of the unsymmetrical phthalocyanine that would modify the interfaces with this sublayer. Under NH3, these MSDIs exhibit a current decrease (Fig. 6 and S12†), as expected for a p-type MSDI heterojunction prepared from a p-type sublayer.31 This reveals that compounds 5 and 6 are capable of transporting majority charge carriers positive in nature, even though their density remains very low. This is not obvious when considering the presence of twelve fluorine atoms on the phthalocyanine ring. It indicates that the four electron-donating alkoxy groups compensate, at least partly, the withdrawing effect of fluorine atoms. It is worth noting that, with a n-type sublayer, as found with perfluorinated phthalocyanines, NH3 induces a current increase resulting from the existence of energy barriers between the two molecular materials and between the electrodes and the sublayer.47Measurements were carried out under controlled relative humidity (rh). At 30% rh, the current reached 13 μA under a polarization of 10 V for the 5/LuPc2 MSDI (Fig. 6). The response to NH3 depends on its concentration. For example, for the 5/LuPc2 MSDI, the response to 60 ppm NH3 was −0.5 μA, which corresponds to a relative response, defined as (Ion − Ioff)/Ioff, of −3.8%. This value is lower than the one obtained with a n-type MSDI device combining a perfluoro-phthalocyanine and LuPc2, about +25% at 60 ppm,47 but very close to the one obtained with other p-type MSDIs, e.g. between −2% and −4% for Cu(tBu4Pc)/LuPc2 MSDIs prepared by different processes.16 In the same conditions, in particular at a same relative humidity value, the 6/LuPc2 MSDI exhibits a response of −3.3% at 60 ppm NH3 (Fig. S12†), i.e. very close to the one of MSDIs prepared from 5. It is worth noting that the response to NH3 increases with the rh value, from −2.5 at 10% rh up to 5.4 at 50% rh.
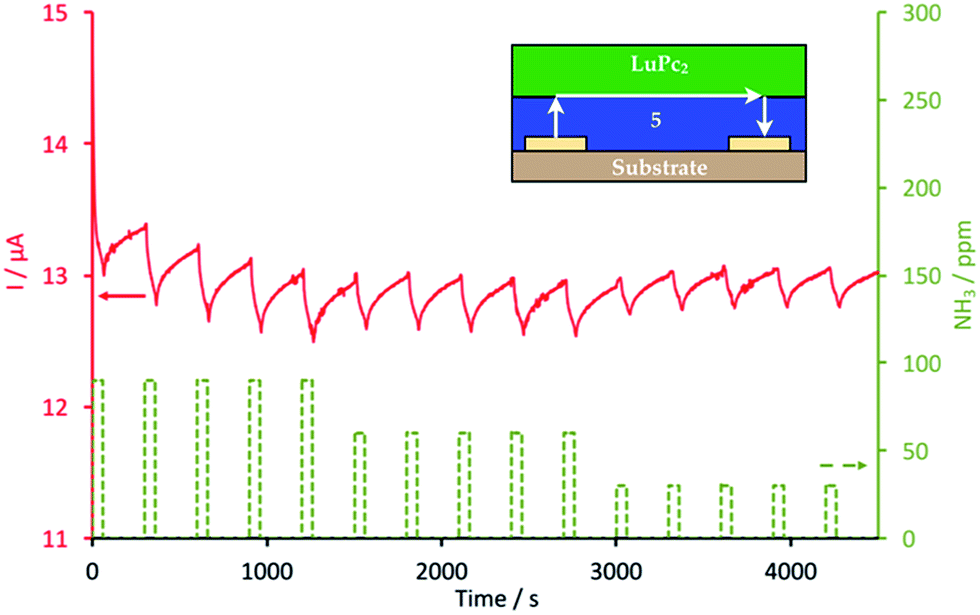 | ||
| Fig. 6 Response of a 5/LuPc2 MSDI heterojunction to ammonia (NH3, 90, 60 and 30 ppm), at 30% relative humidity, during exposure/recovery cycles (1 min/4 min). | ||
Experimental
Reagents and instrumentation
Elemental analyses were performed on an Analyzer CHNS/O Thermo Electron Flash EA 1112 Series.
The UV-visible absorption spectra of solutions were obtained from a Varian UV-vis spectrophotometer Cary 50 using quartz cells and these of thin films were recorded using a Shimadzu UV-2600 Spectrometer with a bare floated glass substrate as a reference.
The infrared spectra were recorded on a Bruker Vector 22 using KBr pellets, in transmission mode.
NMR spectra were recorded using a BRUKER 300 MHz Avance III Nanobay spectrometer. 1H NMR spectra were calibrated with respect to TMS on the basis of the relative chemical shift of the residual non-deuterated solvent as an internal standard and 19F NMR spectra were calibrated with respect to CFCl3.
MS experiments were performed on a hybrid electrospray quadrupole time-of-flight mass spectrometer (Synapt G2 HDMS, Waters, Manchester, U.K.) coupled to an automated chip-based nanoelectrospray device (Triversa Nanomate, Advion Biosciences, Ithaca, U.S.A.) operating in the positive ion mode. Denatured MS analysis was performed on the Synapt G2 HDMS instrument only, with external calibration using the multiply charged ions produced by 2 μM horse heart myoglobin solution diluted in water/acetonitrile/formic acid (50 v/50 v/1 v), and classical interface tuning parameters of the mass spectrometer (Vc = 40 V, Pi = 2.1 mbar). MALDI-TOF mass spectra were obtained on a Bruker ProFLEX III spectrometer, using dithranol as a matrix.
Single clear light colorless prism-shaped crystals of 1a were obtained by slow crystallization from a toluene solution. A suitable crystal (0.68 × 0.28 × 0.28 mm3) was selected and mounted on a MITIGEN holder oil on a Bruker D8 VENTURE diffractometer. The crystal was kept at T = 100 K during data collection. Using Olex2,50 the structure was solved with the ShelXT structure solution program,51 using the Direct Methods solution method. The model was refined with version 2014/7 of ShelXL52 using Least Squares minimization. All non-hydrogen atoms were refined anisotropically. Hydrogen atom positions were calculated geometrically and refined using the riding model. The thiophene substituent was found disordered over two positions (75%/25%) and some EADP constraints were employed to maintain a reasonable model.
Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1509, 1485 and 1379 (C–F), 1317, 1161 (C–O), 1113, 1033, 713 (CC). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.20 (1H, dd, JHe–Hd 5.1 Hz, JHe–Hc 1.2 Hz, He), 6.96 (1H, dd, JHd–He 5.1 Hz, JHd–Hc 3.5 Hz, Hd), 6.92 (1H, m, JHc–Hd, JHc–He, Hc), 4.65 (2H, tt, JHa–Hb 6.4 Hz, JHa–F1,3 1.6 Hz, Ha), 3.36 (2H, t, JHb–Ha 6.4 Hz, Hb). 19F NMR (282.4 MHz, CDCl3): δ (ppm) −120.1 (1F, dd, JF1–F3 13.8 Hz, JF1–F4 9.7 Hz, F1) −127.6 (1F, dd, JF4–F3 19.9 Hz, JF4–F1 9.7 Hz, F4), −137.8 (1F, dd, JF3–F4 20.0 Hz, JF3–F1 13.8 Hz, F3). m/z (HR-ESMS): 309.0297 [M + H]+, calculated 309.0309.:1) as eluent, to yield compound 4 as a colorless solid (1 g, 46%) after solvent evaporation. Mp 52.6 °C, crystallization peak 23.2 °C (a 10.700 mg sample was heated from −20 °C to 70 °C, and cooled down to −20 °C, both at a rate of 1 °C min−1), dec. 260 °C (a 8.029 mg sample was heated from 30 °C to 650 °C at a rate of 20 °C min−1). Elemental analysis: found: C, 60.95; H, 4.42; N, 4.42. Calc for C32H28F3N2O4S4 C, 60.74; H, 4.46; N, 4.43. FTIR (KBr pellet): ν (cm−1) [3103, 3085 and 3071 (Ar–H)], [2953, 2915, 2891 and 2856 (aliph. C–H)], 2232 (CN), 1560 (CC), [1468, 1455, 1443 and 1433 (C–F)], 1115, [1042, 1027 and 1014 (C–O)], 851, 832, 697. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.15 (4H, td, He), 6.93 (4H, m, Hd), 6.83 (4H, m, Hc), 4.28 and 4.15 (2 × 4H, 2 t, JHa–Hb 6.9 Hz, Ha ortho and meta (CN)), 3.28 and 3.09 (2 × 4H, 2 t, JHb–Ha 6.4 Hz, Hb ortho and para (CN)). m/z (MALDI-ToF): 671.0654 [M + K]+, calculated 671.0569.ε 4.85/dm3 mol−1 cm−1), 666 (sh), 625 (logε 4.14) and 363. FTIR (KBr pellet): ν cm−1 3110 and 3070 (Ar–H), 2958 and 2925 (aliph. C–H), 1616 (CC), 1506, [1476, 1463, 1437 and 1380] (C–F), 1306, 1145 (C–O), 995, 700 (CC). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.20 (12H, m, Hc–e), 5.04 (4H, s, Ha), 4.53 (4H, s, Ha), 3.72 (4H, s, Hb). 19F NMR (282.4 MHz, DMSO-d6): δ (ppm) −133.2 (F1), −142.5 (F4), −146.3 (F3). m/z (HR-ESMS) 1297.0381 [M + H]+, calculated 1297.0294.:1) and ethyl acetate as eluents, allowing the elution of green and blue fractions, respectively. The green fraction was further purified on a preparative chromatography plate with CHCl3 as eluent. The product was dried at 100 °C under reduced pressure for 2 h to yield a green solid (3.6 mg, 0.6%). Elemental analysis: found: C, 51.20; H, 2.42; N, 7.86. Calc for C56H28F12N8O4S4Zn: C, 51.8; H, 2.17; N, 8.63. λmax (CH2Cl2)/nm 690, 650 (sh), 619 (sh) and 350. FTIR (KBr pellet): ν (cm−1) 3115 and 3073 (Ar–H), 2924 and 2854 (aliph. C–H), 1636 and 1521 (CC), [1487, 1455 and 1418] (C–F), 1369, 1349, 1333, 1313, 1270, 1146 and 1127 (C–O), 1011, 955, 693 (CC). m/z (MALDI-ToF) 1297.0806 [M + H]+, calculated 1297.0289. The blue fraction was identified as Zn(F16Pc). m/z (MALDI-ToF), 863.7301 [M]+, calculated 863.9282.925 reflections measured, 3059 unique (Rint = 0.0482) which were used in all calculations. The final ωR2 was 0.1323 (all data) and R1 was 0.0532 (I > 2σ(I)).
C), 1509, 1485 and 1379 (C–F), 1317, 1161 (C–O), 1113, 1033, 713 (CC). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.20 (1H, dd, JHe–Hd 5.1 Hz, JHe–Hc 1.2 Hz, He), 6.96 (1H, dd, JHd–He 5.1 Hz, JHd–Hc 3.5 Hz, Hd), 6.92 (1H, m, JHc–Hd, JHc–He, Hc), 4.65 (2H, tt, JHa–Hb 6.4 Hz, JHa–F1,3 1.6 Hz, Ha), 3.36 (2H, t, JHb–Ha 6.4 Hz, Hb). 19F NMR (282.4 MHz, CDCl3): δ (ppm) −120.1 (1F, dd, JF1–F3 13.8 Hz, JF1–F4 9.7 Hz, F1) −127.6 (1F, dd, JF4–F3 19.9 Hz, JF4–F1 9.7 Hz, F4), −137.8 (1F, dd, JF3–F4 20.0 Hz, JF3–F1 13.8 Hz, F3). m/z (HR-ESMS): 309.0297 [M + H]+, calculated 309.0309.:1) as eluent, to yield compound 4 as a colorless solid (1 g, 46%) after solvent evaporation. Mp 52.6 °C, crystallization peak 23.2 °C (a 10.700 mg sample was heated from −20 °C to 70 °C, and cooled down to −20 °C, both at a rate of 1 °C min−1), dec. 260 °C (a 8.029 mg sample was heated from 30 °C to 650 °C at a rate of 20 °C min−1). Elemental analysis: found: C, 60.95; H, 4.42; N, 4.42. Calc for C32H28F3N2O4S4 C, 60.74; H, 4.46; N, 4.43. FTIR (KBr pellet): ν (cm−1) [3103, 3085 and 3071 (Ar–H)], [2953, 2915, 2891 and 2856 (aliph. C–H)], 2232 (CN), 1560 (CC), [1468, 1455, 1443 and 1433 (C–F)], 1115, [1042, 1027 and 1014 (C–O)], 851, 832, 697. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.15 (4H, td, He), 6.93 (4H, m, Hd), 6.83 (4H, m, Hc), 4.28 and 4.15 (2 × 4H, 2 t, JHa–Hb 6.9 Hz, Ha ortho and meta (CN)), 3.28 and 3.09 (2 × 4H, 2 t, JHb–Ha 6.4 Hz, Hb ortho and para (CN)). m/z (MALDI-ToF): 671.0654 [M + K]+, calculated 671.0569.ε 4.85/dm3 mol−1 cm−1), 666 (sh), 625 (logε 4.14) and 363. FTIR (KBr pellet): ν cm−1 3110 and 3070 (Ar–H), 2958 and 2925 (aliph. C–H), 1616 (CC), 1506, [1476, 1463, 1437 and 1380] (C–F), 1306, 1145 (C–O), 995, 700 (CC). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.20 (12H, m, Hc–e), 5.04 (4H, s, Ha), 4.53 (4H, s, Ha), 3.72 (4H, s, Hb). 19F NMR (282.4 MHz, DMSO-d6): δ (ppm) −133.2 (F1), −142.5 (F4), −146.3 (F3). m/z (HR-ESMS) 1297.0381 [M + H]+, calculated 1297.0294.:1) and ethyl acetate as eluents, allowing the elution of green and blue fractions, respectively. The green fraction was further purified on a preparative chromatography plate with CHCl3 as eluent. The product was dried at 100 °C under reduced pressure for 2 h to yield a green solid (3.6 mg, 0.6%). Elemental analysis: found: C, 51.20; H, 2.42; N, 7.86. Calc for C56H28F12N8O4S4Zn: C, 51.8; H, 2.17; N, 8.63. λmax (CH2Cl2)/nm 690, 650 (sh), 619 (sh) and 350. FTIR (KBr pellet): ν (cm−1) 3115 and 3073 (Ar–H), 2924 and 2854 (aliph. C–H), 1636 and 1521 (CC), [1487, 1455 and 1418] (C–F), 1369, 1349, 1333, 1313, 1270, 1146 and 1127 (C–O), 1011, 955, 693 (CC). m/z (MALDI-ToF) 1297.0806 [M + H]+, calculated 1297.0289. The blue fraction was identified as Zn(F16Pc). m/z (MALDI-ToF), 863.7301 [M]+, calculated 863.9282.925 reflections measured, 3059 unique (Rint = 0.0482) which were used in all calculations. The final ωR2 was 0.1323 (all data) and R1 was 0.0532 (I > 2σ(I)).Conclusions
The reaction of tetrafluorophthalonitrile with 2-(2-thienyl)-ethanol in the presence of NaH allowed us to identify all the possible substituted derivatives, from mono- to tetraalkoxy phthalonitriles. The optimization of the reaction conditions and in particular the decrease of the reaction temperature and a very slow addition of the alcoholate solution, allowed us to obtain the monoalkoxy derivative 1a with a very good yield (93%). Its reaction with zinc acetate in chloronaphthalene, during two days, led to the formation of the tetraalkoxy-dodecafluorinated zinc phthalocyanine 5. The unsymmetrical analogue 6 was also synthesized by the mixed condensation of F4PN with the tetraalkoxyphthalonitrile 4. MSDI heterojunctions were prepared by coating these zinc phthalocyanines, used as poor conducting sublayers, with LuPc2 (50 nm) as a top layer. These MSDI devices exhibit symmetrical but nonlinear I–V characteristics, which brings to light the existence of an energy barrier at the interface between the materials. Chemosensing experiments under ammonia allowed us to demonstrate the positive nature of the majority charge carriers in 5 and 6. Work is currently in progress to modify the electron-donating/withdrawing balance in the phthalocyanine macrocycle in order to get an ambipolar material that is expected to exhibit original chemosensing properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Agence Nationale de la Recherche for funding through the ANR projects CAP-BTX 2010 and OUTSMART 2015 and the MENESR for a PhD grant (A. W.). Financial support from the European Union and the Conseil Régional de Bourgogne through the FABER and the PARI SMT 08 and CDEA programs is gratefully acknowledged. We would like to thank the European Union for fundings (FEDER and short term missions through the COST action TD1105 EuNetAir). Dr Yohann Rousselin (ICMUB) is thanked for the X-ray structure determination of 1a, and Jean-Marc Strub (Univ. Strasbourg) for electrospray high resolution mass spectrometry analyses.Notes and references
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 15259–15278 CrossRef CAS PubMed.
- Y. Chen, D. Li, N. Yuan, J. Gao, R. Gu, G. Lu and M. Bouvet, J. Mater. Chem., 2012, 22, 22142–22149 RSC.
- H. Sirringhaus, Adv. Mater., 2005, 17, 2411–2425 CrossRef CAS.
- H. Usta, A. Facchetti and T. J. Marks, Acc. Chem. Res., 2011, 44, 501–510 CrossRef CAS PubMed.
- R. Schlaf, B. A. Parkinson, P. A. Lee, K. W. Nebesny and N. R. Armstrong, J. Phys. Chem. B, 1999, 103, 2984–2992 CrossRef CAS.
- W. Chen, D. C. Qi, Y. L. Huang, H. Huang, Y. Z. Wang, S. Chen, X. Y. Gao and A. T. S. Wee, J. Phys. Chem. C, 2009, 113, 12832–12839 CAS.
- M. Aghamohammadi, R. Rödel, U. Zschieschang, C. Ocal, H. Boschker, R. T. Weitz, E. Barrena and H. Klauk, ACS Appl. Mater. Interfaces, 2015, 7, 22775–22785 CAS.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- T. Lei, J.-Y. Wang and J. Pei, Chem. Mater., 2014, 26, 594–603 CrossRef CAS.
- H. E. Katz, J. Johnson, A. J. Lovinger and W. Li, J. Am. Chem. Soc., 2000, 122, 7787–7792 CrossRef CAS.
- M. Berggren, D. Nilsson and N. D. Robinson, Nat. Mater., 2007, 6, 3–5 CrossRef CAS PubMed.
- J. Peet, A. J. Heeger and G. C. Bazan, Acc. Chem. Res., 2009, 42, 1700–1708 CrossRef CAS PubMed.
- Y. S. Rim, S.-H. Bae, H. Chen, N. De Marco and Y. Yang, Adv. Mater., 2016, 28, 4415–4440 CrossRef CAS PubMed.
- N. B. Chaure, J. L. Sosa-Sanchez, A. N. Cammidge, M. J. Cook and A. K. Ray, Org. Electron., 2010, 11, 434–438 CrossRef CAS.
- J. Gao, G. Lu, J. Kan, Y. Chen and M. Bouvet, Sens. Actuators, B, 2012, 166–167, 500–507 CrossRef CAS.
- Y. Chen, M. Bouvet, T. Sizun, G. Barochi, J. Rossignol and E. Lesniewska, Sens. Actuators, B, 2011, 155, 165–173 CrossRef CAS.
- Y. Chen, M. Bouvet, T. Sizun, Y. Gao, C. Plassard, E. Lesniewska and J. Jiang, Phys. Chem. Chem. Phys., 2010, 12, 12851–12911 RSC.
- R. D. Yang, J. Park, C. N. Colesniuc, I. K. Schuller, J. E. Royer, W. C. Trogler and A. C. Kummel, J. Chem. Phys., 2009, 130, 164703–164709 CrossRef PubMed.
- H. Wang, J. Wang, H. Huang, X. Yan and D. Yan, Org. Electron., 2006, 7, 369–374 CrossRef CAS.
- I. Muzikante, V. Parra, R. Dobulans, E. Fonavs, J. Latvels and M. Bouvet, Sensors, 2007, 7, 2984–2996 CrossRef CAS.
- O. A. Melville, B. T. H. Lessard and T. P. Bender, ACS Appl. Mater. Interfaces, 2015, 7, 13105–13118 CAS.
- N. Kilinc, D. Atilla, A. G. Gürek, Z. Z. Öztürk and V. Ahsen, Talanta, 2009, 80, 263–268 CrossRef CAS PubMed.
- S. Casilli, M. De Luca, C. Apetrei, V. Parra, Á. A. Arrieta, L. Valli, J. Jiang, M. L. Rodríguez-Méndez and J. A. De Saja, Appl. Surf. Sci., 2005, 246, 304–312 CrossRef CAS.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296–1323 CrossRef CAS PubMed.
- H. Usta, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 8580–8581 CrossRef CAS PubMed.
- J. Obirai and T. Nyokong, Electrochim. Acta, 2005, 50, 5427–5434 CrossRef CAS.
- W. M. Sharman and J. E. van Lier, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Elsevier, San Diego, 2003, vol. 15, pp. 1–60 Search PubMed.
- S. V. Kudrevich, H. Ali and J. E. van Lier, J. Chem. Soc., Perkin Trans. 1, 1994, 2767–2774 RSC.
- V. N. Nemykin, S. V. Dudkin, F. Dumoulin, C. Hirel, A. G. Gürek and V. Ahsen, ARKIVOC, 2013, 2014, 142–204 Search PubMed.
- N. B. McKeown, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Elsevier, San Diego, 2003, vol. 15, pp. 61–124 Search PubMed.
- V. Parra, J. Brunet, A. Pauly and M. Bouvet, Analyst, 2009, 134, 1776–1778 RSC.
- M. Bouvet, H. Xiong and V. Parra, Sens. Actuators, B, 2010, 145, 501–506 CrossRef CAS.
- W. Eberhardt and M. Hanack, Synthesis, 1997, 95–100 CrossRef CAS.
- T. Tanabe and N. Ishikawa, J. Synth. Org. Chem., Jpn., 1971, 29, 792–795 CrossRef CAS.
- M. Tian, T. Wada, H. Kimura-Suda and H. Sasabe, J. Mater. Chem., 1997, 7, 861–863 RSC.
- K. Jones and E. F. Mooney, in Annual Reports on NMR Spectroscopy Volume 3, Elsevier, 1970, vol. 3, pp. 261–421 Search PubMed.
- M. M. Ahmida and S. H. Eichhorn, ECS Trans., 2010, 25, 1–10 CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- J. L. Brédas, R. Silbey, D. S. Boudreaux and R. R. Chance, J. Am. Chem. Soc., 1983, 105, 6555–6559 CrossRef.
- L. Leonat, G. Sbârcea and I. V. Branzoi, UPB Scientific Bulletin, Series B: Chemistry and Materials Science, 2013, 75, 111–118 CAS.
- B. Schöllhorn, J. P. Germain, A. Pauly, C. Maleysson and J. P. Blanc, Thin Solid Films, 1998, 326, 245–250 CrossRef.
- S. Hiller, D. Schlettwein, N. R. Armstrong and D. Wöhrle, J. Mater. Chem., 1998, 8, 945–954 RSC.
- M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS.
- A. W. Snow, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Elsevier, San Diego, 2003, pp. 129–176 Search PubMed.
- M. Bouvet and A. Pauly, in The Encyclopedia of Sensors, ed. C. A. Grimes, E. C. Dickey and V. Pishko, American Scientific Publishers, 2006, vol. 6, pp. 227–270 Search PubMed.
- M. Bouvet and V. Parra, Patent US8450725 B2, 2013.
- M. Bouvet, P. Gaudillat, A. Kumar, T. Sauerwald, M. Schüler, A. Schütze and J.-M. Suisse, Org. Electron., 2015, 26, 345–354 CrossRef CAS.
- I. S. Kirin, P. N. Moskalev and Y. A. Makashev, Russ. J. Inorg. Chem., 1965, 10, 1065–1066 Search PubMed.
- C. Clarisse and M. T. Riou, Inorg. Chim. Acta, 1987, 130, 139–144 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, SHELXL-97 and SHELXS-97, Program for X-ray Crystal Structure Solution and Refinement, University of Göttingen, 1997 Search PubMed.
- P. Gaudillat, A. Wannebroucq, J.-M. Suisse and M. Bouvet, Sens. Actuators, B, 2016, 222, 910–917 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 19F NMR spectrum of the crude mixture obtained during the synthesis of 1a at room temperature; 1H NMR spectrum of 4; electrospray spectrum of 5 and MALDI-ToF spectrum of 6; UV-visible absorption spectra of 6; DSC thermograms of 1a and 4, thermogravimetric analyses of 1a, 4 and 5; I–V characteristics of the 5/LuPc2 and 6/LuPc2 MSDI heterojunctions and the response of the 6/LuPc2 MSDI heterojunction to NH3. CCDC 1497477. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra05325h |
| This journal is © The Royal Society of Chemistry 2017 |